Аналитическая химия
Аналитическая химия
Вариант 8
1.
Охарактеризуйте прямые методы фотометрического анализа: метод добавок и
сравнения
Ответ: Фотометрический метод анализа основан на способности определяемого
вещества поглощать электромагнитное излучение оптического диапазона.
Концентрацию поглощающего вещества определяют, измеряя интенсивность
поглощения. Поглощение при определенной длине волны является информацией о
качественном и количественном составе определяемого вещества и составляет
аналитический сигнал.
Фотометрический анализ относится к молекулярному абсорбционному анализу,
т.е. анализу основанному на поглощении света молекулами анализируемого вещества
и сложными ионами в ультрафиолетовой (УФ), видимой и инфракрасной (ИК) областях
спектра.
В настоящих указаниях рассматриваются метода анализа, основанные на
избирательном поглощении электромагнитного излучения в видимой и
ультрафиолетовой областях спектра: фотоколориметрия и спектрофотометрия.
Спектрофотометрический метод анализа - основан на поглощении монохроматического
излучения, т. е. излучения с одной длиной волны в видимой и УФ областях
спектра.
Фотоколориметрический метод анализа - основан на поглощении
полихроматического (немонохроматического) излучения, т. е. пучка лучей с
близкими длинами волны в видимой области спектра. Фотоколориметрию используют в
основном для анализа окрашенных растворов.
Оба метода основаны на общем принципе - пропорциональной зависимости
между светопоглощением и концентрацией определяемых веществ.
С помощью фотометрического анализа можно определять малые количества
вещества, например, содержание примесей не ниже 5 ´ 10-5% (спектрофометрически) и 1 ´
10-4 % (фотоколориметрически)
при погрешности определения 1-3 %.
Концентрация исследуемого вещества может быть определена методом
фотометрии в том случае, если в спектре поглощения раствора этого вещества
имеются ясно выраженные полосы поглощения в УФ и видимой областях спектра. В
основе количественного определения лежит закон Бугера - Ламберта-Бера, который
устанавливает прямопропорциональную зависимость между оптической плотностью и
концентрацией вещества в исследуемом растворе. С помощью фотометрии можно
проводить анализ, как индивидуальных веществ, так и их смесей.
Метод добавок представляет собой разновидность метода сравнения.
Определение концентрации раствора этим методом основано на сравнении оптической
плотности исследуемого раствора и этого же раствора с добавкой известного
количества определяемого вещества.
Метод добавок применяют для устранения мешающего влияния посторонних
примесей и чаще всего, для оценки правильности методики фотометрического
анализа. Этот метод позволяет создать одинаковые условия для фотометрирования
исследуемого и стандартного (с добавкой) окрашенных растворов, поэтому его
целесообразно применять для определения малых количеств различных элементов и
веществ в присутствии больших количеств посторонних веществ (воздух рабочей
зоны, атмосферный воздух, вода, почва, биосреды).
Метод добавок требует обязательного соблюдения основного закона
светопоглощения.
Неизвестную концентрацию вещества Сх находят либо графическим
способом, либо расчетным методом по формуле:
Сх = (а+Ах) / (Ах+а + Ах);
(1.1.)
Где, a - добавка стандартного вещества, мкг;
Ах - оптическая плотность измеряемого раствора;
Ах + а - оптическая плотность измеряемого раствора с известной
добавкой стандартного вещества а.
Наибольшая прецизионность результатов определения с использованием метода
добавок достигается при Ах + а = 2Ах.
Метод добавок обычно применяют для устранения мешающего действия
посторонних примесей, а также в ряде случаев для оценки правильности методики
определений. Этот метод позволяет создать одинаковые условия для
фотометрирования исследуемого раствора и раствора с добавкой, поэтому его
целесообразно применять для определения небольших количеств различных
соединений в присутствии больших количеств посторонних веществ. Метод добавок
требует обязательного соблюдения основного закона светопоглощения.
При определении неизвестной концентрации вещества в растворе графическим
способом (рис. 1.) на оси ординат откладывают значение оптической плотности
исследуемого раствора, Ах, а на оси абсцисс концентрации
добавляемого вещества в растворе. Из точек Са1 и Са2, на
оси абсцисс проводят перпендикуляры, на которых откладывают соответствующие
значения оптической плотности Ах+а1 и Ах+а2 растворов с
добавками а1 и а2. Через полученные 3 точки Ах,
Ах + а1; Ах + а2 проводят прямую линию до пересечения ее
с продолжением оси абсцисс в точке Сх. Абсолютное значение отрезка
ОСх выражает неизвестную концентрацию исследуемого раствора.
фотометрический хроматография титрование радиоактивный
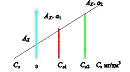
Рис. 1. Графическое определение концентрации раствора методом добавок
Рассмотрим расчет неизвестной концентрации вещества по методу сравнения.
При соблюдении основного закона светопоглощения и постоянной толщине слоя
измеряемого раствора, отношение оптических плотностей исследуемого раствора и
исследуемого раствора с добавкой, будет равно отношению их концентраций:
Ах / Ах + а = Сх / (Сх + Са);
(1.2)
Откуда,
Сх = (Са*Ах) /(Ах + а - Ах);
(1.3.)
Где, Ах - оптическая плотность исследуемого раствора;
А х + а - оптическая плотность исследуемого раствора с
добавкой;
Са - концентрация добавки в исследуемом растворе, мкг.
Добавки следует брать в таких количествах, чтобы не происходило
уменьшения точности, минимальная разность Ах + а - Ах
должна быть не менее 0,1.
. Какое явление называют тушением
люминесценции? Укажите его виды
Ответ: люминесце́нция (от лат. lumen, родительный падеж luminis - свет
и -escent - суффикс, означающий слабое действие), свечение, избыточное над
тепловым излучением тела и продолжающееся после прекращения возбуждения в
течение времени, значительно превышающего период световой волны (по определению
С.И. Вавилова). Т.е. люминесценция - процесс неравновесный и не относится к
тепловому равновесному излучению тел. Но люминесценция не относится и к таким
практически безинерционным неравновесным процессам, как отражение и рассеяние
света и тормозное излучение. Для наблюдения люминесценции вещество необходимо
вывести из состояния термодинамического равновесия, т. е. возбудить. При
люминесценции акты возбуждения и излучения света разделены во времени
промежуточными процессами, что обусловливает относительно длительное время
существования свечения вещества после прекращения возбуждения.
Тушение люминесценции - уменьшение выхода люминесценции, вызываемое
различными причинами. Тушение люминесценции может происходить при добавлении в
люминофор посторонних примесей, при увеличении в нём концентрации самого
люминесцирующего вещества (концентрационное тушение), при нагревании, под
действием инфракрасного света, электрического поля и др. воздействий на
люминесцирующее вещество.
В результате действия этих факторов относительно возрастает вероятность
безызлучательных переходов люминесцирующих молекул из возбуждённого состояния в
основное по сравнению с вероятностью их излучательных переходов. В случае
рекомбинационной люминесценции кристаллофосфоров тушение люминесценции объясняется
безызлучательной рекомбинацией носителей заряда с центрами тушения, которыми
могут служить дефекты кристаллической решётки или атомы примеси.
Отличие выхода люминесценции от единицы обусловлено такими процессами
тушения - различают концентрационное, внутреннее, температурное, внешнее
статическое и динамическое тушение.
Внутреннее тушение обусловлено безызлучательными переходами внутренней
конверсии и колебательной релаксации. Наиболее ярко оно проявляется в
симметричных структурах с большим числом сопряженных связей, конформационно
нежёстких структурах.
Температурное тушение является разновидностью внутреннего. Под влиянием
температуры способность молекулы деформироваться растёт, и, как следствие,
растёт вероятность безызлучательных переходов.
Внешнее статическое тушение основано на взаимодействии люминесцирующего
соединения с другой молекулой и образованием неизлучающего продукта.
Динамическое тушение наблюдается, когда возбуждённая молекула люминофора
вступает в постороннюю реакцию и теряет свои свойства. Концентрационное тушение
- результат поглощения молекулами вещества собственного излучения.
В широком смысле слова под тушением возбужденных состояний понимают любые
процессы их дезактивации, являющиеся результатом взаимодействия возбужденных молекул
с компонентами системы. Выход люминесценции очень чувствителен к различным
внутримолекулярным и межмолекулярным взаимодействиям, которые вызывают его
уменьшение и приводят к развитию процессов тушения люминесценции. К числу
наиболее активных тушителей люминесценции относятся:
тяжелые анионы и катионы I−, Br−, Cs+,
Cu2+ (при этом облегчается S1 → T1
переход);
парамагнитные ионы и молекулы O2, Mn2+,
нитроксильные радикалы;
молекулы растворителя. Наибольшим тушащим действием обладают обычно
полярные растворители, такие, как вода;
акцепторы электронной энергии возбуждения.
Согласно С. И. Вавилову, тушитель может быть статическим (тушение первого
рода) и динамическим (тушение второго рода).
Тушение первого рода. К тушению первого рода были отнесены все те процессы,
в которых уменьшение выхода люминесценции не сопровождается уменьшением средней
длительности возбуждённого состояния. Тушение первого рода вызывается быстрыми
химическими или физико-химическими процессами в возбужденных молекулах
исследуемого вещества. В этом случае часть энергии света, поглощенного
молекулами, расходуется на их диссоциацию, ионизацию или на увеличение энергии
их колебания и вращения. Такие процессы развиваются с большой скоростью и
происходят за время, соизмеримое со временем собственных колебаний молекул (~10-13÷10-14 с), что значительно меньше времени
жизни молекул в возбуждённом состоянии, 10-9 с. Статическое тушение
связано также с образованием нефлуоресцирующих комплексов НК флуоресцирующих
молекул Ф с молекулами тушителя Q:
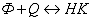 ; (2.1.)
; (2.1.)
Отношение концентраций свободного флуоресцирующего и связанного
нефлуоресцирующего вещества [Ф]/[HK] может быть найдено из уравнения
равновесия:
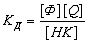 ; (2.2.)
; (2.2.)
Где, ДК - константа диссоциации комплекса. Если поглощение
вещества Ф и комплекса не различаются, то отношение квантовых выходов
люминесценции вещества Ф в присутствии и отсутствии комплекса будет равно:
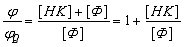 ; (2.3.)
; (2.3.)
Используя предыдущее уравнение, получаем:
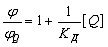 ; (2.4.)
; (2.4.)
Если поглощение комплекса отлично от поглощения флуоресцирующего
вещества, то уравнение (2.3.) не соблюдается. Однако при низких оптических
плотностях растворов будет справедливо отношение:
 ; (2.5.)
; (2.5.)
Частным случаем статического тушения является так называемое
концентрационное тушение, которое связано с образованием нефлуоресцирующих
димеров и более крупных ассоциатов молекул при высокой концентрации
флуоресцирующего вещества:
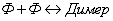 (не флуоресцирует); (2.6.)
(не флуоресцирует); (2.6.)
В этом случае димеризация может сопровождаться деформацией электронного
спектра поглощения молекул растворенного вещества.
Концентрационное тушение является обратимым процессом - выход свечения
полностью восстанавливается при разбавлении концентрированного раствора.
Тушение второго рода. К тушению второго рода были отнесены все те
процессы, в которых уменьшение выхода люминесценции вызывается воздействием на
возбужденные молекулы исследуемого вещества за времена, соизмеримые со временем
жизни возбуждённого состояния. В этом случае происходит безызлучательная
дезактивация возбужденных молекул, которая развивается, либо вследствие
передачи энергии от возбужденных молекул к невозбужденным, либо благодаря
переходу энергии возбуждения в энергию колебания ядер, либо из-за протекания
химических реакций с участием возбужденных молекул.
Вследствие того, что при тушении первого рода все воздействия
осуществляются на невозбужденные молекулы, то это никак не может сказаться на
величине τ, так как в возбужденное состояние переходят лишь те молекулы,
которые избежали этих воздействий. Напротив, в случае тушения второго рода во
всех взаимодействиях принимают участие возбужденные молекулы. Поэтому при
развитии тушения такого вида значение τ должно существенно изменяться, т.е.
постоянство τ или его изменения являются надежным критерием, позволяющим
однозначно установить природу тушения. В случае тушения второго рода, при
экспоненциальном законе затухания свечения и экспоненциальном ходе тушения
люминесценции, выполняется важное соотношение между выходом свечения и средней
длительностью возбужденного состояния исследуемых молекул:
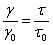 ; (2.7.)
; (2.7.)
Где, γ0 γ, а также τ0 и τ - соответственно выход люминесценции и
средняя длительность возбужденного состояния молекул в случае отсутствия и при
наличии тушения. Таким образом, при выполнении указанных условий между выходом
люминесценции и τ должна осуществляться пропорциональная зависимость. В
ряде случаев это соотношение хорошо выполняется на опыте.
Факторы, стимулирующие люминесценцию, при определенных условиях могут
дать обратный эффект, т.е. уменьшить интенсивность свечения или совсем
прекратить его. Это явление называют уменьшением люминесценции. Повышение
температуры, изменение влажности, ИК-облучение, электрическое поле, изменение
внешнего давления, наличие некоторых газов - все эти факторы могут привести к
тушению люминесценции. Так, например, присутствие кислорода, бензохинона или
йода уменьшает интенсивность фотолюминесценции, в тоже время как присутствие
молекул воды увеличивает ее. Наличие электрического поля, перпендикулярного
поверхности люминофора, тушит радикалолюминесценцию, изменение же направления
поля на обратное - усиливает свечение.
Обычно тушение люминесценции нежелательно, поэтому к чистоте
люминесцирующих веществ предъявляются очень высокие требования. Однако
специальные виды люминофоров, в которых происходит быстрое Тушение
люминесценции при повышении температуры или под действием инфракрасного
излучения, применяются в качестве чувствительных индикаторов длинноволновых
излучений.
. Дайте характеристику метода тонкослойной
хроматографии
Адсорбционный метод основан на различии степени сорбции-десорбции
разделяемых компонентов на неподвижной фазе. Адсорбция осуществляется за счет
вандервальсовских сил, являющейся основой физической адсорбции,
полимолекулярной и хемосорбцией. Для эффективных процессов сорбции-десорбции
необходима большая площадь, что предъявляет определенные требования к
адсорбенту. При большой поверхности разделения фаз происходит быстрое
установление равновесия между фазами компонентов смеси и эффективное
разделение. Впервые метод тонкослойной хроматографии заявил о себе как
"Бумажная тонкослойная хроматография", которая основывалась на распределительном
методе разделения компонентов.
В этом методе хроматографирование веществ происходит в тонком слое
сорбента, нанесенного на твердую плоскую подложку. Разделение в этом методе в
основном происходит на основе сорбции-десорбции.
Использование различных сорбентов, позволило значительно расширить и
улучшить этот метод.
Современная хроматографическая пластинка представляет собой основу из
стекла, алюминия или полимера (например, политерефталат). В связи с тем, что
стеклянная основа становится менее популярной (часто бьется, нельзя разделить
пластинку на несколько частей не повредив слой сорбента, тяжелая по весу),
наибольшее распространение получили пластины, в качестве основ которых
используют алюминиевую фольгу или полимеры.
Для закрепления сорбента применяют гипс, крахмал, силиказоль и др.,
которые удерживают зерна сорбента на подложке. Чем меньше диаметр зерен
сорбента, тем качественнее (идентификаци дороже) пластинка. Толщина слоя может
быть различна (100 и более мкм), но самый важный критерий - слой должен быть
равномерный по толщине в любом месте хроматографической пластинки.
Тонкослойная
хроматография имеет несколько способов, связанных, в основном, с видом движения
растворителей:
· Восходящая тонкослойная хроматография
· Нисходящая тонкослойная хроматография
· Горизонтальная тонкослойная хроматография
· Радиальная тонкослойная хроматография.
Восходящая
тонкослойная хроматография - этот вид хроматографии наиболее распространен и
основан на том, что фронт хроматографической системы поднимается по пластинке
под действием капиллярных сил. Для этого метода используется наиболее простое
оборудование, так как в качестве хроматографической камеры можно использовать
любую емкость с плоским дном и плотно закрывающейся крышкой, в которую свободно
помещается хроматографическая пластинка.
Метод восходящей тонкослойной хроматографии имеет ряд своих недостатков.
Например, скорость поднятия фронта по пластинке происходит неравномерно, т.е. в
нижней части она самая высокая, а по мере поднятия фронта уменьшается. Это
связано с тем, что в верхней части камеры насыщенность парами растворителя
меньше, поэтому растворитель с хроматографической пластинки испаряется
интенсивнее, следовательно уменьшается его концентрация и скорость движения
замедляется. Для устранения этого недостатка по стенкам хроматографической
камеры до начала хроматографирования прикрепляют полоски фильтровальной бумаги,
по которым поднимающаяся хроматографическая система насыщает парами камеру по
всему объему.
Недостатком также можно считать необходимость следить за фронтом
растворителя, так как возможно "убегание" лини фронта растворителя до
верхнего края. В таком случае определить значение Rf уже невозможно.
Радиальная тонкослойная хроматография заключается в том, что в центр
пластинки наносится исследуемое вещество и туда же подается система, которая
движется от центра к краю пластинки.
Нисходящая тонкослойная хроматография - этот метод хроматографии основан
на том, что фронт хроматографической системы опускается по пластинке в основном
под действием сил тяжести, т.е. фронт подвижной фазы движется сверху вниз. Для
этого метода в верхней части хроматографической камеры крепится кювета с
хроматографической системой, из которой с помощью фитиля на хроматографическую
пластинку поступает растворитель, который стекает, и происходит
хроматографирование исследуемого образца.
К недостаткам этого метода можно отнести усложнение оборудования. Этот
метод используется в основном в бумажной хроматографии.
Горизонтальная тонкослойная хроматография - этот метод наиболее сложен в
аппаратурном оформлении не наиболее удобен. Так, в хроматографической камере
пластинка размещается горизонтально и подача системы происходит на один край
пластинки с помощью фитиля. Фронт растворителя движется в противоположную
сторону. Есть еще один прием, позволяющий предельно упростить камеру. Для этого
хроматографическую пластинку на алюминиевой основе слегка изгибают и помещают в
камеру. В данном случае система будет поступать с двух сторон одновременно. Для
этой цели подходят только пластины с алюминиевой подложкой, так как пластиковая
и стеклянная основа "несгибаема", т.е. не сохраняет форму.
К достоинствам этого метода можно отнести то, что в горизонтальной кювете
насыщение парами системы происходит гораздо быстрее, скорость движения фронта
постоянная. А при хроматографировании с двух сторон, фронт не
"убегает".
4. В чем заключается сущность амперометрического
титрования? Какие электроды используют в этом методе
Ответ: Амперометрическое титрование представляет собой электрометрический
метод анализа, основанный на измерении величин предельного диффузионного тока,
наблюдаемого на отдельном электроде в процессе титрования.
Момент эквивалентности устанавливается на основании резкого изменения
величины предельного диффузионного тока.
Величина предельного диффузионного тока прямо пропорциональна
концентрации деполяризатора находящегося в растворе и участвующего в
электрохимическом процессе. Эта зависимость, может быть выражена уравнением
Ильковича:
id = cCo, c=
605zD1/2m2/3e1/6; (4.1.)
Где, id - предельный ток (мкА),
c - константа уравнения Ильковича,
Со - концентрация деполяризатора в растворе (моль/л),
D -
коэффициент диффузии деполяризатора (см2/с),
z -
число электронов, принимающих участие в электродной реакции,
m -
скорость вытекания ртути из капилляра (мг/с),
e - время жизни ртутной капли (сек).
Такая зависимость будет соблюдаться, если электролиз проводится в
присутствии избытка постороннего сильного электролита (фона).
Значение потенциала поляризуемого (индикаторного) электрода задается
экспериментатором и должно соответствовать предельному диффузионному току
восстановления или окисления одного из следующих веществ: определяемого
вещества, реагента (титранта), продукта взаимодействия определяемого вещества с
титрантом, или же специально введенного до титрования «полярографического
индикатора».
В зависимости от того, какое из перечисленных веществ окисляется или
восстанавливается на электроде при выбранном значении потенциала, кривые
титрования могут быть различного типа.
1 Электрохимически активно только
определяемое вещество, т.е. только оно восстанавливается или окисляется на
электроде. Пример: титрования солей Fe2+ окислителями при потенциале вращающегося платинового электрода +0,8В
(нас. кал. э.). В этом случае на электроде протекает реакция окисления: Fe2+ - e- ® Fe3+ и, следовательно, величина
предельного тока будет зависеть от концентрации в растворе соли Fe2+.
2 Электрохимически активен только
титрант (реагент), т.е. он окисляется или восстанавливается на электроде:
например, титрование солей Zn2+, Cd2+, Mn2+, Pb2+, Cu2+ ферроцианидом при значении
потенциала платинового вращающегося электрода, равном +0,8 В (нас.к.э.). В этом
случае на электроде протекает реакция окисления ферроцианид-ионов, предельный
ток пропорционален концентрации ферроцианида в растворе.
3 Восстанавливаются или окисляются на
электроде два вещества - определяемое соединение и титрант. Например,
титрование солей Cu2+, Cd2+, Zn2+ ортооксихинолином при значении
потенциала ртутного капающего электрода равном -1,6 В (нас.к.э.). В этом случае
на электроде до момента эквивалентности восстанавливаются ионы Cu2+, Cd2+, Zn2+, а ортооксихинолин - после момента
эквивалентности. Т.о., в этом случае величина предельного тока будет прямо
пропорциональна концентрации определяемых ионов Cu2+, Cd2+, Zn2+ в растворе - до точки эквивалентности - и концентрации в
растворе ортооксихинолина - после точки эквивалентности.
4 Электрохимически активны как
определяемое вещество, так и титрант, причем одно восстанавливается на электроде,
другое - окисляется. Например, титрование соли Fe3+ раствором TiCl3 при
значении потенциала ртутного электрода, равном -0,25 В (нас.к.э.). Точка
эквивалентности обнаруживается вследствие различия угла наклона прямых id - v мл. реагента, описывающих изменение тока до и после момента
эквивалентности; это связано, с различным числом электронов, принимающих
участие в электродных реакциях определяемого вещества и титранта, а также с
различием в коэффициентах диффузии этих веществ. В приведенном примере до момента
эквивалентности на электроде восстанавливаются ионы Fe3+, величина предельного тока пропорциональна концентрации соли
Fe3+ в растворе. После момента
эквивалентности на электроде протекает процесс окисления соли титана (III), величина предельного тока пропорциональна
концентрации последней в растворе.
5 Электрохимически активен только
продукт химической реакции, т.е. на электроде протекает восстановление или
окисление образующегося в результате химической реакции соединения. Пример:
титрование соединений пятивалентного мышьяка иодидами в кислой среде: в
результате химической реакции образуется иод, который восстанавливается на
вращающемся платиновом электроде. Предельный ток в этом случае прямо
пропорционален концентрации иода, образующегося в растворе.
6 Реагирующие вещества и продукты
реакции электрохимически неактивны. Тогда специально в раствор вводится
электрохимически активное вещество - «полярографический индикатор». Пример:
титрование с «полярографическим индикатором» - солью Fe3+, вводимым перед титрованием соединений алюминия, магния или
циркония, раствором фторида; электрод - вращающийся платиновый, значение
потенциала равно 0,0 В (нас.к.э.) и отвечает предельному току восстановления
активированных ионов Fe3+. В этом случае до точки эквивалентности
происходит взаимодействие ионов фтора с определяемым веществом с образованием
прочных соединений; только после точки эквивалентности ионы фтора смогут
взаимодействовать с Fe3+ (полярографическим индикатором),
поскольку образующийся фторидный комплекс менее устойчив. В результате падения
концентрации ионов Fe3+ после точки эквивалентности величина
предельного тока начинает убывать.
Последний вид титрования основан на использовании различной прочности
соединений: определяемое вещество - титрант, «полярографический индикатор» -
титрант, образующихся в процессе титрования.
Можно использовать Fe(II) в качестве «полярографического
индикатора» при амперометрических титрованиях с ЭДТА. При потенциале +0,4 В на
платиновом электроде окисляется не Fe(II), а хелат железа (II) с ЭДТА, и, т.о., после достижения
точки эквивалентности предельный ток растет. Резкий подъем тока наблюдается при
рН 4 для большинства ионов металлов, образующих с ЭДТА хелаты, константы
устойчивости которых больше, чем 1018.
7 В отдельных, редких случаях можно
проводить титрование, когда на поляризуемом (индикаторном) электроде сначала
происходит восстановление (или окисление) определяемого вещества, а после
момента эквивалентности - титранта. При этом излом на кривой титрования в точке
эквивалентности вызван различием в величинах коэффициентов диффузии этих
веществ, а также в числе электронов. Примером такого вида титрования может
служить определение ванадия (IV)
раствором Се(IV). До точки эквивалентности V(IV), взаимодействуя с раствором Ce (IV), переходит в V(V), который, являясь электрохимически активным,
восстанавливается на электроде. После момента эквивалентности на электроде
протекает восстановление ионов Се(IV).
С помощью амперометрического титрования можно:
1 определить концентрацию изучаемого
соединения в растворе;
2 устанавливать стехиометрические
соотношения, при которых образуются соединения в результате химического
взаимодействия между определяемым веществом и титрантом;
3 определять величину произведения
растворимости осадка, образующегося в процессе титрования.
В амперометрических титрованиях могут быть использованы реакции
комплексообразования, окисления-восстановления и осаждения.
В качестве поляризующегося индикаторного электрода в амперометрии можно
использовать различные металлы, чаще всего применяют ртутный капающий,
платиновый и графитовый электроды.
Выбор материала электрода определяется в первую очередь тем, какую
электродную реакцию предполагается использовать для титрования. Ртутный
капающий электрод применяется в тех случаях, когда нужно восстанавливать ион
какого-либо электроотрицательного металла или восстановить органические
соединения. На ртути перенапряжение выделения водорода велико: последний будет
выделяться при потенциале (Е) около -1,1 В в кислых растворах, -1,5 В в
нейтральных и -1,9 В в щелочных.
На платиновом электроде перенапряжение выделения водорода мало и
выделение водорода протекает при Е = 0,0 В в кислых растворах, -0,4 В в
нейтральных и -0,8 В в сильнощелочных.
Отсюда следует, что на ртутном электроде процессу восстановления многих
электроотрицательных ионов не мешает водород. На платиновом электроде
восстановления этих веществ не происходит, т.к. не может быть достигнут
достаточно отрицательный потенциал.
С другой стороны на платиновом электроде могут протекать такие реакции,
какие не могут быть проведены на ртути. Платина обладает высоким положительным
потенциалам, она индифферентна к большинству окислителей: при использовании
платины в качестве анода она практически в большинстве случаев анодно не растворяется.
Графитовый электрод находит применение для изучения процессов как
окисления, так и восстановления. Чаще всего для работы применяется графит,
предварительно пропитанный воском, парафином или клеем БФ-2. На пропитанном
электроде наблюдается меньший остаточный ток. Это можно объяснить способностью
пропитывающих веществ снижать емкостной ток.
Титрование следует проводить при таком потенциале индикаторного
электрода, который соответствовал бы области предельного тока иона или
молекулы, участвующих в электродной реакции. Для определения потенциала
индикаторного электрода необходимо снять полярограмму (вольтамперную кривую)
соответствующего иона (молекулы) на этом электроде в той среде, в которой будет
проводиться титрование. При снятии полярограмм необходимо учитывать возможность
восстановления кислорода (как на ртутном, так и на платиновом электроде). Для
удаления кислорода из раствора предварительно через раствор в течение 20-30
минут пропускается струя инертного газа (водород, азот, аргон, гелий).
В амперометрическом титровании поляризация индикаторного электрода
осуществляется относительно неполяризуемого электрода, в качестве которого
используют электроды сравнения: каломельные, меркуриодидный, хлор - серебряный
и др., а также платиновый электрод в случае трехэлектродной ячейки.
. Как проводят количественное определение веществ в газовой
хроматографии методами нормировки
Ответ: Хроматография - процесс, основанный на многократном повторении
актов сорбции и десорбции вещества при перемещении его в потоке подвижной фазы
вдоль неподвижного сорбента. Разделение сложных смесей хроматографическим
способом основано на различной сорбируемости компонентов смеси. В процессе
хроматографирования так называемая подвижная фаза (элюент), содержащая
анализируемую пробу, перемещается через неподвижную фазу. Обычно неподвижная
фаза представляет собой вещество с развитой поверхностью, а подвижная - поток
газа или жидкости, фильтрующейся через слой сорбента. При этом происходит
многократное повторение актов сорбции - десорбции, что является характерной
особенностью хроматографического процесса и обуславливает эффективность
хроматографического разделения.
Количественный хроматографический анализ проводят обычно на хроматографе.
Метод основан на измерении различных параметров хроматографического пика,
зависящих от концентрации хроматографируемых веществ - высоты, ширины, площади
и удерживаемого объема или произведения удерживаемого объема на высоту пика.
Газовая хроматография универсальный метод разделения смесей разнообразных
веществ, испаряющихся без разложения. При этом компоненты разделяемой смеси
перемещаются по хроматографической колонке с потоком газа-носителя. По мере
движения разделяемая смесь многократно распределяется между газом-носителем
(подвижной фазой) и нелетучей неподвижной жидкой фазой, нанесенной на инертный
материал (твердый носитель), которым заполнена колонка. Принцип разделения
неодинаковое сродство веществ к летучей подвижной фазе и стационарной фазе в
колонке. Компоненты смеси селективно задерживаются последней, поскольку
растворимость их в этой фазе различна, и таким образом разделяются (компонентам
с большей растворимостью требуется большее время для выхода из жидкой фазы, чем
компонентам с меньшей растворимостью). Затем вещества выходят из колонки и регистрируются
детектором. Сигнал детектора записывается в виде хроматограммы автоматическим
потенциометром (самописцем) или же регистрируется компьютером.
В количественной газовой хроматографии применяют методы абсолютной
градуировки и внутренней нормализации, или нормировки. Используется также метод
внутреннего стандарта. При абсолютной градуировке экспериментально определяют
зависимость высоты или площади пика от концентрации вещества и строят
градуировочные графики или рассчитывают соответствующие коэффициенты. Далее
определяют те же характеристики пиков в анализируемой смеси, и по
градуировочному графику находят концентрацию анализируемого вещества. Этот
простой и точный метод является основным при определении микропримесей.
При использовании метода внутренней нормализации принимают сумму
каких-либо параметров пиков, например сумму высот всех пиков или сумму их
площадей, за 100%. Тогда отношение высоты отдельного пика к сумме высот или
отношение площади одного пика к сумме площадей при умножении на 100 будет
характеризовать массовую долю (%) компонента в смеси. При таком подходе
необходимо, чтобы зависимость величины измеряемого параметра от концентрации
была одинаковой для всех компонентов смеси.
. Какой вид имеет кривая кондуктометрического титрования соляной
кислоты раствором гидроксида аммония? Объясните вид кривой титрования. Как
определяют объем в конечной точке титрования и рассчитывают массу определяемого
вещества? Какие приборы и электроды можно использовать для данного титрования?
Ответ: При сливании 2-х растворов в результате протекающей химической
реакции изменяется ионный состав раствора, а, следовательно, и его
электропроводность. Измеряя электропроводность можно найти эквивалентную точку.
Посторонние ионы в этом случае являются фоном и не оказывают влияния на
результат анализа. Точка эквивалентности будет выглядеть тем отчетливее, чем
заметнее разница между электропроводностями титруемого раствора и титранта.
Рассмотрим несколько случаев:
Титрование слабой кислоты сильным основанием рассмотрим на примере
титрования уксусной кислоты гидроксидом натрия:
CH3COOH + NaOH => CH3COONa + H2O3COOН + Na+ + OH- => Na+ + CH3COO-
+ H2O
CH3COOН + OH- => H2O+ CH3COO-
В результате титрования образуется ацетат натрия, соль, которая
подвержена гидролизу: CH3COONa + HOH => CH3COOH +
NaOH, сдвигающая рН раствора в сторону щелочной среды. Кроме того, присутствие
CH3COONa способствует уменьшению диссоциации слабой кислоты
(буферное действие). Эти противоположные эффекты дают кривые с минимумом,
положение которого зависит как от концентрации так и силы кислоты.
Титрование слабой кислоты сильным основанием рассмотрим на примере
титрования уксусной кислоты гидроксидом аммония:
CH3COOH + NH4OH => CH3COONH4
+ H2O3COOH +NH4OH => CH3COO- +
NH4+ + H2O
Т.к. концентрация ионов водорода очень мала, то наклон будет небольшой до
точки эквивалентности. Также мала концентрация ионов гидроксила вследствие
диссоциации NH4OH, поэтому после точки эквивалентности резкого
возрастания кривой наблюдаться не будет. Этот случай практически не
используется из-за размытости точки эквивалентности.
Титрование смеси сильной и слабой кислот сильным основанием:
В этом случае ход кривой изменяется и на кривой титрования наблюдается 2
точки эквивалентности: первая точка будет соответствовать титрованию сильной
кислоты, например соляной, если взята смесь соляной и уксусной кислот. Объем,
пошедший на титрование слабой кислоты определяется по формуле:
V (CH3COOH) = V2 - V(HCl); (6.1.)
Кривые кондуктометрического титрования. Если отложить
по оси абсцисс значения объемов прибавляемого титрованного раствора кислоты (в
миллилитрах), а по оси ординат - величины электропроводности, получится
характерная кривая кондуктометрического титрования (рис. 6). Точка эквивалентности
при этом титровании совпадает с точкой минимума электропроводности.
Другой вид имеет кривая кондуктометрического
титрования нитрата серебра хлоридом бария (рис. 6.1.). В этом случае в процессе
титрования не наблюдается заметного изменения электропроводности, но после
достижения точки эквивалентности прибавление даже незначительного избытка
хлорида бария вызывает повышение электропроводности. Кривые третьего типа
получаются при титровании слабых кислот сильными основаниями или слабых оснований
сильными кислотами (рис. 6.2.). Здесь электропроводность до точки
эквивалентности возрастает менее резко, чем после ее достижения.
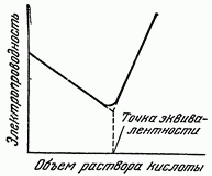
Рис. 6. Кривая кондуктометрического титрования NaOH раствором
HCl.
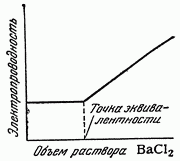
Рис. 6.1. Кривая кондуктометрического титрования AgNO3
раствором BaCl2.

Рис. 6.2. Кривая кондуктометрического титрования
слабой кислоты сильным основанием.
Таким образом, форма получаемой кривой
кондуктометрического титрования зависит от типа титрования, а изменения
электропроводности обусловлены различной подвижностью ионов (в см/сек) в
одинаковых условиях опыта.
Из вышесказанного можно сделать следующие выводы:
. Электропроводность кислот и щелочей намного выше, чем эквивалентных им
по концентрации растворов солей.
. Прямой кондуктометрический метод не пригоден для анализа
многокомпонентных систем, что связано с аддитивным вкладом каждого иона в
электропроводность системы в целом, но может быть применен для контроля систем,
содержащих один электролит.
. Так как величина электрической проводимости сильно зависит от
температуры, измерительную ячейку следует помещать в термостат и проводить
анализ при одной и той же определенной температуре.
Схема установки для проведения кондуктометрического анализа включает в
себя измерительную ячейку, состоящую из двух электродов, закрепленных на
фиксированном расстоянии, которые погружены в анализируемый раствор, помещенный
в сосуд произвольной формы. Для уменьшения поляризации используют электроды с
сильной развитой поверхностью из химически стойких материалов. Это обычно
платина, покрытая платиновой чернью или графит.
Измерение электропроводности растворов электролитов практически сводится
к измерению их сопротивления по мостовой схеме Уитстона обычно на приборе
Кольрауша.
Наибольшее значение имеет метод, основанный на применении переменного
тока. Для питания схемы применяют источники с частотой 50 - 1000 Гц, чтобы
исключить побочные процессы на границе гальванического контакта электрод -
анализируемая система, такие как электролиз, поляризация, которые приводят к
искажению результатов измерения.
Электрическим мостом в технике измерений называют электрический прибор
для измерения сопротивлений, емкостей, индуктивностей и других электрических
величин, представляющих собой измерительную мостовую цепь, действие которой
основано на методике сравнения измеряемой величины с образцовой мерой. Как
известно, метод сравнения дает весьма точные результаты измерений, вследствие
чего мостовые схемы получили широкое распространение как в лабораторной, так и
в производственной практике.
Классическая мостовая цепь состоит из четырех сопротивлений Z1,
Z2, Z3, Z4, соединенных последовательно в виде
четырехугольника (рис. 6.3.), причем точки А, Е, В, D называют вершинами. Ветвь АВ, содержащая источник питания Un, называется диагональю питания, а
ветвь ЕD, содержащая сопротивление нагрузки ZH, - диагональю нагрузки. Сопротивления Z1,
Z2, Z3, Z4, включенные между двумя соседними
вершинами, называются плечами мостовой цепи.
Название «мостовая цепь» объясняется тем, что диагонали, как мостики,
соединяют две противолежащие вершины (диагональ нагрузки, например, ранее так и
называлась - мост). Схема, представленная на рис. 6.3., известна в литературе
как четырехплечный мост, или мост Уитстона.

Рис. 6.3. Схема мостика Уитстона
R-мост Уитстона
предназначен для измерения величин сопротивлений. Он состоит из реохорда АВ,
чувствительного гальванометра  и двух
резисторов - известной величины R и неизвестной - Rх (рис. 6.4.).
и двух
резисторов - известной величины R и неизвестной - Rх (рис. 6.4.).
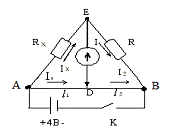
Рис. 6.4. R-мост Уитстона
для измерения величин сопротивлений
Реохорд представляет собой укрепленную на линейке однородную проволоку,
вдоль которой может перемещаться скользящий контакт D. Рассмотрим схему без
участка ЕD. Замкнем ключ К. Тогда по проволоке АВ потечет ток и вдоль нее будет
наблюдаться равномерное падение потенциала от величины ja (в точке А) до величины jb (в точке В). В цепи АЕВ пойдет ток и
будет наблюдаться падение потенциала от ja до je
(на резисторе Rх) и от je до jb (на резисторе R). Очевидно, в точке Е потенциал имеет промежуточное значение
je между значениями ja и jb. Поэтому на участке АВ всегда можно найти точку D,
потенциал которой равен потенциалу в точке Е: jD=je.
Если между точками Е и D включен гальванометр, то в этом случае ток через него
не пойдет, т.к. φe - φD= 0.
Мост
Уитстона может быть также использован для определения внутреннего сопротивления
гальванометра r , причем гальванометр в этом
случае включается, как показано на рис. 6.5.
, причем гальванометр в этом
случае включается, как показано на рис. 6.5.
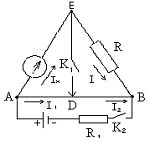
Рис. 6.5. Мост Уитстона для определения внутреннего сопротивления
гальванометра
Если потенциалы je
и jD равны, то сила тока в диагонали ЕD
равна нулю, а поэтому замыкание и размыкание ключа К1 не будут
вызывать изменения силы тока в ветвях мостовой схемы, в том числе и в ветви
гальванометра.
. Какие индикаторные электроды можно использовать в реакциях
комплексообразования при потенциометрическом титровании? Приведите пример,
обоснуйте выбор электродов
Ответ: Для осуществления потенциометрического титрования необходимо
правильно выбрать индикаторный электрод. Выбор индикаторных электродов можно
осуществить экспериментальным путем, проверяя различные типы электродов из
разных материалов.
Наилучшим будет тот, на котором возникнет наибольший скачок потенциала (ΔЕ). Такой путь трудоемок и требует
много времени.
Выбор индикаторных электродов можно спрогнозировать. Прежде всего, выбор
электрода зависит от типа химической реакции, используемой в
потенциометрическом титровании (кислотно-основные реакции, осаждения, осаждения
- комплексообразования, образования растворимых комплексных соединений,
окисления - восстановления), констант равновесия соответствующих реакций,
оптимальных условий их протекания, природы материла индикаторных электродов и
их типов.
В статье обсуждаются только классические обратимые электроды,
подразделяющиеся на две группы:
электроды, возникновение потенциала на которых обусловлено протеканием на
них электродных электрохимических реакций с участием материала электродов и
электролитов; электроды, возникновение потенциала на которых определяется не
электрохимическими реакциями. К первым относятся электроды I, II и III родов
различных типов (из металлов, металлоидов, оксидов, газов). Ко вторым относятся
ионоселективные и окислительно-восстановительные электроды [1, c. 144].
По записям электрохимических систем электродов (полуэлементов) в учебной
и научной литературе имеются для металлических электродов I рода четыре
варианта: 1) Mn+, M; 2) Mn+/M; 3) M/Mn+;4) M, Mn+, для электродов II рода -
семь вариантов. Для самого используемого хлорид серебряного электрода:
) Cl-/AgCl, Ag; 2)
Ag/AgCl/Cl-;
3) Ag, AgCl, Cl-;
4) AgCl, Ag, Cl-; 5) AgCl/Cl-, Ag; 6) AgCl, Cl-/Ag;
7) Cl-, AgCl/Ag.
Для электродов III рода - 9 вариантов.
С участием хлорид ионов такие же варианты, как для электродов II рода, но
чаще встречаются Pb2+/PbCl2, AgCl, Ag; Pb2+, PbCl2, AgCl/Ag и у Лайтинена
единственный вариант для ртутнокомплексонатного электрода Mn+, MY2-, HgY2-,
Hg2+/Hg, т.е. Pb2+, PbCl2, AgCl, Ag+/Ag. Но во всех этих вариантах нет
обоснования форм записи. Для электродов I рода единственно приемлемой является
запись электрохимической системы электрода Ag+/Ag, так как все электродные
системы являются окислительно-восстановительными. А они записываются как
отношение окисленной формы к восстановленной, т.е. Ag+/Ag. Для электродов II
рода запись такая же, как для электродов I рода, но добавляется ионная форма
окисленного серебра. Так как на поверхности серебра существует его окисленная
форма, но ее концентрация лимитируется произведением растворимости AgCl и
концентрацией Cl- ионов, т.е. Cl-, AgCl, Ag+/Ag.
Для электродов III рода запись электрохимической системы электродов такая
же, как для электродов II рода, только добавляется еще и форма малорастворимого
соединения «чужого» иона относительно материала электрода (Ag) и «чужой» ион.
Если рассматривать прогноз выбора индикаторных электродов в
потенциометрических титрованиях в зависимости от типа химических реакций, то он
будет разным в зависимости от конкретного типа реакции.
Реакции осаждения, осаждения - комплексообразования. Реакции осаждения и
осаждения - комплексообразования сопровождаются образованием малорастворимых
солей или малорастворимых внутрикомплексных соединений. Константа равновесия
образования этих солей называется произведением растворимости (ПР). В литературе
для описания констант равновесия малорастворимых внутрикомплексных соединений
используют понятие «ионное произведение», которое отличается от ПР тем, что
прочность комплексных солей зависит от равновесной концентрации иона
комплексообразователя и лиганда. При определении растворимости
внутрикомплексного соединения необходимо учитывать ионную и молекулярную
растворимость, а также равновесные концентрации ионов комплексообразователя и
лиганда, которые больше по величине, чем для малорастворимых солей, при растворимости
которых образуются только ионы.
При потенциометрических титрованиях прогнозируется использование
электродов из металлов, ионы которых образуют наиболее прочные соединения с
ионами титранта, и чем прочнее эти соединения, тем больше величина скачка
потенциала. При этом электроды из этих металлов могут образовывать
электрохимические системы электродов I рода Ag+/Ag (Hg2+/Hg, Hg2 2+/Hg,
Pd2+/Pd) до точки стехиометричности (т.с.). В этом случае титруются ионы,
одноименные с материалом электрода. За т.е. происходит смена электродных
реакций и образуется система электрода II рода, обратимого относительно аниона
титранта A-, AgA, Ag+/Ag; A-, HgA2, Hg2+/Hg и т.д.
Пример. В растворе протекает химическая реакция
Cu 2+ + 2A- = CuA2↓
Используется индикаторный электрод из серебра.
. До начала титрования и до т.с. электрод работает как электрод I рода
Ag+/Ag.
. За т.с. происходит смена электродной реакции, и электрод работает как
электрод II рода A-, AgA, Ag+/Ag.
При образовании электродов I и II рода возникают наибольшие скачки
потенциала в процессе потенциометрического титрования.
Титрование ионов, не одноименных с материалом электрода, приводит к
образованию системы электрода I рода до начала титрования, в процессе
титрования до т.с. - системы электрода III рода, а за т.с. - II рода.
Возникновение системы электрода I рода до начала титрования объясняется
тем, что в любом электролите на поверхности металлического электрода всегда
присутствует его окисленная форма. В случае электрода из серебра - Ag+/Ag. С начала
титрования и до т.с. образуется электрохимическая система электрода III рода,
обратимая относительно «чужого» иона по отношению к материалу электрода. При
титровании соли меди (II) с использованием вышеуказанного индикаторного
электрода из серебра до т.с. образуется электрохимическая система Cu2+, CuA2,
AgA, Ag+/Ag, а за т.с. система электрода II рода (при избытке А-) - A-, AgA, Ag+/Ag.
Пример. В растворе протекает химическая реакция
2 Cu 2+ + 2A- = CuA2↓
опр. титр.
Используется индикаторный электрод из серебра.
. До начала титрования в растворе присутствуют только ионы Cu2+ и более
электроактивные ионы серебра, присутствующие на поверхности электрода, образуют
электрохимическую систему электрода I рода - Ag+/Ag.
. С момента начала титрования и до т.с. в растворе присутствует избыток
ионов Cu2+, осадок CuA2 и небольшое количество ионов A-, образующиеся в
результате диссоциации комплексного соединения↔ Cu2+ + 2 А-. Поэтому на
поверхности электрода образуется пленка из Ag+ + А- ↔ AgA и возникает
электрохимическая система электрода III рода - Cu2+,CuA2, AgA, Ag+/Ag.
. За т.с. в растворе находится избыток ионов титранта и возникает
электрохимическая система электрода II рода - A-, AgA, Ag+/Ag.
При использовании в качестве потенциометрического титранта диэтилдитиокарбамината
наряду с индикаторным электродом из серебра можно применять электрод из ртути,
так как ртутный комплекс диэтилдитиокарбамината более устойчив, чем комплекс
серебра. Следовательно, скачок потенциала на электроде из ртути будет больше, чем
на электроде из серебра. Кроме того, можно использовать индикаторный платиновый
окислительно-восстановительный электрод, так как реагент является слабым
восстановителем. Реагент в своем составе всегда содержит следы окисленной формы
и при хранении его концентрация увеличивается, так как он окисляется
атмосферным кислородом. Диэтилдитиокарбаминат - ионы окисляются по следующему
уравнению:
(C2H5)2NC(S)S- - 2e ↔ (C2H5)2NC(S)S-S(S)CN(C2H5)2
восстановленная форма окисленная форма
Реакционноспособной является восстановленная форма реагента. При
титровании ионов Cu2+ и всех сульфидобразующих ионов происходит медленное
накопление окисленной формы реагента, а восстановленная форма появляется после
т.с. Потенциал платинового индикаторного электрода до начала титрования и до
т.с. мало меняется. За т.с. при избытке реагента резко изменяется соотношение
окисленной формы к восстановленной форме (избыток восстановленной формы резко
возрастает) и возникает скачок потенциала ΔЕpt.
В таких случаях выбирают электрод, на котором возникает наибольший скачок
ΔЕинд., его можно рассчитать
теоретически или определить экспериментально.
. Что называют абсолютным и относительным показателями преломления в
рефрактометрии? Для анализа каких веществ используют этот метод в пищевой промышленности
Ответ: Показатель преломления зависит от оптических свойств и той среды,
из которой луч падает, и той среды, в которую он проникает. Показатель
преломления, полученный в том случае, когда свет из вакуума падает на
какую-либо среду, называется абсолютным показателем преломления данной среды.
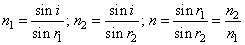 ; (8.1.)
; (8.1.)
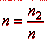 ; (8.2.)
; (8.2.)
Наоборот, при переходе из второй среды в первую имеем относительный
показатель преломления (рис. 8):
 ; (8.3.)
; (8.3.)
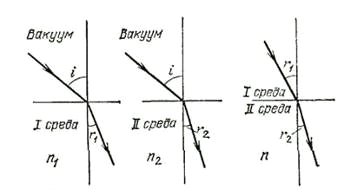
Рис. 8. Относительный показатель преломления двух сред
Установленная связь между относительным показателем преломления двух сред
и их абсолютными показателями преломления могла бы быть выведена и
теоретическим путем, без новых опытов, подобно тому, как это можно сделать для
закона обратимости.
Среда, обладающая большим показателем преломления, называется оптически
более плотной. Обычно измеряется показатель преломления различных сред
относительно воздуха. Абсолютный показатель преломления воздуха равен 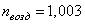 . Таким образом, абсолютный
показатель преломления какой-либо среды
. Таким образом, абсолютный
показатель преломления какой-либо среды  связан с ее показателем преломления
относительно воздуха
связан с ее показателем преломления
относительно воздуха 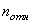 формулой:
формулой:
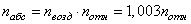 ; (8.4.)
; (8.4.)
В таблице 6 содержатся относительные показатели преломления, найденные
для ряда случаев преломления света на границе воздуха и соответствующей среды.
Таблица 6. Показатель преломления различных веществ относительно воздуха
|
Жидкости
|
Твердые вещества
|
|
Вещество
|

|
Вещество
|
|
|
Вода
|
1,333
|
Сахар
|
1,56
|
|
Спирт этиловый
|
1,362
|
Алмаз
|
2,417
|
|
Сероуглерод
|
1,632
|
Рубин
|
1,76
|
|
Глицерин
|
1,47
|
Стекло (легкий крон)
|
1,57
|
|
Жидкий водород
|
1,12
|
Стекло (тяжелый флинт)
|
1,80
|
|
Жидкий гелий
|
1,028
|
Лед
|
1,31
|
Показатель преломления зависит от длины волны света, т. е. от его цвета.
Различным цветам соответствуют различные показатели преломления. Это явление,
называемое дисперсией, играет важную роль в оптике. Данные, приведенные в табл.
6, относятся к желтому свету.
Интересно отметить, что закон отражения может быть формально записан в
том же виде, что и закон преломления. Вспомним, что мы условились всегда
измерять углы от перпендикуляра к соответствующему лучу. Следовательно, мы
должны считать угол падения  и угол отражения
и угол отражения 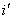 имеющими противоположные знаки, т.е.
закон отражения можно записать в виде:
имеющими противоположные знаки, т.е.
закон отражения можно записать в виде:
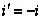 ; (8.5.)
; (8.5.)
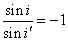 ; (8.6.)
; (8.6.)
Сравнивая (8.6.) с законом преломления, мы видим, что закон отражения
можно рассматривать как частный случай закона преломления при 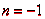 . Это формальное сходство законов
отражения и преломления приносит большую пользу при решении практических задач.
. Это формальное сходство законов
отражения и преломления приносит большую пользу при решении практических задач.
В предыдущем изложении показатель преломления имел смысл константы среды,
не зависящей от интенсивности проходящего через нее света. Такое истолкование
показателя преломления вполне естественно, однако в случае больших
интенсивностей излучения, достижимых при использовании современных лазеров, оно
не оправдывается. Свойства среды, через которую проходит сильное световое
излучение, в этом случае зависят от его интенсивности. Как говорят, среда
становится нелинейной. Нелинейность среды проявляется, в частности, в том, что
световая волна большой интенсивности изменяет показатель преломления.
Зависимость показателя преломления от интенсивности излучения  имеет вид:
имеет вид:
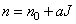 ; (8.7.)
; (8.7.)
Здесь 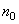 - обычный показатель преломления, а
- обычный показатель преломления, а 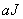 - нелинейный показатель преломления,
- нелинейный показатель преломления,
 - множитель пропорциональности.
Добавочный член в этой формуле может быть как положительным, так и
отрицательным.
- множитель пропорциональности.
Добавочный член в этой формуле может быть как положительным, так и
отрицательным.
Относительные изменения показателя преломления сравнительно невелики. При
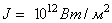 нелинейный показатель преломления
нелинейный показатель преломления 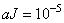 . Однако, даже такие небольшие
изменения показателя преломления ощутимы: они проявляются в своеобразном
явлении самофокусировки света.
. Однако, даже такие небольшие
изменения показателя преломления ощутимы: они проявляются в своеобразном
явлении самофокусировки света.
Прямой рефрактометрический анализ широко используется для определения
сухих веществ в продуктах кондитерской промышленности, соках овощей и плодов. В
спиртовой промышленности рефрактометры применяют для определения содержания
спирта в воде, а в комбинации с пикнометрическим анализом (по плотности)
определяют содержание различных спиртов в воде. В некоторых случаях
определяемое вещество извлекают из анализируемого объекта подходящим
растворителем и о концентрации определяемого вещества судят по изменению
коэффициента преломления растворителя. Таким путем определяют содержание жира в
продуктах кондитерского производства и в растениях, фенолов и оснований в
каменноугольной смоле и ее продуктах. Например, для определения жира продукты
кондитерского производства обрабатывают α-монобромнафталином. По изменению
коэффициента преломления вычисляют содержание жира. Аналогичным способом
определяют влагу в ячмене при контроле пивоваренного производства. Навеску
ячменя растирают с глицерином, и содержание влаги определяют по изменению
величины показателя преломления глицерина (изменение происходит вследствие
разбавления глицерина извлеченной из ячменя водой).
В ряде случаев определение показателя преломления используют для
идентификации органических веществ. Так, например, он является важным
показателем, характеризующим природу жиров, эфирных масел и других органических
веществ. При этом показатель преломления совместно с другими физико-химическими
характеристиками, например температурой плавления, плотностью, часто используют
для анализа смесей разных веществ.
. Какова природа происхождение атомных спектров
Ответ: Атомно-эмиссионный спектральный анализ состава вещества, в свою
очередь, основан на двух фундаментальных принципах:
. спектр, испускаемый предварительно возбужденными атомами и ионами
данного химического элемента, строго индивидуален (т.е. характерен только для
данного химического элемента);
. интенсивность линий этого спектра зависит от концентрации этого
элемента, определение которой и является целью анализа.
Спектр представляет собой распределение мощности излучения по длинам волн
и характеризуется зависимостью интенсивности от длины волны I (lambda).
Для получения эмиссионного спектра атомам анализируемого вещества
необходимо придать дополнительную энергию так, чтобы электроны перешли на более
высокие орбиты, т.е. перевести атомы в «возбужденное» состояние.
С этой целью анализируемую пробу вводят в источник возбуждения спектров,
где она подвергается абляции (т.е. «вырыванию» с поверхности микрочастиц),
нагреву и испарению. Источник возбуждения спектров тем или иным способом
формирует насыщенную энергией область пространства с достаточно высокой
температурой.
Попавшие в эту высокотемпературную область пространства микрочастицы
анализируемой пробы распадаются на атомы. Эти атомы пробы при столкновениях с
другими частицами переходят в возбужденное и ионизированное состояния. В таком
состоянии атомы и ионы могут находиться очень короткое время (10-8-10-7
с). Самопроизвольно возвращаясь в нормальное или промежуточное состояние, они
испускают избыточную энергию в виде фотонов, совокупность которых и образует
эмиссионный спектр.
Измеряя интенсивность линий спектра атомов (или ионов) того или иного
химического элемента, определяют концентрацию этого химического элемента в
анализируемой пробе.
В качестве источников возбуждения спектров в атомно-эмиссионном
спектральном анализе (АЭСА) наиболее широко применяют плазму (однако это не
исключает применения и пламён).
Напомним, что плазма является четвертым состоянием вещества (наряду с
твердым, жидким и газообразным), характерной особенностью которого является
обязательное наличие нейтральных частиц (атомов или молекул), положительно
заряженных ионов (атомарных или молекулярных, одно- или многозарядных) и
отрицательно заряженных электронов.
Для образования плазмы в какой-либо области пространства (заполненной
чаще всего некоторым газом, хотя это может быть и жидкость) к ней надо
приложить электрическое поле. Если поле имеет достаточную напряженность, чтобы
имеющиеся в пространстве свободные электроны на длине свободного пробега
приобрели энергию, достаточную для ионизации атомов (или молекул) окружающей
среды, то ускорившиеся таким образом электроны ионизируют атомы окружающей
среды с образованием ионов и вырванных из атомов дополнительных электронов.
Эти дополнительные электроны, в свою очередь, набирают энергию в поле и
тоже ионизируют атомы окружающей среды. Развивается лавинообразный процесс
образования ионов и дополнительных электронов.
Естественно, что идет и обратный процесс нейтрализации ионов при
столкновениях с электронами (рекомбинация ионов). В некоторый момент наступает
равновесие между этими процессами и образуется стационарная равновесная плазма
с определенным соотношением между концентрациями нейтральных атомов,
положительных ионов и отрицательных электронов.
Для плазм, применяемых в атомно-эмиссионном спектральном анализе,
характерными являются температуры от 3000-4000 K и выше вплоть до более
10 000 K.
Из сказанного очевидно, что существование плазмы возможно до тех пор,
пока в области пространства действует приложенное (внешнее) электромагнитное
поле, передающее свою энергию заряженным частицам. С прекращением воздействия
внешнего электромагнитного поля процесс рекомбинации ионов становится
превалирующим, плазма начинает распадаться и исчезает. Остается несветящийся
нагретый газ (если исходная область пространства была наполнена газом).
В плазме идут процессы не только ионизации атомов и рекомбинации ионов,
но и процессы возбуждения атомов и ионов (т.е. переходы внутренних орбитальных
электронов в более высокие энергетические состояния с поглощением энергии) и
процессы релаксации возбужденных атомов и ионов (т.е. обратные переходы из
более высоких энергетических состояний в более низкие с выделением энергии).
Вот эта выделившаяся в виде фотонов энергия в процессе релаксации возбужденных
атомов и ионов и формирует эмиссионный спектр.
10. Каким из методов - кондуктометрией,
потенциометрией или инверсионной вольтамперометрией можно определить ионы
цинка, концентрация которых в растворе равна 1·10-8 моль/дм3
Ответ: Инверсионная вольтамперометрия является одним из вариантов
электрохимических методов анализа, основанных на предварительном
концентрировании определяемого компонента. Предварительное концентрирование
осуществляется за счет перевода определяемого компонента из большого объема
раствора с малой концентрацией на поверхность или в малый объем электрода.
Перевод определяемого компонента из раствора на поверхность или в объем
электрода может быть осуществлен за счет протекания соответствующей
электрохимической реакции или за счет процесса адсорбции. После накопления на
поверхности или в объеме электрода определяемое вещество подвергается
электрохимическому превращению (восстановлению или окислению), причем этот
процесс можно проводить в разных режимах:
Существенными преимуществами инверсионных электрохимических методов
(ИЭАМ) перед другими методами определения следовых количеств неорганических и
органических веществ в растворах являются:
¾ возможность определения значительного числа химических
элементов Периодической системы и многих органических веществ;
¾ низкие пределы обнаружения, достигающие для некоторых
элементов (Cd, Bi, Tl, Pb, Sb, Ni) и органических веществ уровня 10-8
- 10-10 М;
¾ высокая селективность ИЭАМ и хорошие метрологические
характеристики методик на их основе;
¾ легкость компьютеризации и автоматизации аналитических
определений;
¾ относительная простота и сравнительная дешевизна приборов для
ИЭАМ.
В качестве рабочих электродов в инверсионной вольтамперометрии чаще всего
используют ртутный капельный электрод (висящая ртутная капля), ртутный
пленочный электрод, платиновый, стеклоуглеродный электроды, электрод из
графитовой пасты и другие. На ртутном электроде удобно проводить определение
концентрации катионов некоторых металлов, которые обратимо восстанавливаются в
определенной области потенциалов (от потенциала окисления ртути до потенциалов
восстановления фоновых катионов) и образуют со ртутью амальгамы. К таким
элементам относятся, в частности, Cu, Cd, Zn и Pb. Накопление определяемых
компонентов на электроде проводят при постоянном значении потенциала электрода.
Значение потенциала предварительного накопления должно быть таким, чтобы
процесс электровосстановления протекал на предельном токе (Id). При этом с
помощью перемешивания раствора поддерживают условия стационарной диффузии, т.е.
постоянные гидродинамические условия. В результате этого количество
восстановившегося за определенное время компонента оказывается прямо
пропорционально его исходной концентрации в растворе. По окончанию стадии
предварительного катодного накопления перед съемкой анодных вольтамперных
кривых перемешивание раствора прекращают, и в течение 10-15 сек осуществляют
стадию успокоения раствора. Процесс окисления образовавшейся амальгамы проводят
в неперемешиваемом электролите в потенциодинамическом режиме. Для увеличения
чувствительности метода при съемке вольтамперограмм используют дифференциальную
импульсную квадратно-волновую методику, которая позволяет исключить влияние
емкостного и фонового токов на получаемые зависимости.
11. Укажите типы радиоактивных превращений,
используемых в радиометрических методах анализа
Ответ: Радиометрический анализ, метод анализа химического состава
веществ, основанный на использовании радиоактивных изотопов и ядерных
излучений. В радиометрическом анализе для качественного и количественного
определения состава веществ используют радиометрические приборы. Различают
несколько способов радиометрического анализа. Прямое радиометрическое
определение основано на осаждении определяемого иона в виде нерастворимого
осадка избытком реагента известной концентрации, содержащего радиоактивный
изотоп с известной удельной активностью. После осаждения устанавливают
радиоактивность осадка или избытка реагента.
Радиометрическое титрование основано на том, что определяемый в растворе
ион образует с реагентом малорастворимое или легкоэкстрагируемое соединение.
Индикатором при титровании служит изменение, по мере введения реагента,
радиоактивности раствора (в 1-м случае) и раствора или экстракта (во 2-м
случае). Точка эквивалентности определяется по излому кривой титрования,
выражающей зависимость между объёмом введённого реагента и радиоактивностью
титруемого раствора (или осадка). Радиоактивный изотоп может быть введён в
реагент или определяемое вещество, а также в реагент и определяемое вещество.
Радиоактивность
(от лат. radio - излучаю, radius - луч и activus - действенный) -
самопроизвольное (спонтанное) превращение неустойчивого изотопа химического
элемента в другой изотоп (обычно - изотоп другого элемента). Сущность явления
радиоактивности состоит в самопроизвольном изменении состава атомного ядра,
находящегося в основном состоянии либо в возбуждённом долгоживущем
(метастабильном) состоянии. Такие превращения сопровождаются испусканием ядрами
элементарных частиц либо других ядер, например ядер 2He (α-частиц). Все известные типы радиоактивных превращений
являются следствием фундаментальных взаимодействий микромира: сильных
взаимодействий (ядерные силы) или слабых взаимодействий. Первые ответственны за
превращения, сопровождающиеся испусканием ядерных частиц, например α-частиц, протонов или осколков деления ядер: вторые
проявляются в β-распаде ядер. Электромагнитные взаимодействия
<http://dic.academic.ru/dic.nsf/bse/153572/%D0%AD%D0%BB%D0%B5%D0%BA%D1%82%D1%80%D0%BE%D0%BC%D0%B0%D0%B3%D0%BD%D0%B8%D1%82%D0%BD%D1%8B%D0%B5>
ответственны за квантовые переходы между различными состояниями одного и того
же ядра, которые сопровождаются испусканием гамма-излучения. Эти переходы не
связаны с изменениями состава ядер и поэтому, согласно современной
классификации, не принадлежат к числу радиоактивных превращений. Понятие
«Радиоактивности» распространяют также на β-распад нейронов.
Радиоактивность
следует отличать от превращений составных ядер, образующихся в процессе ядерных
реакций в результате поглощения ядром-мишенью падающей на него ядерной частицы.
Время жизни такого ядра значительно превышает время пролёта падающей частицей
расстояния порядка ядерных размеров (10-21-10-22 сек) и
может достигать 10-13-10-14 сек. Поэтому условно нижней
границей продолжительности жизни радиоактивных ядер считается время порядка 10-12
сек.
Типы
радиоактивных превращений. Все известные виды радиоактивности можно разделить
на две группы: элементарные (одноступенчатые) превращения и сложные
(двухступенчатые). К первым относятся: 1) Альфа-распад
<http://dic.academic.ru/dic.nsf/bse/63176/%D0%90%D0%BB%D1%8C%D1%84%D0%B0>,
2) все варианты Бета-распад
<http://dic.academic.ru/dic.nsf/bse/69035/%D0%91%D0%B5%D1%82%D0%B0>а (с
испусканием электрона, позитрона или с захватом орбитального электрона), 3) спонтанное
деление ядер, 4) протонная радиоактивность, 5) двупротонная радиоактивность, 6)
двунейтронная радиоактивность. В случае β-распада достаточно большое время жизни ядер обеспечивается природой
слабых взаимодействий. Все остальные виды элементарных радиоактивных процессов
обусловлены ядерными силами. Замедление таких процессов до промежутков времени ≥
10-12 сек вызвано наличием потенциальных барьеров (кулоновского и
центробежного), которые затрудняют вылет ядер или ядерных частиц.
К
двухступенчатым радиоактивным превращениям относят процессы испускания т. н.
запаздывающих частиц: протонов, нейтронов, α-частиц, ядер трития и 3He, а также запаздывающее спонтанное
деление. Запаздывающие процессы включают в себя β-распад как предварительную стадию, обеспечивающую
задержку последующего, мгновенного испускания ядерных частиц. Т. о., в случае
двухступенчатых процессов критерий радиоактивности относительно времени жизни
удовлетворяется только для первой стадии, благодаря её осуществлению за счёт
слабых взаимодействий.
Список
используемой литературы
1. Аверяскина Е.О., Ермаков С.С., Москвин Л.Н.
Определение ртути в воздухе методом инверсионной вольтамперометрии c
электрогенерированием йода / Тезисы VII конференции "Аналитика Сибири и
Дальнего Востока - 2004";
2. Алесковский В.Б., Бардин В.В., Булатов М.И.
Физико-химические методы анализа. Практическое руководство.- Л.: Химия, 1998.;
3. Баталова В.Н., Э.А. Захарова, Г.Б. Слепченко, В.М.
Пичугина, О.В. Рихерт. Аналитические проблемы определения ртути в пищевых продуктах
методом инверсионной вольтамперометрии / Тезисы VII конференции "Аналитика
Сибири и Дальнего Востока - 2004";
4. Березина Е.С., Киселева А.А., Филиппова Ю.В. /
Вестник Пермской государственной фармацевтической академии №2 - Пермь, 2007 г.,
с.123-125;
. Будников Г.К., Майстренко В.Н., Вяселев М.П. Основы
современного электрохимического анализа. М: Мир, 2003;
6. Булгакова О.Н., Халфина П.Д. Определение мышьяка в
воде / Тезисы VII конференции "Аналитика Сибири и Дальнего Востока -
2004";
7. Васильева В.П. Аналитическая химия. Книга 2.
Физико-химические методы анализа. Под ред. В.П. Васильева, - М.: Дрофа, 2004. -
384с.;
. Виноградова Е.Н., Галлай З.А., Финогенова З.М.
«Методы полярографического и амперометрического анализа», МГУ, 2003г.;
9. Глушко И.А. Применение инверсионной вольтамперометрии
в анализе кислотных вытяжек из почв для определения Cu, Pb и Cd. // Вестник
удмуртского университета / Химия 2005 №8 с. 88 - 101.;
. Гиндуллина Т.М. Хроматографические методы анализа:
учебно-методическое пособие /Т.М. Гиндуллина, Н.М. Дубова - Томск:Изд-во
Томского политехнического университета, 2010. - 80с.;
11. Дорохова Е.Н. Аналитическая химия. Физико-химические
методы анализа. Под ред. Е.Н. Дорохова, Г.В. Прохорова, - М.: Высш. шк.., 2001.
- 256с.;
. Дорохова Е.Н. Прохорова Г.В. Задачи и вопросы по
аналитической химии. - М.: Мир, 2001. - 267с. ил.;
. Захаров И.А., Тимофеев В.Н. Люминесцентные методы
анализа. - Л., 2008. - 95 с.;
. Зотов Ю.А., Дорохова Е.Н. Основы аналитической
химии. В 2 кн. Ю.А. Золотов, Е.Н. Дорохова, В.И. Фадеева и др.; Под ред. Ю.А.
Золотова. - М.: Высш. шк., 2006.;
15. Кальвода Р. Электроаналитические методы в контроле
окружающей среды/ Р. Кальвода, Я. Зыка, К- Штулик и др. Пер. с англ. Под ред.
Е.Я. Неймана. -М.: Химия, 1990. -240 с.;
. Каплан Б.Я., Ф. Выдра, К. Штулик, Э. Юлакова.
Инверсионная вольтамперометрия / пер. с чешского. В.А. Немова. // Под ред. Б.
Я. Каплана. М.: Мир, 2000. с. 257 - 265.;
. Каплин А.А., Катюхин В.E., Стромберг А.Г. Завод.
лаб., 2000, 36с.;
. Кнорре Д.Г., Крылова Л.Ф., Музыкантов В.С.
Физическая химия. - М.: Высш. шк., 2000. с. 190.;
. Кнунянц И.Л. Краткая химическая энциклопедия. Гл.
редактор И. Л. Кнунянц М.: Советская энциклопедия, 1961.;
. Комиссаренков А.А., Пругло Г.Ф. Кондуктометрия и
высокочастотное титрование: учебно-методическое пособие / Комиссаренков А.А.,
Пругло Г.Ф.; ГОУ ВПО СПбГТУРП СПб., 2009.-42с.;
. Курс аналитической химии: Учеб. для с.-х. вузов. -
6-е изд., испр. и доп. - М.: Высш. шк. 2004. - 495с.;
. Лакович Дж. Основы флуоресцентной спектроскопии. -
М.: Мир, 2006. - 496 с.;
. Патяковский В.М. Гигиенические основы питания и
экспертизы продовольственных товаров. - Новосибирск: Издательство
Новосибирского Университета, 2009. -431с.;
24. Плембэк Д. Электрохимические методы анализа. - М.:
Мир, 2005 - 496с.;
. Полторанина Т.Н., Соколова О.Ю., Халфина П.Д.
Инверсионно-вольтамперометрическое определение цинка, кадмия, свинца и меди при
совместном присутствии в различных объектах окружающей среды.;
26. Сизых А.Г., Слюсарева Е.А. Тушение люминесценции в жидких
растворах: Метод. указания / Красноярск. гос. ун-т.- Красноярск, 2003. - 26с.;
. Сонгина О.А. «Амперометрическое титрование», Химия,
2006. - 230с.;
. Столяров К.П., Григорьев Н.Н. Введение в
люминесцентный анализ неорганических веществ. - Л., 2007. - 364 с.;
. Чакчир Б.А., Алексеева Г.М. Фотометрические методы
анализа: Методические указания. - СПб.: Изд-во СПХФА, 2002.-44с.;
. Чеботарев В.К., Пасека А.Е., Терентьев Р.А.,
Полякова И.Ю., Шапоренко К.В. Прогнозирование выбора индикаторных электродов в
потенциометрических титрованиях. - М.: Мир, 2009. - 195с., ил.