Молекулярная физика
Причины возникновения поверхностных явлений в дисперсных
системах. Приведите различные типы классификации дисперсных систем и примеры их
представителей
Поверхностные явления - процессы, происходящие на границе раздела фаз, в межфазном
поверхностном слое, и возникающие в результате взаимодействия сопряженных фаз,
имеющих различный состав и строение.
Поверхность раздела фаз - это граничная область между фазами, конечный по
толщине слой, в котором происходит изменение свойств от значений, характерных
для одной фазы, до значений, характерных для другой.
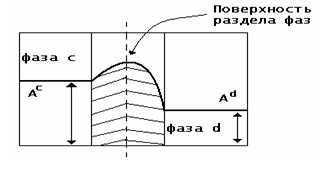
Ас и Аd - значение свойства соответственно в фазах
а и d
На границе раздела фаз формируется поверхностный слой толщиной в один или
несколько молекулярных размеров. Состояние вещества в поверхностных слоях - это
коллоидное состояние вещества.
Пусть из двух соседних фаз в первой межмолекулярные взаимодействия
сильнее, чем во второй. Тогда в этой фазе важнейшее свойство поверхностного
слоя состоит в том, что находящиеся в нем молекулы обладают избыточной энергией
Гиббса (по сравнению с молекулами внутренней части той же фазы).
Для внутренних молекул равнодействующая всех межмолекулярных
взаимодействий равна нулю, а для поверхностных молекул она направлена
перпендикулярно поверхности внутрь фазы. Следовательно, для выведения
молекул из объема на поверхность надо преодолеть эту силу, т. е. совершить
работу и сообщить молекулам определенную энергию. Увеличение площади
поверхности приводит к увеличению числа поверхностных молекул и поверхностная
энергия возрастает.
Поверхностные явления присущи всем системам, имеющим поверхность раздела
фаз, но сильнее всего они проявляются в дисперсных системах, которые являются
гетерогенными и имеют высокоразвитую поверхность.
Классификация дисперсных систем:
. Кл. в зависимости от размера коллоидных частиц:
) Грубодисперсные - Naт>1018, D< 105, m>10-5
(Сахар-песок, капли дождя)
2) Среднедисп. - Naт>109, D=105-107, m=10-7-10-5
(эритроциты крови, сажа)
3) Высокодисп. - Naт<109 - 103, D=107-109,
m=10-9-10-7 (наночастицы Ме, золи)
- ультрадисперсные - Naт=сотни, D=108-107, m=10-8-5∙10-8
(10-7) (Нитевидные кристаллы, сверхтонкие пленки)
- наноразмерные - Naт=десятки, D=108-109, m=10-9-10-8
(наночастицы металлов)
. Кл. по числу характеристических размеров частиц ДФ или по
топографическому признаку:
1) Одномерная (ламинарная) - Пленки, мембраны
) Двухмерная (фибриллярная) - Нити, волокна, капилляры, поры
) Трёхмерная (корпускулярная) - Твердые частицы, капли, пузырьки
. Кл. по характеру взаимодейтсивя между веществами дисперсной фазы и
дисперсионной среды (пригодна лишь для систем с жидкой дисперсионной средой).
1) Лиофобные - слабо выражено взаимодействие частиц ДФ с ДС, принудительное
образование в результате диспергирования и конденсации, термодинамически
агрегативно неустойчивы: Золи, суспензии, эмульсии
) Лиофильные - сильное взаимодействие со средой, самопроизвольное
диспергирование, т\д агрегатно устойчивы: Критические эмульсии, мицеллярные
растворы ПАВ, некоторых ВМС, (белков и др.)
**В том случае, когда дисперсионной средой является вода, эти два класса
можно называть соответственно гидрофильными и гидрофобными системами.
. Классификация дисперсных систем по агрегатному состоянию дисперсной
фазы и дисперсионной среды:
1) Т-Ж: грубодисперсные (суспензии, пасты), высокодисперсные (золи)
2) Ж-Ж: г\д (эмульсии), в\д (микроэмульсии)
3) Г-Ж: г\д, в\д (пены)
) Т-Т: г\д (сплавы), в\д (твёрдые коллоидные растворы )
) Ж-Т: Пористые тела, заполненные жидкостью, капиллярные тела, гели
) Г-Т: Пористые и капиллярные системы, ксерогели
) Т-Г: г\д (пыли, дымы), в\д (аэрозоли)
8) Ж-Г: г\д (туманы), в\д (аэрозоли)
) Г-Г
5. Классификация по степени взаимодействия частиц ДФ:
) Свободнодисперсные - частицы ДФ не связаны между собой и могут
свободно перемещаться; не оказывают сопротивления сдвиговому усилию, обладают
текучестью и всеми остальными свойствами, характерными для обычных жидкостей;
вязкость определяется в основном вязкостью дисперсионной среды. Это аэрозоли,
разбавленные суспензии и эмульсии, лиозоли.
) Связнодисперсные - одна из фаз структурно закреплена и не может
перемещаться свободно. Частицы образуют сплошной пространственный каркас. К
этому классу относятся гели и студни, пены, капиллярно-пористые тела, твердые
растворы.
. Кл\, основан на распределении частиц дисперсной фазы по размерам:
) Полидисперсные - характеризуются достаточно широким
распределением частиц ДФ по размерам. (большинство известных коллоидных систем)
) Монодисперсные - состоят из близких по форме, размеру и
характеру взаимодействия между собой частиц. В таких системах может наблюдаться
явление, не характерное для полидисперсных систем - коллоидная
кристаллизация. Этот процесс самопроизвольного упорядочения частиц в
периодические пространственные структуры во многом аналогичен происходящему при
образовании атомных или молекулярных кристаллов. Нижняя размерная граница
используемых для этого частиц определяется высокой интенсивностью броуновского
движения частиц размером меньше нескольких нанометров, препятствующего
стабилизации протяженных упорядоченных структур; верхняя - слишком малой
подвижностью частиц микронного размера, препятствующей залечиванию дефектов
формируемой структуры.
Выведите уравнение, описывающее диффузионно-седиментационное
равновесие и поясните, как его можно использовать для анализа дисп. систем
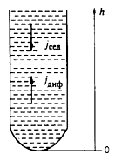
Оседание частиц создает градиент концентрации частиц: их концентрация при
приближении ко дну заметно увеличивается. Соответственно, возникает
диффузионный поток jдиф, направленный противоположно потоку
седиментации jсед, т. е. к верху пробирки.
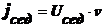 ,
,
где
ν - концентрация частиц в ДФ.
 ,
,
где
h - высота.
Каков
же результат конкуренции этих двух потоков? Возможны 3 варианта:
Œ  , т. е.
, т. е.  , т. е.
, т. е. 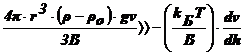
Чтобы
выполнилось это неравенство, значения Т и  должны
быть малы, а (ρ-ρо) и ν - велики. В реальных условиях эти параметры заметно
изменить сложно, а радиус частиц в ДС изменяется в широком интервале: от 10-7
до 10-2 см и именно радиус частиц является определяющим.
Установлено, что данное неравенство соблюдается, когда r≥10-5
см. В этих случаях диффузией можно пренебречь, идет быстрая седиментация -
система является седиментационно неустойчивой.
должны
быть малы, а (ρ-ρо) и ν - велики. В реальных условиях эти параметры заметно
изменить сложно, а радиус частиц в ДС изменяется в широком интервале: от 10-7
до 10-2 см и именно радиус частиц является определяющим.
Установлено, что данное неравенство соблюдается, когда r≥10-5
см. В этих случаях диффузией можно пренебречь, идет быстрая седиментация -
система является седиментационно неустойчивой.
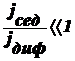 , т. е.
, т. е. 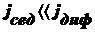 , т. е.
, т. е. 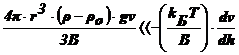
Это
условие должно выполнятся, когда Т и велики,
а (ρ-ρо) и ν - малы. Но и здесь решающую роль играет радиус
частиц. Установлено, что это неравенство выполняется при r≤10-5
см. в этом случае можно пренебречь седиментацией, диффузия приведет к
равномерному распределению частиц по всему объему сосуда. ДС является
седиментационно устойчивой.
Ž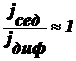 , т. е.
, т. е.  , т. е.
, т. е. 
В
системе имеет место диффузионно-седиментационное равновесие.
разделив
переменные, получим:
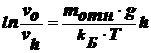
гипсометрический
закон распределения частиц (закон Лапласса-Перрена), характеризующий
распределение частиц по высоте в условиях диффузионно-седиментационного
равновесия и позволяющий определить молекулярные массы частиц:
 ,
,
где
М - мольная масса коллоидной частицы. В этом случае система является
седиментационно устойчивой, но распределение частиц в ней не равномерное, а
равновесное. Это распределение наблюдается, когда 10-5‹r‹10-3 см.
Историческое значение закона:с его помощью впервые было найдено значение числа
Авогадро
Диспергационные методы получения лиофобных дисперсных систем
Если частицы ДФ состоят из вещества, слабо взаимодействующего со средой,
системы являются лиофобными (от греч. лиос - жидкость, фобо - ненавижу).
Лиофобная система: характер образования - принудительное образование в
результате диспергирования и конденсации, термодинамически агрегативно
неустойчивы, слабое взаимодействие между фазами, представители - золи,
суспензии, эмульсии.
Для того, чтобы получить коллоидный раствор или золь, необходимо
выполнить два условия: 1) создать в жидкости твердые или жидкие нерастворимые
частицы коллоидной степени дисперсности; 2) обеспечить устойчивость этих
частиц, предохранив их от слипания друг с другом (от коагуляции), т.е.
стабилизировать систему. Стабилизация коллоидных систем может производиться
путем введения в систему нового компонента - стабилизатора, который
адсорбируется на поверхности коллоидных частиц и придает частицам заряд и/или
образует защитную оболочку.
Свободнодисперсные системы (порошки, суспензии, эмульсии, золи) можно
получить двумя способами: диспергированием и конденсацией.
Диспергирование основано на получении из сплошного и крупного по размерам
тела более мелких частиц дисперсной фазы. Диспергирование может быть
самопроизвольным и несамопроизвольным. Самопроизвольное диспергирование
характерно для лиофильных систем. Несамопроизвольное диспергирование характерно
для лиофобных систем. Здесь процесс диспергирования осуществляется за счет
внешней энергии. При несамопроизвольном диспергировании (нсд): ΔH > 0, ΔS > 0, ΔG > 0, ΔH >TΔS, Wад < Wк,
где Wк - работа по преодолению когезии внутри тела в пределах одной
фазы(связь между молекулами, атомами или ионами) для разрушения существующих
связей внутри тела, Wад - работа адгезии по отношению к окружающей
среде после образования новых поверхностей раздела фаз. Здесь процесс
диспергирования осуществляется за счет внешней энергии. Несамопроизвольное
диспергирование бывает: - механическое, - физическое (диспергирование
ультразвуком, электрическими методами), - физико-химическое (пептизация).
Механическое диспергирование в зависимости от агрегатного состояния дисперсной
фазы: - измельчение, истирание, раздавливание и т. д.; - распыливание, -
барботаж. Резкое изменение свойств вещества с повышением дисперсности связано с
быстрым увеличением суммарной поверхности раздела между частицами и средой.
Измельчение проводят в мельницах различной конструкции, например в шаровых (а)
или коллоидных (б) мельницах.



В шаровых мельницах получают частицы размером 6·104 нм при
сухом помоле и менее 103 нм при мокром; в коллоидных - 100 нм и
менее.
Измельчением получают системы типа т/г, т/ж, распылением - ж/г, ж/ж,
барботажем - г/ж. Ребиндер и Щукин в своих работах показали, что развитие
микрощелей под действием внешних деформирующих сил может происходить
значительно легче при адсорбции различных веществ из среды, в которой ведется
диспергирование. Адсорбироваться могут как ионы электролитов, так и молекулы
поверхностно-активных веществ. Образуя на адсорбировавшей их поверхности двухмерный
газ в результате нелокализованной адсорбции, они под давлением этого газа
проникают в устья возникших щелей и стремятся раздвинуть каждую микрощель,
содействуя таким образом внешним деформирующим силам и способствуя
диспергированию. Разрушение материалов в процессе диспергирования может быть
облегчено при использовании эффекта Ребиндера - адсорбционного понижения
прочности твердых тел. Этот эффект заключается в уменьшении поверхностной
энергии с помощью поверхностно-активных веществ.
Диспергирование ультразвуком высокой частоты эффективно лишь в том
случае, если диспергируемое вещество обладает малой прочностью. При действии на
суспензию ультразвука возникают механические колебания (порядка нескольких
тысяч в 1 с), которые разрывают частицы на более мелкие. Таким путем получают
органозоли хрупких металлов, гидрозоли серы, графита, гидроксидов металлов,
различных полимеров и т. п.
При диспергировании в электрических аппаратах избыток электрических
зарядов сообщается распыляемой жидкости, и в результате отталкивания
одноименных зарядов происходит дробление жидкости на капли.
К физико-химическому диспергированию относится метод пептизации.
Пептизацией называют переход осадков под действием пептизаторов в состояние
коллоидного раствора. Пептизировать можно только “свежие” (свежеприготовленные)
осадки, в которых частицы коллоидного размера соединены в более крупные
агрегаты через прослойки ДС. По мере хранения осадков происходят явления
рекристаллизации и старения, приводящие к сращиванию частиц друг с другом, что
препятствует пептизации. Выделяют адсорбционную; диссолюционную пептизацию и
промывание осадка растворителем.
Выведите уравнение седиментации в дисперсных системах
Седиментация - характерное свойство эмульсий, суспензий, аэрозолей.
Рассмотрим силы, действующие на коллоидную частицу во взвешенном
состоянии:
1. сила тяжести -

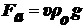
сила
Архимеда,
где
ρ - плотность ДФ, ρо -
плотность ДС.
Сила, вызывающая седиментацию:
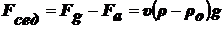
 (1),
(1),
где
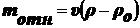 - относительная масса.
- относительная масса.
В
определенный момент частица начинает двигаться равномерно:
 (2),
(2),
где
U - скорость, В - коэффициент Стокса:  .
.
Приравняем
(1) и (2):
 (3).
(3).
Если
ρ ‹ ρо, то получится отрицательное выражение для скорости -
происходит не оседание, а всплывание частиц. Так, в частности, происходит в
случае эмульсии масла в воде.
Кроме
того, на скорость оседания влияет вязкость среды (чем меньше вязкость, тем
меньше скорость) и радиус частиц:
Если
экспериментально определить скорость оседания, можно легко рассчитать радиус
частицы:
 (4).
(4).
Т.
к. величины η, g, ρ, ρо
характеризуют систему и от дисперсности не зависят, можно записать:
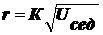 (5).
(5).
Это
условие справедливо только для условий, при которых выполняется закон Стокса, а
именно:
Ø Частицы имеют сферическую форму;
Ø Движутся ламинарно, независимо друг от друга и с постоянной
скоростью;
Ø Трение является внутренним для дисперсионной среды.
Конденсационные методы получения лиофобных дисперсных систем
Если частицы ДФ состоят из вещества, слабо взаимодействующего со средой,
системы являются лиофобными (от греч. лиос - жидкость, фобо - ненавижу).
Лиофобная система: характер образования - принудительное образование в
результате диспергирования и конденсации, термодинамически агрегативно
неустойчивы, слабое взаимодействие между фазами, представители - золи,
суспензии, эмульсии.
Для того, чтобы получить коллоидный раствор или золь, необходимо
выполнить два условия: 1) создать в жидкости твердые или жидкие нерастворимые
частицы коллоидной степени дисперсности; 2) обеспечить устойчивость этих
частиц, предохранив их от слипания друг с другом (от коагуляции), т. е.
стабилизировать систему. Стабилизация коллоидных систем может производиться
путем введения в систему нового компонента - стабилизатора, который
адсорбируется на поверхности коллоидных частиц и придает частицам заряд и/или
образует защитную оболочку.
Свободнодисперсные системы (порошки, суспензии, эмульсии, золи) можно
получить двумя способами: диспергированием и конденсацией.
Конденсация - метод получения дисп. систем, в основе которого лежит
процесс укрупнения мельчайших частиц ДФ (молекулярных размеров) до частиц с
размерами определённого класса дисп. систем. К конденсационным методам
относятся конденсация, десублимация и кристаллизация. Конденсация бывает
гомогенная и гетерогенная. Гомогенная конденсация предполагает формирование
новой фазы на зародышах, самопроизвольно возникающих в результате флуктуаций
плотности и концентрации в системе, а гетерогенная - формирование новой фазы на
уже имеющихся поверхностях (ядрах конденсации - стенки сосудов, частицы
примесей). Необходимое условие конденсации - пересыщение и неравновесное
распределение вещества в объеме, а также образование центров конденсации или
зародышей. Различают конденсацию химическую; физико-химическую; физическую. К
физической конденсации относятся метод Брэдига и метод Рогинского и Шальникова.
К физико-химической конденсации относится метод замены растворителя. Химический
метод конденсации основан на реакциях, приводящих к возникновению твердого
продукта.
К физической конденсации относятся метод Брэдига и метод Рогинского и
Шальникова. Метод Брэдига: два электрода из металла погружают в жидкость (она
станет дисперсионной средой золя), концы их сближают и пропускают электрический
ток. Возникающий дуга отчасти распыляет металл до коллоидных частиц, отчасти
испаряют его, и пар конденсируется в холодной жидкости также в виде коллоидных
частиц. Таким способом получают золи многих металлов.
Вещество, находящееся в молекулярно-дисперсионном состоянии, можно
перевести в колл. состояние при замене одного растворителя другим - т.н.
методом замены растворителя.
Пример: Канифоль не растворяется в воде, но хорошо растворима в этаноле.
При постепенном добавлении спиртового раствора канифоли к воде происходит
резкое понижение растворимости канифоли, в результате чего образуется гидрозоль
канифоли.
Колл. растворы можно получать также и методом химической конденсации,
основанном на проведении хим. реакций, сопровождающихся образованием
нерастворимых или малорастворимых веществ. Используются различные типы реакций
- разложения, гидролиза, окислительно-восстановительные и т.д. Так, красный
золь золота получают восстановлением натриевой соли золотой кислоты
формальдегидом:
2
+ HCOH + Na2CO3 → Au(колл.) + HCOONa + H2O
Также лиофобных дисп. системы могут образовываться путём конденсации
пересыщенного пара.
Химический метод конденсации основан на реакциях, приводящих к
возникновению твердого продукта. Это реакции восстановления, окисления,
гидролиза, обмена.
Примером получения коллоидных систем кристаллизацией является
кристаллизация из пересыщенного раствора сахарозы в производстве сахара.
Процесс десублимации имеет место при образовании облаков, когда в условиях
переохлажденного состояния из водяных паров образуются сразу кристаллики, а не
капли воды.
Адсорбция газов на твердой поверхности. Теория Ленгмюра
Адсорбция газов на твердой поверхности
С точки зрения термодинамики адсорбция - самопроизвольное выравнивание
химических потенциалов в объеме фазы и на межфазной поверхности.
Для твердых адсорбентов адсорбцию удобно рассматривать как взаимодействие
адсорбата с активными центрами поверхности адсорбента. В зависимости от типа
взаимодействий различают физическую и химическую адсорбцию (хемосорбция).
Сравнительная характеристика физической адсорбции и хемосорбции
|
Признакотличия
|
Физическая адсорбция
|
Хемосорбция
|
|
Природадействующихсил
|
Ван-дер-Ваальсовы
|
химические
|
|
Тепловойэффект, кДж/моль
|
4 - 40
|
› 40
|
|
Обратимость
|
Обратима
|
Необратима
|
|
Способность молекул адсорбата к перемещению
|
Локализована, нелокализованная
|
Локализованная
|
Пример химической адсорбции - образование оксидной пленки на поверхности
твердых тел.
Для характеристики процесса адсорбции с ориентацией вводится понятие
работа адсорбции - работа, которую совершает система при обратимом
изотермическом переносе вещества из объема фазы на МФП. По Ребиндеру:
 ,
,
где Wc - работа по переносу полярной части, ∆W - работа
по переносу одной СН2- группы, n - число групп.
Ленгмюр предложил
 ,
,
где
Сv - равновесная концентрация в объеме.
Если
адсорбция происходит в стандартных условиях, то  .
.
Теория мономолекулярной адсорбции Ленгмюра
Ее основные положения:
 адсорбция локализована
адсорбция локализована
адсорбционные центры энергетически эквивалентны
адсорбированные молекулы не взаимодействуют друг с другом, и
каждый активный центр может взаимодействовать только с одной молекулой адсорбата
поверхность адсорбента ограниченна и по мере увеличения
концентрации адсорбента происходит адсорбционное насыщение поверхности
адсорбированные молекулы только определенное время
удерживаются на поверхности адсорбента, они могут покинуть поверхность и их
место займут другие
адсорбция носит квазихимический характер.
Примем,
что при адсорбции происходит реакция между распределяемым компонентом В и
адсорбционными центрами поверхности  :
:
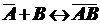 ,
,
где
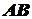 - комплекс, образующийся на поверхности.
- комплекс, образующийся на поверхности.
Введем:
θа - доля
поверхности, покрытая адсорбированными молекулами,
ν - число молекул на 1 м2 адсорбционного слоя при максимально
плотной упаковке,
1-
θа- доля
свободной незанятой поверхности,
ка-
константа адсорбции.
Скорость
адсорбции пропорциональна числу ударов молекул адсорбата о незанятую
поверхность адсорбента:
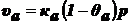 ,
,
где
р - давление.
Скорость процесса десорбции пропорциональна числу молекул на единицу
площади поверхности в адсорбционном слое:
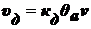 .
.
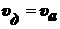 ,
,
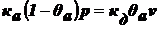 .
.
Выразим θа:
 .
.
 .
.
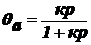 .
.
Обозначим величину адсорбции а:
 .
.

уравнение
Ленгмюра.
Оно было получено для газов, но работает и для растворов.
Капиллярное давление. Закон Лапласа. Капиллярное поднятие
жидкости, уравнение Жюрена. Примеры капиллярных явлений
Капиллярные явления - физические явления, обусловленные действием поверхностного
натяжения на границе раздела несмешивающихся сред. К капиллярным явлениям
относят обычно явления в жидких средах, вызванные искривлением их поверхности,
граничащей с др. жидкостью, газом или собственным паром. Кривизна возникает за
счет изменения площади и положения поверхности. Искривление поверхности ведёт к
появлению в жидкости дополнительного капиллярного давления Dp,
величина которого связана со средней кривизной r поверхности уравнением
Лапласа:
Dp= p1- p2= 2s12/r,
где (s12- поверхностное натяжение на границе двух сред;p1иp2-
давления в жидкости 1 и контактирующей с ней среде (фазе) 2. В случае вогнутой
поверхности жидкости (r < 0) давление в ней понижено по сравнению с
давлением в соседней фазе: p1< p2и Dp<
0. Для выпуклых поверхностей (r > 0) знак Dp меняется на
обратный. Капиллярное давление создаётся силами поверхностного натяжения,
действующими по касательной к поверхности раздела. Искривление поверхности
раздела ведёт к появлению составляющей, направленной внутрь объёма одной из
контактирующих фаз. Для плоской поверхности раздела (r = ¥) такая составляющая отсутствует и Dp=
0.
Высота поднятия жидкости в капиллярной трубке h
определяется уравновешиванием лапласовского и гидростатичесого давлений:
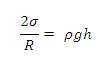
Высота подъёма (опускания) уровня жидкости в капилляре
будет равна:
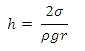 ,
,
ρ - плотность жидкости
· σ - поверхностное натяжение
· R - радиус сферической формы мениска
Капиллярное давление можно рассматривать как добавку, которая в
зависимости от кривизны поверхности увеличивает или уменьшает внутреннее
молекулярное давление по сравнению с молекулярным давлением при наличии плоской
поверхности.
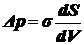
закон
Лапласа.
Из уравнения следует, что фазы, разделенные искривленной поверхностью,
могут находиться в равновесии только при разных давлениях внутри фаз.
· Для сферы
 ;
;
· Для цилиндра
 ;
;
· В общем случае
 ,
,
где
r1 и r2 - главные радиусы кривизны.
Следовательно: чем меньше r, тем больше капиллярное давление Δр.
При равновесии лаплассовское давление равно гидростатическому давлению
столба жидкости высотой h:
 ,
,
· где ρ - плотность жидкости; ρо - плотность газовой фазы; g - ускорение свободного
падения; r - радиус мениска.
Чтобы высоту капиллярного поднятия связать с характеристикой смачивания,
радиус мениска необходимо выразить через угол смачивания θ и радиус капилляра rо.rо=r∙cos
θ, тогда высоту капиллярного поднятия
можно представить в виде формулы Жюрена:
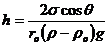 .
.
þ При отсутствии смачивания θ › 90º, cos θ ‹ 0, уровень жидкости в капилляре
опускается на величину h.
þ При полном смачивании θ = 0, cos θ = 1, в этом случае радиус мениска
равен радиусу капилляра.
Примеры капиллярных явлений:
) Жидкость не может вытечь из капилляра под действием силы тяжести;
2) Жидкость не переполняет капилляр, хотя высота капилляра меньше h,
определенной по формуле Жюрена.
Роль капиллярных явлений в природе и технике огромна. Ими обусловлено проникновение
жидкости по тонким каналам в почвах и растениях, пропитка бумаги, тканей,
появление сырости в подвалах зданий. Водонепроницаемость тканей обеспечивается
их гидрофобностью и, как следствие, отрицательным капиллярным поднятием
Уравнение двухмерного состояния вещества в адсорбционном
слое. Типы поверхностных пленок. Изотермы двухмерного давления. Весы Ленгмюра
Для исследования строения адсорбционных слоев, образованных
нерастворимыми ПАВ, Ленгмюром был сконструирован прибор (весы Ленгмюра),
позволяющий с необходимой точностью измерить двухмерное давление адсорбционных
слоев нерастворимых ПАВ. На поверхности плоской кюветы находится легкий
плавающий барьер, соединенный с динамометром (весами). Чтобы предотвратить миграцию
молекул ПАВ из рабочей области, зазор между измерительным прибором и стенками
прибора перегораживают тонкими золотыми листочками или парафинированными
нитками. Второй барьер, который можно передвигать, служит для изменения площади
рабочей поверхности прибора. Малое количество очень разбавленного раствора ПАВ
в легколетучем растворителе наносят на поверхность воды между измерительным и
вспомогательным барьерами.

Адсорбцию можно изменять, передвигая вспомогательный барьер и тем самым
меняя площадь, на которую нанесено заданное количество ПАВ. После испарения
летучего растворителя уравновешивают силу F, действующую на измерительный
барьер. Полученное значение делят на ширину барьера и находят двухмерное
давление π. Повторяя измерения при разных значениях адсорбции, получают
зависимость двухмерного давления π от адсорбции Г (или от площади на
молекулу в адсорбционном слое sм).
При более высоких значениях адсорбции начинают проявляться эффекты,
связанные с взаимодействием молекул ПАВ в адсорбционном слое - их взаимным
притяжением и отталкиванием. Если притяжение молекул выражено слабо, то
зависимость двухмерного давления от площади на молекулу может быть описана
выражением:
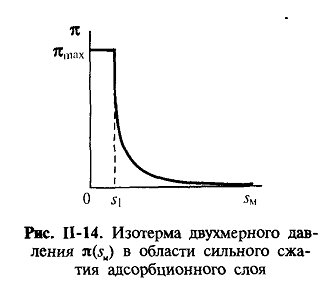
π(sм - s1)=kT,
(П.19)-уравнение двухмерного состояния вещества в адсорбционном слое.
При этом, однако, рост двухмерного давления при сжатии адсорбционного
слоя ограничен некоторой величиной πmax которая называется давлением
коллапса (рис. П-14). При πmax адсорбционный слой теряет
устойчивость, на его поверхности образуются складки и возникают
полимолекулярные слои.
Предельное значение площади на молекулу s1может
рассматриваться как собственное сечение молекулы ПАВ; для некоторых классов ПАВ
эта величина приблизительно одинакова и не зависит от длины цепи (это
подтверждает высказанное выше соображение о вертикальной ориентировке молекул в
таком плотном адсорбционном слое). Независимость же этой величины и от природы
полярной группы свидетельствует о том, что она определяется сечением
углеводородной цепи.

Для определения s1из экспериментально полученной зависимости
двухмерного давления от площади на молекулу в поверхностном cлое
экспериментальные данные удобно представить в координатах πsм - π. В отсутствие заметных сил притяжения
между молекулами экспериментальные данные в этих координатах ложатся на прямую
линию (рис, 11-16), угол наклона которой дает значение площади на молекулу в
плотном адсорбционном слое. В самом общем случае в адсорбционном слое действуют
как силы отталкивания, так и силы межмолекулярного притяжения. Состояние пленки
определяется, с одной стороны, природой веществ, а с другой стороны,
концентрацией пленкообразующего вещества.
А. Адамсонразличает следующие типы поверхностных пленок:
) Газообразные G-пленки, которые приближенно подчиняются уравнению
состояния идеального двухмерного газа или газа с молекулами конечного размера.
Такие пленки образуют, например, жирные кислоты при низких значениях
двухмерного давления или достаточно высоких температурах. Иногда выделяют еще
парообразные пленки при температурах ниже температуры конденсации адсорбционных
слоев.
2) Жидкорастянутые L2-пленки. Жидкорастянутые пленки
образуют многие вещества, в частности кислоты, с не очень большой длиной цепи
при повышенных двухмерных давлениях. Особенно характерно образование
жидкорастянутых пленок для веществ с разветвленной цепью. В таких пленках
площадь на молекулу значительно выше площади сечения углеводородной цепи.
Вместе с тем в них углеводородные цепи, несомненно, находятся в
конденсированном состоянии, образуя пленку, толщина которой меньше длины цепи
молекул ПАВ и увеличивается с ростом двухмерного давления. Образование такого
состояния, не имеющего объемного аналога, может быть связано с взаимным
притяжением углеводородных цепей при одновременном умеренном отталкивании
полярных групп.
) Жидкие L1-пленки. Эти пленки образуются из
жидкорастянутых при высоких значениях двухмерного давления. Такие вещества, как
высшие жирные кислоты (начиная с тридециловой), дают при повышенных
температурах жидкие пленки без образования жидкорастянутых. Между жидкими и
жидкорастянутыми состояниями может существовать переходная область сравнительно
высокой сжимаемости, которая отвечает так называемым промежуточным пленкам;
природа этого состояния не вполне ясна.
) Твердые, или S-пленки, сжимаемость которых еще ниже, чем у
жидких. Наиболее важные отличия твердых поверхностных пленок обнаруживаются при
сопоставлении их реологических свойств. В жидких пленках течение происходит уже
при малых напряжениях сдвига и скорость сдвига линейно связана с напряжением,
тогда как твердые пленки способны выдержать значительные напряжения сдвига без
остаточной деформации и затем разрушаются. Качественным тестом, позволяющим
различать жидкие и твердые адсорбционные пленки, может служить метод сдувания:
поверхность жидкости, несущая адсорбционный слой, посыпается тонким порошком
(обычно тальком), и на нее под углом направляется струя воздуха. При этом в
случае жидких поверхностных слоев заметно движение частиц, тогда как на твердых
этого не происходит, но иногда наблюдается откалывание отдельных больших
«льдин», которые движутся как единое целое.
Теория броуновского движения и её значение для естествознания
В 1828 году ботаник Броун при наблюдении в микроскопе взвешенных в воде
частиц цветочной пыльцы и спор обнаружил, что они находятся в непрерывном
беспорядочном движении, не затухающем во времени.
В 1903 г. Зигмонди и Зидентопф сконструировали ультрамикроскоп и
установили, что мелкие частицы движутся более интенсивно, чем крупные. Вначале
это явление объяснялось с позиций витализма, но уже сам Броун установил, что
оно свойственно любым мельчайшим частицам как орг., так и неорг. происхождения
и проявляется тем интенсивнее, чем выше температура и чем меньше масса частицы
и вязкость среды.
В 1888г Гуи и Экснер высказали предположение, что источник движения -
тепловое движение молекул дисперсионной среды. Впоследствии теоретически
обоснованная интерпретация БД была дана Эйнштейном (1905) и Смолуховским
(1906). Правильность полученных ими соотношений была экспериментально
подтверждена Сведбергом и Перреном для систем с жидкой ДС, а де Бройлем и
Малликеном для азрозолей.
Тепловое движение молекул ДС сопровождается ударами молекул о поверхность
частиц ДФ и приводит к смещению последних под действием этих ударов.
Следовательно, броуновское движение есть результат флуктуаций числа ударов,
которые получает коллоидная частица.
Теория БД подтвердила существование атомов и молекул, послужила
доказательством статистического характера II закона термодинамики, позволила
рассчитать NA и kБ.

За одну секунду коллоидная частица может изменить свое направление свыше
1020 раз. Поэтому, определить фактическое перемещение частиц
затруднительно. В 1905 - 1906 гг. А. Эйнштейн и М. Смолуховский предложили
движение частиц ДФ характеризовать при помощи сдвига. Сдвиг - это изменение
координаты частицы за определенный отрезок времени.
 ,
,
где
∆1, ∆2, ∆n- отдельные проекции
смещения частиц на ось х; n- число таких проекций, взятых для расчета.
 -
уравнение Эйнштейна-Смолуховского.
-
уравнение Эйнштейна-Смолуховского.

Рассмотрим
цилиндр, заполненный коллоидной системой и разделенный полупроницаемой
перегородкой МN. Расстояние от стенок цилиндра слева и справа до перегородки -  , ν1, ν2 - частичная концентрация справа и слева, ν1 › ν2.
, ν1, ν2 - частичная концентрация справа и слева, ν1 › ν2.
Перенос
кол частиц в одну и другую сторону равновероятен.
Кол-во
вещества, перенесенное за время τ через перегородку МN направо: 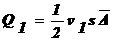 . Справа
налево:
. Справа
налево:  .
.
Количество
вещества, перенесенное за время τ через поперечное сечение:
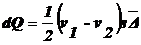 .
.
Выразим
градиент концентрации:
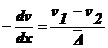
 .
.
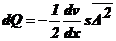 .
.
По
Фику:
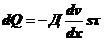 .
.
Приравняем:

.
 зависит
от температуры, вязкости среды, размера частиц. БД пренебрегают, если размеры
частиц больше 5 мкм.
зависит
от температуры, вязкости среды, размера частиц. БД пренебрегают, если размеры
частиц больше 5 мкм.
Модели строения ДЭС (теории Гельмгольца-Перрена, Гуи-Чепмена,
Штерна). Электрокинетический потенциал
ДЭС - пространственное разделение зарядов противоположного знака в
межфазном слое. ДЭС - тонкий поверхностный слой из пространственно-разделенных
эл. зарядов противоположного знака, образующихся на границе раздела фаз. ДЭС -
поверхностный и пограничный. Он образуется самопроизвольно при стремления системы
снизить энергию Гиббса поверхностного слоя. Следствие самопроизвольного
формирования ДЭС - электрокинетические (относительное движение фаз обусловлено
эл. разностью потенциалов) и электрокапиллярные явления (взаимосвязь м/д
поверхн. натяжением и разностью потенциалов на границе раздела фаз). ДЭС -
важнейший фактор, обеспечивающий агрегативную устойчивость дисперсных систем.
Он возникает в результате адсорбции ионов на поверхности частиц ДФ и
представляет собой конденсатор, обкладки которого состоят из противоположных
зарядов т. е. ДЭС - слой ионов одного знака + слой ионов второго знака).
Для объяснения строения ДЭС были предложены 3 теории:

Теория Гельмгольца - Перрена. Они представляли ДЭС в виде плоского
конденсатора, обкладки которого состоят из противоположных зарядов и
расположены на расстоянии порядка молекулярного диаметра.
График зависимости ψ(х) имеет вид:
- резкое падение потенциала.
Недостатки теории:граница скольжения расположена от поверхности на
большем расстоянии, чем молекулярные размеры; согласно теории ξ=φ, но экспериментально установлено, что
ξ ‹ φ;т. к. по этой теории ξ =φ, то они одинаково зависят от
присутствия индифферентных электролитов. φ не зависит от присутствия
электролитов, а ξ зависит достаточно сильно.
Теория Гуи и Чемпена


На твердой поверхности адсорбируются ионы определенного знака и, помимо
электростатических сил, на их распределение влияет тепловое движение. Вследствие
этого слой против ионов размыт. Распределение в нем зарядов подчиняется закону
Больцмана:
 ,
,
где Wi - работа против сил электростатического притяжения к
поверхности. Т. е. облако зарядов с экспоненциально убывающей плотностью.
Чем больше концентрация, тем круче кривая.
Недостатки теории:
· не учитываются размеры ионов, они рассматриваются как точечные заряды;
· не объясняется явление перезарядки;
· согласно этой теории, различные ионы одинакового заряда
должны одинаково сжимать ДЭС и снижать ξ-потенциал, но реально этого нет: чем
больше радиус иона, тем сильнее он сжимает ДЭС и понижает ξ-потенциал.
· Работает только для разбавленных растворов.
Достоинства теории:ξ-потенциал рассматривается как часть φ-потенциала;позволила понять действие
индифферентных электролитов на ξ-потенциал.
Теория Штерна. Штерн объединил две предыдущие теории.
Ионы - не точечные заряды, они имеют размеры и не могут находится на
расстоянии от поверхности меньшем, чем радиус иона.Помимо электростатического
взаимодействия с твердой поверхностью существует специфическое адсорбционное.
Эти силы действуют на небольших расстояниях, имеют адсорбционную природу и
убывают с расстоянием быстрее, чем электростатические. Согласно этой теории
существует два слоя противоионов:
I-ый - адсорбционный слой (слой Гельмгольца, Штерна). Он удерживается на
твердой поверхности электростатическими и адсорбционными силами;
II-ой - диффузный слой (слой Гуи). Это размытый из-за диффузии слой
оставшихся противоионов, который удерживается только электростатическими
силами.
При введении электролитов в систему ионы из диффузионного слоя переходят
в адсорбционный слой из-за чего ξ-потенциал уменьшается и может достичь
нуля. При разбавлении системы ξ-потенциал увеличивается.
На размер ДЭС сильно влияет природа противоионов:
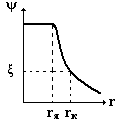
чем больше валентность противоиона, тем ДЭС меньше и меньше ξ-потенциал. Если валентность
одинакова, то толщина диффузного слоя и ξ-потенциал определяются специфической
адсорбцией - увеличение адсорбции вызывает сжатие слоя;
- чем больше радиус иона (чем меньше гидратируемость), тем меньше ДЭС;
- чем больше поляризуемость, тем меньше ДЭС.
График зависимости ψ(r) имеет вид:

Факторы, влияющие на ξ-потенциал:
· Температура. Повышение температуры приводит к расширению диффузного слоя
d даже при прежнем содержании в нем противоионов и ξ-потенциал увеличивается. Но если
температура велика, то происходит десорбция потенциалопределяющих ионов иξ-потенциал повышается.

Разбавление. При разбавлении ξ-потенциал увеличивается (т.к.
снижается ионная сила раствораºувеличивается толщина диффузного слоя противоионов и
усиливается десорбция противоионов с частицы), при сильном разбавлении происходит
десорбция и потенциалопределяющие ионы, ξ-потенциал понижается.
Добавление электролитов. Индифферентные электролиты- электролиты, не
имеющих ионов, способных достраивать кристаллическую решетку коллоидной
частицы. Электрокинетический потенциал такие электролиты снижают при увеличении
концентрации противоионов и сжатия ДЭС. При введении индифферентных
электролитов - два случая: 1) в систему вводится электролит, один из ионов
которого одинаков с противоионами, 2) в систему вводится электролит, не имеющий
общих ионов с электролитом - стабилизатором.
Первый случай. При увеличения электролита толщина ДЭС стремится стать
равной толщине адсорбционного слоя за счет сжатия диффузного слоя. ξ-потенциал понижается, пока не станет
равным нулю, что отвечает изоэлектрическому состоянию систему.
Второй случай: обменпротивоионов коллоидной частицы на эквивалентное
число одинаковых по знаку ионов введенного электролита. Наиболее простой обмен
ионов происходит, когда на поверхности твердой фазы имеется ДЭС типа Гуи - Чэпмена,
т. е. когда можно пренебречь специфическим адсорбционным потенциалом ионов.
Обмен определяться только валентностью ионов. Также повышении ионной силы
оказывает два действия: уменьшение толщины диффузного слоя противоионов (d),
сдвиг противоионов между плотным и диффузным слоями в сторону плотного слоя
(уменьшение х - заряда частиц). И то, и другое приводит к снижению ξ-потенциала (кривая 1). Если это
снижение достаточно сильное, дисперсные частицы начинают коагулировать.
Коагуляция - важный процесс.

Правило Шульце-Гарди: коагулирующим действием обладают ионы, заряженные
противоположно заряду частицы; сила коагулирующего действия возрастает с
увеличением заряда ионов. Но в случае многозарядных ионов возможен и такой
эффект, как перезарядка частиц (кривая 2). Добавляемые ионы могут обмениваться
с противоионами, замещая их и в диффузном, и в плотном слоях. При этом если
многозарядный ион является маленьким (Al3+, Тh4+), он
замещает на поверхности частиц неэквивалентное по заряду количество прежних
ионов (сверхэквивалентную адсорбцию).При дальнейшем повышении концентрации
многозарядных ионов вновь начнет сказываться эффект возрастания ионной силы - с
уменьшением (по модулю) ξ-потенциала и устойчивости частиц.
Влияние неиндифферентных электролитов. Влияние электролита, один из ионов
которого способен достраивать кристаллическую решетку ДФ т.к.
потенциалопределяющий ион электролита может повышать потенциал φ0, а находящийся с ним в паре ион, одноименный с
зарядом противоиона, способен сжимать ДЭС. При малых концентрациях
неиндифферентного электролита проявляется первая тенденция, связанная с
поверхностным действием иона, способного достраивать кристаллическую решетку.
При больших концентрациях вторая тенденция. Поэтому при введении в систему все
возрастающих количеств неиндифферентного электролита ξ-потенциал сначала возрастает, а потом
падает, проходя через максимум. При введении неиндифферентного электролита
возможна перезарядка коллоидных частиц.
Влияние концентрации. При разбавлении коллоидной системы ξ-потенциал возрастает, т.к. толщина
ДЭС увеличивается при уменьшении концентрации противоионов в растворе . При
разбавлении может наблюдаться десорбция потенциалопределяющего иона с
поверхности ДФ, что должно приводить к падению φ0-потенциала и ξ-потенциала. Концентрирование -
противоположное действие.
Электрофорез - перемещение под действием электрического поля
неэлектропроводных частиц ДФ относительно ДС. Граница м/д коллоидной частицей и
диффузионным слоем - граница скольжения (поверхностное скольжение). В мицелле
эта граница м/д адсорбционным и диффузным слоями. Граница скольжения обозначает
ту геометр. поверхность, по которой происходит разделение мицеллы на коллоидную
частицу и диффузный слой в случае ее перемещения относительно ДС. Скорость
движения частиц ДФ пропорциональна ξ-потенциала. Величина ξ-потенциала связана со скоростью
электрофореза заряженных частиц уравнением Гельмгольца-Смолуховского:ξ=kπην/εx, где к - коэффициент, зависящий от
формы частиц, η - вязкость среды, ν - линейная скорость перемещения
частиц, х - напряженность элект. поля. Электроосмосом- протекание жидкости ч/з
неподвижную пористую перегородку под действием наложенного электрического поля.
Вокруг частиц возникает ДЭС, ионы одного знака удерживаются на частицах, а ионы
другого знака сохраняют подвижность.
Диффузия и ее особенности в кол. системах. Уравнение
Эйнштейна.
Диффузией(от лат. diffusio - распространение) называют самопроизвольно
протекающий в системе процесс выравнивания концентрации молекул, ионов или
коллоидных частиц под влиянием их теплового хаотического движения.

Процесс диффузии необратим. Схематически он представлен на рисунке. В
нижней части коллоидной системы концентрация частиц (с1) больше, чем
в верхней (с2), т. е. с1>с2.
Законы диффузии установил Фик по аналогии с переносом тепла или
электричества:
 (1),
(1),
где
dQ - количество продиффундировавшего вещества; D - коэффициент диффузии; 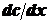 - градиент концентрации; s - площадь, через которую
идет диффузия; τ - продолжительность диффузии.
- градиент концентрации; s - площадь, через которую
идет диффузия; τ - продолжительность диффузии.
Это
уравнение можно представить в др. виде:
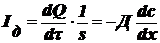 - I
закон Фика,
- I
закон Фика,
где Iд - диффузионный поток (характеризует количество
вещества, переносимое в результате диффузии за единицу времени через сечение,
равное единице площади.зависит от свойств диффундирующих частиц и среды единицы
измерения - м2/сек.
Физ. смысл: D - количество вещества, переносимое через единицу площади в
единицу времени при единичном градиенте концентрации
Коэффициенты диффузии для различных частиц в водной среде отличаются:
Если
диффузия нестационарна , то выполняется II закон Фика:
, то выполняется II закон Фика:
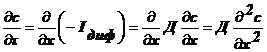 (2).
(2).
Диффузия
возникает, если есть градиент химического потенциала grad μ, который обусловлен градиентом концентрации.
 (3).
(3).
Для
разбавленных растворов 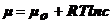 .
.
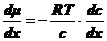 (4),
(4),
где
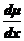 - сила, действующая на моль частиц.
- сила, действующая на моль частиц.
Выразим
силу F1, которая действует на одну частицу:
 (5)
(5)
и скорость перемещения υ в предположении сферических частиц:
 (6),
(6),
Где
В - коэффициент трения Стокса.
 (7),
(7),
-радиус
сферической частицы;η - вязкость среды.
Подставим
(5) в (6):
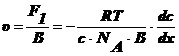 (8).
(8).
Учитывая
(7), (8), I закон Фика и  , получим уравнение Эйнштейна:
, получим уравнение Эйнштейна: 
Реологические модели: упругость,
вязкость, пластичность
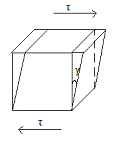
В теле - кубик с
единичным ребром. К противопол. сторонам приложим силу F, кот. создает
напряжение сдвига τ (Н/м). В рез-те - деформация кубика (смещение верхней
границы относит. нижней на γ (м)). Связь между величинами τ, γ и из изменениями во времени -
механич. поведение.
3 реологические модели:
. Упругое поведение.

Харак-тся линейн. зави-стью τ и γ - по закону Гука: τ=G γ (модуль упругости G(Н/м2)
-это модуль сдвига).График - прямая линия ч/з начало координат, ctg угла
наклона кот. относит. оси абсцисс равен модулю G2.Особ-ти: полная
механич. и термодин. обратимость, при снятии назрузки медленно восстанавл-ся
первонач.форма, не происходит рассеяние энергии в процессах нагружения и
разгружения тела. Энергия, запасаемая единицей объема:
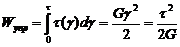
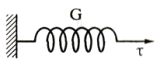
Модель - пружина жесткость кот. численно равна модулю упругости данного
тела. Упругая деформация наблюд-ся до некот. предельного значения τс, выше кот. происходит разрушение твердых тел
(характериз. их прочность).
2.Вязкое
поведение. Лин. завис-ть м/у τ и
скоростью сдвига d γ/dt по з-ну Ньютона  [η-вязкость(Н*с/м2)]. Полностью механ.
итермодин. не обратимо, исх.форма не восстан-ся. Вся соверш. работа- в тепло.
[η-вязкость(Н*с/м2)]. Полностью механ.
итермодин. не обратимо, исх.форма не восстан-ся. Вся соверш. работа- в тепло.

График
аналогичен, но котангенс угла наклона в оси абсцисс равен вязкости η. Скорость диссипации энергии  .Модель: цилиндр, заполн. некой вязкой средой с
неплотно прилегающим к стенкам цилиндра поршнем.
.Модель: цилиндр, заполн. некой вязкой средой с
неплотно прилегающим к стенкам цилиндра поршнем.
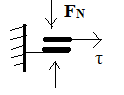

.Пластичность.отсутствует
пропорциональность м/у воздействиями и деформациямиями. При напряжениях,
меньших предельного напряжения сдвига τ*, деформация не происходит. При τ=τ* наступает деформация с заданной скоростью. Мех. и
термодин. необратимо. Скорость диссипации энергии 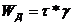 . Модель: две поверхности, прижатые друг к другу с
такой силой FN, что возникающая касательная сила Fтр
числен.равна предельному напряжению сдвига материала. Отношение F к скорости
перемещения поршня F/[dl/dt] числен. равно вязкости рассматр. жидкости.
. Модель: две поверхности, прижатые друг к другу с
такой силой FN, что возникающая касательная сила Fтр
числен.равна предельному напряжению сдвига материала. Отношение F к скорости
перемещения поршня F/[dl/dt] числен. равно вязкости рассматр. жидкости.
Адсорбция газов и паров на однородной твердой поверхности.
Потенциальная теория Поляни, её возможности и отличие от теории БЭТ
Адсорбция - самопроизвольное перераспределение компонентов между
поверхностным слоем и объемной фазой. С точки зрения термодинамики адсорбция -
самопроизвольное выравнивание химических потенциалов в объеме фазы и на межфазной
поверхности.
При адсорбции в условиях Т‹Ткр имеет место конденсация, и
мономолекулярный слой не компенсирует поверхностную энергию è образуется несколько адсорбционных
слоев (полимолекулярная адсорбция).
Полимолекулярная адсорбция реализуется в двух случаях:

Для первого случая была предложена потенциальная теория адсорбции Поляни,
которая дает термодинамическое описание процесса адсорбции.
Теория Поляни:
Ее основные постулаты:
все адсорбированное вещество находится в конденсированном
состоянии;
адсорбат у поверхности адсорбента образует адсорбционный
объем V.
 ,
,
где Vm - мольный объем адсорбата в конденсированном состоянии.
Физический смысл ε - это изотермическая работа по переносу 1 моля пара
адсорбата из достаточно удаленного от поверхности объема в адсорбционный объем
или это изотермическая работа по сжатию адсорбата от давления р до давления
насыщения рs.
 ,
,
где
рs - давление насыщенного пара адсорбата в отсутствие адсорбента; р
- равновесное давление.

Каждой
точке изотермы адсорбции соответствуют определенные значения а и р/рs,
которые позволяют получить значения V и ε, т. е. найти зависимость адсорбционного потенциала от объема адсорбата
на адсорбенте - потенциальную кривую адсорбции.
 -
потенциальная характеристическая кривая данного адсорбента
-
потенциальная характеристическая кривая данного адсорбента
 - т. е.
адсорбционный потенциал от температуры не зависит.
- т. е.
адсорбционный потенциал от температуры не зависит.
Для
данного адсорбента и адсорбата, зная изотерму адсорбции при данной температуре,
можно рассчитать величины aи pи построить изотерму для другой температуры.
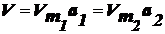 .
.
Рассчитаем
а2 и р2 для другой температуры:



Важная
особенность потенциальных кривых была обнаружена Дубининым. Она заключается в
том, что характеристические кривые для одного и того же объема адсорбента и
разных адсорбатов при всех значениях объемов адсорбата в поверхностном слое
находятся в постоянном соотношенииβ:
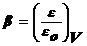 ,
,
где
β - коэффициент аффинности, ε - адсорбционный потенциал для одного адсорбата, εо - для
другого.
Для
данного адсорбента, имея βи зная изотерму адсорбции, можно рассчитать изотерму
любого другого адсорбата на данном адсорбенте.
Эта теория не дает уравнения изотермы адсорбции, только ТД описание.
Для 2го случая исп. теорию БЭТ (теория Брунауэра, Эмета и Теллера).
Согласно этой теории каждый адсорбционный центр sх связывает
по несколько молекул адсорбента Х, образуя цепочки:
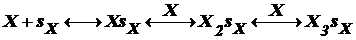 .
.
Уравнение,
описывающее адсорбцию:
 ,
,
где
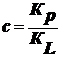 ; Кр - константа адсорбции; КL -
константа конденсации.
; Кр - константа адсорбции; КL -
константа конденсации.
Для
нахождения констант этого уравнения строят график:

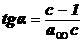 .
.
Это
уравнение работает только в интервале  , в
котором предполагается отсутствие взаимодействия между цепочками
адсорбированных молекул.
, в
котором предполагается отсутствие взаимодействия между цепочками
адсорбированных молекул.
При - большая степень заполнения, возникают боковые
взаимодействия меду молекулами.
- большая степень заполнения, возникают боковые
взаимодействия меду молекулами.
При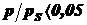 - малая степень заполнения, сильно влияют
энергетические неоднородности поверхности.
- малая степень заполнения, сильно влияют
энергетические неоднородности поверхности.
Теория
имеет практическое значение, т. к. можно определить удельную поверхность
адсорбента.
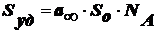 ,
,
о - площадь, занимаемая
молекулой адсорбата.
Межфазная поверхность, ее силовое поле. Поверхностное
натяжение как характеристика этого поля (энергетическая и силовая трактовка, ТД
определение, методы измерения на различных границах)
Поверхность раздела фаз - это граничная область между фазами, конечный по
толщине слой, в котором происходит изменение св-в от значений, характерных для
одной фазы, до значений, характерных для другой. На границе раздела фаз
формируется поверхностный слой толщиной в один или несколько молекулярных
размеров.
На межфазной поверхности сущ. поле нескомпенсированных межмолярных сил
из-за различия в составе и растворе контактирующих фаз, избыточные значения
плотностей ТД функций, их “сгущение”.
Пусть из 2х соседних фаз в 1й межмолекулярные взаимодействия сильнее, чем
во второй. Тогда в этой фазе важнейшее Вещество поверхностного слоя состоит в
том, что находящиеся в нем молекулы обладают избыточной Е.Гиббса. Для выведения
молекул из объема на поверхность надо преодолеть эту силу, т. е. совершить
работу и сообщить молекулам определенную энергию. Увеличение площади
поверхности приводит к увеличению числа поверхностных молекул и поверхностная
энергия возрастает.
Молекулы, прилегающие к поверхности по энергетическому состоянию отличны
от находящихся в объеме.
Для внутренних молекул равнодействующая всех межмолекулярных
взаимодействий равна 0, а для поверхностных молекул она направлена
перпендикулярно поверхности внутрь фазы с большим межмолекулярным взаимодействием.
Поверхностные молекулы втягиваются в глубь жидкости и возникает
внутреннее давление.
Следствием этого является поверхностное натяжение - важная характеристика
поверхности. Существует две трактовки σ - силовая и энергетическая:
|
Силовой подход
|
Энергетический подход
|
Основываясь
на законах механики, величину σ
рассматривают как следствие внутреннего давления и в частности как силу,
приложенную к единице длины контура на поверхности раздела, стремящуюся
сократить эту поверхность или препятствующую растяжению. Величина σ - мера стремления поверхности к сокращению, следствие
межмолекулярных сил. Отсюда:  - сила,
приложенная к единице длины контура поверхности раздела фаз, действующая
перпендикулярно контуру и тангенциально (вдоль) поверхности.При ↑
поверхности (напр, диспергирование), то необходимо вывести молекулы из объема
на поверхность s. Надо совершить работу против рвн. Она тем больше,
чем больше рвн, и мера этой работы - величина σ.
- сила,
приложенная к единице длины контура поверхности раздела фаз, действующая
перпендикулярно контуру и тангенциально (вдоль) поверхности.При ↑
поверхности (напр, диспергирование), то необходимо вывести молекулы из объема
на поверхность s. Надо совершить работу против рвн. Она тем больше,
чем больше рвн, и мера этой работы - величина σ.
Если
осуществлять обратимый процесс увеличения площади поверхности s на величину ds,
то полезная работа будет равна:  .
.
В
обратимом процессе полезная работа max и равна изменению энергий Гиббса или
Гельмгольца, взятых с обратным знаком. Тогда σ можно представить в виде:
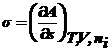 и
и 
|
В общем случае σ - частная производная любого ТД
потенциала по площади межфазной поверхности
|
|
Опыт Дюпре


Демонстрирует единство энергет. и силового подходов:
На
проволочной рамке (рис. а) образуем мыльную пленку. Нижняя сторона рамки -
подвижная и, если ничем не нагружена, поднимается вверх из-за стремления пленки
сократиться, т. е. На рамку действует сила поверхностного натяжения Fп.
Эту силу можно уравновесить грузиком весом Р = Fп. При увеличении
веса груза на бесконечно малую величину происходит перемещение подвижной
стороны рамки на dh (рис. б). Груз при этом совершает работу против силы Fп:
 .
.
Одновременно
из-за ↑ поверхности пленки возрастает поверхностная энергия: 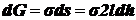 (к-нт 2 учитывает двусторонность пленки).
(к-нт 2 учитывает двусторонность пленки).
Так
как 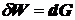 , получаем:
, получаем:
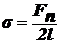
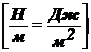 .
.
Величина
σ может рассматриваться не только как удельная
поверхностная энергия, но и как сила, отнесенная к единице длины контура,
огранич. поверхность.
Сила
поверхностного натяжения Fп имеет ту же природу, что и показанная
ранее равнодействующая сил, приложенных к поверхностным молекулам, но направлена
не ┴ поверхности, а вдоль нее (тангенциально к поверхности). Это хорошо
видно из модели Дюпре. Именно такое направление силы Fп и означает
стремление к уменьшению площади поверхности.
Методы
измерения поверхностного натяжения
Статическими
методами опред. поверхностное натяж. практически неподвижных поверхностей,
образованных задолго до начала измерений и поэтому находящихся в равновесии с
объемом жидкости. К этим м-дам относится м-д капиллярного поднятия (основан на
ф-ле Жюрена, исп тонкие капилляры), м-д вращающейся капли (измерение оч низких
значений σ на границах 2х жидкостей), м-д уравновешивания
пластинки (м-д Вильгельми). Динамические методы основаны на том, что некоторые
виды механических возд-вий на жидкость сопровождаются периодическими растяжениями
и сжатиями ее поверхности, на к-рые влияет поверхностное натяж. Этими м-дами
определяется неравновесное значение. К динамическим методам относятся методы
капиллярных волн и колеблющейся струи. Полустатическими называются м-ды
определения поверхностного натяжения границы раздела фаз, возникающей и
периодически обновляемой в процессе измерения (м-д максимального давления
пузырька и сталагмометрический м-д(определение веса капли)), а также м-ды
отрыва кольца (измеряется усилие необходимое для отрыва от поверхности жидкости
тонкого кольца, хорошо смачиваемого жидкостью) и втягивания пластины. Эти м-ды
позволяют опред. равновесное значение поверхностного натяжения, если измерения
производятся в таких условиях, что время, в течение к-рого происходит формирование
поверхности раздела, знач. больше времени установления равновесия в системе.
Экспериментальное
опред. поверхностной энергии тт (в случае тт о натяжении обычно не говорят).
Трудная
задача из-за медленного (по сравнению с жидкостью) протекания релаксационных
процессов и большой диссипации энергии при разрушении и обр-нии новой
поверхности, что обычно затрудняет проведение этого процесса как
изотермического обратимого. Сущ. несколько м-дов измерения поверхностной
энергии, из которых наиболее достоверные результаты дает м-д нулевой ползучести
(Таммана-Удина), основанный на наличии у тела вязкой ползучести, то есть
способности при достаточно высокой температуре медленно течь под действием
приложенной силы.
Для
хрупких твёрдых тел с хорошо выраженной спайностью (напр, слюда) подходит метод
расщепления по плоскости спайности (предложил Обреимов). Измеряется сила,
которую необходимо приложить чтобы заранее образованная в твёрдом теле трещина
стала развиваться далее. Связь этой силы с поверхностным натяжением, проявляющемся
в этом случае как работа образования новой поверхности, с длиной трещины l, а
также толщиной h, шириной d и модулем Юнга Е отщепляемой пластинки даётся
уравнением:  .
.
Используется
также зависимость растворимости от размера частиц с привлечением уравнения
Томсона (Кельвина). Ограничение: повышенная растворимость частиц, полученных
при механическом измельчении, связана также с появлением в них многочисленных
дефектов: упругих и неупругих искажений решётки в результате механического
воздействия.
Основы
термодинамики поверхностных явлений. Метод избыточных термодинамических функций
поверхностного слоя (метод Гиббса) и метод слоя конечной толщины
В
любой гетерогенной системе между двумя соприкасающимися фазами существует
область, свойства которой отличаются от свойств составляющих систему фаз -
поверхностный слой, величина которого порядка нескольких нм.
Для
описания термодинамики поверхностных явлений используют два метода: метод
избыточных величин Гиббса и метод «слоя конечной толщины».
Гиббс
предложил относить все изменения термодинамических параметров в слое в
сравнении с параметрами объемной фазы к разделяющей поверхности, не имеющей
объема или толщины. В соответствии с этим методом поверхность характеризуется
избыточными термодинамическими параметрами, а объемные фазы считаются
однородными вплоть до разделяющей поверхности. В соответствии с методом
избыточных величин (I) энергия Гиббса системы равна сумме энергий Гиббса
объемных фаз и поверхностной энергии Гиббса σs, которая является избыточной: 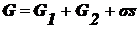 .
.
В
м-де «слоя конечной толщины» рассматривается слой, имеющий определенные размеры
(II). Энергия Гиббса той е системы выразится уравнением:
 ,
,
где
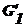 и
и  -
энергия Гиббса соответственно фазы 1 и фазы 2 до границы поверхностного слоя;
-
энергия Гиббса соответственно фазы 1 и фазы 2 до границы поверхностного слоя; 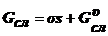 - энергия Гиббса поверхностного слоя;
- энергия Гиббса поверхностного слоя;  - энергия объема поверхностного слоя; σs - избыточная поверхностная энергия.
- энергия объема поверхностного слоя; σs - избыточная поверхностная энергия.
В
этом уравнении все параметры отвечают реальному строению системы. Но этот метод
использует довольно сложные уравнения, т. к. термодинамические параметры
изменяются нелинейно по толщине слоя. С математической точки зрения метод
избыточных величин проще, поэтому далее будем пользоваться им.
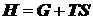 .
.
Аналогично
 , где индекс s означает отнесение потенциалов к
единице поверхности. Зная, что
, где индекс s означает отнесение потенциалов к
единице поверхности. Зная, что  и
и 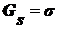 получим:
получим:
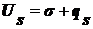 (),
(),
где
qs - теплота образования единицы поверхности.
Из
объединенного уравнения первого и второго начал термодинамики, в которое входят
все виды энергии
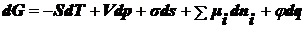
при
постоянстве всех параметров, кроме температуры Т, имеем:
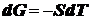 или
или  .
.
Подставляя
это выражения в уравнение (), получим:

уравнение
Гиббса-Гельмгольца, которое связывает полную поверхностную энергию с
поверхностным натяжением.
 -
температурный коэффициент поверхностного натяжения.
-
температурный коэффициент поверхностного натяжения.
Рассеяние и поляризация света в коллоидных системах. Закон
Релея и условия его применимости
При падении луча света на дисперсную систему могут наблюдаться следующие
явления:
1. Прохождение света через систему;
2. Преломление света частицами дисперсной среды;
. Отражение света частицами дисперсной фазы;
. Рассеяние света;
. Поглощение света дисперсной фазой.
· Если длина волны l
много больше размера коллоидных частиц а, то свет проходит через систему, не
меняя направления.
· Если l » а, то происходит релеевское и
комбинационное рассеивание, поглощение света. Возможно появление окраски.
· При l << а происходит отражение света
Явление рассеяния света коллоидными системами наблюдал
еще
Фарадей (1857 г.), исследовавший золи
золота. Подробно это явление было описано Тиндалем в 1868 г. В проходящем свете
золи не отличаются от истинных растворов, они ведут себя как прозрачные тела.
Тиндаль установил, что светорассеяние удобно наблюдать на темном фоне при
пропускании пучка лучей через золь сбоку. Особенно четко оно заметно при
фокусировании световых лучей внутри коллоидной системы, когда наблюдается
светящийся конус (конус Тиндаля). Это явление часто называют эффектом Тиндаля.
Рассеяние света происходит по схеме:
Падающий свет + молекулы (атомы) + hn è поляризация молекул (атомов) è возникновение диполей è излучение кванта света hn1
Световая волна вызывает поляризацию молекул, не проводящих и не
поглощающих свет. При этом образуется дипольный момент. Возникающие диполи
колеблются и создают вторичное излучение во всех направлениях
Так как частицы коллоидных систем имееют размеры не
более 10-7м, а длина лучей видимого света находится в пределах от
4·10-7м (фиолетовая область) до 7 10-7м (красная
область), то в коллоидных системах наблюдается только светорассеяние, а не
отражение световых лучей (как в грубодисперсных системах). Интенсивность
светорассеяния зависит не только от длины световой волны и размеров коллоидных
частиц, но и от ряда других факторов.
Зависимость интенсивности света, рассеянного в
результате дифракции светового луча от внешних и внутренних факторов системы
можно выразить формулой Релея:
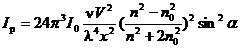 ,
,
если принять что расстояние от частицы до
наблюдателя(x) постоянная величина, угол между направлениями падающего и
рассеянного света(α) равен 90◦:
 ,
,
К
- константа, зависящая от интенсивности падающего света(I0) и
разности показателей преломления дисперсной фазы(n) и дисперсионной среды(n0),ν - число частиц в единице объема, V -объем частицы
дисперсной фазы, λ-длина волны падающего света.
Из уравнения можно сделать выводы:
· Если сравнить два одинаковых золя с одинаковой концентрацией, но разной
степенью дисперсности и принять I0 и λ постоянными, то увидим, что
интенсивность рассеяния прямо пропорциональна кубу радиуса частиц (обратно
пропорциональна кубу степени дисперсности)
· Если сравнить два одинаковых золя с одинаковой степенью
дисперсности, то увидим, что большую интенсивность рассеяния дает свет с более
короткими волнами.
· Если принять равными λ, I0 и степень
дисперсности, то решающим становится разность (n-n0), чем она
больше, тем больше светорассеяние.
Применимость формулы Рэлея:
· Не применима к металлическим золям, т.к. кроме рассеяния в них решающую
роль играет поглощение света. Протекает в них и ряд других физ-хим процессов:
меняется степень поляризации рассеянного света, световые волны в частице
индуцируют электродвижущую силу, возникает переменный электрический ток.
наблюдается переход электрической энергии в тепловую и т.д.
· Применимо только для золей с определенной степенью
дисперсности(от 5 до 100 нм).Чем больше размер частиц, тем меньше
светорассеяние зависит от длины волны, так как природа рассеяние станет иной,
обязанной не дифракции, а простому отражению света.
· З-н Рэлея пригоден только для сферических частиц.
· для разбавленных растворов;
· n1 ¹ n2;
· длина световой волны больше размера частиц: l = 10 - 20 r;
· среда не взаимодействует с фазой;
· частицы не проводят электрический ток, иначе световая энергия
переходит в электрическую (фотоэффект), а затем в тепловую - частица двигается.
Эмульсии, микроэмульсии, критические
эмульсии
Эму́льсия - дисп
сис-ма
<#"669641.files/image156.gif">. Специфика: быстрое дефор-ние до γ0, а затем сохранение
на этом уровне γ=γ0=const.Внач.момент
времени вся деформация сосредоточена в упругом элементе, начальное напряжение
равно τ0=Gγ0. Под его
действием - деформирование вязкого элемента. Т.к. общая деформация постоянна,
происходит спад деформации упругого элемента. τ=τ0e-t/tp.
Величина tp= η/G - период релаксации.
Б)
Модель Кельвина. Паралл. соединение упругости и вязкости.Деф-ции обоих
элементов одинаковы, напряжение сдвига суммируется τ =τG+τη.Вязкий элемент не позволяет немедленно
реализовываться деформации упругого тела. В рез-те общая деформация лишь
постепенно развивается во времени. Этому соотв.постепенно замедляющ. нарастание
деформации вплоть до γmax= τ0/G. Такой
процесс называют упругим последействием. Эластическое поведение механически
обратимо.
В)Моделью,
опис. возникновение внутренних напряжений, является паралл. сочетание
упругого элемента и сухого трения. Если приложенное τ превышает предел текучести(τ>τ*), в
теле возникает  , обусловл. накопление энергии упругим элементом.
Если же при этом τ< 2 τ*, то после снятия напряжений вследствие действия элемента сухого
трения в теле остается «замороженное» напряжение, равное τ - τ* и
противоположное по знаку исходному.
, обусловл. накопление энергии упругим элементом.
Если же при этом τ< 2 τ*, то после снятия напряжений вследствие действия элемента сухого
трения в теле остается «замороженное» напряжение, равное τ - τ* и
противоположное по знаку исходному.
Г)Модель
Бингама. Параллельное соединение вязкого ньютоновского элемента и
кулоновского элемента сухого трения. Деформации одинаковы, а напряжение на
элементах складываются. При этом на кулонов. элементе напряжение не может
превышать τ*. При τ<τ*
деформация не происходит. Скорость деформации вязкого элемента пропорциональна
разности действующего напряжения и предельного напряжения сдвига 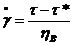 .
.
Краткий
исторический обзор развития коллоидной химии
.
Чисто практическое использование
дисперсных материалов и связанных с ними процессов и поверхностных явлений -
было известно в первоначальной ремесленной химии еще во времена глубокой
древности (краски и окрашивание тканей, керамика и глазури, цветное стекло,
изготовление мыл и др.).
.
Свою историю коллоидная химия
отсчитывает с 1861 года, с открытия Т.Грэмом коллоидных растворов. (Коллоидами
Грэм назвал вещества, которые образуют водные растворы с клееподобными
свойствами и не проходят через мембраны. Вещества, дающие молекулярные или
ионные растворы, Грэм назвал кристаллоидами).
3.
Далее следовал “химический” период
развития коллоидной химии, и до конца ХIХ в. усилия исследователей были
направлены на синтез различных коллоидов - неорганических, органических,
белковых. Стала понятна условность названия «коллоиды» и пришло понимание того,
что большинство вешеств могут быть получены в коллоидном состоянии.
.
Исследования зависимости свойств
систем от размера частиц составили содержание следующего этапа в развитии
представлений об объектах коллоидной химии. Главным результатом этого этапа
явился вывод о том, что к признакам объектов коллоидной химии - относится не
только дисперсность, но и гетерогенность (многофазность).
5.
В начале ХХ в. в коллоидную химию
вошли идеи и методы физики и физической химии. Они быстро привели к
фундаментальным открытиям: установлена гетерогенная природа коллоидных
растворов; открыто седиментационно-диффузионное равновесие в суспензиях и
эмульсиях; разработан метод измерения размера высокодисперсных частиц и
макромолекул с помощью ультрацентрифуг; создана кинетическая теория адсорбции и
строения адсорбционных слоев поверхностно-активных веществ. В сочетании с
термодинамикой поверхностных явлений Гиббса эти работы составили теоретический
фундамент коллоидной химии.
Теория
электролитной коагуляции. Порог коагуляции. Правило Шульце-Гарди
Электролитную
коагуляцию дисперсных систем вызывают: механическое воздействие (ультразвук);
сильное разбавление; концентрирование; замораживание; нагревание; введение электролитов.
Для коагуляции необходима определенная минимальная концентрация
электролита-коагулятора, соответствующая порогу коагуляцииγк данной
системы. Обычно это небольшие количества. Иногда используют обратную величину  , которую называют коагулярующей способностью. В
соответствии с природой электролитов различают 2 возможных механизма их
действия:
, которую называют коагулярующей способностью. В
соответствии с природой электролитов различают 2 возможных механизма их
действия:
Индифферентные электролиты вызывают концентрационную коагуляцию: Исходным
здесь является повышение ионной силы, что приводит к уменьшению толщины
диффузного слоя противоионов и переходу части противоионов из диффузного слоя в
плотный. И то, и другое приводит к уменьшению ζ-потенциала.
Неиндифферентные электролиты вызывают нейтрализационную (адсорбционную)
коагуляцию: Данный механизм реализуется лишь тогда, когда заряд частиц невелик.
При этом слои противоионов малы и возможна непосредственная адсорбция
коагулирующих ионов на поверхности частиц. Может быть два варианта:
специфическая адсорбция по правилу Панета-Фаянса, т. е ионы достраивают
кристаллическую структуру твердой фазы частиц; замещение в плотном
(адсорбционном) слое одних противоионов другими, у которых плотность заряда
больше.
В обоих случаях происходит нейтрализация / уменьшение заряда ч-ц.
Правила коагуляции: коагуляцию вызывают любые электролиты; в ряду
органич. ионов коагулирующее действие возрастает с повышением адсорбционной
способности; в ряду неорганич. Ионов с одинак. зарядом их коагулирующая
активность возрастает с уменьшением гидратации.
Для коагулирующего действия электролитов используется правило
Шульце-Гарди: Коагулирующим действием обладает ион с зарядом,
противоположным заряду частицы и сила коагулирующего действия увеличивается с
ростом заряда иона.
Используя понятие порога коагуляции, правило записывается:γ1: γ2 : γ3 = 500:25:1,где γ1, γ2, γ3 - порог коагуляции для 1, 2 и 3-хзарядных ионов.
По теории ДЛФО пришли к зависимости порога коагуляции от заряда:
 ,
,
т.е.
порог коагуляции обратно пропорционален 6-й степени заряда иона коагулятора z.
Cоотношение:γ1 : γ2 : γ3 =
73~0:11:1.
Понятие порога коагуляции позволяет прийти к количественной оценке еще
одного явления - коллоидной защиты. Суть: добавление белка или иного ВМС
повышает устойчивость ДС к электролитам. Характеризуют данное защитное действие
белка с помощью защитного числа -min концентрация белка, предотвращающая
коагуляцию частиц в присутствии пороговой концентрации коагулирующего
электролита.
Пены: получение, свойства, применение
Пена - дисперсия газа (чаще всего воздуха) в жидкой ДС - представ собой
лиофобную сис-му. Различ разбавленные дисперсии газа в жидкости, которые за их
сходство с разбавленными эмульсиями обычно называют газовыми эмульсиями, и
собственно пены с содержанием газовой фазы более 70 % по объему. В качестве
хар-ки конц-ции П часто использ отнош объема пены к объему сод-ся в ней жид-ти,
эту величину называют кратностью пены К. Вследствие седиментационной
неустойчивости большинства газовых эмульсий, при всплывании (обратной
седиментации) пузырьков сверху образуется слой конц-ной пены, и именно в ней
происходят затем процессы разруш сис-мы.
В пенах заполненные газом ячейки разделены пленками ДС. Характерной
«идеализированной» фигурой ячеек пен является пентагональный додекаэдр
(двенадцатигранник с пятиугольными гранями, имеющий 30 ребер и 20 вершин).
Однако эти фигуры не могут непрерывно заполнять пространство, и в реальной пене
среднее число пленок, окружающих ячейку, близко к 14.
Ребрами пенной ячейки служат заполненные дисперсионной средой каналы
Гиббса - Плато. Плато показал, что в одном канале могут сходиться только три
пленки, располож под углами 120 . Пов-ть канала имеет сложную вогнутую форму,
опис условием постоянства сумм двух главных кривизн; капиллярное давление под
вогнутой поверхностью обусловливает пониженное давление в канале Гиббса -
Плато.
В пенах с кратностью до 100 - 1000 (в зависимости от дисперсности и
толщины пленок) основная часть ДС сод-ся в каналах и лишь ее малая доля -в
пленках. Сод-ние жид-ти в узлах наиболее велико в очень низкократных пенах (при
кратности, приближающейся к трем).
Реальные пены, как правило, полидисперсны . Это влечет изменение формы
ячеек пен. Однако правила Плато (три пленки образуют канал, четыре канала
образуют вершину) соблюдаются во всех случаях. Дисперсность пены можно хар-ть
ее удельной поверхностью. Чаще измеряют некоторые средние значения
геометрических параметров ячейки, например, сред число ячеек в_ед объема или
средний эквивалентный радиус.
Пены широко исп-ют в различ областях соврем техники: при тушении пожаров,
во флотации, в произ-ве хлебопекарных и кондитерских изделий (хлеб-пример
отвержденной пены), теплоизоляционных мат-лов (пенобетоны, пенопласты,
микропористые резины) и т. д. Получ пен, осущ-тся диспергированием воздуха
(реже другого газа) в жидкости, сод какое-либо ПАВ -пенообразователь. Иногда
вводят добавки стабилизаторов пены, также являющихся ПАВ, которые усилив
действие пенообразователя.
Диспергирование газа может осуществляться пропусканием воздуха через слой
жидкости (барботажные пены) или с помощью мешалок в объеме жид-ти. Прим и
пеногенераторы разных конструкций, во многих из которых образование пены
происходит на сетке. При этом, задавая расход воздуха и раствора
пенообразователя, можно получать пену необходимой кратности. Для обеспечения
требуемой дисперсности пены на пути пенного потока устанавливается ряд сеток,
на которых происходит диспергирование ячеек пены. Такие пеногенераторы могут
обеспечить быстрое получение больших кол-в пены, необходимых при тушении
пожаров, особенно горящих топлив и других органических жидкостей.
Пены, в которых происходит отверждение ДС (хлеб, пеноматериалы), образ
конденсационным путем, основ на выделении какого-либо газа, чаще всего С02
в результате хим р-ции или биохим процесса.
Разруш пены сопровожд изменением во времени параметров, хар-щих строение
пены, и связано с протеканием процессов утоньшения и прорыва пленок,
изотермической перегонки газа от мелких ячеек к более крупным, а также
синерезиса - вытекания ДС из каналов Гиббса - Плато под действием силы
тяжести..
Утоньшение пленок может заканчив их разрывом или же образованием
мета-стабильно-равновесного состояния, когда расклинивающее давление в пленк
равно по абсолютной величине капиллярному давлению, определяемому кривизной
поверхности окружающего пленку мениска. Эту величину можно изменять, отсасывая
жидкость из канала Гиббса - Плато.
Изменение дисперсности пены во времени может быть связано как с
протеканием изотермического переноса газа через пленки, так и с разрывом самих
пленок. Измерение дисперсности пены и ее изменений во времени проводится
подсчетом числа ячеек, контак-щих со стенкой сосуда, в кот наход пена (по
микрофотографиям). Изотермический перенос газа от малых ячеек с более высоким
давлением воздуха к крупным, в которых давление воздуха ниже, особенно
существен для высокодисперсных полидисперсных пен, т. е. на начальных стадиях
их разрушения. Т к толщина пленок в пенах в состоянии, близком к
метастабильно-равновесному, не изменяется во времени, кинетика роста ячеек пен
при изотермическом переносе газа.
Разрыв пленок в пенах носит случайный хар-р, при этом вероятность прорыва
пленок и объединения соседних ячеек пропорциональна числу пленок в данный
момент времени.
Измен-е кратности пены К во времени связано с вытеканием дисперсионной
среды по системе каналов Гиббса - Плато под действием силы тяжести (синерезис).
Этому противостоит капиллярное давление в каналах Гиббса - Плато, так что может
возникнуть гидростатически равновесное состояние.
В высокократных высокодисп пенах нач значение кратности может превышать
эту величину. Дальнейшее падение дисперсности пены приведет к уменьшению
количества жидкости, которое может содержаться в пене, из нее начнет вытекать
дисперсионная среда и возрастать кратность. При этом увеличение во времени
кратности пены целиком определяется падением ее дисперсности: разрыв пленок
приводит к уменьшению числа каналов в единице объема пены, сечение же каналов
(при заданной высоте) остается неизменным. В низкократных пенах, наоборот,
именно в первые моменты времени происходит быстрое истечение дисперсионной
среды и рост кратности вплоть до достижения состояния, близкого к
гидростатически равновесному. В дальнейшем ход изменения кратности оказывается
таким же, как и для высокократных пен. Удобным методом определения кратности
пены является измерение ее электрической проводимости (кратность обратно
пропорциональна электропроводности пены).
Высокоустойчивые пены, стабилизированные ПАВ третьей и четвертой групп,
используются в пожаротушении, особенно при горении нефти и жидких топлив. В
этом случае важными характеристиками пен являются скорость растекания по
поверхности горящего нефтепродукта и их изолирующая способность - время
предотвращения выхода паров горючей жидкости. Для получения подобных
высокоустойчивых пен используют сложные составы, включающие, помимо основного
пенообразователя, добавки других ПАВ, дополнительно стабилизирующих пену.
Значительные перспективы открывает применение фторзамещенных соединений.
Важной задачей во многих производствах является также разрушение
(гашение) пены или предотвращение пенообразования. Так, в микробиологическом
производстве иногда начинает образовываться обильная пена, затрудняющая
проведение процесса. Обильная пена может образовываться в химических процессах,
связанных с выделением газов или их взаимодействием с жидкостями. Чрезмерное
пенообразование мешает и работе стиральных машин, поэтому стиральные порошки
для машин содержат большее количество неионогенных ПАВ, которые являются более
слабыми пенообразователями по сравнению с алкилсульфатами, особенно при
повышенных температурах.
Образовавшуюся пену можно разрушить с помощью перегретого пара,
ультразвука либо введением так называемых пеногасителей. По Ребиндеру,
пеногасителями являются ПАВ, имеющие более высокую поверхностную активность,
чем пенообразователи, и поэтому вытесняющие пенообразователь с поверхности
пузырьков пены, но не способные сами к ее стабилизации. Пеногасителями могут
служить низшие спирты, а также кремнийорганические соединения. Последние
особенно эффективно предотвращают пенообразование.
Седиментационный анализ. Основан на различной скорости
движения ч-ц ДФ относительно ДС в поле в поле действия внешних сил
Характерное свойство эмульсий, суспензий, аэрозолей. Простой вид движение
- оседание под действием силы тяжести. Седиментацию ускоряют с помощью
центростремительного ускорения и магнитного поля. Простой способ проведения
анализа - наблюдение за скоростью оседания ч-ц. Сущ-ет несколько методов -
микроскопический метод, м-д отбора проб, весовой, гидростатический (Вигнером:он
основан на измерении зависимости гидростатического давления столба от
времени.).
.
Если
ρ ‹ ρо, то получится отрицательное выражение для скорости -
происходит не оседание, а всплывание частиц. Так, в частности, происходит в
случае эмульсии масла в воде. Кроме того, на скорость оседания влияет вязкость
среды (чем меньше вязкость, тем меньше скорость) и радиус частиц:
Если
экспериментально определить скорость оседания, можно легко рассчитать радиус
частицы:
Это условие справедливо только для условий, при которых выполняется закон
Стокса,: частицы имеют сферическую форму; Движутся ламинарно, независимо друг
от друга и с постоянной скоростью; Трение является внутренним для ДС. Скорость
седиментации:
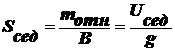 .
.
Константа
седиментации в сек или в Сведбергах10-13с =1 S.
Если
частицы небольшие и не очень сильно отличаются от среды по плотности, то их оседание
происходит очень медленно или вообще не происходит из-за противодействия со
стороны диффузии. Увеличение скорости седиментации путем центрифугирования.
Создавается ускорение до 105 - 106g и происходит
седиментация коллоидных ч-ц и седиментационное разделение макромолекул разной
массы.
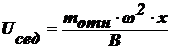 ,
,
где
х - расстояние от частицы до оси вращения центрифуги,ω - угловая скорость вращения центрифуги,
 ,
,
-
число оборотов в минуту.
Диапазон
размеров ч-ц, при котором можно использовать центрифугальный метод определения
дисперсного состава, составляет 0,05 - 1 мкм.
Существует
два метода контроля системы на разных расстояниях от оси вращения: скорость
седиментации оценивают по изменению со временем градиента показателя
преломления; по оптической плотности р-ров
Известно несколько принципов, лежащих в основе методов седиментационного
анализа:
1. наблюдение за скоростью оседания частиц в спокойной жидкости;
2. взмучивание суспензии с разделением ДФ на фракции па размерам
частиц в струе текущей жидкости;
. разделение порошков на фракции с помощью воздушной сепарации.
Весовой метод
Экспериментальное получении кривой седиментации, т. е. зависимости массы
осадка m ДФ от времени осаждения τ. Прибор -седиментометр Фигуровского.
Седиментационная кривая, т. е. зависимость m- τ имеет вид:

tgα- скорость седиментации; Q - общая
масса ДФ;
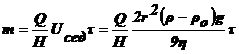
это
уравнение описывает кинетику седиментации в монодисперсной системе;
 - масса
частиц в объеме, приходящемся на единицу высоты.
- масса
частиц в объеме, приходящемся на единицу высоты.
В бидисперсной системе. Оседание частиц бидисперсной системы
(суспензии), имеющей две фракции частиц - мелкие и крупные. Идет одновременное
оседание двух монодисперсных суспензий. Кинетика более крупных частиц
выражается прямой OВ, а более мелких - прямой ОС, то график кинетики оседания
бидисперсной суспензии получается суммированием ординат этих прямых и
представляет собой ломаную линию ОСD с двумя точками перегиба D и С и абсциссы
этих точек τ1
и τ2соответсвуют времени полного оседания крупных и мелких
ч-ц.

ОО1=Аτ1 -содержание крупных ч-ц;
О1О2=Вτ2 - содержание мелких частиц;
ОО2 - характеризуют суммарную массу частиц.
На кривой седиментации бидисперсной системы 2 излома (ломаная ОСД),
трехдисперсной 3 и т. д.
Реальная кривая седиментации полидисперсной системы - плавная. Уравнение
касательной в любой точке кривой седиментации имеет вид:

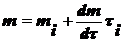
уравнение
Одена - обоснованием графического метода расчета распределения частиц по
размерам в полидисперсных системах.
В
начале этой кривой имеется прямолинейный участок ОА, т.к. в начальный период времени
на чашечку седиметометра оседают равномерно частицы всех размеров, до тех пор,
пока не осядут все самые крупные частицы (точка А). С этого момента времени τm скорость
накопления осадка уменьшается, и прямая переходит в кривую.
По
результатам, полученным при обработке кривой седиментации, обычно строят
дифференциальную и интегральную кривые распределениия.

Интегральная или суммарная кривая распределения Q(r) показывает
зависимость от радиуса суммарного количества частиц с размерами, превышающими
радиус r.
Важным с практической стороны свойством интегральной кривой распределения
является возможность быстрого определения содержания в данной суспензии любой
фракции частиц.

Интегральная кривая обычно имеет S-образную форму, с характерной точкой
перегиба, соответствующей наиболее вероятному размеру частиц, содержащихся в
данной дисперсной системе.
Дифференциальная кривая распределения показывает изменение весового
кол-ва при изменении радиуса частиц на единицу вблизи данного значения радиуса
 .
.
Максимум,
соответствующий наибольшей по весу фракции и наиболее вероятному размеру частиц
в данной суспензии. Площадь, ограниченная дифференциальной кривой и осью
абсцисс дает общее весовое количество частиц всех размеров, а площадь,
ограниченная двумя значениями радиусов rn и rm -
процентное содержание в суспензии частиц в интервале радиусов от rm
до rn. Метод весовой недостаточно точен. Длявысокая точность
результатов, используют аналитический метод.
Адгезия,
растекание, смачивание, правили антонова
В
соответствии со вторым началом термодинамики самопроизвольными являются
процессы, для которых dGs‹0.

Исходя
из этого, все поверхностные явления можно разделить на две большие группы.
Одни
процессы приводят к самопроизвольному уменьшению поверхности раздела фаз: ds‹0,
σ = const. (приобретение каплями жидкости сферической
формы, укрупнение частиц в процессах коагуляции, коалесценции, изотермическая
перегонка.)
Другие
процессы сопровождаются снижением межфазного натяжения:
dσ ‹0, s = const.( Это процессы адгезии, адсорбции, смачивание,
растекание.)
Адгезия
- это слипание двух разнородных твердых тел или жидких поверхностей за счет
межмолекулярных сил.
Если
две взаимно нерастворимые жидкости либо жидкость и твердое тело, либо, наконец,
два твердых тела приведены в тесный контакт, то под действием межмолекулярных
сил они прочно прилипают друг к другу, так что для их разделения нужно
произвести определенную работу.
Работа
адгезии Wa - это работа обратимого разрыва адгезионной связи,
отнесенная к единице площади, Дж/м2.

Предположим, что две жидкости 1 и 3 соприкасаются друг с другом и находятся
в среде 2. мысленно отделим их друг от друга. Тогда образуются две поверхности:
жидкости 1 с поверхностным натяжением σ1-2, жидкости 3 с поверхностным
натяжением σ2-3, а поверхность раздела с поверхностным натяжением σ1-3 исчезнет.
Работа адгезии будет равна:
 (5)-
уравнение Дюпре.
(5)-
уравнение Дюпре.

Работа
когезии - работа, необходимая для разрыва однородной объемной фазы Wк.
 (6),
(6),
коэффициент
2, т. к. образуется две новые поверхности.
Смачивание
- разновидность адгезии, относящаяся к взаимодействию типа ж-т. Оно может быть
контактным (при контакте трех фаз) и иммерсионным (при полном погружении
твердого тела в жидкость).
Характеристики
процесса смачивания - интегральная и дифференциальная теплоты смачивания.
Дифференциальная
теплота - теплота, которая выделяется при нанесении на поверхность при данной
степени ее заполнения бесконечно малого количества жидкости. Это характеристика
поля поверхностных сил.
Интегральная
теплота смачивания - теплота, которая выделяется при нанесении определенного
количества жидкости на единицу площади поверхности.
Ребиндер предложил за критерий гидрофильности поверхности взять отношение
теплот смачивания поверхности водой и каким-либо углеводородом.
Если
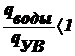 поверхность гидрофобна;
поверхность гидрофобна; 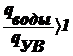 поверхность гидрофильна.
поверхность гидрофильна.
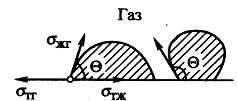
Жидкость
при контакте с твердым телом принимает такую форму, при которой по ее контуру
устанавливается равновесие сил поверхностного натяжения. Возьмем любую точку
контура капли. Здесь действуют три силы поверхностного натяжения. Каждая из них
направлена тангенциально к соответствующей поверхности и стремиться уменьшить
эту поверхность. При этом сила σтг стремиться, по существу, «растянуть» каплю по
поверхности (для уменьшения взаимодействия типа тг), а две другие силы
стремятся «сжать» каплю. Отсюда - уравнение Юнга для баланса сил:
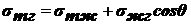 ,
,
где используется силовая трактовка σ. Угол θ между твердой поверхностью и
касательной к капле в точке контура - это краевой угол смачивания.
Случай, когда θ = 0°, т. е. жидкость растекается и покрывает всю
поверхность, называется полным смачиванием; когда θ = 180° - полным несмачиванием.
Подставив в уравнение Юнга (5) и (6), получим:
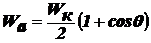 .
.
Для характеристики процесса растекания вводится понятие работы Wp
или коэффициента f растекания:
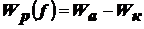 .
.
Чтобы
улучшить растекание необходимо увеличить Wa или уменьшить Wк.
Если
жидкость контактирует с другой жидкостью (например, бензол с водой), то
вследствие их взаимного насыщения f уменьшается.
σбензола ‹ σводы.
Жидкость
с меньшим σ растекается по жидкости с большим σ.
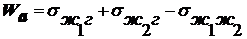 и
и  .
.
Тогда
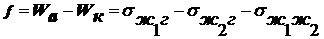 .
.
Пусть
будет неограниченное растекание, т. е. f=0:
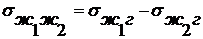
иллюстрирует
эмпирическое правило Антонова:
межфазное
натяжение на границе двух жидкостей равно разности поверхностных натяжений этих
жидкостей (на границах с воздухом) в условиях их взаимного насыщения.
Явление
смачивания можно наблюдать и тогда, когда вместо воздуха взята вторая жидкость,
не смешивающаяся с первой и имеющая большую плотность. Между ними будет
происходить конкуренция и нетрудно видеть, что из двух жидкостей смачивать
поверхность будет та, значение полярности которой ближе к полярности твердого
тела. О жидкости, лучше смачивающей поверхности, говорят, что она облает
большим избирательным смачиванием по отношению к данной поверхности.
На
угол влияют:
следы
в-в загр. поверхность
плёнка
оксидов
шероховатость
пов-ти
гистерезис
смачивания
Устойчивость гидрофобн. систем, ф-ция агрегативной устойчивости
Устойчивость дисперсных систем - это способность системы сохранять во
времени средний размер частиц и их равномерное распределение в среде. Иногда
сюда добавляют также условие постоянства состава частиц, исключая тем самым
возможные химические превращения.
Различают два вида неустойчивости (устойчивости седиментационная
агрегативная.
Седиментационная устойчивость - устойчивость ДФ по отношению к силе
тяжести за счет поддержания седиментационно-диффузионного равновесия.
Агрегативная устойчивость - способность системы к сохранению дисперсности
и индивидуальности частиц ДФ.
Они связаны между собой: агрегативная неустойчивость приведет к
седиментационной неустойчивости.
Процессами разрушения дисперсных систем, приводящими к уменьшению
свободной поверхностной энергии, служат изотермическая перегонка вещества от
малых частиц к более крупным, коалесценция (слияние частиц, капель) и
коагуляция (агрегирование частиц при их слипании). При изотермической перегонке
и коалесценции уменьшение свободной поверхн. энергии обусловлено уменьшением
площади поверхности раздела фаз, при коагуляции - частичное насыщение
нескомпенсированных на поверхности частиц молекулярных сил. Роль этих процессов
в нарушении агрегативной устойчивости различна и зависит прежде всего от фазового
состояния дисперсионной среды. Коагуляция, коалесценция - в системах с
легкоподвижной (жидк или газ) дисперсионной средой, изотермическая перегонка -
в любом состоянии ДС, в т.ч. и в твердом (единственный механизм). Если ДС
легкоподвижна, то роль изотермической перегонки мала, но если же коагуляция или
коалесценция затруднена - именно перегонка определяет разрушение дисперсной
системы. При колебаниях температуры (в реальных системах) процесс перегонки
может значительно ускоряться.
Факторы агрегативной устойчивости лиофобных дисперсных систем:
. Эффективная упругость пленок с адсорбционными слоями ПАВ. Увеличение
размеров пленки, связанное (например) с ее деформацией, приводит к изменению
условий равновесия адсорбционного слоя с раствором ПАВ с объемной части пленки.
Если деформация медленная, а толщина пленки мала, то растяжение обуславливает
выход части молекул ПАВ из объема на поверхность пленки. При этом уменьшается
концентрация ПАВ в объеме, уменьшается равновесная адсорбция и растет
поверхностное натяжение (эффект Гиббса). Т.е. с ростом площади пленки растет
поверхностное натяжение - возникает зависимость, которая эквивалентна
существованию эффективного модуля упругости.
Эффект Марангони-Гиббса - при высоких скоростях деформации не успевает
установиться равновесие между адсорбционным слоем и объемной частью пленки, и
модуль эффективной упругости оказывается завышенным - приводит к большему
увеличению устойчивости пленок. При быстром и локальном деформировании
нарушается также равномерное распределение вещества по поверхности - еще
большее увеличение модуля упругости.
. Электростатическое отталкивание диффузных частей двойных
электрических слоев. Вокруг частиц образуется ДЭС, состоящий из адсорбционного
слоя ионов одного знака и диффузного слоя противоионов. А т.к. противоионы в
обоих случаях одинаковы и имеют одинаковый заряд, кулоновский барьер в таком
случае не дает частицам сблизиться.
3. Гидродинамическое сопротивление прослойки среды вытеканию -
снижение скорости агрегации частиц вследствие образования на поверхности слоев
с повышенной вязкостью.
. Структурно-механический барьер (по Ребиндеру). Сильный фактор
стабилизации, связанный с образованием на границах раздела фаз адсорбционных
слоев низко- и высокомолекулярных ПАВ. Структура и механические свойства таких
слоев способны обеспечить весьма высокую устойчивость прослоек дисперсионной
среды между частицами дисперсной фазы. Структурно-механический барьер возникает
при адсорбции молекул ПАВ, которые способны к образованию гелеобразного
структурированного слоя на межфазной границе (защитные коллоиды).
Стабилизированные таким путем лиофобные системы приобретают свойства дисперсий
данного стабилизатора, т.е. становятся лиофилизированными.
Лиофильные системы. Мицеллярные растворы ПАВ. Критическая
концентрация мицеллообразования (ККМ). Термодинамика мицеллообразования
Лиофильные ДС Под вз-вием фаз ДС подразумевают пр-сы сольватации
(гидратации в случае водных систем), т. е. обр-ние сольватных (гидратных)
оболочек из молекул ДС вокруг частиц ДФ. По интенсивности взаимодейтсивя между
веществами ДФ и ДС (только для с-м с жидкой ДС), классификация Г. Фрейндлиха,
различают следующие ДС: лиофильные и лиофобные.
Лиофильные (гидрофильные, если ДС - вода): Для них характерно сильное
вз-вие ч-ц ДФ с мол-ми ДС. В предельном случае набл-ся полное растворение.
Лиофильные ДС обр-тся самопроизвольно вслед-ие пр-сса сольватации.
Термодинамически агрегативно устойчивы. Возрастание энергии в пр-ссе
диспергирования, связанное с увел-ем поверхности, компенс-ся уменьш-ем энтропии
в пр-се сольватации и ростом энтропии из-за участия ч-ц в поступательном
броуновском (тепловом) движении. Т/д-ски агрегативно устойчивы, т.е. уст-вы в
отношении обр-ния агрегатов коллоидных ч-ц. Пр-с слипания коллоидных ч-ц наз-т коагуляцией,
а в случае ч-ц жидкости - коалесценцией. В более широком смысле термин
коагуляция исп-ся для обозначения явления потери золями уст-сти, т.е. полного
разделения фаз в золе.
Представители: критические эмульсии, микроэмульсии, мицеллярные
растворыПАВ, растворынекоторых природных ВМС, (белков = яичный белок, желатин и
полисахариды = крахмал)