Химия древесины
СОДЕРЖАНИЕ
ВВЕДЕНИЕ
ОБЩИЕ ПОНЯТИЯ О ВМС. КЛАССИФИАЦИЯ ПОЛИМЕРОВ.
ПОЛЯРНЫЕ И НЕПОЛЯРНЫЕ ПОЛИМЕРЫ
.1 Общие понятия о ВМС
.2 Классификация полимеров
.3 Полярные и неполярные полимеры
. ХИМИЧЕСКИЙ СОСТАВ ДРЕВЕСИНЫ. ХОЛОЦЕЛЛЮЛОЗА И МЕТОДЫ
ЕЕ ВЫДЕЛЕНИЯ
.1 Химический состав древесины
.2 Холоцеллюлоза и методы ее выделения
. СТРОЕНИЕ ДРЕВЕСИНЫ. АНТОМИЧЕСКИЕ ЭЛЕМЕНТЫ ХВОЙНОЙ И
ЛИСТВЕННОЙ ДРЕВЕСИНЫ
.1 Строение древесины
.2 Антомические элементы хвойной и лиственной древесины
. ХИМИЧЕСКОЕ СТРОЕНИЕ МАКРОМОЛЕКУЛ ЦЕЛЛЮЛОЗЫ. СРЕДНИЕ
И КОНЦЕВЫЕ ЗВЕНЬЯ. КОНФОРМАЦИИ ЭЛЕМЕНТАРНЫХ ЗВЕНЬЕВ
.1 Химическое строение макромолекул целлюлозы
.2 Средние и концевые звенья
.3 Конформации элементарных звеньев
. СОДЕРЖАНИЕ И СОСТАВ ГЕМИЦЕЛЛЮЛОЗ ХВОЙНОЙ И
ЛИСТВЕННОЙ ДРЕВЕСИНЫ. ПОСТРОИТЬ СТРУКТУРНЫЕ ФОРМУЛЫ ОСНОВНЫХ ПРЕДСТАВИТЕЛЕЙ
ГЕКСОЗАНОВ И ПЕНТОЗАНОВ
.1 Содержание и состав гемицеллюлоз хвойной и
лиственной древесины
.2 Структурные формулы основных представителей
гексозанов и пентозанов
. ОБЩИЕ ПОНЯТИЯ О ЛИГНИНЕ КАК АРОМАТИЧЕСКОМ ПОЛИМЕРЕ.
СТРУКТУРНЫЕ ЕДИНИЦЫ ЛИГНИНА. ДИМЕРНЫЕ СТРУКТУРЫ ЛИГНИНА
.1 Общие понятия о лигнине как ароматическом полимере
.2 Структурные единицы лигнина
.3 Димерные структуры лигнина
. РАСТВОРИТЕЛИ ЦЕЛЛЮЛОЗЫ, ИХ ЗНАЧЕНИЕ
. ГИДРОЛИТИЧЕСКАЯ ДЕСТРУКЦИЯ ЦЕЛЛЮЛОЗЫ. КАТАЛИЗАТОРЫ И
СХЕМЫ ГИДРОЛИЗА
.1 Гидролитическая деструкция целлюлозы
.2 Катализаторы и схемы гидролиза
. ГИДРОЛИТИЧЕСКАЯ ДЕСТРУКЦИЯ ЛИГНИНА, АЦИДОЛИЗ И
ЭТАНОЛИЗ ЛИГНИНА
.1 Гидролитическая деструкция лигнина под действием
водных растворов кислот и оснований (гидролиз)
.2 Ацидолиз и этанолиз лигнина
. ХИМИЧЕСКИЕ РЕАКЦИИ ЛИГНИНА ПРИ СУЛЬФИТНОЙ ВАРКЕ.
АКТИВНЫЕ ГРУППЫ ЛИГНИНА
.1 Сульфирование лигнина
.2 Гидролитическая деструкция лигнина
.3 Кондеснация лигнина
ХИМИЯ СУЛЬФАТНОЙ ВАРКИ. ОСНОВНЫЕ ОТЛИЧИЯ ОТ НАТРОННОЙ
ВАРКИ И ХИМИЧЕСКИЕ РЕАКЦИИ ЛИГНИНА
.1 Химия сульфатной варки. Основные отличия от
натронной варки
.2 Химические реакции лигнина
. НИТРАТЫ ЦЕЛЛЮЛОЗЫ, ИХ ПОЛУЧЕНИЕ, СВОЙСТВА И
ПРИМЕНЕНИЕ
.1 Нитраты целлюлозы, их получение
.2 Свойства нитратов целлюлозы и их применение
СПИСОК ИСПОЛЬЗОВАННЫХ ИСТОЧНИКОВ
ВВЕДЕНИЕ
Химия древесины и синтетических полимеров - теоретическая основа
технологий химической и химико-механической переработки древесины. Древесина
является уникальным сырьем, постоянно возобновляемым в процессе фотосинтеза, и
квалифицированное комплексное использование всей ее биомассы представляет
важнейшую задачу с позиции экономики и экологической безопасности. Возрастание
роли древесины в связи с сокращением запасов традиционного сырья химической промышленности:
угля, нефти и газа - определяет основную перспективность исследований в области
химии и химической технологии древесины и других растительных источников сырья.
Несмотря на все большее широкое развитие производства различных синтетических
полимерных материалов, древесина как промышленное сырье для механической
технологии не теряет своего значения.
Для химической переработки древесина интересна своим комплексом природных
органических полимеров - целлюлозы, нецеллюлозных полисахаридов, лигнина, а также
разнообразных низкомолекулярных соединений - экстрактивных веществ.
Химия древесины является теоретической основой процессов химической
технологии производства целлюлозы, бумаги, лесохимических продуктов, продуктов
гидролиза, древесных пластиков и плит. Химия древесины как наука изучает:
строение, состав и свойства древесных тканей; строение и свойства химических
компонентов древесины; методы аналитического определения и выделения из
древесины ее компонентов; сущность технологических процессов химической
переработки древесины и отдельных ее компонентов с целью управления этими
процессами.
целлюлоза древесина полимер деструкция
1. ОБЩИЕ ПОНЯТИЯ О ВМС. КЛАССИФИКАЦИЯ ПОЛИМЕРОВ. ПОЛЯРНЫЕ И
НЕПОЛЯРНЫЕ ПОЛИМЕРЫ
1.1 Общие понятия о ВМС
Высокомолекулярные соединения (ВМС), или полимеры, получили свое название
благодаря их высокой молекулярной массе. По своим свойствам они отличаются от
других соединений. ВМС составляют главную часть сухого вещества растительных и
животных организмов и играют исключительную роль в жизни и деятельности
человека.
Структурные компоненты древесины (целлюлоза, гемицеллюлоза, лигнин) - это
высокомолекулярные соединения, тогда как экстрактивные вещества представляют
собой главным образом низкомолекулярные соединения (НМС).
К высокомолекулярным соединениям (ВМС) относятся вещества, состоящие из
больших молекул. Принято считать высокомолекулярными соединения с молекулярной
массой более 5000, но нередко она может достигать нескольких миллионов
(особенно у природных ВМС). Резкой границы между высокомолекулярными и
низкомолекулярными соединениями нет. Так, некоторые таннины с молекулярной
массой около 1000 ведут себя, как типичные низкомолекулярные соединения, а
парафины с такой же молекулярной массой обладают всеми свойствами высокомолекулярных
соединений. Переход от низкомолекулярных соединений к высокомолекулярным связан
не с самим изменением молекулярной массы, а с обусловленным им качественным
изменением свойств (переход количества в качество).
Размеры молекул ВМС по сравнению с размерами обычных молекул очень
велики. Так, длина молекул целлюлозы достигает 25∙10-5 - 50∙10-5
см (25 000 - 50 000 Ǻ) при размере в поперечнике 3,5∙10-8
- 7∙10-8 см (3,5 - 7 Ǻ).
Всем полимерам присущи некоторые общие свойства, которыми они существенно
отличаются от НМС:
. Они не могут существовать в газообразном состоянии; вместо
испарения при нагревании они разлагаются.
2. Полимеры, способные плавиться, не имеют точек плавления, а
размягчаются постепенно в определенном температурном интервале.
. Только у полимеров существует особое физическое состояние -
высокоэластическое, отсутствующее у НМС.
. Полимеры проявляют особые механические свойства: ведут себя
одновременно как твердые вещества и как жидкости и могут выдержать большие
напряжения.
. При растворении они проявляют характерную особенность:
растворению всегда предшествует набухание (явление, не известное у НМС).
. Растворы полимеров имеют высокую вязкость.
. Химическая реакционная способность у ВМС в значительной мере
зависит от их физической структуры.
Все полимеры являются ВМС, но не все ВМС могут быть полимерами. В
молекуле полимера должны быть химически связаны и закономерно повторяться
остатки исходного вещества - мономера. Большая молекула полимера называется
макромолекулой, или полимерной цепью, а сами остатки - элементарными
(мономерными, повторяющимися) звеньями, или просто звеньями. Звенья связаны
между собой химическими (ковалентными) связями. В отличие от молекулы
низкомолекулярного соединения макромолекула не является наименьшей частицей -
носителем химических свойств вещества, так как при разрыве макромолекулы на
более короткие цепи эти свойства сохраняются.
Число звеньев в цепи называется степенью полимеризации (обозначается n, P или СП).

При
написании эмпирических формул полимеров вследствие их большой молекулярной
массы (М) концевые звенья не принимают во внимание (эмпирическая формула
целлюлозы (С6Н10О5)n,
каучука (С5Н8)n).
Каждый полимер всегда состоит из макромолекул различной длины. Поэтому в
химии высокомолекулярных веществ введено понятие о полимергомологах, под
которыми понимают соединения одинакового химического строения, отличающиеся по
молекулярным массам. Любое полимерное соединения представляет собой смесь
полимергомологов - соединения с различным числом звеньев в макромолекуле, т.е.
с различной длиной цепи.
Полимергомологи образуют полимергомологический ряд, т.е. ряд соединений,
в котором каждый последующий член отличается от предыдущего на группу атомов -
элементарное звено.
В случае НМС молекулярная масса каждого соединения является постоянно
величиной, характерной для каждого соединения.
У полимеров можно определить только среднюю молекулярную массу и среднюю
СП.
Средняя молекулярная масса - это произведение среднего числа звеньев
(средней СП) на молекулярную массу звена.

Кроме
того, значения молекулярной массы у полимеров зависит от метода ее определения,
т.е. от измеряемого свойства, обуславливающего характер усреднения.
Различают
среднечисленную молекулярную массу (Mn) и среднемассовую
молекулярную массу (Mw), причем Mw/Mn ˃ 1.
 .
.
Определяют
также средневзвешенную молекулярную массу (Mz) и
средневязкостную молекулярную массу (Mv)
 ;
;
 ,
,
Где
ni - число макромолекул, wi -
масса макромолекул с молекулярной массой Mi.
Средневязкостную
молекулярную массу рассчитывают по данным вискозиметрии с использованием
уравнения Марка-Куна-Хаувинка:
 ,
,
Где
η - характеристическая вязкость, К -
вязкостно-молекулярная константа, a - параметр формы макромолекул,
зависящий от природы полимера и растворителя, обычно 0,5 ˂ а ˂ 1.
Все
методы определения молекулярной массы полимера можно подразделить на
абсолютные, которые не требуют калибровки, и косвенные. В косвенных
определяется какая-либо характеристика полимера, например вязкость раствора, по
которой с помощью констант рассчитывают М и СП.
По
измеряемым свойствам методы определения молекулярной массы подразделяют на
химические (метод концевых групп) и физические (физико-химические). К
физико-химическим методам относятся термодинамические (криоскопический,
эбулиоскопический и осмометрический), гидродинамические (метод диффузии, метод
ультрацентрифугирования и вискозиметрический метод), а также метод
светорассеяния.
Неоднородность
ВМС по молекулярной массе называется молекулярной неоднородностью,
полидисперсностью или полимолекулярностью. Ее определяют методом
фракционирования ВМС по молекулярной массе. Многие физико-механические свойства
ВМС - способность к набуханию и растворению, свойства растворов - в
значительной мере зависят не только от средней молекулярной массы, но и от их
полидисперсности.
Отношение
Mw/Mn называется показателем полидисперсности полимера.
Полную
характеристику полидисперсности дают кривые молекулярно-массового распределения
(ММР), которые строят по результатам фракционирования полимеров по молекулярной
массе.
Кроме
неоднородности по молекулярной массе многим полимерам свойственна химическая
(композиционная) неоднородность. Структурная формула макромолекулы всегда
идеализирована. Макромолекула может содержать группы, отличающиеся от основных
звеньев. В результате в образце могут содержаться различные по составу и
строению макромолекулы.
Свойства
ВМС определяются химическим составом, строением молекул, средней молекулярной
массой и молекулярной неоднородностью, формой макромолекул, их взаимным
расположением (физической структурой) и т.д.
1.2 Классификация полимеров
Из-за большого разнообразия ВМС единая система их классификации
практически оказалась невозможной. Поэтому классифицируют ВМС по самым
разнообразным признакам. Выбор признаков для классификации условен, так как
полимеры однотипные по одной классификации могут относиться к совершенно
различным типам - по другой.
Основные категории признаков для классификации полимеров следующие:
химический состав макромолекул, происхождение, пространственная структура
макромолекул, тип и взаимное расположение мономерных звеньев в цепях
макромолекул, физическое состояние полимеров, электрические свойства и пр.
По происхождению ВМС подразделяют на три типа:
. Природные, выделенные из природных материалов. ( В последнее время
природные полимеры, обладающие биологической активностью, - белки, нуклеиновые
кислоты, некоторые полисахариды и смешанные полимеры выделяют в отдельную
группу биологических полимеров, или биополимеров.)
2. Искусственные, полученные химической модификацией природных полимеров.
. Синтетические, синтезируемые из низкомолекулярных соединений. В
зависимости от способа получения они подразделяются на полимеризационные и
поликонденсационные полимеры.
В соответствии с пространственной структурой макромолекул ВМС делятся на:
· Линейные, или цепные (называемые также нитевидными);
· Разветвленные;
· Пространственные (трехмерные).
В ряде случае линейные макромолекулы могут приобретать сферическую форму
наподобие клубка; такие полимеры называют глобулярными.
По типу звеньев в цепях полимеры подразделяются на:
· Гомополимеры, макромолекулы которых состоят из одинаковых элементарных
звеньев;
· Гетерополимеры, или сополимеры (совместные полимеры),
макромолекулы которых состоят из двух и более различных элементарных звеньев
Сополимеры, состоящие из линейных макромолекул, которые содержат
относительно длинные участки элементарных звеньев одного состава, чередующиеся
с участками из звеньев другого состава называют блок-сополимерами.
Для макромолекул привитых, или графт-сополимеров, характерно, что вдоль
основной цепи через некоторые интервалы расположены боковые цепи второго
полимера.
По взаимному расположению мономерных звеньев в цепях макромолекул
полимеры разделяют на:
· Регулярные, если соблюдается правильное чередование звеньев,
дальний порядок расположения звеньев по цепи (каучук, целлюлоза);
· Нерегулярные.
Большое значение имеет стереорегулярность полимеров. Стереорегулярными
называют полимеры, у которых все звенья и все заместители расположены в
пространстве в определенном порядке (изотактические и синдиотактические
полимеры).
Полимеры, макромолекулы которых имеют преимущественное расположение цепей
вдоль некоторых направлений - осей ориентации, называются ориентированными.
Регулярность строения отражается на механических, физических и других
свойствах полимеров, так как при регулярном строении гораздо легче достигается
плотная упаковка макромолекул и максимальное сближение цепей и тем самым
обеспечивается наиболее эффективное действие межмолекулярных сил и водородных
связей.
В зависимости от гибкости макромолекулы и области применения полимеры
делятся на:
· Эластомеры (каучуки и каучукоподобные эластические вещества);
· Пластомеры (пластмассы);
· Волокнообразующие и пленкообразующие полимеры.
Для эластомеров характерны большие обратимые деформации.
В технологии полимеры делят на:
· Термореактивные, способные переходить при нагревании в
неплавкие и нерастворимые материалы;
· Термопластичные, не обладающие таким свойством и не теряющие
во время обработки своей способности формоваться (пластичности).
По электрическим свойствам полимеры подразделяются на:
· Диэлектрики (полимеры, которые не содержат легко
диссоциирующих на ионы групп и сопряженных двойных связей вдоль цепи
макромолекулы);
· Полупроводники (полимеры, для которых характерно наличие
сопряженных двойных связей или комплексов с переносом заряда);
· Электропроводящие материалы (в композицию обычно входит
токопроводящий наполнитель).
Наиболее важное значение имеет классификация полимеров по химическому
составу. В соответствии с химическим составом и химическим строением
макромолекул различные исследователи предлагают различные системы
классификации. Рассмотрим наиболее широко применяемую классификацию. В
соответствии с этой классификацией ВМС в зависимости от состава молекул делятся
на органические, неорганические и элементоорганические. Разграничение этих трех
классов очень затруднительно, так как существует много различных промежуточных
соединений.
К органическим ВМС обычно относят соединения, которые состоят из атомов
углерода, а также могут содержать атомы водорода, кислорода, азота, серы и
галоидов.
К элементоорганическим ВМС относят соединения, цепи которых построены из
атомов углерода и каких-то гетероатомов (помимо кислорода, азота и серы). А
также разветвленные полимеры с главными цепями из атомов углерода и
неорганическими боковыми цепями или, наоборот, неорганическими главными цепями
и боковыми цепями из атомов углерода.
К неорганическим полимерам относят соединения, не содержащие атомов
углерода.
Высокомолекулярные органические соединения в соответствии с принятой в
органической химии классификацией делятся на два класса: карбоцепные и
гетероцепные полимеры.
1. Карбоцепные полимеры имеют цепи, состоящие только из атомов
углерода. Эти полимеры подразделяются на классы в соответствии с
классификацией, принятой в органической химии.
. Гетероцепные полимеры содержат в цепях гетероатомы. Они
подразделяются на кислородсодержащие, азотсодержащие и серосодержащие полимеры.
В отдельную группу принято выделять полиацетилен. К ним относятся различные
полисахариды (крахмал, целлюлоза и ее производные, гемицеллюлозы и др.) и
полиуроновые кислоты.
Например, целлюлоза - полисахарид, макромолекулы которого состоят из
звеньев D глюкопиранозы, соединенных
глюкозидными связями 1 - 4.

Рис. 1.1. Структурная формула целлюлозы
1.3 Полярные и неполярные полимеры
Полярными полимерами называются высокомолекулярные соединения,
макромолекулы которых имеют полярные группы. У таких полимеров плотность
электронных облаков, образующих связь между атомами, распределена
несимметрично. Степень полярности зависит от полярности групп, входящих в
состав полимера, частоты и симметрии их расположения вдоль цепи молекулы.
К полярным группам относятся группировки:
≡ С - OH, ≡ C - COOH, ≡ C - NH2, ≡ C - Cl и др.
В этих группах имеется полярная связь, т.е. связь атомов с разной
электроотрицательностью (целлюлоза, лигнин).
Полимер, содержащий полярные связи, будет полярным, если только нет
компенсации диполей за счет симметрии молекул.
Степень полярности оценивается значением дипольного момента, зависящим от
расстояния между зарядами.
В макромолекулах, построенных по типу углеводородов (полиэтилен,
полипропилен и др.), связи неполярны, так как дипольный момент их равен нулю.
Среди полярных полимеров в отдельную группу выделяют полиэлектролиты -
полимеры, у которых звенья содержат ионогенные группы. При их ионизации
образуется полиион и низкомолекулярные ионы (соли полиакриловой кислоты).
2. ХИМИЧЕСКИЙ СОСТАВ ДРЕВЕСИНЫ. ХОЛОЦЕЛЛЮЛОЗА И МЕТОДЫ ЕЕ
ВЫДЕЛЕНИЯ
.1 Химический состав древесины

Рис. 2.1. Заготовка древесины

Рис. 2.2. Круглый лес
Древесина - это продукт биологического (растительного) происхождения. Она
представляет собой сложный комплекс, как в анатомическом, так и в химическом
отношении.
Древесина состоит из комплекса органических веществ, в состав которых
входит углерод (49,5%), кислород (44,1%), водород (6,3%) и азот (0,1%), которые
образуют сложные органические вещества. Основные из них - целлюлоза, лигнин,
гемицеллюлозы - пентозаны и гексозаны,- составляющие 90-95% массы абсолютно
сухой древесины и образующие клеточную оболочку.
Как любой биологический организм древесина, состоит из клеток. Клеточные
стенки примерно на 99% состоят из органических соединений (для тропических
пород примерно на 95%), которые можно подразделить на углеводную часть,
ароматическую и так называемые экстрактивные вещества. Последние не являются
составной частью клеточной стенки, а могут лишь ее пропитывать. В основном же
они содержаться в полостях клетки и в межклеточных пространствах. Такое
название эти компоненты получили потому, что их можно извлекать
(экстрагировать) из древесины нейтральными растворителями (водой или
органическими растворителями).
Наибольшее значение из них имеют дубильные вещества и смолы.
Углеводная часть древесины состоит из различных полисахаридов, которые
могут гидролизоваться кислотами. В хвойной древесине она составляет около 70%, в
лиственной - около 80%.
В углеводную часть входят:
· Целлюлоза - полисахарид, который является основным
компонентом древесины;
· Гемицеллюлозы - нецеллюлозные полисахариды.
Всю углеводную часть в целом называют холоцеллюлозой. Ее можно выделить
из древесины обработкой окислителями, при этом вещества ароматического
характера переходят в растворимые продукты, а в остатке получается волокнистый
материал - холоцеллюлоза.
Углеводную (гидролизуемую) часть в свою очередь можно подразделить на:
· Трудногидролизуемые вещества.
Представитель - целлюлоза, она гидролизуется лишь при действии
концентрированных кислот (например, 72%-ной H2SO4). При ее гидролизе получается D-глюкоза.
· Легкогидролизуемые вещества.
Представитель - гемицеллюлоза. Она гидролизуется при действии
разбавленных кислот (например, 2 - 5%-ной HCl или H2SO4). Продуктами гидролиза являются различные
моносахариды - пентозы (D-ксилоза
и L-арабиноза), гексозы (D-манноза и D-галактоза) и некоторые гексуроновые кислоты (D-глюкуроновая и D-галактуроновая).
Подразделение на легко- и трудногидролизуемые полисахариды в известной
степени условно. Не вся целлюлоза целиком трудно гидролизуется, часть ее
гидролизуется легко, т.е. при действии разбавленных кислот, и, наоборот,
некоторая часть гемицеллюлоз оказывается трудногидролизуемой, гидролизующейся
только совместно с целлюлозой при действии концентрированных кислот. Такое
различие в поведении полисахаридов обусловлено не различиями в химическом
строении, а особенностями надмолекулярной структуры целлюлозы и гемицеллюлоз.
В соответствии с получающимися продуктами гидролиза гемицеллюлозы условно
подразделяются на:
· Пентозаны (ксиланы, арабинаны);
· Гексозаны (маннаны, галактаны).
В действительности почти все гемицеллюлозные полисахариды являются
смешанными. В большинстве древесных пород содержатся следующие гемицеллюлозы:
глюкуроноксилан, арабиноглюкуроноксилан, глюкоманнан, арабиногалактан и др.
К веществам ароматической природы относится лигнин (смесь полимеров
родственного строения). Эта часть древесины является негидролизуемой. Она
получается в остатке после обработки древесины концентрированной кислотой.
Отдельные органические соединения очень трудно выделить из древесины в чистом
виде, так как они, как правило, изменяются при выделении.
По высоте ствола химический состав древесины меняется мало. Однако есть
некоторое различие в химическом составе между стволовой древесиной и древесиной
ветвей. Например, в древесине ветвей сосны, ели и осины содержится меньше
целлюлозы (44- 48%), но больше лигнина и пентозанов. Кора по химическому
составу значительно отличается от древесины. В коре содержится больше золы,
экстрактивных веществ и лигнина, но значительно меньше целлюлозы и пентозанов.
Кроме органических веществ, в древесине есть минеральные соединения,
дающие при сгорании золу, количество которой колеблется в пределах 0,2-1,7%;
однако у отдельных пород (саксаула, ядра фисташки) количество золы достигает
3-3,5%.
У одной и той же породы количество золы зависит от части дерева,
положения в стволе, возраста и условий произрастания. Больше золы дают кора и
листья; так, стволовая древесина дуба дает 0,35%, листья - 3,5% и кора - 7,2%
золы. Древесина ветвей содержит золы больше, чем древесина ствола; например,
ветви березы и ели дают при сгорании 0,64 и 0,32% золы, а стволовая древесина -
0,16 и 0,17% золы. Древесина верхней части ствола дает золы больше, чем нижняя;
это указывает на большое содержание золы в древесине молодого возраста; так,
древесина бука в возрасте 10, 20 и 50 лет давала при сгорании 0,56; 0,46 и 0,36%
золы.
Наибольшее количество золы приходится на кору и листья. По данным
профессора РГУ им. С.А. Есенина Козлова В.Н. в золе из древесины сосны, ели и
березы содержится свыше 40% солей кальция, более 20% солей калия и натрия и до
10% солей магния. Часть золы (10-25%) растворима в воде (главным образом,
щелочи - К2СО3 и сода). В прежнее время К2СО3,
употребляемый в производстве хрусталя, жидкого мыла и других веществ, добывали
из древесной золы. Зола от коры содержит больше солей кальция (до 50% у ели),
но меньше солей калия, натрия и магния.

Рис. 2.3. Содержание органических веществ в древесине разных пород.
Из этих данных видно, что древесина хвойных пород содержит повышенное
количество целлюлозы и гексозанов, а для древесины лиственных пород характерно
высокое содержание пентозанов. В клеточной оболочке целлюлоза находится в
соединении с другими веществами; особенно тесная связь, характер которой до
настоящего времени не ясен, наблюдается между целлюлозой и лигнином. Ранее
считали, что лигнин лишь механически примешан к целлюлозе; однако в последнее
время становится очевидным тот факт, что между ними существует химическая
связь.
Смолы
Эту группу веществ принято делить на нерастворимые в воде смолы (жидкие и
твердые) и камедесмолы, содержащие растворимые в воде камеди. Среди жидких смол
наибольшее значение имеет живица, которую получают из древесины (иногда из
коры) хвойных пород в результате подсочки.
Дубильные вещества, или танниды обладают свойствами дубить сырую кожу,
придавая ей стойкость против гниения, эластичность, способность не разбухать.
Наиболее богата дубильными веществами древесина ядра дуба (6 - 11%) и каштана
(6-13%). В коре дуба, ели, ивы, лиственницы и пихты содержится от 5 до 16%
таннидов.
Из древесных растений можно получать также эфирные масла, лакторезины и
красящие вещества.
Определение химического состава древесины и выделение ее отдельных
компонентов в чистом виде имеет большое теоретическое и практическое значение.
Однако это связано с большими трудностями из-за сложности строения клеточных
стенок и существования тесной связи между отдельными компонентами древесины.
.2 Холоцеллюлоза и методы ее выделения
Холоцеллюлозой называется весь углеводный комплекс древесины, т.е.
остаток древесины после удаления из нее экстрактивных веществ и лигнина. В
холоцеллюлозу входят целлюлоза и гемицеллюлозы - пентозаны, гексозаны и
связанные с ними уроновые кислоты.
Для выделения холоцеллюлозы необходимо удалить из древесины экстрактивные
вещества и обработать древесину (после экстракции органическими растворителями)
окислителями с целью удаления из нее лигнина (делигнификации), не допуская при
этом потерь и химических изменений полисахаридов.
Теоретически сумма холоцеллюлозы, лигнина, экстрактивных веществ и золы должна
составлять 100%. В практике препараты холоцеллюлозы всегда содержат остаточный
лигнин; гемицеллюлозы в процессе делигнификации частично теряются; целлюлоза и
присутствующие в холоцеллюлозе гемицеллюлозы химически несколько изменяются.
Выход холоцеллюлозы, содержание в ней гемицеллюлоз и примесь остаточного
лигнина зависят от древесной породы, используемого для делигнификации реагента
и условий обработки. Применяемые методы выделения холоцеллюлозы должны
удовлетворять следующему требованию: достижение низкого содержания остаточного
лигнина при минимальных потерях полисахаридов и минимальной гидролитической и
окислительной деградации целлюлозы.
Методы выделения холоцеллюлозы:
Впервые холоцеллюлозу получил американский исследователь Риттер,
подвергая древесину попеременной обработке влажным хлором и смесью спирта с
пиридином. При такой обработке лигнин хлорируется, окисляется и переходит в
раствор. Оставшуюся углеводную часть назвали холоцеллюлозой.
Выделить углеводный комплекс можно также при переменной обработке опилок
на холоду слабыми растворами (0,3 - 0,5%) двуокиси хлора и сульфита натрия.
В настоящее время для получения препаратов холоцеллюлозы и определения ее
содержания в древесине используют главным образом попеременную обработку
древесных опилок влажным хлором и горячим 3%-ным раствором моноэтаноламина в
95%-ном спирте, растворяющем продукты окисления и хлорирования лигнина. Этот
растворитель в отличие от водного раствора сульфита натрия, позволяет сохранить
гемицеллюлозы. Конец процесса делигнификации определяют по окраске препарата.
При данной обработке опилки лиственных пород окрашиваются в красный цвет,
хвойной - в коричневый. Хлорирование и промывку опилок раствором
моноэтаноламина проводят до тех пор, пока остаток не перестанет окрашиваться.
Вместо спиртового раствора моноэтаноламина можно использовать также
диоксановый раствор (5%-ный раствор моноэтаноламина в диоксане).
Часто применяют метод делигнификации, заключающийся в обработке древесины
хлоритом натрия в уксусной кислоте (метод Уайза). Однако, по мнению многих
исследователей, холоцеллюлоза при такой обработке частично изменяется, поэтому
в отличие от холоцеллюлозы, выделяемой по методу хлорирования, данный препарат
назван хлоритной холоцеллюлозой.
В последнее время предложен метод делигнификации древесины перуксусной
(надуксусной) кислотой CH3 - COOOH, которую получают из уксусного ангидрида окислением
перекисью водорода.
Выход холоцеллюлозы при выделении ее из древесины различными способами
составляет в среднем для хвойной древесины 70 - 73%, для лиственной 72 - 79%.
Холоцеллюлоза имеет большое теоретическое и практическое значение. Она
является хорошим объектом для выделения отдельных фракций гемицеллюлоз путем
последующих экстракций водой, растворами соды и едкого натра.
Гемицеллюлозы извлекаются щелочью из холоцеллюлозы легче, чем из
древесины: они менее загрязнены и менее разрушены.
Определение холоцеллюлозы в древесине служит одновременно косвенным
способом определения содержания лигнина. Кроме того, холоцеллюлоза включает всю
целлюлозу древесины и может использоваться для определения ее содержания.
3. СТРОЕНИЕ ДРЕВЕСИНЫ. АНТОМИЧЕСКИЕ ЭЛЕМЕНТЫ ХВОЙНОЙ И
ЛИСТВЕННОЙ ДРЕВЕСИНЫ
.1 Строение древесины

Рис. 3.1. Главные разрезы ствола: 1 - поперечный, или торцовый; 2 -
радиальный; 3 - тангенциальный

Рис. 3.2. Поперечный разрез стволика
сосны.
В середине разреза расположена сердцевина, окруженная концентрическими
кольцами (годовые слои), светлое периферийное кольцо в древесине - заболонь,
более темная центральная часть - ядро; снаружи древесина одета корой, в которой
заметны два слоя: более светлый, прилегающий к древесине - луб, и наружный -
темного цвета - корка. Между лубом и заболонью находится тонкий слой клеток
камбия (на рисунке не виден).
При рассмотрении поперечного разреза ствола дерева можно отметить
следующие части: сердцевину, собственно древесину (ксилему), живой слой -
камбий, кору (флоэму), в которой можно выделить внутренний слой, или луб, и наружный
слой.
При изучении древесину рассматривают по отношению к оси ствола в трех
направлениях: поперечном (торцевом), идущем перпендикулярно оси ствола;
радиальном, идущем через сердцевину и секущем дерево по его диаметру; и
тангенциальном, плоскость которого проходит вдоль ствола по хорде. Вследствие
того, что клетки древесины имеют преимущественно вытянутую форму и
располагаются слоями, вид древесины изменяется в зависимости от направления
разреза.
На торцевом разрезе ствола различают сердцевину диаметром 2 - 3 мм,
древесину, состоящую из концентрических колец - годичных слоев, и кору. У
некоторых пород годичный слой состоит из внутренней, более светлой, мягкой и
более пористой части - ранней, или весенней, древесины и наружной, более
тонкой, твердой и плотной части - поздней, или летней, древесины.
Поскольку годичные кольца представляют ежегодное нарастание древесины,
количество их на поперечном срезе у основания ствола определяет возраст дерева.
Древесина большинства пород имеет светлую окраску, но у некоторых, кроме
светлой, прилегающей к коре части, называемой заболонью, имеется
темноокрашенная центральная часть - ядро. В соответствии с этим породы делятся
на ядровые (лиственница, сосна, дуб, тополь и др.) и безъядровые. Из группы
безъядровых пород выделяют спелодревесные породы (ель, пихта, осина и др.), у
которых центральная часть древесины, не отличаясь по цвету, содержит
значительно меньше воды, чем периферийная. Остальные безъядровые породы, не
имеющие спелой древесины, называются заболонными (береза, ольха, липа).
В раннем возрасте древесина всех пород состоит только из заболони.
Заболонь служит для проведения воды от корней к кроне и для отложения
питательных веществ. Она содержит больший процент живых клеток по сравнению с
ядром.
Процесс образования ядра заключается в отмирании живых элементов
древесины, закупорке водопроводящих путей, отложении смол, пропитке дубильными
и красящими веществами, в результате чего цвет ядровой древесины изменяется, ее
плотность (объемный вес) увеличивается, повышаются механические свойства и
стойкость против гниения.
На разрезах некоторых пород, например, дуба, хорошо видны светлые,
расходящиеся от сердцевины к коре по радиусам линии - сердцевидные лучи (Рис.
3.1.). Их составляют клетки, расположенные горизонтально и образующие
лентообразные структуры. Сердцевидные лучи служат для проведения воды и
питательных веществ в горизонтальном направлении и для хранения запасных
питательных веществ зимой.

Рис. 3.3. Поперечный разрез ствола
Древесина корней по своему строению несколько отличается от древесины
ствола: хорошо развиты проводящие элементы. Древесина корней имеет меньший
объемный вес, меньшую прочность и большую водопроницаемость, чем древесина
ствола.
Древесина снаружи покрыта корой, которая состоит из двух слоев6 наружного
мертвого - внешней коры или корки, и внутреннего живого - луба, или флоэмы. При
росте дерева слои флоэмы образуются из года в год снаружи слоя камбия. Корка
защищает дерево от внешних влияний, а луб выполняет защитные и проводящие
функции. Кора составляет от 7 до 20% объема ствола и служит сырьем, например в
производстве экстрактов для дубления кожи.
.2 Анатомические элементы хвойной и лиственной древесины
Древесина состоит из отдельных клеточных элементов, среди которых можно
выделить два основных типа:
. Прозенхимные клетки (мертвые) - длина которых во много раз
больше ширины. Эти клетки придают древесине волокнистое строение; они в
основном мертвые. Они имеют сильно вытянутую, напоминающую волокно форму.

Рис 3.4. Прозенхимные клетки
Диаметр прозенхимных клеток 0,01 - 0,07 мм, длина 0,5 - 3 мм, иногда до 8
мм. К ним относятся главным образом трахеиды, клетки либриформа и сосуды.
. Паренхимные клетки (живые) - короткие, длина и ширина их
примерно одинаковы; они в основном живые. Они имеют округлую или многогранную
форму, в большинстве случаев тонкие стенки и примерно одинаковые размеры в трех
направлениях (от 0,01 до 0,1 мм).

Рис 3.5. Паренхимная клетка (а - паренхима, б - запасающая паренхима
сердцевинных лучей)
Анатомические элементы древесины в основном (90 - 95%) являются уже
отмершими клетками. Живая клетка имеет оболочку, протоплазму (протопласт) и
ядро. В мертвой клетке протоплазма и ядро отсутствуют, остается оболочка
(клеточная стенка) и полость, заполненная водой или воздухом, а иногда
экстрактивными веществами.

Рис. 3.6. Анатомическое строение древесины
Клетки одинакового строения имеют, выполняющие одну и ту же функцию,
образуют ткани. Различают три основных типа тканей: механические, проводящие и
запасающие.
Древесина хвойных пород имеет более простое строение (Рис. 3.7). Она
почти целиком состоит из клеток одного типа - трахеид (Рис. 3.8). Это мертвые
длинные веретенообразные клетки (длиной от 1,5 до 5 мм) со стенками различной
толщины. В стенках трахеид имеются простые и окаймленные поры (рис. 3.9). Поры
не являются свободными отверстиями: в них сохраняется тонкая первичная стенка
(мембрана). Окаймленная пора имеет мембрану с торусом. Окаймление образуется
нависающим выступом оболочки, а торус играет роль клапана, который может
закрывать пору. Каждой поре в оболочке одной клетки соответствует пора соседней
клетки, т.е. всегда существует пара пор. Мембрана, разделяющая поры, не
является сплошной. Она пронизана мельчайшими отверстиями, ведущими из клетки в
клетку. В живых клетках эти отверстия заполнены тончайшими нитями протоплазмы,
при помощи которых содержимое всех живых клеток соединяется в одно целое.

Рис. 3.7. Схема микроскопического строения древесины сосны (1 - годичный
слой, 2 - сердцевинный луч, 3 - вертикальный смольный ход, 4 - ранние трахеиды,
5 - поздние трахеиды, 6 - окаймленная пора, 7 - лучевые трахеиды, многорядный
луч с горизонтальным смоляным ходом)

Рис. 3.8. а - весенняя трахеида сосны, б - осенняя трахеида сосны

Рис. 3.9. Строение пор (а - простая пора, б - окаймленная пора, в -
полуокаймленная пора, / - канал, 2 - мембрана, 3 - торус)
Трахеиды, образующиеся весной имеют широкие полости и тонкие стенки, а
осенние трахеиды - более узкие полости и толстые стенки. У весенних трахеид
отношение диаметра к длине составляет 1 : 1000, у осенних - 1 : 400.
Широкополостные весенние трахеиды являются водопроводящими элементами
древесины, а толстостенные осенние трахеиды придают механическую прочность
стволу.
Живая ткань хвойной древесины - паренхима. Паренхимные клетки находятся в
хвойной древесине в сердцевинных лучах (клетки лучевой паренхимы). В небольшом
количестве имеется и так называемая древесная (вертикальная) паренхима,
располагающаяся поблизости от камбия. Кроме того, паренхимные клетки выстилают
поверхность смоляных ходов. Такие эпителиальные клетки принимают участие в
образовании смолы. Смоляные ходы существуют только в древесине хвойных пород.
Они представляют собой межклеточные каналы, образованные паренхимными клетками.
Различают вертикальные и горизонтальные смоляные ходы, которые, пересекаясь,
образуют единую смолоносную систему.
Объемная доля паренхимной ткани в хвойной древесине составляет всего 3 -
5%. Живые паренхимные клетки служат для хранения в зимний период запасов
крахмала и жиров, которые потребляются весной. У хвойных деревьев часть запасов
хранится в хвое, поэтому у них мало паренхимных клеток.
Анатомическое строение лиственной древесины более сложное, чем у хвойной.

Рис. 3.10. Схема микроскопического строения древесины дуба (1 - мелкие
сосуды, 2 - крупные сосуды, 3 - либриформ, 4 - сердцевинные лучи)

Рис. 3.11. Схема микроскопического строения древесины березы (1 - сосуды,
2 - либриформ, 3 - сердцевинные лучи)
Древесина лиственных пород включает: проводящие элементы (сосуды,
трахеиды), механические (волокна, либриформа) и запасающие, представленные
древесной и лучевой паренхимами.

Рис. 3.12. Анатомические элементы древесины лиственных пород (а - членик
сосуда: ПП - простая пора, ОП - окаймленная пора, б - сосудистая трахеида, в -
тяж древесной паренхимы, г - клетка веретеновидной паренхимы, д - волокнистая
трахеида, е - волокно либриформа, ж - клетки сердцевинных лучей.
Сосуды - типичные водопроводящие элементы только лиственных пород
представляют собой длинные тонкостенные трубки, образовавшиеся из длинного
вертикального ряда коротких клеток, называемых члениками сосудов, путем
растворения перегородок между ними. После соединения клеток, образующих сосуд,
протопласт их отмирает, и сосуды превращаются в мертвые капиллярные трубки,
заполненные водой.
Трахеиды у лиственных пород бывают двух типов: сосудистые и волокнистые.
Сосудистые трахеиды - преимущественно водопроводящие элементы, длина которых
0,5 мм. От волокон либриформа волокнистые трахеиды отличаются несколько меньшей
толщиной оболочек, но главным образом наличием окаймленных пор, в то время как
у либриформа поры простые.
Либриформ (волокна либриформа) - это механическая ткань лиственной
древесины. Клетки либриформа веретенообразной формы, имеют толстые стенки с
простыми щелевидными порами. Полости клеток либриформа очень узкие, поэтому они
очень плохо проводят воду. У лиственной древесины клетки либриформа довольно
короткие (0,3 - 0,8 мм), но у некоторых растений, таких как лен, конопля,
механические волокна могут достигать нескольких сантиметров (до 12 см, у рами -
до 30 - 50 см).
Живых паренхимных клеток в лиственной древесине больше, чем в хвойной
(около 10% и даже более). Живые паренхимные клетки здесь также образуют
сердцевинные лучи, которые в лиственной древесине развиты сильнее, чем в
хвойной.
Сердцевинные лучи в древесине лиственных пород развиты значительно
сильнее, чем в хвойной.
а  б
б 
Рис. 3.13. Сердцевинные лучи в
древесине лиственных и хвойных пород (а - лиственница, б - сосна)
Смоляные ходы встречаются у хвойных пород и служат для накопления и
выделения смолы. Они бывают вертикальными, тянущимися параллельно оси ствола, и
горизонтальными, встречающимися только в сердцевинных лучах. Диаметр
вертикальных смоляных ходов обычно бывает равен сумме диаметров четырех трахеид
и равен в среднем 0,1 мм (изменяемость от 0,06 до 0,13 мм). Длина смоляных
ходов колеблется от 10 до 80 см. Смоляные ходы располагаются главным образом в
летнем слое годового кольца. Они занимают от 0,1 до 0,17% объема древесины.
4. ХИМИЧЕСКОЕ СТРОЕНИЕ МАКРОМОЛЕКУЛ ЦЕЛЛЮЛОЗЫ. СРЕДНИЕ И КОНЦЕВЫЕ ЗВЕНЬЯ.
КОНФОРМАЦИИ ЭЛЕМЕНТАРНЫХ ЗВЕНЬЕВ
.1 Химическое строение макромолекул целлюлозы
Целлюлоза представляет собой линейный полисахарид - поли - 1-4-β-D - глюкопиранозил-D-глюкопиранозу и является гликозидом глюкозы. Макромолекула целлюлозы
образована глюкозными остатками в пиранозной форме, соединенными β-гликозидной (в данном случае β-глюкозидной) связью в положении 1 -
4. (Гликозид - это типовое название, общее для алкильных и других ацетальных
производных сахаров, глюкозид - термин, обозначающий такие производные для
глюкозы).
Чистая целлюлоза имеет эмпирическую формулу (С6Н10О5)n, которую с учетом трех гидроксилов в
элементарном звене можно представить в виду [C6H7O2(OH)3]n.
Строение макромолекулы целлюлозы изображается формулой, предложенной
Хеуорсом, которая сейчас является общепринятой.

Рис. 4.1. Строение макромолекулы целлюлозы (формула Хеуорса)
В данной формуле отражены концевые звенья макромолекулы и показан
целлобиозный структурный элемент.
4.2 Средние и концевые звенья
Средние звенья
Элементарные звенья β-D - глюкопиранозы, из которых построены макромолекулы
целлюлозы, называются глюкозными остатками, а также ангидро-D-глюкозой.
Согласно правилам номенклатуры углеводов ангидромоносахаридом называется
внутримолекулярный ангидрид, образующийся при выделении элементов воды за счет
двух гидроксильных групп молекулы моносахарида.
Чистая целлюлоза состоит из остатков глюкозы и не содержит каких-либо
других элементарных звеньев, т.е. является гомополимером.
Каждое среднее β-D -
глюкопиранозное звено целлюлозы содержит три свободных гидроксила, из которых
один первичный - при шестом и два вторичных - при втором и третьем углеродных
атомах. Гидроксилы придают целлюлозе слабокислые свойства и обусловливают
различные полимеранологичные превращения, например реакции этерификации с
получением сложных эфиров и реакции алкилирования с получением простых эфиров
целлюлозы. Наличие только трех гидроксилов подтверждается возможностью
получения при полной этерификации целлюлозы только трехзамещенных эфиров.
В настоящее время большинство исследователей склонно признать гипотезу,
что чистая природная целлюлоза не содержит карбоксильных групп и
карбоксилсодержащие вещества следует относить к гемицеллюлозам.
Концевые звенья
Из формулы целлюлозы (Рис. 4.1.) видно, что два концевых элементарных
звена макромолекулы отличаются от средних, многократно повторяющихся, и друг от
друга. Левое (условно) концевое звено содержит по сравнению со средними
звеньями дополнительный вторичный гидроксил и имеет, таким образом, четыре
гидроксила у углеродных атомов 2, 3, 4 и 6 - три вторичных и один первичный.
Правое (условно) концевое звено имеет у первого С-атома редуцирующую
полуацетальную группу и его называют редуцирующим (в водной среде может
существовать в альдегидной форме) в отличие от левого нередуцирующего.
Гидроксил при первом С-атоме, называемый глюкозидным (гликозидным) или
полуацетальным гидроксилом, отличается от остальных гидроксилов своими особыми
свойствами. Он один способен метилироваться метанолом (с образованием
метилгликозида), при этом исчезает альдегидная функция целлюлозы, которая вновь
восстанавливается при удалении метила гидролизом. Из всех гидроксилов только
один гидроксил у первого С-атома соединен с углеродным атомом, при котором
имеется другой кислородный заместитель, и представляет собой гидроксил
полуацеталя. Углеродный атом, с которым связан полуацетальный гидроксил (иначе
называемый гликозидным гидроксилом), получил название гликозидного или
аномерного центра. Высокая реакционная способность полуацетального гидроксила
объясняется, с современной точки зрения, стабилизацией образующегося при его
отщеплении карбониевого иона за счет свободной пары электронов соседнего
кислородного атома.
Наличие только одной альдегидной группы на молекулу целлюлозы позволяет
примерно судить о числе и размерах молекул в различных целлюлозных материалах
путем определения восстановительной способности материала, которую
характеризуют так называемым медным числом. Медным числом целлюлозы называют
количество меди в граммах, восстанавливаемой из двухвалентного состояния в
одновалентное и осаждаемой в виде закиси меди при взаимодействии 100г абсолютно
сухой целлюлозы с реактивом Фелинга в стандартных условиях. Реакция идет по
схеме:
R - C - OH + 2Cu(OH)2 → RCOOH + ↓Cu2O
+ 2H2O
При этом альдегидные группы целлюлозы окисляются до карбоксильных, а
окись меди восстанавливается до закиси. Жидкость Фелинга представляет собой
щелочной раствор гидроокиси меди и виннокислого калия-натрия (сегнетовой соли),
с которым нагревают образец целлюлозы. Сегнетова соль не участвует в реакции
окисления, она лишь препятствует выпадению осадка гидроокиси меди и удерживает
окисную медь в растворе.
Обычно чем больше коротких макромолекул в данном целлюлозном материале,
тем выше его медное число.
Число средних повторяющихся звеньев в макромолекуле целлюлозы по
сравнению с двумя крайними весьма велико, без большой погрешности можно
формулой целлюлозы считать (C6H10O5)n.
Основные сведения о строении целлюлозы:
1. Целлюлоза - жесткоцепной полимер стереорегулярного строения.
2. Элементарным звеном макромолекулы целлюлозы является остаток
глюкозы, что доказывается ее полным гидролизом. При котором D-глюкоза получается с выходом до 99%
от теоретического.
. Каждое среднее элементарное звено макромолекулы содержит три
гидроксила, один первичный и два вторичных. Это подтверждается возможностью
получения в качестве продуктов полной этерификации только трехзамещенных эфиров
целлюлозы.
. Элементарное звено макромолекулы целлюлозы имеет пиранозную, а
не фуранозную форму. Оно представляет собой шестичленный цикл, содержащий
амиленоксидный (1-5) кислородный мостик. Этот вывод является следствием из
положений о циклической структуре глюкозного остатка, о наличии в нем
гидроксильных групп при 2, 3 и 6-м атомах углерода и глюкозидной связи 1-4
между элементарными звеньями. Очевидно, связь внутри элементарного звена может
быть только между 2 и 5-м атомами углерода. Таким образом, элементарным звеном
целлюлозы является -D-глюкопираноза.
. Элементарные звенья соединены друг с другом -глюкозидной связью 1-4. Это
подтверждается получением при частичном гидролизе целлюлозы дисахарида
целлобиозы 4-О--D-глюкопиранозил-D-глюкозы, в которой два остатка
глюкозы соединены -глюкозидной
связью. Все связи между элементарными звеньями макромолекулы целлюлозы
практически равноценны.
. В целлюлозе остаток D-глюкозы, находящийся на одном из концов макромолекулы, содержит
восстанавливающую группу, наличие которой доказывается рядом реакций, в
частности способностью целлюлозы восстанавливать окисные соединения меди до
закисных и окисляться йодом в щелочной среде.
.3 Конформации элементарных звеньев
Перспективная формула целлюлозы по Хеуорсу (Рис. 4.1.) не отражает
истинной геометрической формы макромолекулы целлюлозы.
Пиранозные кольца, составляющие макромолекулу, не являются плоскими, а
могут для уменьшения внутренних напряжений принимать конформации кресла,
обозначается 1С и С1 (от слова chair
- кресло) или ванны (лодки), обозначается 1В и В1 (от слова boat - лодка).
В более выгодной, более устойчивой конформации кресла (обладает меньшей
энергией) атомы углерода расположены по три в двух параллельных плоскостях,
перпендикулярно которым проходит ось симметрии третьего порядка. Двенадцать
связей С-Н циклогексана подразделяются на два класса. Связи, параллельные оси
симметрии (и друг другу), называются аксиальными (а), другие шесть атомов
водорода связаны экваториальными связями (е), направленными под углом 109,5º к оси симметрии.
Молекула циклогексана может принимать две во всех отношениях
эквивалентные креслообразные конформации. При переходе из одной в другую
аксиальные атомы водорода становятся экваториальными и обратно. В замещенном
циклогексане, например метилциклогексане, две креслообразные конформации уже
неравноценны, поскольку в одной из них метил занимает аксиальное, а в другой
экваториальное положение:



Конформация с наименьшим числом аксиальных заместителей является наиболее
устойчивой. Устойчивость конформации следует понимать в статистическом смысле.
Макромолекулы могут принимать различные конформации; однако в каждый момент
большая часть молекул представлена наиболее энергетически выгодной
конформацией.
С повышением температуры содержание менее выгодных конформаций
увеличивается. Напротив, понижение температуры повышает содержание наиболее
устойчивой конформации, поскольку становится менее возможным преодолеть
потенциальный барьер за счет энергии, взятой из среды. При достаточно низкой
температуре молекулы принимают наиболее выгодную конформацию.
Согласно исследованиям Ривза глюкопиранозное кольцо может существовать в
восьми конформациях - двух креслообразных и шести ваннообразных. Конформация
типа кресла, соответствующая минимуму энергии, более вероятна и
предпочтительна.
В зависимости от конформации пиранозного цикла меняется положение
гидроксилов в цикле. В конформации кресла С1 гидроксилы находятся в
элементарном звене целлюлозы в экваториальном положении. Гидроксилы,
находящиеся в аксиальном и экваториальном положениях, обладают различной
реакционной способностью; этот вывод экспериментально подтвержден при
тозилировании углеводов. Этерифицируются в первую очередь гидроксилы,
находящиеся в экваториальном положении, так как для них стерические условия
осуществления реакции более благоприятны.
Для D-глюкопиранозы и ее производных (в
том числе для целлюлозы) наиболее энергетически выгодной формой является
конформация кресла С1, где все гидроксильные группы находятся в экваториальном
положении. Макромолекула целлюлозы состоит из большого числа остатков D-глюкопиранозы, находящихся в
конформации кресла С1, соединенных между собой 1-4-гликозидными связями. Однако
различные химические и физические воздействия могут привести к образованию и
других конформационных форм.
Конформационные превращения в макромолекуле целлюлозы не исчерпываются
изменениями пространственных форм глюкопиранозного кольца. Изменения формы
макромолекулы могут быть связаны с вращением глюкопиранозных звеньев вокруг
гликозидных связей.
Макромолекулы целлюлозы и ее производных являются наиболее жесткими среди
высокополимеров. По мнению Херманса, переход креслообразной конформации
глюкопиранозного кольца в ваннообразную должен увеличивать гибкость цепи.
Хенлей получил экспериментальные доказательства того, что в некоторых
растворителях молекула целлюлозы обнаруживает свойства гибкого полимера.
Характерной особенностью целлюлозы, определяющей в значительной степени
ее механические, физико-химические и химические свойства, является линейная
конформация ее макромолекул, закрепленная внутримолекулярными водородными
связями. Линейные макромолекулы целлюлозы, располагаясь почти параллельными
пучками, образуют в отдельных областях волокон за счет дополнительных
межмолекулярных водородных связей структуры, регулярные в трех измерениях, что
свойственно кристаллам.
5. СОДЕРЖАНИЕ И СОСТАВ ГЕМИЦЕЛЛЮЛОЗ ХВОЙНОЙ И ЛИСТВЕННОЙ ДРЕВЕСИНЫ.
ПОСТРОИТЬ СТРУКТУРНЫЕ ФОРМУЛЫ ОСНОВНЫХ ПРЕДСТАВИТЕЛЕЙ ГЕКСОЗАНОВ И ПЕНТОЗАНОВ
.1 Содержание и состав гемицеллюлоз хвойной и лиственной древесины
К гемицеллюлозам относят группу нецеллюлозных полисахаридов клеточных
стенок высших растений, способных извлекаться из них водными растворами щелочей
и гидролизоваться разбавленными кислотами при кипячении. Вследствие меньшей, по
сравнению с целлюлозой, устойчивости к гидролизу гемицеллюлозы относят к легкогидролизуемым
полисахаридам. При кипячении с кислотами гемицеллюлозы гидролизуются и
переходят в простейшие сахара, а при действии щелочей (холодных или горячих
водных растворов) растворяются и извлекаются из древесины.
В состав гемицеллюлоз входят полисахариды, содержащие элементарные звенья
из пяти и шести атомов углерода, т.е. звенья пентоз и гексоз.
В древесине большая часть гемицеллюлоз, т.е. нецеллюлозных полисахаридов,
представляет собой не однородные (гомополимеры), а смешанные (комбинированные)
полисахариды (сополимеры), которые состоят из остатков различных моносахаридов,
соединенных гликозидными связями в различных положениях. В некоторые из них
входят остатки уроновых кислот.
Содержание гемицеллюлоз в древесине составляет от 17 до 43%. В лиственных
породах гемицеллюлоз примерно в 1,5 раза больше, чем в хвойных.
Таблица 1 Содержание гемицеллюлоз в древесине
|
Пихта
|
22,6
|
Осина
|
23,3
|
|
Кедр
|
21,0
|
Береза европейская
|
27,8
|
|
Ель обыкновенная
|
20,6
|
Дуб
|
29,1
|
|
Сосна
|
20,5
|
Бук
|
36,9
|
|
Ель черная
|
16,1
|
Береза тянь-шаньская
|
41,2
|
В ветвях и вершинной части ствола обычно находят несколько больше
гемицеллюлоз; содержание и свойства гемицеллюлоз меняются с возрастом дерева.
Состав гемицеллюлоз лиственной и хвойной древесины различен:
Таблица 2 Состав гемицеллюлоз лиственной и хвойной древесины (%)
|
Хвойная древесина
|
Лиственная древесина
|
|
Ксиланы
|
19-23
|
79-82
|
|
Глюкоманнаты
|
64-65
|
11-12
|
|
Арабиногалактан
|
15-18
|
7-8
|
Лиственные породы содержат больше пентозанов (17-25%, иногда до 30%), а
хвойные породы меньше (от 5 до 13%). Гемицеллюлозы лиственных пород почти
целиком состоят из глюкуроноксилана (4-О-метилглюкуроноксилан), а хвойных имеют
более сложный состав. К ним относятся арабиноглюкуроноксилан
(4-О-метилглюкуроноарабоксилан), глюкоманнан, а также галактоманнан и арабиногалактан.
Содержание гексозанов в хвойных породах составляет 8-14%, а в лиственных - лишь
0,5-6%.
.2 Структурные формулы основных представителей гексозанов и пентозанов
Гексозаны
К гексозанам относятся полисахариды, молекулы которых построены главным
образом из звеньев гексоз. Упрощенная эмпирическая формула гексозанов (C6H10O5)n. Цепи гексозанов, как и пентозанов, значительно
короче цепей целлюлозы.

Рис. 5.1. Продукты гидролиза маннана. Структурная формула маннана

Рис. 5.2. Продукты гидролиза галактана. Структурная формула галактана

Рис. 5.3. Продукты гидролиза фруктана
Главнейшим представителем гексозанов в древесине являются маннаны, дающие
при гидролизе в качестве основного моносахарида D-маннозу, и галактаны, которые дают при гидролизе в качестве
основного моносахарида D-галактозу.
Менее изучен фруктан, или левулан, при гидролизе которого получается D-фруктоза. Гексозаны, как и
пентозаны, являются в основном смешанными полисахаридами.
Пентозаны
Пентозаны - это полисахариды, главными звеньями макромолекулы которых
являются звенья пентоз. Упрощенная эмпирическая формула пентозанов (С5Н8О4)n.
Из пентозанов наиболее распространены и более изучены ксиланы, дающие при
гидролизе в качестве основного моносахарида D-ксилозу. В меньших количествах в древесине содержится
арабинан, который дает при гидролизе L-арабинозу.

Продукты гидролиза арабинана. Структурная формула арабинана.
6. ОБЩИЕ ПОНЯТИЯ О ЛИГНИНЕ КАК АРОМАТИЧЕСКОМ ПОЛИМЕРЕ. СТРУКТУРНЫЕ
ЕДИНИЦЫ ЛИГНИНА. ДИМЕРНЫЕ СТРУКТУРЫ ЛИГНИНА
.1 Общие понятия о лигнине как ароматическом полимере
Лигнины представляют собой аморфные полуфункциональные высокомолекулярные
соединения ароматической природы, построенные из фенилпропановых структурных
единиц. Для макромолекул лигнинов характерны нерегулярность строения и
поливариантность связей.
Макромолекула лигнина образована не из однородных структурных единиц, как
целлюлоза, а из нескольких различающихся производных фенилпропана. Многообразие
связей, возникающих между структурными элементами при образовании лигнина в
растениях, приводит к полимеру с нерегулярным строением.
Лигнины различных растений заметно отличаются друг от друга, и их можно
разделить на три обширные класса: лигнины хвойной древесины, лигнины лиственной
древесины и лигнины травянистых растений. Лигнины хвойной древесины более
изучены, они образуют довольно однородную группу веществ, причем лигнины разных
хвойных пород очень близки по свойствам. Лигнины же лиственной древесины и
травянистых растений менее изучены и, по-видимому, отличаются большим
разнообразием.
Лигнины накапливаются в растениях в процессе лигнификации, в срединной
пластинке и в клеточной стенке. Окончание процесса лигнификации обычно
совпадает с прекращением жизнедеятельности клетки. Установлено, что лигнин
является необратимым конечным продуктом обмена (метаболизма) в растении.
Лигнины, содержащиеся в растительных тканях - природные (или
протолигнины), представляют весьма лабильные вещества, которые легко изменяются
под влиянием повышенной температуры и различных реагентов. Даже мягкое
химическое воздействие на природный лигнин приводит к его изменению. Поэтому до
сих пор нет метода, позволяющего полностью выделить лигнин из растения, не
изменив при этом его химических и физических свойств, и проблема получения
препаратов лигнина, аналогичных природному, продолжает оставаться нерешенной.
Препараты лигнина, мало измененные по сравнению с природным, иногда называют
нативными. Таковыми являются, например, лигнин Бьеркмана (лигнин механического
размола, ЛМР) и лигнин Браунса.
Сильно измененными лигнинами являются препараты, выделяемые кислотными
методами, а также все виды технических лигнинов, являющихся отходами химической
переработки древесины.
Природные лигнины обладают рядом специфических свойств:
. Легко окисляются (используется при отбелке и при получении
ванилина, ванилиновой кислоты и пр.).
. Легко взаимодействуют с хлором (это свойство используется при
отбелке для разрушения макромолекул лигнина с целью повышения растворимости и
удаления их из целлюлозы).
. Растворимость в щелочах при нагревании (служит основой щелочной
делигнификации - натронного и сульфатного способов производства целлюлозы).
. Способность растворяться при нагревании в водных растворах
сернистой кислоты и ее кислых солей (используется для делигнификации древесины
сульфитным способом).
Лигнин устойчив, в отличие от полисахаридов, к гидролизующему действию
растворам минеральных кислот. При действии кислот в лигнине могут
гидролизоваться углерод-кислородные связи, но сохраняются существующие в нем
углерод-углеродные связи и образуются новые.
. Содержит большое количество метоксильных групп, которые служат
источником образования метанола и неприятно пахнущих метилсернистых соединений:
метилмеркаптана, диметилсульфида, диметилдисульфида при сульфатной варке
древесины.
. Способность образовывать при этанолизе мономеры Гибберта -
низкомолекулярные ароматические производные, преимущественно кетоны,
моделирующие собой структурные звенья лигнина, например ванилин (по мнению
Браунсов).
Массовая доля лигнина в древесине хвойных пород составляет в среднем 27 -
30%, а в древесине лиственных пород - 18 - 24%. В отличие от целлюлозы и других
полисахаридов выделенный из древесины лигнин не является индивидуальным
веществом, а представляет собой смесь нерегулярных разветвленных полимеров
родственного строения, в основе которых лежат близкие по строению ароматические
вещества.
6.2 Структурные единицы лигнина
Лигнин как полимер состоит из фенилпропановых структурных единиц
(мономерных составляющих звеньев) - ФПЕ, обозначаемых сокращенно С6-С3,
или единицы С9. Для лигнинов хвойной древесины основными являются
структуры гваяцилпропана - 4-окси-3метоксифенилпропана (G-единицы - I), а для лиственных наряду с ними
структуры производных сирингилпропана - 4-окси-3,5-диметилоксифенилпропана
(S-единицы - II). В лигнины однолетних растений
наряду с вышеуказанными структурами входят производные n-оксифенилпропана (Н-единицы - III).
Элементарные звенья макромолекулы лигнина хвойных можно рассматривать как
производные пирокатехина (IV), а
лиственных - как производные пирокатехина и пирогаллола (V).

I II III
а)  б)
б)
Рис. 6.1. а) пирокатехин, б)
пирогаллол
Предшественниками образования этих структур в растениях наряду с
конифериловым спиртом (VI)
считают соответственно синаповый (VII) и n-кумаровый (n-оксикоричный) (VIII) спирты. Строение пропановой цепочкиу этих спиртов аналогично, однако в
элементарных звеньях макромолекул лигнина пропановая цепочка приобретает
различные варианты. Некоторые варианты строения элементарных звеньев лигнина
хвойных можно представить формулой (XI). Она не включает звеньев, содержащих карбонильные группы, и некоторых
других вариантов. Позиции 2, 5, 6 ароматического ядра могут быть замещены
водородом или связаны с C-атомами
других звеньев макромолекулы.
Наличие вышеуказанных структур в макромолекулах лигнинов подтверждено
окислением лигнинов нитробензолом в щелочной среде при 160ºC по Фрейденбергу. Этот метод имеет
важное значение для идентификации лигнинов. Из лигнина хвойных при этом
получают ванилин (X), немного n-оксибензальдегида (XII) и следы сиреневого альдегида (XI), в примерном полярном соотношении
1:3.

Рис. 6.2. Элементарные звенья лигнина и их молекулы (VI-IX)

Рис. 6.3. Элементарные звенья лигнина и их молекулы (X-XII)
n-Оксифенилпропановые
звенья, которых много в лигнинах однолетних растений, дают при нитробензольном
окислении значительное количество n-оксибензальдегида.
Лигнины древесины лиственных пород изучены менее, чем хвойных.
Характерное различие между лигнинами древесины лиственных и хвойных пород
состоит в количестве конденсированных структурных звеньев. С помощью ядерного
магнитного резонанса ЯМР определено, что лигнин березы содержит 43%
сирингиловых единиц, 11% неконденсированных гваяциловых и 46% конденсированных
гваяциловых единиц. При окислении древесины березы пушистой нитробензолом был
получен ванилин и сиреневый альдегид в отношении 1:3,5, что тоже соответствует
12% неконденсированных гваяциловых и 43% сирингиловых структурных единиц.
Исследование ИК-спектров ЛМР березы указало на присутствие в нем
несопряженных сложноэфирных связей, которые не были обнаружены в лигнине ели. В
продуктах окисления нитробензолом проэкстрагированной древесины березы
хромотографическим анализом установлено присутствие гомованилиновой (XIII, а) и гомосиреневой (XIII, б) кислот.

Рис. 6.4. Гомованилиновая (XIII,
а) и гомосиреневая (XIII, б) кислоты
Сопоставление элементарного состава образцов ЛМР березы, выделенных
Клемола и Бьеркманом, и образцов ЛМР ели указывает на различия в структуре
боковых цепей лигнина березы и ели. Эти авторы получили примерно 9 атомов Н на
единицу С9 в ЛМР березы, в то время как в ЛМР хвойных количество
атомов Н на С9 меньше. Большое количество атомов Н на С9
березы может обусловливаться менее конденсированной структурой его боковых
цепей.
Следует отметить, что лигнины лиственных пород древесины значительно
больше отличаются друг от друга, чем лигнины хвойных. Различия имеются также в
составе и в содержании функциональных групп. Формулы элементарного звена
лигнинов вычислены на основе данных элементарного анализа и содержания
метоксилов.

Рис. 6.5. Состав элементарного звена макромолекул лиственных лигнинов
Соотношение структур I : II : III для лигнина бука 49:46:5.
Кроме того, Пирл установил, что древесина осины (Populus tremula) при варке по сульфитному методу или при кипячении с
разбавленной щелочью дает значительные количества n-оксибензойной кислоты. Исследования показали, что n-оксибензойную кислоту образуют
только лигнины семейства Salicaceae, что указывает на различия между лигнинами этого семейства и лигнинами
других лиственных пород.
Исследование биосинтеза лигнинов древесины лиственных пород и их строения
показывает, что они являются более сложными, чем лигнины хвойных.
До сих пор не установлено, является этот лигнин смесью полимеров
сирингиловых и гваяциловых единиц или сложным полимеров этих единиц.
Все лигнины, даже в пределах одной клеточной стенки, гетерогенны. У
травянистых растений доли сирингильных и гидроксифенильных единиц колеблются в
широких пределах.
В отличие от полисахаридов в лигнине отсутствует единый вид связи между
структурными единицами. Для него характерно многообразие связей, в результате
чего он имеет макромолекулы с высокой степенью разветвленности. Предполагают,
что лигнин в древесине является пространственным полимером, т.е. имеет сетчатую
структуру.
6.3 Димерные структуры лигнина
Образование лигнина из n-оксикоричных
спиртов происходит по радикальному механизму. Из образующегося первоначального
радикала ароксила и его мезомерных форм образуются в первую очередь димеры, или
дилигнолы (по Фрейденбергу). В состав лигнина хвойной древесины входят
следующие димерные структуры: гваяцилглицерин--конифериловый эфир (XIV) (арилглицерин--арилэфирная структура) - около 30%,
дегидродиконифериловый спирт XV или
альдегид (XVI) (фенилкумарановые структуры) - 18-20%,
D, L-пинорезинол XVII
(пинорезинольная структура) - около 20%, дегидродиванилин XVIII (дифенильная структура) - около 25%.
Нимц выделил из лигнина 1,2-диарилпропандиол XIX, что указывает на присутствие дилигнолов с
отщепленной пропановой цепочкой.

Рис. 6.6. Основные димерные структуры лигнинов (XIV-XVI)
Фрейденберг ввел в в схему фрагмента макромолекулы лигнина ели структуры XIX (звенья 13b - 14b) и
циклолигнанолида (XX) (звенья 13a - 14a).

Рис. 6.7. Основные димерные структуры
лигнинов (XVII-XIX)
Ранее он описал структуру циклолигнана XXI. Структура циклолигнанолида ответственна за
образование бензолпентакарбоновой кислоты при окислении лигнина перманганатом
калия.
Количество арилглицерин-арилэфирных структур типа (XIV) было рассчитано Адлером на
основании выходов продуктов ацидолиза лигнина и соответствующих модельных
соединений. По его данным, по меньшей мере 25% структурных единиц лигнина
связаны с соседними структурными единицами за счет b-алкиларильной эфирной связи. В ЛМР из ели содержится на
фенилпропановое звено С9 структур типа (XXII, a) до
0,02 моля, структур типа (XXII,
б) - 0.05-0.07 моля.

Рис. 6.8. Основные димерные структуры лигнинов (XX-XXII)
Адлер показал, что фенилкумарановые структуры при ацидолизе переходят в
фенилкумароновые. Дегидродиконифериловый спирт образует кумарон (XXIII) с выходом 75%. На основании
спектрального исследования Адлер считает, что 20 фенилпропановых звеньев из 100
участвуют в образовании фенилкумарановых структур лигнина.
Присутствие структур пинорезинольного типа было показано Фрейденбергом,
который выделил этот димер из промежуточных продуктов при биосинтезе лигнина и
в небольших количествах из нативного елового лигнина. Содержание
пинорезинольных структур в лигнине составляет примерно 10%.
Наличие в лигнине дифенильных структур показано Пью, который обнаружил
дегидродиванилин в продуктах нитробензольного окисления. На основании
спектральных данных Пью пришел к заклчению, что до 25% фенилпропановых единиц
лигнина связаны в дифенильные структуры типа дегидродиванилина.
В лигнине установлено наличие арилалкиловых (бензилариловых) эфиров,
которые могут перегруппировываться в структуры с C-C связью между -углеродным атомом одного
фенилпропанового звена и пятым положением бензольного кольца другого (Рис.
6.9.).

Рис. 6.9. Перегруппировка арилалкиэфирных
структур в структуру с С-С связью
Бензиларилэфирные структуры в значительной степени несут ответственность
за исключительную лабильность природного лигнина и его склонность к конденсационным
превращениям.
В лигнине лиственных возможно наличие всех основных типов структур,
подобных структурам хвойного лигнина. Например, присутствие в лиственном
лигнине структур пинорезинольного типа подтверждается выделением
сирингарезинола (XXIV).
Нимц провел мягки перколяционный гидролиз древесины бука, причем 40%
древесины, в том числе и лигнин, перешли в раствор. В перколяте были обнаружены
мономерные, димерные и олигомерные фенолы - продукты расщепления лигнина. Среди
них присутствовал сирингарезинол, но главным продуктом был
диметилпирогаллоглицерин XXV,
три 1,2-диарилпропандиола (XXVI),
что говорит о присутствии в буковом лигнине диарилпропановых структур.

Рис. 6.10. Основные димерные структуры лигнинов (XXIII-XXVI)
7. РАСТВОРИТЕЛИ ЦЕЛЛЮЛОЗЫ, ИХ ЗНАЧЕНИЕ
Целлюлоза растворима в сравнительно ограниченном числе растворителей:
. В щелочи:
При действии растворов щелочей на целлюлозу может происходить не только
ограниченное, но частично и неограниченное набухание, т.е. растворение
целлюлозы в щелочи.
Растворимость целлюлозы в щелочи зависит от степени полимеризации
целлюлозы. С уменьшением степени полимеризации растворимость в щелочи
повышается. На растворимость целлюлозы оказывает влияние также характер
целлюлозного материала, его доступность (плотность упаковки и морфологическая
структура).
Максимальная растворимость хлопковой целлюлозы при обычной температуре
наблюдается при концентрации щелочи около 12%, древесной целлюлозы - около 10%.

Рис. 7.1 Растворимость в водных растворах NaOH раличных целлюлоз: 1 - хлопковой; 2 - перебеленной
хлопковой; 3 - сульфитной; 4 - регенерированной
С понижением температуры растворимость целлюлозы в щелочи повышается и
требуется меньшая ее концентрация. Понижая температуру щелочного раствора,
можно осуществить фракционирование целлюлозы. Особенно повышается растворимость
целлюлозы после ее замораживания в щелочи.
Растворимость целлюлозы зависит от природы щелочи. Растворяющая
способность LiOH несколько выше, чем NaOH, КОН, наоборот, ниже. Значительно
повышают растворимость целлюлозы в щелочи некоторые добавки, например цинкат
натрия, гидроокись бериллия.
Растворение целлюлозы в щелочи применяют для производства искусственного
волокна.
При характеристике качества древесной целлюлозы, в частности целлюлозы
для производства искусственного волокна, определяют устойчивость целлюлозы к
растворяющему действию щелочей. Процесс мерсиризации при производстве
вискозного волокна сопровождается растворением низкомолекулярных фракций
целлюлозы и сопутствующих ей гемицеллюлоз. В зависимости от содержания этих
компонентов в технической целлюлозе меняется выход вискозного волокна.
Показателем, определяющим пригодность целлюлозы для производства
вискозного волокна, является содержание альфа-целлюлозы.
Альфа-целлюлозой условно называют часть целлюлозы, не растворимую
17,5%-ной NaOH при 20ºС. Она не является индивидуальным химическим
соединением. Это чисто техническое понятие, позволяющее судить о пригодности
целлюлозы для тех или иных промышленных целей, характеризующее степень
деструкции (т.е. степень разрушения) технической целлюлозы. Считают, что в
17,5%-ной щелочи не растворяются молекулы целлюлозы большой длины, наиболее
длинные молекулы маннана и ксилана, совместно ориентированные с целлюлозой
гемицеллюлозы и некоторая часть остаточного лигнина.
В раствор переходит низкомолекулярная фракция целлюлозы, а также
гемицеллюлозы (с СП ниже 200).
Фракцию. Переходящую в щелочной раствор, но способную высаживаться при
подкислении уксусной кислотой, называют бета-целлюлозой, а фракцию, остающуюся
в растворе - гамма-целлюлозой.
Бета-целлюлоза представляет собой главным образом низкомолекулярную
разрушенную целлюлозу. В древесине она, по видимому, не содержится, а
образуется во время варки и отбелки. Во фракции бета-целлюлозы также содержатся
и полисахариды неглюкозного характера. Ее содержание качество волокна.
Гамма-целлюлоза - это низкомолекулярная фракция гемицеллюлоз с примесью
продуктов распада целлюлозы.
. Водных смесях комплексных соединений гидроксидов переходных
металлов (Сu, Cd, Ni) с NH3 и аминами;
а) Реакция с реактивом Швейцера:
В процессе протекания реакции идет образование комплексного соединения в
результате частичного вытеснения целлюлозой молекул NH3 из координационной сферы меди по схеме:

или

Растворы
целлюлозы в медноаммиачном реактиве (куоксаме) имеют большое практическое
значение. Их применяют для определения вязкости технических целлюлоз, а также
для определения СП. При этом следует иметь в виду, что растворы целлюлозы в
медноаммиачном реактиве очень чувствительны к окислению кислородом воздуха и
поэтому необходимы меры предосторожности для защиты от окисления.
В
технике растворение целлюлозы в медноаммиачном растворе применяют в
производстве одного из видов искусственного волокна - медноаммиачного (примерно
с 1900г.).
Медноаммиачные
растворы целлюлозы используют для формования гидратцеллюлозных волокон и
пленок.
б)
Кроме медноаммиачного раствора целлюлозу растворяет ряд других комплекных
оснований. Из них наибольшее применение получил раствор гидрата окиси меди в
этилендиамине - куприэтилендиамин (кущксен) [Cu(NH2CH2CH2NH2)2](OH)2,
обозначаемый сокращенно [Cu(en)2](OH)2
или Cuen.
Растворы
целлюлозы в куприэтилендиамине широко применяют для определения вязкости
целлюлозы и СП. Эти растворы более устойчивы к кислороду воздуха, чем
медноаммиачные.
в)
Дают прозрачные растворы и мало деструктируют целлюлозу такие растворители, как
цинкоксен (гидроокись цинкэтилендиамина) [Zn(en)3](OH)2,
кадоксен [Cd(en)3](OH)2,
ниоксен [Ni(en)3](OH)2,
ниоксам [Ni(NH3)6](OH)2,
кооксен [Со(en)3](OH)2,
а также щелочной раствор железовиннонатриевого комплекса (ЖВНК) [(C4H3O6)3Fe]Na6.
Из
них наибольшее применение для определения СП целлюлозы получили кадоксен и
ЖВНК. Раствор целлюлозы в кадоксене устойчив к окислению кислородом воздуха и
бесцветен, что позволяет использовать его при исследовании растворов целлюлозы
оптическими методами. ЖВНК, ро Джайме, имеет следующее строение:

3. Концентрированных солевых растворах
Кроме комплексных оснований целлюлоза способна набухать и в определенных
условиях растворяться в концентрированных растворах некоторых солей.
а) хлористого цинка
Обработку концентрированным раствором хлористого цинка применяют для
получения фибры.
б) целлюлоза способна также постепенно набухать в горячих
концентрированных растворах AlCl3, SnCl4, KI, BaI2, Ca(CNS)2 и др. При определенных
для каждой соли условиях (концентрации и температуре) целлюлоза не только
набухает, но и переходит в раствор. Растворимость целлюлозы зависит также и от
ее свойств. Установлено, что все легкорастворимые соли, обладающие склонностью
к гидратации, способны более или менее растворять целлюлозу.
Целлюлоза легко растворяется при температуре 120-130ºС в растворе роданистого кальция Ca(CNS)2, насыщенном при комнатной температуре.
. Некоторых минеральных кислотах (H2SO4, Н3РО4);
а) в серной кислоте H2SO4:
При обработке на холоду концентрированной серной кислотой целлюлоза
растворяется в ней, образуя вязкий раствор. Если этот раствор вылить в избыток
воды, выделяется белый хлопьевидный продукт, так называемый амилоид,
представляющий собой частично гидролизованную целлюлозу. Он сходен с крахмалом
по реакции с йодом (синее окрашивание; целлюлоза не дает этой реакции). Если не
проклеенную бумагу опустить на короткое время в концентрированную серную
кислоту и затем сейчас же промыть, то образующийся амилоид склеивает волокна
бумаги, делая ее более плотной и прочной. Так изготавливается пергаментная
бумага.
При продолжительном действии на целлюлозу концентрированных растворов
минеральных кислот она при нагревании подвергается гидролизу, конечным
продуктом которого является глюкоза.
б) в ортофосфорной кислоте Н3РО4:
Весьма перспективной растворяющей системой для целлюлозы и ее смесей с
другими полимерами является водный раствор ортофосфорной кислоты.
Концентрированная ортофосфорная кислота - прямой растворитель целлюлозы, она
пригодна для получения прядильных растворов и отвечает основным требованиям,
предъявляемым в промышленности к растворителям целлюлозы. Кроме того она
растворяет многие ВМС различной химической природы, что дает возможность
получать композиционные материалы, придавая целлюлозе дополнительные полезные
свойства.
Учитывая экологичность и простоту приготовления водных растворов УБГУ
«Научно исследовательским институтом физико-химических проблем» было проведено
систематическое исследование процесса растворения в них как самой целлюлозы,
так и ее смесей с хитозаном, полиакрилонитрилом, ацетатом целлюлозы,
поливиниловым спиртом, поликапроамидом, меланином,
полипарафенилен-1,3,4-оксидиазолом и некоторыми дргими полимерами. Для
растворения целлюлозы необходимо использовать высококонцентрированные растворы
ортофосфорной кислоты в температурном интервале от 60 до -10ºС. Волокна, сформированные в
стандартные водно-спиртовые осадительные ванны из растворов в ортофосфорной
кислоте обладают хорошими физико-механическими характеристиками (прочность 15 -
25 сН/текс при удлинении 8 - 20%) при высоком модуле упругости в сухом
состоянии (500 - 9100 сН/текс).
Процесс получения гидратцеллюлозных нитей из растворов целлюлозы в
ортофосфорной кислоте включает значительно меньшее количество технологических
стадий. В будущем процессе отсутствуют кислотный и щелочной стоки, а также
шламонакопители.
Новый процесс представляет собой замкнутый производственный цикл, что
позволяет практически полностью ликвидировать стоки, загрязняющие водоемы.

Рис. 7.2. Схема замкнутого процесса получения гидратцеллюлозных волокон
из растворов целлюлозы в ортофосфорной кислоте
При производстве вискозных нитей тратится огромное количество воды.
Процесс получения гидратцеллюлозных нитей из растворов целлюлозы в
ортофосфорной позволит сократить водопотребление в 25 раз.
8. ГИДРОЛИТИЧЕСКАЯ ДЕСТРУКЦИЯ ЦЕЛЛЮЛОЗЫ.
КАТАЛИЗАТОРЫ И СХЕМЫ ГИДРОЛИЗА
Для целлюлозы характерны все особенности химического поведения,
свойственный ВМС.
Для целлюлозы, как для высокомолекулярного соединения можно выделить
четыре основных типа реакций:
. Деструкции;
. Функциональных групп;
. Сшивания цепей;
. Внутримолекулярных перегруппировок.
Рассмотрим реакции деструкции.
Для целлюлозы характерны реакции деструкции. При реакциях деструкции
происходит разрыв глюкозидных связей в цепных макромолекулах целлюлозы с
понижением ее степени полимеризации, а в некоторых случаях и разрыв углеродных
связей. Деструкция может протекать под физическим воздействием (физическая
деструкция) и под действием химических реагентов (химическая деструкция).
Основными типами деструкции являются:
. Гидролиз целлюлозы под действием гидролизующих агентов;
. Щелочная деструкция;
. Термическая деструкция;
. Фотохимическая деструкция;
. Биохимическая деструкция;
. Механохимическая деструкция.
.1 Гидролитическая деструкция целлюлозы
При гидролитической деструкции целлюлозы в результате воздействия
гидролизующих агентов - водных растворов кислот происходит разрыв глюкозидных
связей между звеньями макромолекул с присоединением по месту разрыва
водородного и гидроксильного ионов воды. В результате такого процесса
происходит понижение степени полимеризации целлюлозы по схеме 8.1.:

Схема 8.1. Гидролитическая деструкция целлюлозы
В месте разрыва глюкозидной связи 1-4 у одного звена образуется свободный
глюкозидный гидроксил, т.е. альдегидная группа (у 1-го атома С), а у другого
звена - спиртовой гидроксил (у 4-го атома С). В качестве конечного продукта
глубокой деструкции целлюлозы (как и других полисахаридов) образуются простые
сахара.
Кинетику гидролитической деструкции можно проследить по увеличению
редуцирующей способности реакционной смеси или по уменьшению средней СП
целлюлозы.
8.2 Катализаторы и схемы гидролиза
Гидролиз целлюлозы и других полисахаридов водой протекает медленно,
поэтому для ускорения реакции применяют катализаторы - кислоты. Реакция
гидролиза катализируется ионом водорода, который в водной среде существует в
виде неустойчивого иона оксониевого соединения Н3О+ -
иона гидроксония.
Чем больше в растворе содержится ионов водорода, тем быстрее идет
гидролиз. Поэтому в качестве катализаторов гидролиза применяют сильные
минеральные кислоты с большой степенью диссоциации. Скорость реакции гидролиза
растет пропорционально активности кислоты.
Скорость реакции гидролиза целлюлозы резко увеличивается с повышением
температуры. Варьируя концентрацию кислоты и температуру, можно обеспечить
различные скорости реакции и выход сахара.
Реакцию гидролиза целлюлозы можно в общем виде представить уравнением:
(С6Н10О5)n + (n - 1)H2O = nC6H12O6
При гидролитической деструкции целлюлозы происходит расщепление
ацетальных ( - глюкозидных) связей под действием
кислот по схеме 8.1.
Гидролиз целлюлозы до глюкозы разбавленной кислотой происходит
постепенно, через ряд промежуточных продуктов с уменьшающейся степенью
полимеризации: целлюлоза → гидроцеллюлоза → целлодекстрины →
олигосахариды → глюкоза.
Гидролитическая деструкция значительно изменяет свойства целлюлозного
материала: снижается прочность, увеличивается гигроскопичность, содержание
альдегидных групп и растворимость в щелочи. Продукт частичного поверхностного
гидролиза целлюлозы разбавленной кислотой, представляющий смесь малоизмененной
целлюлозы с нерастворимыми в воде продуктами ее частичного гидролиза называется
гидроцеллюлозой (термин был введен Жераром в 1875г.).
Реакция гидролиза целлюлозы разбавленными кислотами имеет гетерогенный
характер и проходит на границе двух фаз. Главная особенность этого процесса -
очень низкая (в десятки раз меньше) скорость реакции по сравнению со скоростью
гомогенного процесса.
Скорость гидролиза целлюлозы непостоянна, степень полимеризации СП
целлюлозы вначале резко падает, а затем почти не изменятся. В результате
быстрого снижения СП исходной целлюлозы вначале гидролиза достигается
постоянная или предельная СП. Величина предельной СП зависит от вида целлюлозы:
для хлопковой целлюлозы около 200, древесной - 150 и регенерированной - 35-40.
Для начального периода гетерогенного гидролиза характерна также
повышенная скорость образования небольшого количества глюкозы.
Исходя из надмолекулярного строения целлюлозы и наличия в ней аморфных -
рыхло упакованных и кристаллических - рыхло упакованных областей, можно
предположить, что катализатор легко проникает в аморфные области, в которых
воздействует на все глюкозидные связи макромолекул, а в кристаллических -
атакует только поверхностные глюкозидные связи.
Реакция гидролиза целлюлозы сопровождается разрушением образовавшейся
глюкозы, которая в результате дегидратации превращается в оксиметилфурфурол и
далее в гуминовые вещества левулиновую и муравьиную кислоты по схеме 8.2.:

Схема 8.2. Разрушение глюкозы при гидролизе
Скорость реакции распада моносахаридов при гидролизе определяется
температурой, концентрацией катализатора и временем (как и реакция их
образования). В зависимости от условий гидролиза может преобладать или реакция
образования моносахаридов, или реакция их распада. При повышении температуры
скорость реакции образования сахара несколько больше скорости его распада, что
способствует увеличению выхода сахара при высоких температурах. Режимы
гидролиза растительного сырья с целью его осахаривания выбирают с таким
расчетом, чтобы распад сахара был наименьшим.
В отличие от гидролиза разбавленными кислотами гидролиз
концентрированными кислотами идет в гомогенной среде, так как целлюлоза
набухает и растворяется в концентрированных кислотах.
Гидролиз полисахаридов концентрированными кислотами идет в условиях
острого недостатка воды, которая связывается как образующимися продуктами
гидролитической деструкции, так и кислотами.
Недостаток воды приводит к тому, что основными продуктами гидролиза
являются не моносахариды, а олигосахариды и декстрины, образующиеся частично в
результате реверсии (реакции противоположной гидролизу). Продукты неполного
гидролиза целлюлозы и продукты реверсии легко гидролизуются разбавленными
кислотами. Для перевода продуктов неполного гидролиза в моносахариды сахарные
растворы, полученные при гидролизе целлюлозы концентрированными кислотами,
разбавляют водой и нагревают, этот процесс называют инверсией.
Гидролиз концентрированными кислотами перед гидролизом разбавленными
кислотами имеет следующие технологические преимущества:
- возможность проведения процесса при температуре 40-60ºС;
- высокая скорость реакции гидролиза в гомогенной среде;
- возможность получения сахаров в кристаллическом виде.
Однако его осуществление связано с рядом технологических трудностей,
вследствие чего главным промышленным методом гидролиза является гидролиз
разбавленной серной кислотой.
9. ГИДРОЛИТИЧЕСКАЯ ДЕСТРУКЦИЯ ЛИГНИНА,
АЦИДОЛИЗ И ЭТАНОЛИЗ ЛИГНИНА
С теоретической и практической точек зрения наибольшее значение имеют
реакции гидролитической деструкции лигнина, происходящие:
- под действием водных растворов кислот и оснований (гидролиз);
- под действием органических растворителей в присутствии
кислотных и щелочных катализаторов, например этанолиз, деструкции под действием
водно-диоксановой смеси (ацидолиз);
- с одновременным гидрированием (гидрогенолиз).
.1 Гидролитическая деструкция под действием водных растворов кислот и
оснований (гидролиз)
Реакция гидролиза лигнина в древесине может идти даже под действием воды
при повышенных температурах за счет катализирующего влияния органических
кислот, отщепляющихся от гемицеллюлозных компонентов древесины. Более сильное
гидролизующее действие оказывают минеральные кислоты. Гидролитическая
деструкция идет по простым эфирным связям лигнина. Причем простая эфирная связь
в α-положении расщепляется значительно
легче α-эфирной связи. При расщеплении этих
связей могут образовываться активные группировки n-оксибензилового спирта.
С увеличением температуры и кислотности среды реакции конденсации
усиливаются и в определенных условиях становятся преобладающими. Поэтому при
гидролизе древесины концентрированными кислотами в растворимо состояние
переходят лишь углеводы, а лигнин получается в виде негидролизуемого остатка,
так как реакции конденсации доминируют и полностью перекрывают эффект
гидролитического расщепления. Однако выделенный из древесины кислотным
гидролизом лигнин лишь по количеству соответствует исходному природному
лигнину, а химическое строении и свойства такого лигнина оказываются
значительно измененными.

Рис. 9.1. Деструкция α-эфирной связи

Рис. 9.2. Деструкция α-эфирной связи
Подобное явление существования двух конкурирующих процессов наблюдается и
при гидролитической деструкции лигнина водными растворами оснований.
Эти особенности поведения лигнина в кислой и щелочной средах проявляются
и в процессах делегнификации древесины. Для их успешного проведения необходима
защита активных группировок лигнина от конденсации. Следует заметить, что при
действии некоторых органических растворителей, например этанола, в присутствии
кислотных и щелочных катализаторов реакции конденсации подавляются в результате
блокирования реакционоспособных бензильных гидроксилов.
8.3
Ацидолиз и этанолиз лигнина
При нагревании древесины с органическими растворителями диоксан, спирты и
т.д.) в присутствии минеральных кислот значительная часть лигнина переходит в
раствор , из которого его можно высадить разбавлением водой. Превращение
лигнина в растворимое состояние происходит вследствие расщепления простых
эфирных связей. Часть лигнина при этом превращается в мономерные продукты.
Полному превращению лигнина в мономеры препятствует углерод-углеродные связи.
Некоторая часть лигнина подвергается конденсации и не переходит в раствор.
Наиболее изучены реакции деструкции лигнина, протекающие при нагревании с
водным диоксаном в присутствии HCl
(ацидолиз) и с этиловым спиртом в присутствии HCl (этанолиз).
Реакцию этанолиза для изучения строения лигнина впервые применил Гибберт,
а реакцию ацидолиза - Адлер, который установил также механизм этих реакций.
Механизм расщепления простой эфирной связи в основной структуре лигнина -
структуре α-арилового эфира арилглицерина Адлер представляет
следующим образом:
Мономерные продукты ацидолиза и этанолиза называют кетонами Гибберта. Из
лиственных лигнинов наряду с гваяцильными производными образуются сирингильные
производные. Таким образом, с помощью ацидолиза доказано наличие в лигнине
простой эфирной связи. Расщепление этих связей приводит к образованию новых
свободных фенольных гидроксилов и в эквивалентных количествах метильных групп в
γ-положении и карбонильных групп в α- и β-положениях. При этанолизе в лигнин
вводятся этоксильные группы, которые защищают образующиеся при деструкции
группировки бензилового спирта от дальнейшей конденсации.
Следует заметить, что при ацидолизе более легко расщепляются простые
эфирные связи в структурах лигнина со свободным фенольным гидроксилом.

Рис. 9.3. Механизм расщепления простой эфирной связи в основной структуре
лигнина - структуре α -арилового эфира арилглицерина
10.
ХИМИЧЕСКИЕ РЕАКЦИИ ЛИГНИНА ПРИ СУЛЬФИТНОЙ ВАРКЕ. АКТИВНЫЕ ГРУППЫ ЛИГНИНА
При делигнификации древесины в процессе сульфитной варки с лигнином
одновременно протекают три основные реакции:
. Сульфирование;
. Гидролитическая деструкция;
. Конденсация.
По мнению большинства исследователей, эти реакции являются
гетеролитическими, протекающими под воздействием нуклеофильных реагентов,
атакующих боковую цепь.
Сульфирование и гидролитическая деструкция способствуют растворению
лигнина. Для Гидролитической деструкции лигнина, а также для расщепления его
связей с углеводами необходимо создание кислой среды. Однако кислая среда
оказывает и противоположное действие, приводя к конденсации лигнина, которая
препятствует его растворению и снижает реакционную способность по отношению к
сульфированию.
Реакции конденсации усиливаются с повышением температуры и кислотности
среды. Кроме химических реакций конденсации под влиянием кислой среды в лигнине
могут наблюдаться и физико-химические (коллоидно-химические) изменения, также
приводящие к снижению растворимости и реакционной способности.
Все процессы, препятствующие растворению лигнина и снижающие его
реакционную способность, называются инактивацией лигнина.
Если инактивация лигнина происходит в очень сильной степени. То лигнин не
сульфируется и не растворяется (т.н. «черная варка»). Инактивация лигнина под
действием кислой среды и повышенной температуры особенно интенсивно идет в тот
период, когда в лигнине еще отсутствуют сульфогруппы. Введение в лигнин даже
небольшого количества сульфогрупп (сульфирование) повышает его устойчивость к
влиянию кислой среды.
Таким образом сульфирование и инактивация являются конкурирующими
процессами. Результаты варки будут зависеть от того, какой из этих процессов
преобладает.
.1 Сульфирование лигнина
При нагревании древесины с растворами бисульфита и свободной сернистой
кислоты в лигнин вводятся сульфогруппы SO3H.
Этот процесс называется сульфированием.
При сульфитной варке реагентом в реакции сульфирования является сернистая
кислота H2SO3, для которой считают возможным существование двух структур:

Средние соли (сульфиты) соответствуют первой форме, кислые соли
(бисульфиты), вероятнее, второй.
В водном растворе SO2 одновременно имеют место следующие
равновесия:

При сульфитной варке, т.е. в кислой среде сернистая кислота ведет себя
как одноосновная.
Ион бисульфита, являющийся нуклеофильным реагентом, может существовать в
обеих таутомерных формах:

В растворе бисульфита существует равновесие:

Слабая сернистая кислота диссоциирует незначительно, поэтому концентрация
ионов бисульфита определяется концентрацией бисульфита NaHSO3, Ca(HSO3)2 и т.д., т.е. содержанием основания.
Сульфирование лигнина начинается в твердой фазе (предположение Хегглунда)
с образованием твердой лигносульфоновой кислоты. Во второй стадии варки в
результате гидролитической деструкции лигносульфоновая кислота превращается в
растворимую форму и переходит в раствор в виде солей - лигносульфонатов.
Окончательно механизм реакции сульфирования лигнина еще не установлен.
По мнению Холмберга, в реакции сульфирования принимают участие группировки
бензилового спирта и его эфира. При этом сульфогруппа встает на место
бензильного спиртового гидроксила:

По современным мировоззрениям, наиболее реакционоспособным к воздействию
нуклеофильных реагентов, в частности ионов HSO3-, является α-положение.
Процесс сульфирования лигнина в древесине сначала протекает довольно
быстро, затем замедляется, причем в тем большей степени, чем выше рН варочного
раствора.
На основании изучения кинетики сульфирования в кислой и нейтральной
средах сульфирующие группы были разделены на два типа (А и В), различающиеся по
реакционной способности (Хегглунд и Эрдтман).
Группы А сульфируются при любом рН: при их сульфировании вводится около
0,3 атома S на одну фенилпропановую единицу. В
кислой среде (при рН меньше 3,5) группы А легче реагируют с фенолами, чем
сульфируются, и легко конденсируются с фенольными экстрактивными веществами
древесины, что делает невозможным проведение кислой сульфитной варки древесины
с высоким содержанием смолистых веществ.
Группы В сульфируются в сильнокислой среде: при рН больше 3,5 с
сульфитным варочным раствором не реагируют или реагируют очень медленно. При их
сульфировании вводится около 0,7 атома S на фенилпропановую единицу.
Дальнейшие исследования показали, что часть групп А в нейтральной и
слабощелочных средах сульфируется быстро, а другая часть - медленно. В
соответствии с эти группы А разделены на X и Z,
которые содержатся примерно в равных количествах (по 0,15 группы).
Группы Х в основном представляют собой группировки бензилового спирта
(или его эфира) со свободным гидроксилом в n-положении.
Группы Z - это группировки бензилового спирта
с этерифицированным фенольным гидроксилом.

Реакция сульфирования группировок бензилового спирта (или его эфира) в
группах Х и Z в кислой среде идет с образованием
промежуточного иона карбония

В нейтральной и щелочной средах (моносульфитная варка) группировки n-оксибензилового спирта (или его эфира)
в группах Х образуют промежуточный хинонметид

В нейтральной и слабощелочной средах быстро сульфируются, т.е. должны
быть отнесены к группам Х. и другие структуры, в том числе структуры,
содержащие реакционоспособный гидроксил в γ-положении

В нейтральной среде могут сульфироваться и группировки кониферилового
альдегида по двойным связям и по альдегидным группам

Двойные связи могут образоваться в процессе варки в результате реакции
элиминирования, например путем отщепления воды.
Группа В представляет собой группировку бензилового эфира с замещенным
фенольным гидроксилом и имеет заместителя в -положении. В результате кислотного
гидролиза группа В переходит в группу В’.

Группа В’ отличается от группы Z строением боковой цепи - наличием заместителя в -положении. Она может сульфироваться
в нейтральном растворе, но из-за наличия заместителя в -положении группа В’ менее
реакционоспособна, чем группа Z.
Реакция сульфирования группы B’
идет подобно реакции сульфирования группы Z, т.е. с образованием промежуточного иона карбония
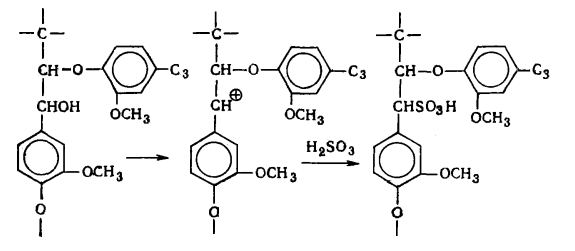
Кроме того, к группам В принадлежат другие структуры с этерифицированными
фенольными гидроксилами, например фенилкумарановые структуры и структуры
пинорезинола (аналогичные структуры со свободными фенольными гидроксилами
являются группами Х):

Таким образом, по реакционной способности сульфирующие группы лигнина
можно расположить в следующий ряд: Х˃Z˃B’˃B.
Эти представления о природе сульфирующихся групп лигнина подтверждены
многими экспериментами, они объясняют основные явления, наблюдающиеся при
сульфитной варке.
10.2 Гидролитическая деструкция лигнина
В первой стадии сульфирования образуется твердая лигносульфоновая
кислота, содержащая около 0,3 сульфогруппы на одну фенилпропановую единицу. При
ее образовании расщепляется очень небольшое число связей между мономерными
единицами, и поэтому твердая лигносульфоновая кислота остается в виде агрегата
с поперечными связями, нерастворимого в варочной жидкости. При варке эта
кислота находится в древесине в виде соли. При сульфитной варке происходит
гидролитическая деструкция связей лигнина с углеводами, а также связей
нециклического бензилового простого эфира и простых эфирных связей в α-положении. При этом макромолекулы
лигнина распадаются на фрагменты, растворимые в варочной жидкости. Возможно,
что деструкция лигнина сопровождается и разрывом некоторых углерод-углеродных
связей.
Какой из процессов - сульфирование или гидролиз - определяет скорость
делигнификации, зависит от условий варки. При низкой кислотности варочного
раствора скорость делигнификации определяется скоростью гидролитической
деструкции, а при высокой кислотности скорость делигнификации может быть
обусловлена и скоростью сульфирования.
При сульфитной варке в производственных условиях кинетика процесса
усложняется частичным расщеплением варочных реагентов и побочными реакциями их
с другими компонентами древесины.
Во время делигнификации в варочный раствор в первую очередь переходят
макромолекулы лигнина с меньшей молекулярной массой, так как они быстрее
диффундируют в раствор, тем более крупные фрагменты. По мере протекания
процесса делигнификации средняя молекулярная масса растворенных
лигносульфонатов возрастает за счет постепенного перехода более крупных
фрагментов в варочный раствор, чему способствует увеличение диаметра
капиллярных пространств в древесины после постепенного удаления гемицеллюлоз.
Максимальная средняя молекулярная масса достигается при удалении из древесины
около 90% лигнина. Растворенные лигносульфонаты подвергаются дальнейшему
гидролизу. При этом средняя молекулярная масса снижается и достигает минимума
после разрыва всех способных к гидролизу связей.
В течение всего процесса делигнификации наряду с реакциями
гидролитической деструкции протекают реакции конденсации. Поэтому после
достижения минимальной средней молекулярной массы становится заметным ее
увеличение. При длительном нагревании в кислой среде могут даже образовываться
водонерастворимые лигносульфонаты. В отработанном сульфитном щелоке наряду с
низкомолекулярными лигносульфонатами(М около 500) содержатся лигносульфонаты с
очень высокой молекулярной массой (800 000 и более). Высокомолекулярные
лигносульфонаты можно выделять из щелока высаливанием, осаждением аминами,
диализом или электродиализом.
10.3 Конденсация лигнина
В процессе сульфитной варки наряду с реакциями сульфирования и
гидролитической деструкции, способствующими переходу лигнина в раствор, идут
конкурирующие реакции конденсации, снижающие реакционную способность лигнина по
отношению к сернистой кислоте и ее солям и препятствующие ее растворению.
Следует заметить, что обработка древесины в кислой среде не прекращает
реакцию лигнина с сульфитным варочным раствором, а лишь замедляет ее. Для
растворения инактивированного лигнина необходимо введение в него большего
количества серы.
В мягких условиях кислотной обработки древесины химических изменений
лигнина может и не быть. В этом случае инактивация лигнина, по-видимому,
связана лишь с коллоидно-химическими изменениями. Частички лигнина под влиянием
кислоты собираются в более групные агрегаты (коалесценция), и поэтому он
становится менее доступным действию сульфитных варочных растворов. При
длительной сульфитной варке такой уплотненный лигнин пентизируется и переходит
в раствор.
В более жестких условиях кислотной обработки древесины решающую роль
играют химические изменения лигнина, обусловленные реакциями конденсации с
образованием поперечных углерод-углеродных связей.
В реакциях конденсации могут участвовать различные группировки в лигнине.
Наиболее активны группировки бензилового спирта и его эфира, особенно со свободным
фенольным гидроксилом в n-положении.
В кислой среде конденсация этих группировок происходит с образованием
промежуточного иона карбония, который реагирует с другой фенилпропановой
единицей в 6-м и 5-м положениях ее ароматического кольца
Реакция конденсации лигнина в кислой среде идет по механизму:
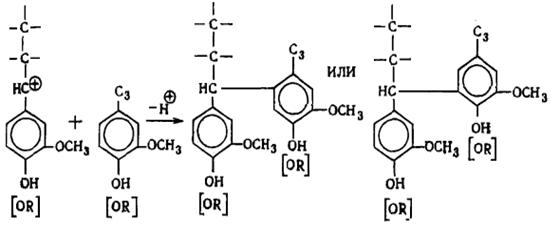
При образовании только одной поперченной связи между двумя молекулами
лигнина молекулярная масса удваивается (элементный состав практически не
изменяется), что резко снижает растворимость продукта конденсации.
Сульфогруппы блокируют активные гидроксилы бензилового спирта и тем самым
препятствуют протеканию реакций конденсации. В определенных условиях, например
при недостаточном сульфировании и высокой кислотности среды, реакции
конденсации могут совершенно затормозить растворение лигнина. Основание в
варочном растворе регулирует кислотность среды и концентрацию ионов бисульфита
и тем самым способствуют протеканию реакций сульфирования, а не конденсации.
Бисульфит кальция, имеющий более высокую степень диссоциации по сравнению
с сернистой кислотой снижая кислотность среды, одновременно увеличивает
концентрацию ионов бисульфита.
При подборе режима варки следует учитывать изменение кислотности
варочного раствора в процессе варки. Во время варки кислотность раствора
повышается за счет образования новых сильных кислот - лигносульфоновой и серной
(за счет окисления сернистой кислоты). Поэтому рН варочного раствора будет
зависеть от скорости образования этих кислот и концентрации основания. Когда
концентрация сильных кислот превышает концентрацию основания, кислотность
раствора начинает увеличиваться.
В процессе варки концентрация ионов бисульфита постепенно снижается за
счет расхода их на реакцию сульфирования лигнина и другие реакции. Поэтому при
отсутствии основания (или небольшой его концентрации), особенно при высокой
температуре, когда константа диссоциации сернистой кислоты становится очень
низкой, концентрация ионов бисульфита будет очень незначительна и сульфирование
замедляется, а высокая кислотность может вызвать инактивацию. Варка с раствором
SO2 без основания возможна лишь при высокой концентрации SO2 и низкой температуре. Высокая температура в этом
случае недопустима, так как уже в начале варки образуется сильная лигносулфоновая
кислота.
Такой механизм (предполагаемый) конденсации лигнина объясняет также
трудности, возникающие при сульфитной варке древесины смолистых пород
(например, сосны), у которых в ядровой части содержатся вещества фенольного
характера.
Группировки бензилового спирта могут конденсироваться с фенолами по схеме

Каждый фенол, имеющий не менее двух свободных положений (орто- или
пара-), способен связать две или более молекулы лигнина. Реакционная
способность наибольшая у многоатомных фенолов м-ряда, например у резорцина,
флороглюцина, так как в этом случае фенольные гидроксилы активируют одни и те
же положения бензольного кольца.
При рН 1,5 - 2 группы А значительно быстрее реагируют с фенолом, чем с
бисульфитом, и образуют нерастворимые продукты конденсации. Фенол
конденсируется с двумя А-группами, и в результате образования поперечных связей
значительно снижается растворимость лигносульфоновых кислот. Если же сульфитную
варку проводить в две ступени, сначала с нейтральным, а затем с кислым
сульфитным раствором, то делигнификация идет нормально, так как в нейтральном
растворе группы А быстрее реагируют с сульфитом, чем с фенолом.
Важную роль играет также реакция взаимодействия лигнина с продуктами
распада углеводов (с фурфуролом, оксиметилфурфуролом). Эти продукты могут
вступать в реакцию конденсации с лигнином, как с фенолом, и приводить к
увеличению молекулярной массы лигнина за счет сшивания его цепей.
11. ХИМИЯ СУЛЬФАТНОЙ ВАРКИ. ОСНОВНЫЕ ОТЛИЧИЯ ОТ НАТРОННОЙ ВАРКИ И
ХИМИЧЕСКИЕ РЕАКЦИИ ЛИГНИНА
Одним из основных способов щелочной варки является сульфатный способ.
.1 Химия сульфатной варки. Основные отличия от натронной варки
Сульфатный способ получил наиболее широкое применение как эффективный и
экономичный способ делигнификации древесины практически всех пород и в том
числе отходов лесопиления.
Натронный варочный процесс имеет ограниченное применение, в основном при
получении целлюлозы из древесины лиственных пород.
При щелочных способах варки древесину в виде щепы обрабатывают варочным
раствором (варочным щелоком) при температуре 170- 175ºС.
В случае натронной варки варочным реагентом является едкий натр NaOH.
При регенерации щелоков потерю NaOH. восполняют добавкой соды Na2CO3 с последующей каустизацией.
Na2CO3 + Ca(OH)2 →
2NaOH + CaCO3
При сульфатной варке (крафт-процесс) варочными реагентам являются едкий
натр NaOH и сульфид натрия Na2S.
Способ получил название сульфатного (а получающаяся целлюлоза -
сульфатной целлюлозой), так как при регенерации для восполнения потерь щелочи
добавляют сульфат натрия Na2SO4, который при сжигании упаренного отработанного щелока
под действием углерода органических веществ восстанавливается в сульфид натрия Na2S:
Na2SO4 + 2C → Na2S + 2CO2
С целью улучшения технологии сульфатного процесса, повышения выхода и
качества получаемой целлюлозы предложены различные модификации этого процесса:
варка с предгидролизом, варка с растворами сульфида и полисульфидов натрия,
ступенчатая варка и др. Большое внимание уделяется щелочным варочным процессам
с использованием восстановителей и окислителей.
Варочными реагентами при сульфатной варке являются едкий натр и сульфид
натрия. В варочном щелоке едкий натр, как сильный электролит, диссоциирует на
ионы натрия и гидроксила:
NaOH
<-> Na+ + OH-
Сернистый натрий диссоциирует на ионы натрия и сульфида
Na2S <-> 2Na+ + S2-
и частично гидролизуется по уравнениям:
Na2S + H2O <-> NaOH + NaSH;+ H2O
<-> NaOH + Na2S
Равновесие этих реакций зависит от рН варочных щелоков. При высоком рН
сернистый натрий гидролизуется практически только до гидросульфида. В процессе
варки щелочность раствора меняется и поэтому меняется и состояние равновесия. В
начале варки при высоком рН в варочном щелоке преобладают ионы сульфида,
которые постепенно с уменьшением рН превращаются в ионы гидросульфида.
В натронном варочном щелоке активными ионами являются ионы ОН-,
а в сульфатном щелоке - ионы OH-, S2- и SH-.
Принципиальной разницы в растворяющем действии на лигнин варочных щелоков
натронного и сульфатного нет. И в том и в другом случае лигнин переходит в
раствор под действием NaOH.
Однако сульфид натрия сульфатного варочного щелока оказывает специфическое
влияние на процесс варки. Скорость делигнификации при сульфатной варке выше,
чем при натронной. Выход и прочность технических сульфатных целлюлоз вследствие
разрушения гемицеллюлоз и самой целлюлозы выше, чем натронных.
Меньшее разрушение и растворение целлюлозы и гемицеллюлоз при сульфатной
варке объясняется более низкой щелочностью сульфатного щелока по сравнению с
натронным при одной и той же концентрации активной щелочи (NaOH + Na2S).
При расходования NaOH при варке
увеличивается степень гидролиза Na2S. Таким образом, Na2S играет как бы роль буфера, из которого образуются все новые
количества NaOH. Кроме того, сульфид натрия как
восстановитель предохраняет углеводы от щелочной деструкции - деполимеризации с
редуцирующего конца макромолекулы. Гидросульфид натрия NaHS, образующийся при гидролизе, также является активным
варочным компонентом, поэтому сульфатный щелок по сравнению с натронным
оказывает более сильное делигнифицирующее действие, что позволяет вести процесс
варки при более низких концентрации щелока и температуре.
Скорость делигнификации увеличивается с повышением температуры и
концентрации щелока. Это допустимо лишь до определенного предела, после
которого начинают сильно снижаться выход и прочность целлюлозы. Тогда
делигнификация становится менее избирательной и усиливается распад
полисахаридов, приводящий к потере выхода и прочности целлюлозы.
В отличие от сульфитного варочного раствора щелочные варочные растворы
почти одинаково проникают в щепу в продольном и поперечном направлениях, причем
скорость их проникновения значительно выше, чем варочной кислоты при сульфитном
способе варки. Этому способствует набухание клеточных стенок древесины под
действием щелочи.
Скорость растворения углевода и лигнина на различных стадиях варочного
процесса не одинакова. Растворение древесины начинается с излечения
гемицеллюлоз. В начале варки при низких температурах скорость химических
реакций невелика, и гемицеллюлозы извлекаются из древесины путем обычной
щелочной экстракции. Дальнейшее растворение гемицеллюлоз связано с химическими
реакциями их деструкции и расщепления связей с лигнином. При растворении
гемицеллюлоз клеточные стенки ослабляются и делаются более доступными для
воздействия варочных реагентов, что способствует удалению лигнина. В процессе
варки скорость удаления лигнина увеличивается. Делигнификация при сульфатной
варке, как и при сульфитной, начинается во вторичной стенке и заканчивается в
срединной пластинке. Лигнин срединной пластинки и лигнин, находящийся в углах
между клетками, начинает интенсивно растворяться только после удаления 50%
всего лигнина. После удаления примерно 90% лигнина скорость делигнификации
резко падает и начинает разрушаться и растворятся целлюлоза со скоростью,
увеличивающейся с уменьшением содержания остаточного лигнина. Продукты реакций
лигнина и углеводов со щелочью, перешедшие в раствор, под действием щелочи
разрушаются далее с образованием различных низкомолекулярных соединений.
При натронной варке в отличие от сульфатной делигнификация практически
прекращается после перехода в раствор половины лигнина, что обусловлено
преобладанием конденсационных процессов лигнина над его деструкцией.
Раствор после варки целлюлозы называют черным щелоком. В нем содержится
растворенный лигнин, называемый щелочным лигнином, и продукты разрушения
углеводов (различные оксикислоты и их лактоны), уксусная и муравьиная кислоты,
метиловый спирт, смолы, жиры и пр. Сложный состав веществ, растворенных в
черном щелоке, говорит о многообразии реакций, протекающих с разрушением всех
составных частей древесины при высокой температуре варки. Вследствие этого
изучение механизма взаимодействия древесины со щелочью при варке связано с
большими трудностями.
10.4 Химические реакции лигнина
Химические превращения лигнина при щелочных способах варки
рассматриваются с точки зрения существования двух конкурирующих между собой
процессов:
. Деградация лигнина, связанная с расщеплением связей в
макромолекуле лигнина и способствующая его растворении;
. Конденсация продуктов расщепления лигнина, сопровождающаяся
превращением их в более крупные фрагменты и тем самым препятствующая его
растворению.
При натронной варке процессы конденсации лигнина доминируют, так как
активные группы лигнина, участвующие в таких реакциях, оказываются
незащищенными. Вследствие этого натронная варка не применяется на практике для
делигнификации хвойной древесины.
Механизм взаимодействия NaOH с
лигнином, как при натронной, так и при сульфатной варке практически одинаков.
Наличие в сульфатном варочном щелоке ионов S2-и SH- усложняет химические реакции,
протекающие при варке с лигнином. Поэтому рассмотрим реакции лигнина с NaOH и Na2S.
Процесс растворения лигнина при щелочных способах варки рассматривается
как комплекс гетеролитических реакций, протекающих под воздействием
нуклеофильных реагентов NaOH и
Na2S. С этой точки зрения растворение лигнина при варке является
следствием определенных ионных реакций фрагментации лигнина с разрывом связей
между структурными единицами.
· Действие едкого натра на лигнин
При действии NaOH на лигнин:
- образуются феноляты за счет взаимодействия со свободными
фенольными гидроксилами;
- происходит деструкция простых эфирных связей с образованием
новых фенольных гидроксилов и уменьшением молекулярной массы: расщепляются
углерод-углеродные связи;
- протекают реакции деструкции фрагментов лигнина с
образованием низкомолекулярных соединений и реакции конденсации с образованием
новых углерод-углеродных связей.
Рассмотрим эти изменения в лигнине более подробно.
Лигнин и его фрагменты переходят в раствор с образованием фенолятов:

При подкислении минеральными кислотами и даже очень слабой угольной
кислотой лигнин (щелочной лигнин) высаживается из черного щелока

или

Осаждаемый щелочной лигнин (в зависимости от способа варки различают
натронный и сульфатный лигнины) значительно изменен по сравнению с природным.
Он не является индивидуальным веществом, а представляет собой смесь продуктов,
различающихся по молекулярной массе и строению.
В процессе щелочной варки в лигнине появляются новые свободные фенольные
гидроксилы. Их образование происходит за счет гидролитической деструкции
алкиларильных простых эфирных связей. К такми реакциям относятся:
1. Расщепление структур с алкиларильной простой эфирной связью -O-4.
При наличии свободного фенольного гидроксила (фенольные структуры, т.е.
группы Х) расщепление может идти через промежуточное соединение - эпоксид,
образование которого идет по механизму:
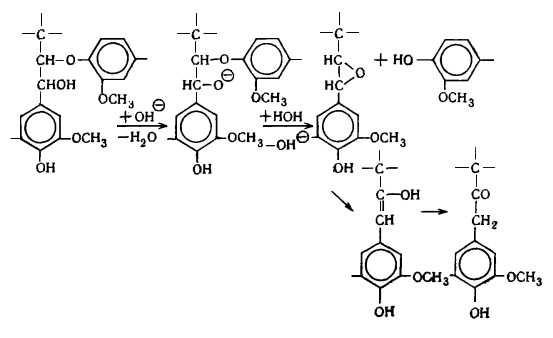
Однако такая реакция идет лишь частично. Одновременно и в большей степени
образуется промежуточный хинонметид, который затем теряет γ-углеродный атом в виде формальдегида

Образовавшаяся структура α-арилового эфира этанола устойчива к
щелочной деструкции. Таким образом, едкий натр расщепляет α-связи в группах Х лишь частично.
В группах с этерифицированными фенольными гидроксилами простая эфирная
связь в -положении может расщепляться лишь
при наличии у соседнего атома углерода свободного гидроксила (группа В’).
Реакция расщепления идет через промежуточный эпоксид по ионному механизму.

. Расщепление структуры с простой эфирной связью О-4. Простая эфирная
связь в αβположении при наличии свободного
фенольного гидроксила (группа Х) расщепляется очень легко. Реакция идет через
промежуточный хинонметид:

В нефенольных структурах (группы Z) расщепление простоя эфирной связи в αβположении идет при наличии свободного
гидроксила у соседнего атома углерода. Реакция идет через промежуточный эпоксид
по ионному механизму.

. Расщепление простой эфирной связи -О-4 в фенилкумарановых структурах
происходит лишь при наличии свободного фенольного гидроксила (группы Х). Оно
идет через промежуточный хинонметид:

При этом γ-углеродистый атом отщепляется в виде формальдегида с
образованием структуры стильбена.
Связи -О-4 в нефенольных фенилкумарановых
структурах устойчивы к действию щелочи в условиях варки. Однако в ходе варки
они постепенно превращаются в фенольные структуры, которые реагируют по
вышеуказанному механизму.
. Расщепление простой эфирной связи О-γ в структурах пинорезинола. Эта связь
в структурах со свободными фенольными гидроксилами расщепляется сравнительно
легко. Реакция идет через промежуточный хинонметид. При этом одновременно могут
отщепляться γ-углеродные атомы в виде формальдегида при сохранении
углерод-углеродной связи. В результате образуется смесь фенольных соединений.
Связи О-γ в нефенольных структурах
пинорезинола устойчивы к щелочной деструкции.
5. Расщепление простой эфирной связи в метоксильных группах.
Простая эфирная связь метоксильных групп более устойчива к щелочному гидролизу
и разрушается полностью лишь при температуре 250ºС (около 300ºС). Однако и при обычной варке в
некоторой степени идет эта реакция, о чем говорит образование метанола с
выходом около 0,7% (11-13кг на 1т целлюлозы) и понижение содержания
метоксильных групп в щелочном лигнине по сравнению с природным (12-13% вместо
16-17%). За счет этой реакции в лигнине появляются группировки пирокатехина

Диарильные простые эфирные связи устойчивы к щелочной деструкции, и
реакции их расщепления при варке имеют второстепенное значение.
Расщепление простых эфирных связей -О-4 и связей -О-4 в открытых структурах приводит к
деградации лигнина с образованием продуктов, растворимых в щелочах. Расщепление
простых эфирных связей только с образованием новых фенольных гидроксилов без
деградации макромолекулы повышает гидрофильность лигнина и тем самым также
способствует его растворению.
Углерод-углеродные связи между структурными единицами лигнина,
по-видимому, устойчивы к щелочной деструкции. Однако под действием щелочи при
повышенной температуре могут расщепляться углеродные связи в боковых пропановых
цепях
За счет реакции расщепления γ образуется формальдегид. Расщепление
связей может привести к образованию ванилина.
Фрагменты лигнина, перешедшие в раствор, подвергаются дальнейшей
деструкции с образованием низкомолекулярных соединений. Поэтому из черного
щелока при подкислении осаждается не весь растворенный лигнин, а только часть
его. Другая часть лигнина (около ¼) остается в растворе
(водорастворимый лигнин). В продуктах разрушения лигнина находят даже димерные
и мономерные соединения.
Образующиеся при деструкции свободные фенольные гидроксилы, с одной
стороны, приводят к увеличению растворимости лигнина в щелочи и, с другой
стороны, активируют группы бензилового спирта, участвующие в реакциях
конденсации.
Основной реакцией конденсации является конденсация за счет гидроксила
бензилового спирта и 5-го или 6-го положения ароматического кольца другой
структурной единицы лигнина с образованием новых углерод-углеродных связей,
т.е.е структур дифиниламина.
Однако процессы конденсации не ограничиваются только этими реакциями.
Образующийся при деструкции лигнина формальдегид может реагировать с водородом
в орто-положении к фенольному гидроксилу ароматического кольца с образованием
метилольного производного. Последнее затем участвует в реакции конденсации с
дургой структурной единицей лигнина (по аналогии с образованием
фенолформальдегидных смол) с образованием дифенилметановой структуры.

Реакции конденсации с участием 5-го положения ароматического кольца идут
более интенсивно с хвойным лигнином, чем с лиственным.
При натронном способе варки в отличие от сульфатного способа активные
группы бензилового спирта не защищены от конденсации. А реакции дегидратации за
счет разрыва простых эфирных связей -O-4 идут лишь
частично. Вследствие этого при натронной варке процессы конденсации оказываются
преобладающими над процессами деструкции.
· Роль сульфида натрия и химические реакции серы
Согласно теории, предложенной шведским и финским исследователями (Адлер,
Гирер, Энквист и др.), гидросульфид натрия (получающийся в результате гидролиза
сульфида натрия) как более сильный нуклеофильный реагент по сравнению с едким
натром вызывает более сильную деградацию лигнина за счет расщепления простых
эфирных связей.
Гидросульфид натрия вступает в химическое взаимодействие с группами
лигнина, способными к конденсации, и, блокируя эти группы, препятствует
развитию этого процесса. Таким образом, сторонники этой теории признают
существование химической связи серы с лигнином. Данная теория является наиболее
распространенной.
Гидросульфид натрия, присутствующий в варочном щелоке, существенно
изменяет поведение фенольных структур с простой эфирной связью O-4. Сульфатный варочный щелок
расщепляет такие структуры практически полностью, а натронный - только на 1/3,
т.е. в реакции расщепления -алкиларильной простой эфирной связи при наличии свободного фенольного
гидроксила NaSH оказывается более эффективным, чем NaOH.
Образующийся в качестве промежуточного соединения хинонметид превращается
в меркаптан (что предотвращает реакцию конденсации), который далее расщепляется
с образованием эписульфида.


За счет димеризации двух групп эписульфида может образоваться дитиана

Являются ли дитианы промежуточными или конечными продуктами реакции, еще
не выяснено. Предполагают, что в начальный период варки образуется лигнин,
богатый серой, по-видимому, за счет структур дитиана, которые затем разрушаются
и содержание серы уменьшается. В лигнине, выделенном из сульфатного черного
щелока, находят обычно 2 - 4% серы.
Если фенольный гидроксил замещен (группы B’), то реакция расщепления простой эфирной связи в -положении идет только через
промежуточные эпоксиды, причем в этой реакции NaOH оказывается более эффективным, чем NaSH, но группировка эписульфида защищает
гидроксил бензилового спирта от конденсации.

Фенилкумарановые структуры при сульфатной варке, как и при натронной,
теряют формальдегид и превращаются в стильбеновые структуры. Простые эфирные
связи O-4 в открытых структурах и O-γ в структурах пенорезинола
расщепляются натронным и сульфатным щелоками одинаково. Ионы гидросульфида во
всех этих реакциях оказывают положительное влияние на ход процесса
делигнификации, защищая группы бензилового спирта от конденсации.
Таким образом, роль серы заключается в блокировании активных групп
лигнина и в участии в реакции расщепления простых эфирных связей.
12. НИТРАТЫ ЦЕЛЛЮЛОЗЫ, ИХ ПОЛУЧЕНИЕ, СВОЙСТВА И ПРИМЕНЕНИЕ
.1 Нитраты целлюлозы, их получение
Нитраты целлюлозы, или азотнокислые эфиры целлюлозы, являются сложными
эфирами целлюлозы и азотной кислоты.
Реакцию нитрования целлюлозы с азотной кислотой можно представить
следующим образом:
[C6H7O2(OH)2]n
+ 3n HNO3 <-> [C6H7O2(ONO2)3]n
+ 3n H2O
Эта реакция обратима. Устанавливается равновесие между целлюлозой (как
спиртом), азотной кислотой, сложным эфиром и водой. Содержание воды в
реакционной смеси является основным условием, определяющим состояние конечного
равновесия.
Азотная кислота этерифицирует целлюлозу слишком медленно и не дает
высокозамещенных стойких азотнокислых эфиров, а при концентрации ниже 75% HNO3 вообще уже не этерифицирует целлюлозу. Поэтому
этерификация одной HNO3 нецелесообразна из-за большого ее
расхода.
Нитрование обычно проводят в присутствии водоотнимающих средств, в
качестве которых в промышленности применяют серную кислоту, а в лабораторной
практике уксусный или фосфорный ангидрид (в смеси с уксусной или фосфорной
кислотой). Нитрование целлюлозы тройными смесями HNO3 - H2SO4 - H2O является основой промышленного
способа производства нитратов целлюлозы.
Серная кислота, входящая в состав нитрующей смеси, связывая выделяющуюся
воду при реакции, сдвигает ее равновесие вправо. Иначе говоря, H2SO4 регулирует количество свободной воды в смеси. Вода,
наоборот, способствует переходу HNO3 в
гидратированную ионную форму:
NO2 - OH + H2O <-> H3O+
+ NO3-
Скорость реакции нитрования зависит от соотношения в смеси между азотной
и серной кислотами. Увеличение содержания H2SO4 с параллельным уменьшением
содержания HNO3 сверх определенного предела понижает скорость реакции
и приводит к деструкции целлюлозы. При слишком высоком содержании H2SO4 целлюлоза растворяется в нитрующей смеси и далее
гидролизуется. Однако необходимо поддерживать в смеси такое количество серной
кислоты, при котором достигается хорошее набухание целлюлозы, облегчающее
доступ азотной кислоты внутрь волокна и тем самым повышающее скорость процесса.
Реакция становится пермутоидной. Поэтому подбирают оптимальное соотношение
азотной и серной кислот. Обычно в практике используют отношение HNO3 к H2SO4 около 1:3.
При получении нитратов целлюлозы происходят побочные реакции:
окислительная и гидролитическая деструкция, а также образуются частично
замещенные сернокислые эфиры (сульфаты) целлюлозы:
C6H9O4(OH) + H2SO4
→ C6H9O4(OSO2OH) + H2O
Образование смешанных сернокислых и азотнокислых эфиров объясняет
невозможность практически достичь теоретического содержания азота,
соответствующего тринитрату целлюлозы. Присутствие сернокислых эфиров (очень
нестойких) уменьшает устойчивость (стабильность) нитратов целлюлозы. Нитраты
целлюлозы уже при умеренных температурах самопроизвольно разлагаются. Скорость
разложения быстро растет с повышением температур и резко возрастает в присутствии
примесей кислот, щелочей и др. разложение нитратов целлюлозы - самоускоряющийся
процесс, который, особенно в присутствии влаги и кислорода, может закончиться
вспышкой и взрывом. Это взывает необходимость стабилизации полученного нитрата
целлюлозы, который для этого промывают горячей водой, разбавленными кислотами.
0,2 - 1%-ным раствором соды и т.д. Сернокислые эфиры при этом гидролизуются.
Повышение температуры увеличивает скорость процесса нитрования, не влияя
на состояние равновесия. Однако при этом возрастают и скорости побочных
процессов - окислительной и гидролитической деструкции. Увеличение температуры
способствует и гидролитической деструкции целлюлозы, особенно при одновременном
повышении содержания воды в кислой смеси. Вода повышает степень ионизации и,
следовательно, гидролизующее действие кислот, особенно H2SO4.
Влияние продолжительности реакции на процесс подобно влиянию температуры.
Так как реакция нитрования идет быстро, процесс осуществляется обычно в течение
30 мин - 1 ч. Дальнейшее увеличение продолжительности способствует
гидролитической деструкции целлюлозы.
Вследствие отрицательного влияния H2SO4 на процесс нитрования (образование
сульфатов, гидролитическая деструкция) в некоторых случаях ее заменяют другими
водоотнимающими агентами. В лабораторной практике обычно применяют смесь
азотной, фосфорной кислот и фосфорного ангидрида или азотной, уксусной кислот и
уксусного ангидрида. В этих случаях нитрование протекает почти в безводной
среде, оно идет более полно (получаются тринитраты почти с теоретическим
содержанием азота) и практически отсутствует деструкция целлюлозы.
Нитраты, полученные таким способом, используются для определения СП
целлюлозы и ее фракционирования.
При нитровании с применением H2SO4 иногда используют разбавители - инертные органические
растворители (например, хлорированные углеводороды). Можно также проводить
нитрование азотной кислотой в присутствии ее солей. Иногда вместо азотной
кислоты применяют другие нитрующие агенты (например, ангидрид азотной кислоты).
Технический процесс получения нитратов целлюлозы состоит из следующих
операций:
. Измельчения и сушки целлюлозы;
. Нитрования смесью кислот (состав смеси подбирается в зависимости
от назначения конечного продукта);
. Удаления отработанной смеси центрифугированием;
. Стабилизации;
. Регулирования вязкости конечного продукта (снижением сп способом
гидролиза);
. Вытеснения воды этиловым спиртом.
Древесная целлюлоза содержит примеси (лигнин и гемицеллюлозы), которые
отрицательно влияют на качество продукта. Нитрат целлюлозы получается
неоднородным и нестойким. Азотнокислые эфиры из хлопковой целлюлозы получаются
более стойкими и имеют большую вязкость, чем эфиры из древесной сульфитной
целлюлозы.
При производстве нитратов целлюлозы выполняют ряд анализов. Определяют
процентное содержание азота (способом Лунге). По процентному содержанию азота [N] рассчитывают степень СЗ:

Анализ
основан на омылении нитрата целлюлозы серной кислотой в присутствии ртути,
восстанавливающей образующуюся азотную кислоту до окиси азота, объем которой
измеряют:
2HNO3 + 3 H2SO4 + 6Hg
→ 2NO + 3Hg2SO4 + 4H2O
Определяют
стойкость нитрата целлюлозы при различной температуре, вязкости и СП,
температуру вспышки и растворимость в различных растворителях.
10.5
Свойства нитратов целлюлозы и их применение
Различают следующие основные виды технических нитратов целлюлозы, которые
в зависимости от СЗ и СП находят различное практическое применение: коллоксилин
(10,7 - 12,5% N), пироколлодий (12,6 ± 0,1% N), пироксилин №2 (12,2 12,5% N), пироксилин №1 (13,0 - 13,5% N).
Нитраты целлюлозы, содержащие 9 - 11% N, растворяются в этиловом спирте, нитраты с содержанием до
13% N - в смеси этилового спирта и эфира.
Все нитраты целлюлозы растворяются в ацетоне. Нитраты целлюлозы любой степени
замещения не растворяются в воде и неполярных растворителях (например, в
бензоле).
Коллоксилин применяют для производства этрола, целлулоида и лаков. Он не
устойчив к действию кислот и щелочей. Разбавленные минеральные кислоты вызывают
медленную денитрацию коллоксилина. Концентрированная серная кислота растворят
коллоксилин. Коллоксилин растворяется в кетонах (ацетон), сложных эфирах
(этилацетат, бутилацетат и др.), фурфуроле, диоксане и уксусной кислоте;
устойчив к действию ароматических и алифатических углеводородов и масел. Из
высококипящих растворителей, применяемых в качестве пластификаторов,
коллоксилин растворяется в камфаре, эфирах фталевой кислоты и др. Этрольные и
целлулоидный коллоксилины с содержанием азота 10,9 - 11,2% растворяются в
спирто-камфарных смесях. Коллоксилин с содержанием азота 11,2 - 12,5%,
предназначенный для изготовления нитролаков, нитроэмалей и нитромастик,
растворяется в смеси растворителей.
Фракционный состав нитратов целлюлозы влияет на их стабильность,
механические свойства пленок и т.п. Основным их недостатком является горючесть
и легкая воспламеняемость.
Целлулоид представляет собой пластическую массу, состоящую из
коллоксилина, пластификатора (камфары), добавок (например, фосфорнокислого
натрия), пигментов и красителей. По существу это твердый раствор нитрата
целлюлозы в камфаре. Широко применяется для изготовления галантерейных изделий,
игрушек, для отделки музыкальных инструментов, изготовления оправ для очков и
др.
Этрол представляет собой термопластический материал, получаемый на основе
пластифицированного нитрата целлюлозы с минеральными и органическими
наполнителями. Из него изготавливают рулевые колеса, рычаги переключения
передач, приборные щитки, детали холодильников, электроизоляционные детали
изделий ширпотреба.
Пироксилин (СП 1000 - 2000) применяется для изготовления взрывчатых
веществ и порохов. Различают три вида порохов, получаемых на основе нитрата
целлюлозы: пироксилиновые (температура горения около 2500ºС), баллистические и кордитные. Два
последних также называют нитроглицериновыми (температура горения около 3500ºС).
При получении порохов применяют желатинирующие добавки (смесь этилового
спирта и диэтилового эфира, нитроглицерин и другие органические растворители),
стабилизаторы (например, дефениламин), флегматизаторы (камфара). Для
изготовления баллистических порохов применяют коллоксилин с высоким содержанием
азота (11,5 - 12,2%). Пороха для ракетных двигателей часто называют твердыми
ракетными топливами.
СПИСОК ИСПОЛЬЗОВАННЫХ ИСТОЧНИКОВ
1. Азаров В.И., Буров А.В., Оболенская А.В. Химия древесины
и синтетических полимеров: Учеб. для вузов. - СПб: СПбЛТА, 1999. - 628 с.
2. Богомолов Б.Д. Химия древесины и основы химии
высокомолекулярных соединений. - М.: Лесная промышленность, 1973. - 400 с.
3. Гончар А.Н., Гришпан Д.Д., Макаревич С.Е., Цыганкова
Н.Г., Шеймо Е.В. Реологические свойства концентрированных растворов целлюлозы и
ее смесей с другими полимерами в ортофосфорной кислоте. - Белоруссия, Минск:
УБГУ «Научно исследовательский институт физико-химических проблем», журнал
«Нефтехимический комплекс» №1 (6), 2011.
4. Евстигнеев Э.И. Химия древесины: Учеб. пособие для
студентов специальности 240406. - СПб: изд-во Политехн. ун-та, 2007. - 148 с.
5. Коротаев В.И., Симонов В.И. Производство
строительных материалов из древесных отходов. - М.: Лесная промышленность,
1972. - 74 с.
. Никитин В.М., Оболенская А.В., Щеголев В.П. Химия
древесины и целлюлозы: Учебное пособие. - М.: Лесная промышленность, 1978. -
368 с.
. Электронная библиотека
. Электронная энциклопедия делопроизводства
. Электронная энциклопедия.