Фармацевтическая химия противоопухолевых препаратов
Вступление
В медицинской практике все большее значение приобретает химиотерапия
злокачественных новообразований, которая возникла не так давно и в настоящее
время стала самостоятельным и перспективным направлением в онкологии.
Во многих странах мира проводятся интенсивные исследования по разработке
методов синтеза, изучению физико-химических и антнбластнческих свойств
соединений различных химических групп. Врачи-онкологи располагают значительным
количеством противоопухолевых препаратов, арсенал которых из года в год
пополняется за счет скрининга, осуществляемого онкологами-экспериментаторами во
многих странах мира. Среди оригинальных отечественных и зарубежных
антибластических средств различной природы большой удельный вес имеют
синтетические соединения.
Проблема злокачественных новообразований, несмотря на значительные
успехи, достигнутые в области изучения опухолевого процесса, разработки методов
профилактики, диагностики и лечения, и до настоящего времени является одной из
важнейших в современной медицине. Среди причин смертности злокачественные
опухоли и опухолевые заболевания кроветворной и лимфоидной тканей, по данным
мировой статистики, занимают второе место в высокоразвитых странах. Хотя
имеются существенные успехи в диагностике и лечении опухолей, количество
онкологических больных довольно велико. Это обусловлено увеличением числа
канцерогенных факторов, содержащихся в окружающей человека среде, недостаточным
знанием фундаментальной природы опухолевого процесса, сущности причин
превращения нормальной клетки в опухолевую, патогенеза новообразований, а также
условий, способствующих их развитию.
Разработка высокоэффективных средств и методов борьбы с опухолевой
болезнью является одной из кардинальных задач здравоохранения. Среди них особое
место занимают создание и внедрение в лечебную практику высокоэффективных
лекарственных средств для лечения онкологических заболеваний.
Применяемые в онкологической практике методы хирургического, лучевого и
лекарственного лечения часто не приводят к полному и стойкому излечению
больных. Поэтому изыскание новых эффективных средств борьбы с этим тяжким
недугом продолжает интенсивно развиваться.
Лекарственное лечение опухолей уже оформилось как самостоятельное и
перспективное направление в онкологии. Противоопухолевые препараты все более
широко используются как самостоятельно, так и в комплексном лечении больных.
Интерес к химиотерапии опухолей обусловлен не только значительными успехами,
которые достигнуты в онкологической практике, но и перспективами использования
ее как метода общего воздействия на опухолевый процесс.
Среди различных групп синтетических химических веществ, используемых для
лечения опухолевой болезни, наиболее важными продолжают оставаться алкилирующие
соединения и антиметаболиты.
Одними из первых в качестве противоопухолевых средств стали применять
вещества из группы алкилирующих соединений - производные бис-(β-хлорэтил)амина.
Знание физико-химических свойств, особенностей каждого препарата,
фармакотоксикологических характеристик, путей и скорости биотрансформации в
организме, фармакокинетики, несомненно, может помочь не только в установлении
зависимости между химическим строением и биологическим действием, разработке
наиболее рациональных лекарственных форм, схем, режимов и методов применения
противоопухолевых препаратов у больных, но и в изыскании новых путей синтеза
малотоксичных и высокоэффективных антибластических веществ.
Цель данной работы - осветить вопросы синтеза, физико-химические и
фармакологические свойства, а также методики идентификации препаратов
производных бис-(β-хлорэтил)амина.
В экспериментальной части работы проведен анализ таблеток циклофосфана
0,05 г, как представителя производных бис-(β-хлорэтил)амина.
Глава 1.
Классификация противоопухолевых препаратов
.1.
Направления развития терапии злокачественных опухолей
Последние несколько десятилетий лечение злокачественных опухолей
продвигалось по пути объединения всех существующих методов. Во многих случаях
главной составляющей лечения онкологических больных была и остается хирургия.
Лучевая и лекарственная противоопухолевая терапия (последняя включает в себя
все виды системного воздействия: химиотерапевтическое, эндокринное, иммунное, а
также генное и таргентное) используются и как дополнение к операции, и в
самостоятельном режиме. Позитивные результаты химиотерапии диссеминированного
рака многих локализаций послужили основанием для использования
противоопухолевых препаратов в адъювантном режиме с целью предупреждения
развития рецидивов и метастазов, а также в предоперационном (индукционном)
режиме, задачей которого является уменьшение первичной опухоли и ее метастазов,
улучшение отдаленных результатов лечения, выживаемости больных и качества их
жизни. Достижения индукционной лекарственной терапии позволяют проводить при
ряде злокачественных новообразований органосохраняющие операции.
Основные тенденции в развитии лекарственного лечения злокачественных
опухолей включают в себя следующие аспекты:
· значительное расширение классификации противоопухолевых агентов;
· нарастание каскада новых противоопухолевых агентов,
направленных на молекулярные и генетические мишени опухолевого роста;
· внедрение средств биологической терапии (новые виды
иммунотерапии, моноклональные антитела, ингибиторы металлопротеиназ, киназ,
ангиогенеза, дифференцирующие агенты и др.);
· изменение традиционной методики испытаний новых
противоопухолевых препаратов (I-III фаза) и оценки их клинической эффективности
(оценка длительности стабилизации);
· расширение исследований по неоадъювантной химиотерапии и
расширение показаний, совершенствование режимов адъювантной химиотерапии;
· предварительное определение наличия или активности мишени
опухолевого роста (ферментов, рецепторов, онкогенов, антигенов и др.) с целью
определения чувствительности к химиотерапии;
· комбинация биологических и цитотоксических агентов;
· путь развития: от неспецифических антипролиферативных
лекарств к целевой (targeted) терапии.
Противоопухолевые лекарственные средства разделяют на ряд групп, исходя
из их химической структуры, механизма действия, источников получения:
алкилирующие вещества, антиметаболиты, антибиотики, агонисты и антагонисты
гормонов, алкалоиды и другие средства растительного происхождения.
1.2
Алкилирующие средства
Одними из первых в качестве противоопухолевых средств стали применять производные
бис-(β-хлорэтил)амина.
Поводом к использованию этих соединений послужили данные, полученные в
40-х годах XX в., во время Второй мировой войны, когда подробно изучалось
влияние на организм боевых отравляющих веществ: иприта (или бис-β-хлорэтилсульфида) и азотистого иприта
(или трихлорэтиламина). Уже ранее, в 1919 г., установили, что азотистый иприт
вызывает выраженную лейкопению и аплазию костного мозга. Дальнейшие
исследования показали, что азотистый иприт оказывает специфическое
цитотоксическое влияние на лимфоидные ткани и обладает противоопухолевой
активностью при лимфосаркоме у мышей. Клинические испытания трихлорэтиламина
были начаты в 1942 г., что явилось началом эры современной химиотерапии
опухолей.
В настоящее время в медицинской практике используются менее токсичные
производные бис-(β-хлорэтил)амина (сарколизин, хлорамбуцил,
циклофосфамид, эмбихин и др.). Вслед за бис-(β-хлорэтил)аминами были получены
цитостатические алкилирующие соединения других химических групп: этиленимины и
этилендиамины (тиофосфамид), алкилированные сульфонаты, производные
метансульфоновой кислоты (бусульфан), триазены, препараты платины,
нитрозомочевины (ломустин, кармустин, фотемюстин, нитрозометилмочевина) и др.
В основе механизма действия алкилирующих веществ лежит образование в
нейтральных или щелочных растворах высокореактивных четвертичных аммониевых
(или им подобных) катионов, образующих ковалентные связи с нуклеофильными
соединениями, в т.ч. с такими биологически важными группами, как фосфатные,
аминные, сульфгидрильные, имидазольные и др. Цитотоксическое действие
алкилирующих соединений обусловлено, в первую очередь, алкилированием
структурных элементов ДНК (пуринов, пиримидинов) и РНК (в меньшей степени), в
результате чего нарушается стабильность, вязкость, целостность нитей ДНК и РНК,
жизнедеятельность клеток, блокируется участие различных групп в метаболических
реакциях, нарушается митотическое деление и репликация клеток. Клетки при
воздействии алкилирующих соединений останавливаются в G1 фазе и не
входят в S-фазу. Высокой чувствительностью к этим веществам обладают ядра
клеток гиперплазированных (опухолевых) тканей и лимфоидной ткани.
Большая часть хлорэтиламинов применяется при гемобластозах
(лимфогранулематоз, лимфо- и ретикулосаркома, хроническая лейкемия). Одним из
первых препаратов данной группы, синтезированных на заре химиотерапии, является
сарколизин, который активно использовался в 60-х годах прошлого века при
метастатической семиноме, саркоме Юинга, миеломной болезни, лимфо- и
ретикулосаркомах. В настоящее время из группы хлорэтиламинов наиболее широко
применяются циклофосфамид и ифосфамид. Эти лекарственные средства нашли свое
место при ряде солидных новообразований - раке яичника, молочной железы,
саркомах мягких тканей, мелкоклеточном раке легкого. Циклофосфамид используется
также при гемобластозах - остром лимфолейкозе, множественной миеломе.
Этиленимины (тиотепа, дипин, бензотэф, имифос и др.) по механизму
действия близки к производным бис-(β-хлорэтил)амина. Они блокируют
митотическое деление клеток, образуя поперечные связи между цепями молекулы
ДНК, что препятствует ее репликации. Область применения этих лекарственных
средств в связи с появлением новых противоопухолевых препаратов несколько
сократилась. Уже вышли из клинической практики отдельные препараты этой группы
- тиодипин и дийодбензотэф. В настоящее время из производных этиленимина в
основном используется тиотепа (рак яичников, молочной железы, опухолевые
серозиты).
Механизм противоопухолевого действия производных платины (карбоплатин,
цисплатин) связан со способностью к бифункциональному алкилированию нитей ДНК,
ведущему к длительному подавлению биосинтеза нуклеиновых кислот и гибели
клеток.
Производные платины не обладают специфичностью в отношении клеточного
цикла, действуют в фазе G0, на первом этапе тормозят синтез ДНК, РНК
и белка, а на втором образуются метаболические продукты, действующие только на
синтез ДНК.
Появление платиновых производных в конце 70-х годов прошлого века
значительно изменило представление о возможностях химиотерапии злокачественных
новообразований.
Лекарственные средства из группы производных нитрозомочевины также
обладают широким спектром цитотоксической активности. Противоопухолевый эффект
(цикло- и фазонеспецифичный) обусловлен переносом алкильных групп на нуклеофильные
центры ДНК, РНК, белков и алкилированием их молекул, что приводит к гибели
опухолевой клетки. Отличительной особенностью препаратов данной группы является
их способность проходить через ГЭБ и отсутствие перекрестной резистентности с
производными хлорэтиламинов и этилениминов. Они эффективны при первичных и
метастатических опухолях головного и спинного мозга.
Производные метансульфоновой кислоты (бусульфан и др.), применяемые при
обострениях хронического миелолейкоза, подвергаются внутримолекулярной
циклизации с образованием иона этиленимина, способного прямо или через
образование карбокатиона переносить алкильную группу на клеточные структуры.
Действуют в первой половине фазы G1, в начале и в конце S и G2
фаз, синхронизируя фазы клеточного цикла раковых клеток. Высокореактивные
группы бусульфана образуют с нуклеофильными центрами ДНК и РНК прочные
ковалентные связи. При этом сшиваются витки и спирали ДНК, утрачивается
возможность ее репликации, происходят разрывы цепей, наступают митотические блоки,
учащаются мутации.
Наряду со специфическим тормозящим влиянием на опухоли, алкилирующие
соединения, как и другие противоопухолевые средства, действуют на различные
ткани и системы организма, что обусловливает их побочные эффекты. Одним из
основных побочных эффектов является угнетающее влияние на кроветворение в
результате взаимодействия с нуклеопротеидами клеточных ядер кроветворных
тканей. Возникает лейкопения, тромбоцитопения, анемия, что требует специального
внимания и точного регулирования доз и режима применения препаратов. С целью
повышения эффективности и переносимости противоопухолевых препаратов используют
дополнительные лекарственные средства. Так для стимуляции кроветворения
применяют цитокины - гранулоцитарно-макрофагальный и гранулоцитарный
колониестимулирующие факторы (филграстим, молграмостим), а также ряд
интерлейкинов и эритропоэтин. Необходимо учитывать, что угнетение гемопоэза
усиливается при комбинированных схемах лечения - сочетании нескольких
противоопухолевых препаратов, лучевой терапии и др.
При применении хлорэтиламинов, платиновых производных часто наблюдается
тошнота, рвота, потеря аппетита, диарея. Для купирования данных эффектов
используют антагонисты серотониновых 5-HT3-рецепторов и блокаторы
дофаминовых D2-рецепторов. При попадании на кожу и слизистые
оболочки возможно раздражающее действие и развитие абсцессов, при введении
растворов под кожу возможна некротизация тканей.
Одной из характерных особенностей противоопухолевых препаратов является
их иммунодепрессивное действие, которое может ослабить защитные силы организма
и облегчить развитие инфекционных осложнений. Для предупреждения возможных
инфекций, обусловленных подавлением иммунитета, возможно использование
антибиотиков.
Следует отметить, что применение алкилирующих соединений, как и других
противоопухолевых препаратов, осуществляется только по назначению
врача-онколога и в специализированном лечебном учреждении. В зависимости от
особенностей заболевания, его течения, эффективности и переносимости
применяемых противоопухолевых препаратов могут меняться: схема их применения,
дозы, сочетание с другими препаратами.
.3
Антиметаболиты
Антиметаболитами называют вещества, близкие по химической структуре к
эндогенным продуктам метаболизма и ингибирующие, в результате конкурентных
отношений, определенные биохимические процессы, что сопровождается нарушением
функции клеток и торможением клеточного роста.
Противоопухолевую активность антиметаболитов обнаружили в начале 60-х гг.
Оказалось, что метотрексат, являющийся антиметаболитом фолиевой кислоты,
эффективен при некоторых опухолях человека, особенно при хориокарциноме у
женщин и при острой лейкемии. Позднее в медицинской практике появились и другие
антиметаболиты - аналоги пурина и пиримидина.
К антиметаболитам, применяемым в качестве противоопухолевых средств,
относят структурные аналоги фолиевой кислоты (метотрексат), пуринов
(меркаптопурин, тиогуанин и др.), пиримидинов (фторурацил, тегафур, цитарабин и
др.).
Цитостатическое действие всех этих соединений связано с нарушением синтеза
нуклеиновых кислот (ДНК и РНК). Антиметаболиты являются фазоспецифичными
средствами - они преимущественно действуют в S-фазе клеточного цикла.
Антагонистом фолиевой кислоты является метотрексат. Он связывается с
активным каталитическим центром и ингибирует активность фермента
дигидрофолатредуктазы, восстанавливающего дигидрофолат до активной формы -
тетрагидрофолата, который является коферментом и играет роль переносчика
одноуглеродных групп (метильная, метиленовая, метенильная и др.) во многих
ферментативных реакциях. Нехватка тетрагидрофолата приводит к нарушению синтеза
тимидилата, пиримидиновых нуклеотидов, аминокислот серина и метионина, в
результате чего происходит ингибирование синтеза ДНК, РНК и белка. Поскольку
метотрексат оказывает действие в S-фазе клеточного цикла, он наиболее активен в
отношении тканей с высокой скоростью пролиферации клеток, таких как опухолевая
ткань, костный мозг, клетки слизистой оболочки ЖКТ, мочевого пузыря и др.
Метотрексат обладает широким спектром противоопухолевой активности. Основными
показаниями к его назначению являются лейкозы, лимфомы, хорионэпителиома матки,
рак молочной железы, рак легких, рак яичника, мочевого пузыря и др.
Аналоги пуринов нарушают биосинтез пуриновых нуклеотидов, входящих в
состав нуклеиновых кислот - аденозин−5`-монофосфата (АМФ) и гуанозин−5`-монофосфата
(ГМФ).
Меркаптопурин - первый из тиопуринов, у которого была обнаружена
противоопухолевая активность. По строению 6-меркаптопурин близок к аденину,
гипоксантину, гуанину.
Цитотоксическая активность проявляется после активации в тканях и
обусловлена конкуренцией с гипоксантином и гуанином за фермент
гипоксантин-гуанинфосфорибозилтрансферазу. Трансформируется в
меркаптопуринфосфорибозил, который под влиянием тиопуринметилтрансферазы
превращается в метилмеркаптопурин. Оба они угнетают
глутамин-5-фосфорибозилпирофосфатамидотрансферазу - первый фермент в синтезе
пуриновых рибонуклеотидов de novo. В результате нарушается синтез нуклеиновых
кислот (более выражено ингибирование синтеза ДНК, чем РНК), нарушается
митотический цикл (S-фаза), особенно в быстро пролиферирующих клетках опухолей
и костного мозга.
Другой антиметаболит из группы аналогов пуринов - тиогуанин - по
структуре и механизму действия близок к меркаптопурину. Наибольшее применение
антагонисты пурина нашли при лейкозах.
Флударабин - фторированный аналог аденина (фторированный нуклеотидный
аналог противовирусного агента видарабина). В организме быстро
дефосфорилируется до 2-фтор-арабинофуранозиладенина, который захватывается
клетками. Внутри клетки фосфорилируется дезоксицитидинкиназой до активного
трифосфата. Этот метаболит ингибирует рибонуклеотидную редуктазу,
ДНК-полимеразу (альфа, дельта и эпсилон), ДНК-праймазу, ДНК-лигазу и блокирует
синтез ДНК. Кроме того, противоопухолеый эффект частично обусловлен связыванием
РНК-полимеразы II и торможением синтеза белка. Широко применяется для лечения
лимфопролиферативных заболеваний (в т.ч. В-клеточный хронический лимфолейкоз,
неходжкинские лимфомы низкой степени злокачественности).
Структурные аналоги пиримидина (фторурацил, тегафур, цитарабин и др.)
являются антиметаболитами пиримидиновых оснований (цитозин, тимин, урацил),
входящих в состав нуклеотидов.
Противоопухолевое действие аналогов пиримидинов обусловлено их
превращением в опухолевых клетках в активные ингибиторы ферментов -
тимидилатсинтетазы (фторурацил и его аналоги, ралтитрексид и др.),
ДНК-полимеразы (цитарабин), рибонуклеотидредуктазы (гидроксикарбамид и др.),
участвующих в синтезе нуклеиновых кислот.
Фторурацил (антиметаболит урацила) был создан в 1962 г. Активность
фторурацила обусловлена его биотрансформацией в тканях в активные формы. В
процессе внутриклеточных превращений 5-фторурацила (5-ФУ) образуются
цитостатически активные метаболиты (5-фтор-2`-дезоксиуридин монофосфат и 5-фторуридина
трифосфат). Возможны два механизма повреждения клеток под действием активных
метаболитов 5-фторурацила. Во-первых, 5-фтор−2`-дезоксиуридин монофосфат
и фолатный кофактор N5-10метилентетрагидрофолат ковалентно
связываются с тимидилатсинтетазой с образованием единого комплекса, что
приводит к подавлению образования тимидилата - предшественника тимидина
трифосфата, необходимого для синтеза ДНК. Во-вторых, в процессе синтеза РНК
транскрипционные ферменты ядра могут ошибочно включить в нее 5-фторуридина
трифосфат вместо уридина трифосфата, что приводит к нарушению процессинга РНК и
синтеза белка.
Вводят фторурацил внутривенно, т.к. он плохо всасывается из ЖКТ. Он имеет
высокую токсичность, проявляющуюся миелосупрессией, язвенным поражением
слизистых оболочек пищеварительного тракта (в т.ч. язвенный стоматит, энтерит и
др.). По спектру противоопухолевого действия фторурацил отличается от аналогов
фолиевой кислоты (метотрексат) и пуринов (меркаптопурин и др.) и проявляет
наибольшую активность при лечении колоректального рака, злокачественных
опухолей молочной железы, желудка, поджелудочной железы и др.
Аналогичными фторурацилу свойствами обладает тегафур (пролекарство),
фтористое производное пиримидина, который в организме гидролизуется с
образованием фторурацила. По сравнению с фторурацилом тегафур является менее
токсичным соединением.
Новой модификацией фторурацила является еще одно производное
фторпиримидина - капецитабин, который, в отличие от фторурацила, применяется
как пероральный цитостатик. В организме под влиянием тимидинфосфорилазы
капецитабин превращается в 5-фторурацил (5-ФУ). Последовательная ферментная
биотрансформация капецитабина в 5-ФУ создает более высокие его концентрации в
тканях опухоли, чем в окружающих здоровых тканях.
Механизм противоопухолевого действия тегафура и капецитабина, таким
образом, обусловлен образованием фторурацила и его последующей
биотрансформацией.
Цитарабин после активации в тканях образует цитарабин-5`-трифосфат,
который конкурентно ингибирует ДНК-полимеразу (фермент, катализирующий реакцию
синтеза ДНК из предшественников - дезоксирибонуклеозид-5`-трифосфатов), что
приводит к ингибированию синтеза ДНК. Кроме того, цитарабин незначительно
влияет на синтез РНК (может встраиваться в ДНК и РНК). Цитарабин обладает противолейкозной
активностью. Особенно активен в отношении миелобластов, лимфобластов,
лимфоцитов, в меньшей степени - гранулоцитов, эритроцитов и тромбоцитов.
Гидроксикарбамид был синтезирован в 1869 г., но его противоопухолевая
активность в клинических исследованиях была доказана только в 80-х гг.
Цитотоксическое действие гидроксикарбамида обусловлено ингибированием фермента
рибонуклеотидредуктазы, в результате нарушается синтез ДНК при отсутствии
эффекта на синтез РНК и белка.
К новым структурным аналогам антиметаболитов, полученным путем
направленного химического синтеза, принадлежат гемцитабин и ралтитрексед.
Гемцитабин является нуклеозидным (дезоксицитидин) аналогом. Имеет фазовую
специфичность действия: останавливает жизнедеятельность клеток в S-фазе и блокирует
опухолевую прогрессию клеток в G1/S-фазе. Гемцитабин подвергается
внутриклеточному метаболизму под действием нуклеозидкиназ с образованием
активных ди- и трифосфатного нуклеозидов. Цитотоксический эффект обусловлен
комбинированным влиянием этих активных метаболитов. Дифосфатные нуклеозиды
ингибируют рибонуклеотидредуктазу, катализирующую реакции образования
дезоксинуклеозидтрифосфатов, необходимых для синтеза ДНК. Трифосфатные
нуклеозиды активно конкурируют с дезоксицитидинтрифосфатом за встраивание в
молекулы нуклеиновых кислот. После встраивания внутриклеточных метаболитов
гемцитабина в цепь ДНК, к ее растущим нитям добавляется еще один дополнительный
нуклеотид, что приводит к полному ингибированию дальнейшего синтеза ДНК и
обусловливает невозможность репарации ДНК. Гемцитабин эффективен при раке
поджелудочной железы, немелкоклеточном раке легких, раке мочевого пузыря и др.
Ралтитрексед специфически ингибирует тимидилатсинтетазу - ключевой
фермент в синтезе тимидинтрифосфата (необходим для синтеза ДНК), вызывает
фрагментацию ДНК и гибель клетки. Применяется при раке толстой кишки.
В целом, антиметаболиты обладают выраженным противоопухолевым действием и
эффективны при ряде злокачественных новообразований. Некоторые из них обладают
иммуносупрессивными свойствами (метотрексат, цитарабин и др.). В настоящее
время уточняются клеточные механизмы действия известных антиметаболитов и
ведется направленный поиск новых соединений этой группы.
.4
Противоопухолевые антибиотики
Первый противоопухолевый антибиотик - дактиномицин - был получен в 1963
году. В последующем скрининг продуктов жизнедеятельности микробов привел к
открытию целого ряда эффективных химиотерапевтических противоопухолевых
препаратов, являющихся продуктами разных видов почвенных грибов или их синтетическими
производными.
В настоящее время из противоопухолевых антибиотиков наибольшее
практическое применение имеют антрациклины (антрахиноновые соединения),
блеомицин, относящийся к флеомицинам, дактиномицин, являющийся актиномицином, и
митомицин - своеобразный антибиотик с алкилирующим механизмом действия.
Антрациклиновые антибиотики (даунорубицин, доксорубицин, идарубицин,
карубицин и эпирубицин) относятся к наиболее эффективным противоопухолевым
средствам.
Структурной основой антрациклиновых противоопухолевых антибиотиков
является тетрагидротетраценхиноновый хромофор, состоящий из шестичленного
алифатического и трех ароматических колец. В химическом отношении они
отличаются друг от друга заместителями в хромофоре и наличием сахарных
остатков.
Механизм цитотоксического действия антрациклиновых антибиотиков связан,
главным образом, с ингибированием синтеза нуклеиновых кислот путем интеркаляции
между парами азотистых оснований, нарушением вторичной спирализации ДНК за счет
взаимодействия с топоизомеразой II, а также связыванием с липидами клеточных
мембран, сопровождающимся изменением транспорта ионов и клеточных функций.
Такой механизм обусловливает высокую антимитотическую активность при низкой
избирательности действия. Антрациклиновые антибиотики оказывают также
иммунодепрессивное (миелосупрессивное) и антибактериальное действие, однако в
качестве антимикробных средств не применяются.
Антрациклиновые противоопухолевые антибиотики применяют при многих
злокачественных новообразованиях - различных гематологических видах рака,
саркомах мягких тканей, карциномах и других сóлидных опухолях. Спектр показаний к
применению конкретного антибиотика определяется его химическим строением,
индивидуальными фармакокинетическими и фармакодинамическими свойствами и степенью
его изученности. Помимо терапевтического действия все противоопухолевые
антрациклиновые антибиотики вызывают ряд побочных эффектов, обусловленных
низкой избирательностью действия. Главным из этих эффектов является
потенциально необратимая кумулятивная дозозависимая кардиотоксичность, которая
предположительно вызвана свободнорадикальным повреждением клеточных мембран
миокарда. Антрациклиновые антибиотики обладают также эмбриотоксическими,
мутагенными и тератогенными свойствами. Применение их в комбинации с другими
противоопухолевыми средствами позволяет уменьшить дозы и снизить частоту и
выраженность токсических эффектов.
Блеомицин представляет собой смесь различных гликопептидов, продуцируемых
Streptomyces verticillus. Он также подавляет синтез нуклеиновых кислот (главным
образом ДНК) и белка, индуцируя фрагментацию ДНК с последующим образованием
свободных радикалов. Более активен на ранних стадиях опухолевого процесса,
относительно мало угнетает костно-мозговое кроветворение, не оказывает
существенного иммуносупрессивного действия. Применяют блеомицин главным образом
при комбинированном лечении тестикулярных видов рака, карцином и лимфом. Так же
как и другие противоопухолевые антибиотики, блеомицин вызывает ряд побочных
эффектов, наиболее тяжелыми из которых являются анафилактический шок,
респираторная токсичность и лихорадка.
Дактиномин, как и антрациклиновые антибиотики, встраивается между парами
азотистых оснований, образуя стойкий комплекс с ДНК, и нарушает ДНК-зависимый
синтез РНК. Применяют его в сочетании с хирургическим вмешательством, лучевой
терапией и/или в комбинации с винкристином, циклофосфамидом и метотрексатом при
лечении опухоли Вильмса, рабдомиосаркомы, хориокарцином и некоторых других
видов опухолей. Основной дозозависимый токсический эффект дактиномицина -
угнетение функции костного мозга, вплоть до развития апластической анемии.
Митомицин, в отличие от других противоопухолевых антибиотиков, проявляет
свойства алкилирующего агента, вызывая избирательное ингибирование синтеза ДНК,
а в высоких концентрациях и супрессию клеточной РНК и синтеза белка. Применяют
его в качестве вспомогательного средства при лучевой терапии и в комбинации с
другими противоопухолевыми средствами (в т.ч. и противоопухолевыми
антибиотиками) при лечении диссеминированных аденокарцином различной
локализации, хронического лимфо- и миелолейкоза. Основным побочным эффектом
митомицина является тяжелая миелосупрессия с относительно поздним токсическим
действием на все три ростковых элемента костного мозга.
Кроме названных антибиотиков противоопухолевой активностью обладает ряд
средств, продуцентами которых являются различные актиномицеты (оливомицин,
руфокромомицин, реумицин).
Взаимодействие между гормонами и гормонально зависимыми опухолями было
выявлено впервые в 1896 г., когда хирург из Глазго J. Beatson опубликовал
данные успешного лечения трех женщин с прогрессирующим раком молочной железы,
которым была произведена двусторонняя овариэктомия.
По механизму действия гормональные препараты отличаются от
цитотоксических противоопухолевых средств. Основная их роль - восстановление
нарушенной гуморальной регуляции функции клеток. Вместе с тем не исключается и
специфическое влияние на опухолевые клетки: они в определенной степени тормозят
деление клеток и способствуют их дифференцировке.
Эстрогены назначают для подавления действия в организме андрогенов
(например при раке предстательной железы), андрогены, напротив, - для
уменьшения активности эстрогенов (при раке молочной железы и др.). При раке
молочной железы и матки используют также прогестины (медроксипрогестерон).
К противоопухолевым гормональным средствам и антагонистам гормонов
относят:
. Андрогенные средства - тестостерон, метилтестостерон, дростанолон (медротестрона
пропионат), пролотестон.
. Эстрогенные средства - фосфэстрол, диэтилстильбэстрол, полиэстрадиола
фосфат, эстрамустин, этинилэстрадиол, хлоротрианизен, полиэстрадиола фосфат,
гексэстрол.
. Гестагенные средства (прогестины) - гестонорона капроат,
медроксипрогестерон, мегестрол и др.
. Антагонисты эстрогенов (антиэстрогены) - тамоксифен, торемифен.
. Антагонисты андрогенов (антиандрогены): бикалутамид, флутамид,
ципротерон и др.
. Гипоталамические факторы («рилизинг-факторы»), высвобождающие гормоны
гипофиза: бусерелин, гозерелин, лейпрорелин, трипторелин и др.
. Ингибиторы ароматазы (аминоглутетимид, анастрозол, эксеместан,
летрозол).
. Ингибиторы биосинтеза гормонов надпочечников (аминоглутетимид,
митотан).
. Глюкокортикоиды (преднизолон, дексаметазон и др.).
. Аналоги соматостатина (октреотид, ланреотид).
Андрогены иногда применяют при метастатическом раке молочной железы.
Назначают их женщинам с сохраненным менструальным циклом и в том случае, когда
продолжительность менопаузы не превышает 5 лет. Нежелательными эффектами
андрогенов, особенно при применении больших доз, являются вирилизация у женщин
(огрубение голоса, чрезмерный рост волос на лице), задержка воды и солей в
организме и др. Начало применения андрогенов (в частности тестостерона) для лечения
рака молочной железы относят к 40-м гг. XX в.
Начиная с 1951 г. при лечении рака молочной железы широко используются
прогестины. Гестагенные препараты применяют также для лечения рака эндометрия и
рака почек, но мало используют для лечения рака простаты.
Основным показанием к назначению эстрогенов, начало использования которых
в онкологической практике также относится к 40-м гг. XX в., является рак
предстательной железы. При раке молочной железы их теперь назначают очень
редко.
Важную роль в механизме действия гормональных препаратов играет их
связывание со специфическими рецепторами, обнаруженными в тканях и некоторых
опухолях.
Антиэстрогены конкурентно связываются с эстрогеновыми рецепторами в
органах-мишенях и препятствуют образованию эстрогенрецепторного комплекса с
эндогенным лигандом - 17-β-эстрадиолом. В результате они тормозят стимулируемый
эстрогенами рост опухоли. Чем больше эстрогенных рецепторов в опухоли - тем
благоприятнее результат лечения антиэстрогенами.
Эффективным антиэстрогеном является тамоксифен - эталонный препарат для
лечения рака молочной железы (особенно у женщин в менопаузе). Клиническое
использование тамоксифена было начато в 1973 г. В настоящее время тамоксифен
является широко используемым препаратом как для проведения адъювантной терапии,
так и при лечении больных с диссеминированным процессом. Показано, что
тамоксифен эффективен при всех стадиях заболевания, хорошо переносится при
приеме в терапевтических дозах. Кроме основного показания - рак молочной железы
у женщин - тамоксифен применяют при лечении рака грудной железы у мужчин, рака
эндометрия, рака предстательной железы, опухолей гипофиза и др.
К антиандрогенам относят ряд соединений стероидной и нестероидной
структур, способных подавлять физиологическую активность эндогенных андрогенов.
Их действие связано с конкурентным блокированием рецепторов андрогенов в
тканях-мишенях, биосинтез и секрецию андрогенов они не нарушают.
Антиандрогенное действие свойственно в той или иной степени ряду эндогенных
стероидных соединений, в т.ч. прогестинам, эстрогенам и их синтетическим
производным, а также некоторым производным самих андрогенов. Из стероидных
антиандрогенов наиболее известен ципротерон. В 70-х гг. XX в. появились
сообщения о высокой антиандрогенной активности нестероидных соединений -
производных карбоксианилида (флутамид и др.). Применяют антиандрогены в
основном при раке предстательной железы. Область их использования включает
также гиперандрогенные состояния у женщин (гирсутизм, облысение и др.), раннее
половое созревание у детей.
Среди антиандрогенов выделяют вещества, которые только блокируют
андрогенные рецепторы (т.н. чистые андрогены) - бикалутамид, флутамид, и
вещества, которые, кроме способности блокировать рецепторы, имеют гонадотропную
активность (т.н. антиандрогены двойного действия) - ципротерон.
Флутамид и бикалутамид блокируют связывание андрогенов с клеточными
рецепторами, вследствие чего препятствуют проявлению биологических эффектов
андрогенов в андрогенчувствительных органах, в т.ч. в клетках предстательной
железы, и таким образом препятствуют росту опухоли. После приема флутамида
отмечается повышение плазменных уровней тестостерона и эстрадиола.
Ципротерон обладает более выраженным андрогенным действием, т.к. помимо
блокирования действия дигидротестостерона на уровне рецепторов, подавляет
высвобождение гонадотропинов и, следовательно, синтез андрогенов. Одновременно
с тестостероном в крови снижается содержание ЛГ и ФСГ.
Особым видом антиандрогенной активности обладают соединения, ингибирующие
5-альфа-редуктазу - внутриклеточный фермент предстательной железы,
способствующий превращению тестостерона в более активный андроген -
дигидротестостерон (ДГТ). Одним из ингибиторов 5-альфа-редуктазы является
финастерид, применяющийся при лечении доброкачественной гиперплазии
предстательной железы.
Гипоталамические рилизинг-факторы - эндогенные пептидные соединения,
оказывающие влияние на высвобождение гипофизом гонадотропных гормонов (в т.ч.
лютеинизирующего и фолликулостимулирующего). В настоящее время в медицинских целях
используют не естественные рилизинг-факторы из гипоталамуса животных (овец,
свиней), а их синтетические аналоги. Аналоги (как агонисты, так и антагонисты)
полипептидных гормонов создают путем присоединения, выделения, замещения или
изменения некоторых аминокислот в полипептидной цепочке природного гормона.
Гонадотропин-рилизинг-гормон (ГнРГ) - гонадорелин, гонадолиберин,
гонадотропин-рилизинг фактор - один из представителей класса рилизинг-гормонов
гипоталамуса. ГнРГ в большей степени влияет на секрецию ЛГ, нежели ФСГ, поэтому
часто его называют также рилизинг-гормоном лютеинизирующего гормона (ЛГРГ).
ГнРГ представляет собой декапептид, состоящий из 10 аминокислот.
Установлено, что аминокислоты в положении 2 и 3 ответственны за биологическую
активность ГнРГ. Аминокислоты в положении 1, 6, 10 имеют структурную
конфигурацию, необходимую для связывания с рецепторами клеток гипофиза.
Замещение молекулы ГнРГ в положении 6 и 10 позволило создать агонисты
рилизинг-гормона.
Синтетические гонадолиберины - нафарелин, гозерелин, гистрелин,
лейпрорелин - аналоги гонадотропин-рилизинг-гормона - содержат D-аминокислоты в
положении 6 и этиламидзамещающий глицин в положении 10. Результатом замены
аминокислотных остатков в молекуле природного гормона является более выраженное
сродство к рецепторам ГнРГ и более продолжительный период полураспада, поэтому
аналоги имеют более сильное и более длительное действие, чем нативный
гонадотропин-рилизинг-гормон. Так, активность гозерелина превышает активность
нативного ГнРГ в 100 раз, трипторелина - в 36 раз, бусерелина - в 50 раз, а
T1/2 синтетических гонадотропинов - 90 - 120 мин - намного превышает T1/2
нативного ГнРГ.
В мировой клинической практике известно более 12 лекарственных
препаратов-аналогов ГнРГ: бусерелин, гистрелин, гозерелин, лейпрорелин,
лутрелин, нафарелин, трипторелин, фертирелин и др. В России зарегистрированы
лишь некоторые из них. Применяемые в России аналоги ГнРГ (гозерелин,
лейпрорелин, трипторелин, бусерелин) сходны по структуре, механизму действия,
основным фармакокинетическим и фармакодинамическим характеристикам, а также
клинической эффективности и безопасности.
Гонадорелин секретируется гипоталамусом не постоянно, а в импульсном
режиме, при этом пики следуют друг за другом с определенными интервалами, различными
у мужчин и женщин: у женщин ГнРГ выделяется каждые 15 мин (фолликулярная фаза
цикла) или 45 мин (лютеиновая фаза цикла и период беременности), у мужчин - 90
мин. ГнРГ обнаружен у всех млекопитающих. Пульсирующее выделение ГнРГ из
гипоталамуса поддерживает выработку гонадотропинов в гипофизе.
Аналоги ГнРГ были предложены для клинического использования в 80-е гг. XX
в. Эти лекарственные средства оказывают двухфазное действие на гипофиз:
взаимодействуя с рецепторами ГнРГ клеток передней доли гипофиза, вызывают
кратковременную стимуляцию с последующей длительной десенситизацией, т.е.
снижением чувствительности рецепторов аденогипофиза к ГнРГ. После однократной
инъекции аналога ГнРГ в результате стимулирующего эффекта усиливается секреция
из передней доли гипофиза ЛГ и ФСГ (проявляется повышением содержания
тестостерона в крови у мужчин и эстрогенов у женщин), обычно этот эффект
наблюдается в первые 7 - 10 дней. При постоянном длительном применении аналоги
гонадорелина подавляют высвобождение ЛГ и ФСГ, снижают функцию яичек и яичников
и, соответственно, содержание половых гормонов в крови. Эффект проявляется
примерно через 21 - 28 дней, при этом концентрация тестостерона в крови у
мужчин снижается до уровня, наблюдаемого после хирургической кастрации (т.н. «лекарственная
кастрация»), а уровень эстрогенов в крови у женщин - до уровня, наблюдаемого в
постменопаузе. Эффект является обратимым и после окончания приема препаратов
физиологическая секреция гормонов восстанавливается.
Применяют аналоги ГнРГ при раке предстательной железы - они способствуют
регрессу опухоли простаты. Женщинам назначают при гормонозависимых опухолях
молочной железы, эндометриозе, фиброме матки, т. к. они вызывают истончение
эндометрия, уменьшение симптоматики и размеров объемных образований. Кроме
того, аналоги ГнРГ применяют при лечении бесплодия (в программах
экстракорпорального оплодотворения).
Побочные эффекты этих ЛС, возникающие в начале лечения и обусловленные
временной стимуляцией гипофиза, проявляются в усилении симптомов, либо появлении
дополнительных симптомов основного заболевания. Эти явления не требуют отмены
препарата. Избежать их при лечении рака предстательной железы позволяет
одновременное назначение на 2 - 4 неделе антиандрогена.
Наиболее часто встречающимися нежелательными эффектами у мужчин являются
«приливы», снижение либидо, импотенция, гинекомастия. У женщин часто отмечаются
«приливы», усиление потоотделения и изменение либидо. При применении аналогов
ГнРГ у женщин существует риск усиления снижения плотности костных трабекул в
позвонках (может быть необратимым). За период 6-месячного лечения это снижение
плотности незначительно, за исключением больных с факторами риска (например
остеопороз).
Аналоги ГнРГ выпускаются в различных лекарственных формах - для п/к, в/м,
интраназального применения. Внутрь эти ЛС не назначаются, т.к. декапептиды
легко расщепляются и инактивируются в ЖКТ. Учитывая необходимость длительного
лечения, аналоги ГнРГ выпускаются также в виде лекформ пролонгированного
действия, в т.ч. микрокапсул, микросфер.
Большая скорость разрушения ГнРГ (2 - 8 мин) не позволяет использовать
его в клинической практике для длительного применения. Для ГнРГ величина T1/2
из крови составляет 4 мин, при п/к или интраназальном введении его аналогов -
примерно 3 ч. Биотрансформация осуществляется в гипоталамусе и гипофизе. При
почечной или печеночной недостаточности коррекции режима дозирования, как
правило, не требуется.
Ингибиторы ароматазы начали применяться в онкологической практике в
70-80-е гг. XX в. Ароматаза - цитохром Р450-зависимый фермент, отвечающий за
превращение синтезирующихся в коре надпочечников андрогенов в эстрогены.
Ароматаза присутствует в различных тканях и органах, включая яичники, жировую
ткань, скелетные мышцы, печень, а также ткань опухоли молочной железы. У женщин
в пременопаузе основным источником циркулирующих эстрогенов являются яичники,
тогда как в постменопаузе эстрогены образуются главным образом вне яичников.
Ингибирование ароматазы приводит к уменьшению образования эстрогенов у женщин
как в пременопаузе, так и в постменопаузе. Однако в пременопаузе снижение
биосинтеза эстрогенов компенсируется усилением синтеза гонадотропинов по
принципу обратной связи - снижение синтеза эстрогенов в яичниках стимулирует
выработку гипофизом гонадотропинов, которые, в свою очередь, усиливают синтез
андростендиона, и уровень эстрогенов вновь повышается. В связи с этим
ингибиторы ароматазы неэффективны у женщин в пременопаузе.
В постменопаузе, когда яичники перестают функционировать, ось гипоталамус
- гипофиз - надпочечники разорвана, и ингибирование ароматазы приводит к
значительному подавлению биосинтеза эстрогенов в периферических тканях, а также
в ткани опухоли молочной железы.
Первым и фактически единственным представителем ингибиторов ароматазы I
поколения является аминоглутетимид - неселективный ингибитор ароматазы.
Поскольку аминоглутетимид ингибирует целый ряд ферментов, участвующих в
стероидогенезе (подавляет секрецию надпочечниками глюкокортикоидов (кортизола)
и используется поэтому при болезни Иценко - Кушинга и др.), при его применении
необходимо следить за функциональным состоянием коры надпочечников (может
развиться ее гипофункция).
Поиски новых средств, обладающих большей селективностью, лучшей
переносимостью и более удобным режимом дозирования, привели к появлению
ингибиторов ароматазы II и III поколений. К настоящему времени созданы новые
нестероидные (летрозол, анастрозол и др.) и стероидные (экземестан) соединения
этой группы.
Основным показанием для ингибиторов ароматаз является рак молочной железы
у женщин в постменопаузе, в т.ч. при резистентности к терапии антиэстрогенами.
К группе ингибиторов биосинтеза гормонов надпочечников, используемых в
онкологии, относят митотан и аминоглутетимид. Они подавляют секрецию
глюкокортикоидов и могут вызывать деструкцию нормальной и опухолевой ткани коры
надпочечников.
Глюкокортикоиды - преднизолон, дексаметазон в связи с их лимфолитическим
действием и способностью угнетать митоз лимфоцитов - применяют при острых
лейкозах (главным образом у детей) и злокачественных лимфомах.
В качестве противоопухолевых средств применяют также некоторые аналоги
соматостатина. Например, октреотид и ланреотид используют для симптоматической
терапии эндокринных опухолей гастроэнтеропанкреатической системы.
.6
Противоопухолевые средства растительного происхождения
Основными средствами этой группы являются винбластин, винкристин,
винорелбин, доцетаксел, иринотекан, паклитаксел, тенипозид, топотекан, этопозид
и др.
Согласно классификации Д.А. Харкевича, противоопухолевые средства растительного
происхождения могут быть представлены следующими группами:
. Алкалоиды барвинка розового - винбластин, винкристин.
. Алкалоиды тисового дерева (таксаны) - паклитаксел, доцетаксел.
. Подофиллотоксины, выделяемые из подофилла щитовидного,- этопозид,
тенипозид.
. Алкалоиды безвременника великолепного - демекольцин (колхамин),
колхицин.
Большинство алкалоидов являются фазоспецифичными противоопухолевыми
средствами, т.е. эффективны в определенных фазах клеточного цикла.
Алкалоиды можно разделить на две группы по точке приложения действия:
действующие на микротрубочки клетки (колхицин, винкаалкалоиды, таксаны);
ингибиторы топоизомераз (этопозид, тенипозид, иринотекан, топотекан).
Винкаалкалоиды - структурно родственные вещества, в химической структуре
которых присутствуют две полициклические единицы - виндолин и катарантин. К
винкаалкалоидам относятся винбластин и винкристин - алкалоиды, выделенные из
растения барвинок розовый (Vinca rosea L.), а также виндезин и винорелбин -
полусинтетические производные винбластина. Винорелбин отличается по структуре
от других алкалоидов барвинка наличием 8-членного кольца катарантина (вместо
9-членного). Противоопухолевое действие этих алкалоидов обусловлено влиянием на
клетки в М-фазе клеточного цикла (фаза митоза).
При нормальном (правильном) течении митоза в стадии профазы начинается
формирование ахроматинового веретена, которое завершается в стадии метафазы. К
концу клеточного деления веретено распадается (митотическое веретено образуется
при каждом делении эукариотической клетки и регулирует ориентацию и
распределение хромосом в двух дочерних клетках). В построении нитей веретена
деления (микротрубочек) участвует цитоплазматический глобулярный белок тубулин.
Тубулин представляет собой димерный белок, состоящий из двух сходных, но
не идентичных субъединиц - альфа-тубулин и β-тубулин. Обе субъединицы имеют
молекулярную массу около 50кД каждая (53 кД и 55 кД) и несколько различаются по
изоэлектрической точке. При определенных условиях, в зависимости от
потребностей клетки, димеры тубулина полимеризуются и образуют линейные
цепочки, состоящие из чередующихся молекул альфа-тубулина и β-тубулина (протофиламенты), из которых
формируются микротрубочки.
Микротрубочки составляют основу митотического аппарата (митотическое
веретено) в период деления клетки, а также являются важным компонентом
цитоскелета клетки. Они необходимы для осуществления многих клеточных функций в
интерфазе, в т.ч. для поддержания пространственной формы клеток,
внутриклеточного транспорта органелл. В нейронах пучки микротрубочек участвуют
в передаче нервных импульсов.
Каждая микротрубочка - это цилиндр с наружным диаметром около 24 нм и
внутренним каналом около 15 нм в диаметре, длина микротрубочки несколько
микрон. Стенки построены из 13 протофиламентов, расположенных в виде спирали
вокруг центральной полости. Микротрубочки представляют собой динамические
полярные структуры с (+)- и (−)-концами. Как полимеризация, так и
деполимеризация тубулина происходят на концах микротрубочек, при этом
наибольшие изменения происходят на (+)-конце.
Антимитотическое действие винкаалкалоидов опосредовано преимущественно
действием на микротрубочки: связываясь с молекулами тубулина микротрубочек
(благодаря выраженному сродству), они препятствуют полимеризации этого белка,
тормозят образование веретена деления (сборку микротрубочек) и останавливают
митоз на стадии метафазы. Винкаалкалоиды могут также изменять метаболизм
аминокислот, цАМФ, глютатиона, активность кальмодулинзависимой Ca2+транспортной
АТФазы, клеточное дыхание, биосинтез нуклеиновых кислот и липидов.
Считают, что в механизме действия разных алкалоидов барвинка имеются
некоторые отличия, что может быть обусловлено различиями в их химической
структуре, взаимодействием с разными участками молекулы тубулина и различным взаимодействием
с белками, ассоциированными с микротрубочками. Эти белки могут изменять
характер взаимодействия алкалоидов с тубулином микротубул, что в результате
также определяет некоторые нюансы в действии разных алкалоидов. Так, в условиях
in vitro, винбластин, винкристин и винорелбин обладают примерно сходной
активностью в отношении сборки тубулина в микротубулы, однако винорелбин не
оказывает специфического действия в отношении индукции образования спиралей.
При экспериментальном сравнительном исследовании действия винбластина,
винкристина и винорелбина на микротрубочки митотического веретена и
микротрубочки аксонов у эмбрионов мышей на ранней стадии развития нейронов было
показано, что винорелбин более избирательно действует на микротрубочки митотического
веретена.
Природные винкаалкалоиды (винкристин, винбластин) применяют для лечения
быстро пролиферирующих новообразований. Один из широко используемых
винкаалкалоидов - винкристин, применяют в основном в комбинированной
химиотерапии острого лейкоза, лимфогранулематоза, а также других опухолевых
заболеваний (вводят в/в 1 раз в неделю). Нейротоксическое действие винкристина
может проявляться нарушением нервно-мышечной передачи, неврологическими
осложнениями, в т.ч. парестезией, двигательными расстройствами, выпадением
сухожильных рефлексов, возможен парез кишечника с возникновением запоров,
вплоть до паралитического илеуса и др.
В отличие от винкристина, другой алкалоид барвинка - винбластин, является
менее нейротоксичным лекарственным средством, но вызывает миелосупрессию, имеет
выраженный раздражающий эффект с риском развития флебита, некроза (при
экстравазальном попадании). Как и винкристин, винбластин применяется в
комплексной терапии ряда опухолевых заболеваний, включая болезнь Ходжкина,
лимфо- и ретикулосаркомы.
К алкалоидам безвременника великолепного (Colchicum Speciosum Stev.)
семейства лилейных (Liliaceae) относятся демекольцин (колхамин) и близкий к
нему по строению колхицин, содержащиеся в клубнелуковицах растения.
В средние века настой семян и клубней безвременника применяли в качестве
средства от подагры, ревматизма, невралгии. В настоящее время демекольцин и
колхицин применяются ограниченно.
Оба алкалоида обладают антимитотической активностью. Механизм действия
колхицина обусловлен, в первую очередь, тем, что, связываясь с тубулином, он
приводит к дезагрегации митотического аппарата и вызывает т.н. К-митоз
(колхициновый митоз) - клеточное деление нарушается на стадии метафазы и
последующей анафазы, при этом хромосомы не могут разойтись к полюсам клетки, в
результате образуются полиплоидные клетки. Колхицин широко используется в
экспериментальных исследованиях в качестве мутагена, а также для получения
полиплоидных форм растений.
Демекольцин, являющийся в 7-8 раз менее токсичным, чем колхицин, применяют
в основном в качестве наружного средства (в виде мази) при опухолях кожи
(ингибирует рост опухолевой ткани, при непосредственном контакте вызывает
гибель опухолевых клеток). Колхицин используют для купирования и предупреждения
приступов подагры. Колхицин, наряду с антимитотической активностью, обладает
способностью препятствовать образованию амилоидных фибрилл и блокировать
амилоидоз, оказывает урикозурическое действие, препятствует развитию
воспалительного процесса (тормозит митотическое деление гранулоцитов и других
подвижных клеток, уменьшает их миграцию к очагу воспаления). Назначают колхицин
при подагре, главным образом при неэффективности НПВС или противопоказаниях к
ним.
К средствам, антимитотическая активность которых преимущественно
обусловлена действием на микротрубочки клеток, относят, кроме винкаалкалоидов и
алкалоидов безвременника великолепного, новую группу алкалоидов - таксаны.
Таксаны - химиотерапевтические средства, получившие широкое
распространение в клинической практике в 90-е гг.
Доцетаксел, близкий к паклитакселу по структуре и механизму действия,
получают путем химического синтеза из природного сырья - игл тиса европейского
(Taxus baccata).
Таксаны относятся к классу препаратов, действующих на микротрубочки. В
отличие от винкаалкалоидов, тормозящих образование митозного веретена, таксаны,
связываясь со свободным тубулином, повышают скорость и степень его
полимеризации, стимулируют сборку микротрубочек, стабилизируют сформировавшиеся
микротрубочки, препятствуют деполимеризации тубулина и распаду микротрубочек.
Таксаны нарушают функционирование клетки при митозе (М-фаза) и в интерфазе.
Образование чрезмерного количества микротрубочек и их стабилизация
приводят к ингибированию динамической реорганизации сети микротрубочек, что в
конечном итоге ведет к нарушению процесса формирования митотического веретена и
ингибированию клеточного цикла в G2 и М-фазах. Изменение
функционирования клетки в интерфазе, в т.ч. нарушение внутриклеточного
транспорта, передачи трансмембранных сигналов и пр., также является следствием
нарушения микротубулярной сети.
Паклитаксел и доцетаксел имеют сходный механизм действия. Однако различия
в химической структуре определяют некоторые нюансы в механизме действия этих
веществ, обнаруженные в эксперименте. Например, доцетаксел обладает более
выраженным эффектом в отношении активирования полимеризации тубулина и
торможения его деполимеризации (примерно в два раза). При действии на клетку
паклитаксела характерны некоторые изменения в строении микротрубочек, не
обнаруженные при действии доцетаксела. Так, в экспериментальных исследованиях
показано, что образовавшиеся в присутствии паклитаксела микротрубочки содержат
только 12 протофиламентов (вместо 13 в норме) и имеют диаметр 22 нМ (в отличие
от 24 в норме).
Кроме того, паклитаксел индуцирует аномальное расположение микротрубочек
в виде пучков на протяжении всего клеточного цикла и образование множественных
звездчатых сгущений (астеров) во время митоза.
Механизмы действия разных препаратов, влияющих на микротрубочки, остаются
до конца не понятыми, несмотря на большое количество накопленной информации.
Установлено, что участки связывания с тубулином различны для природных
винкаалкалоидов, винорелбина, колхицина, таксанов. Так, в экспериментальных
исследованиях паклитаксела показано, что он, преимущественно, связывается с β-субъединицей тубулина, при этом его
способность связываться с микротрубочками выше, чем у димеров тубулина.
Таксаны эффективны при раке молочной железы, яичников, немелкоклеточном
раке легких, опухолях головы и шеи и др.
Подофиллотоксины. К противоопухолевым средствам растительного
происхождения относят подофиллин - смесь природных веществ, выделяемая из
корневищ с корнями подофилла щитовидного (Podophyllum peltatum L.) семейства
барбарисовых (Berberidaceae). Подофиллин содержит не менее 40%
подофиллотоксина, альфа- и β-пельтатины. Экстракт из корневищ
подофилла издавна применялся в народной медицине как слабительное средств при
хронических запорах, в качестве рвотного и противоглистного средства. В
дальнейшем была обнаружена его цитостатическая активность, проявляющаяся
блокадой митоза на стадии метафазы (по действию напоминает колхицин).
Подофиллотоксин применяют местно при лечении папиллом и других новообразований
кожи.
В клинической практике широко используются полусинтетические производные
подофиллотоксина - эпиподофиллотоксины (этопозид и тенипозид), по механизму
действия относящиеся к ингибиторам топоизомераз.
Топоизомеразы - ферменты, непосредственно участвующие в процессе
репликации ДНК. Эти ферменты меняют топологическое состояние ДНК: осуществляя
кратковременные разрывы и воссоединения участков ДНК, они способствуют быстрому
раскручиванию и скручиванию ДНК в процессе репликации. При этом целостность
цепей сохраняется.
Ингибиторы топоизомераз, связываясь с комплексом топоизомераза-ДНК,
воздействуют на пространственную (топологическую) структуру фермента, снижают
его активность и тем самым нарушают процесс репликации ДНК, тормозят клеточный
цикл, задерживая пролиферацию клеток.
Ингибиторы топоизомераз оказывают фазоспецифичное цитотоксическое
действие (в период S и G2 фазы клеточного цикла).
Этопозид и тенипозид являются ингибиторами топоизомеразы II.
Камптотецины - полусинтетические производные алкалоида камптотецина,
выделенного из стеблей кустарника Camptotheca acuminata, представлены
иринотеканом и топотеканом. В соответствии с механизмом действия они относятся
к группе ингибиторов топоизомераз. В отличие от эпиподофиллотоксинов, камптотецины
являются ингибиторами топоизомеразы I. В настоящее время иринотекан является
препаратом первой линии для лечения рака толстой кишки. Топотекан широко
применяется при лечении рака легкого и яичников.
Глава 2.
Препараты производные бис-(β-хлорэтил)амина
.1.
Классификация и история изучения
В зависимости от химической природы носителя бис-(β-хлорэтил)аминиой группы хлорэтиламины
разделяются на производные алифатического ряда (эмбихин; новэмбихин),
жирно-ароматического ряда (новэмбитол), ароматического ряда (хлорбутин;
пафенцил), производные на основе аминокислот и пептидов (сарколизин; асалин,
лофенал, фентирин), вещества, содержащие остатки азотистых гетероциклических
соединений (допан), фосфорилированные производные (циклофосфан), производные
полиспиртов (дегранол).
Синтез производных бис-(β-хлорэтил) амина впервые был
осуществлен в конце XIX в., однако только после второй мировой войны показана
возможность использования этих соединений при заболеваниях кроветворной системы
и практически положено начало новой области медицинской и химической наук -
химиотерапии злокачественных новообразований.
К 1950-1953 гг. было синтезировано около 500 хлорэтиламинов, но в то
время некоторые из них применялись только в качестве дополнительных лечебных
средств при хирургическом вмешательстве. Ограниченное их применение объяснялось
тем, что хлорэтиламины, подавляя рост опухолей, действуют одновременно на все
быстроделящиеся клетки. С 1955 г. исследователи осуществляли синтез
многочисленных производных галоидэтиламинов с целью получения соединений с
большей избирательностью действия в отношении опухолей. Если к 1960 г. было
получено более 2000 производных галоидэтиламинов, то к 1978 г. количество этих
соединений, синтезированных с целью изучения их противоопухолевой и противолейкозной
активности, превысило 3000.
Важнейшими свойствами, характеризующими реакционную способность
хлорэтиламннов, являются степень их основности как аминов, а также скорость
гидролиза.
Хлорэтиламины алифатического ряда. Характерной особенностью алифатических
хлорэтиламннов в водных растворах является быстрая ионизация с циклизацией.
Механизм взаимодействия алифатических хлорэтиламинов с нуклеофильными
агентами зависит от полярности (диэлектрической постоянной) растворителя и от
нуклеофильности атакующей группы.
Алифатические бис-(β-хлорэтил) амины, например эмбихин, при растворении в
воде образуют этилениммоииевый катион с меньшей скоростью, чем монохлорэтил
амины.
Особенностью этих соединений является то, что второй электрофильный центр
в их молекуле может вступать в реакцию только после размыкания первого
этилениммониевого иона.
Первым препаратом из группы хлорэтиламннов алифатического ряда, который
длительное время применялся в онкологической практике нашей страны, был
эмбихин. Вследствие высокой токсичности он был снят с производства и заменен
менее токсичным препаратом- новэмбихином. Во многих зарубежных странах эмбихии
производится и применяется для лечения опухолей и в настоящее время.
С целью получения менее токсичных препаратов с большей избирательностью
действия на солидные опухоли исследователи пошли по пути замены хлора в
галоидалкильных группах алифатических хлорэтиламинов на другие галоиды (бром,
йод, фтор).
Как оказалось, замещение атомов хлора в молекуле алифатических
хлорэтиламинов бромом почти не отражается на биологическом действии соединений
и не приводит к уменьшению токсичности. Полное замещение в молекуле эмбихина
атомов хлора фтором приводит к сиижеиию токсичности, но без усиления
противоопухолевого действия. В то же время монофторпроизводиые алифатических
хлорэтиламинов оказались менее токсичными и в некоторых случаях проявляют
большую противоопухолевую активность.
Хлорэтиламииы ароматического ряда. Свойства ароматических
2-хлорэтиламинов в определенной степени отличаются от свойств хлорэтиламинов
алифатического ряда. Ароматические хлорэтиламины с непосредственной связью арил
- азот типа АrN(СН2СН2С1)2
обладают весьма малой основностью и не способны образовывать устойчивые соли.
Их гидролиз и взаимодействие с тиосульфатом натрия проходят очень медленно.
Гидролиз моно- и бифункциональных N-(2-хлорэтил)анилинов при рН 5,0
протекает по SN1-механизму. Медленной стадией этой
реакции является стадия отщепления иона хлора, в связи с чем константа скорости
образования промежуточного катиона непосредственно определяется по скорости
выделения протона.
Скорость образования этилениммониевого катиона при гидролизе
ароматических хлорэтиламинов зависит от двух факторов: донорно-акцепторных
свойств заместителей ароматического ядра и степени замещения атома азота.
Из ароматических хлорэтиламинов после установления их выраженного
антибластического действия в эксперименте изучались в клинике следующие
соединения: лимфохин, хлоронафтин, N,N-ди(2'-хлорпропил)-2-нафтиламин,
хлорбутин, пафенцил и др.
Однако в настоящее время разрешены к применению в онкологической практике
нашей страны только хлорбутин и пафенцил.
Хлорэтиламины жирно-ароматического ряда общей формулы
Ar(СН2)nN- (СН2СН2С1)2
где п= 1, 2, можно рассматривать как тип, промежуточный между хлорэтиламинами
алифатического и ароматического рядов. Для хлорэтиламинов при n=2 характерна большая основность, чем
для соединений второго типа - при (n=1).
Гидролиз хлорэтиламинов жирно-ароматического ряда с п=2 уже в первые
минуты приводит к значительной неэквивалентности количества образующихся С1-
и Н+-ионов. Преобладание концентрации С1--иона
свидетельствует, очевидно, о высоких скоростях образования аммониевого катиона
за счет внутримолекулярной циклизации. Собственно гидролиз идет с малой
скоростью. Удаление арильного радикала от атома азота на два углеродных атома
(п=2) приводит к снижению гидролитической устойчивости по сравнению с таковой
хлорэтиламинов, содержащих метиленовый мостик между арилом и атомом азота (n=1).
Как показали экспериментальные и клинические исследовании, наиболее
выраженным антибластическим действием из группы хлорэтиламинов
жирно-ароматического ряда обладает эмбитол, представляющий собой смесь пара- и
орто- изомеров ксилил-бис-(β-хлорэтил) амина в соотношении 1:1.
После успешных клинических испытаний препарат был внедрен в медицинскую
практику в 1958 г. для лечения лимфогранулематоза, лимфосаркомы и других
заболеваний. В начале 60-х годов он был заменен синтезированным чистым
орто-изомером, названным новэмбитолом, который оказался в 4 раза менее
токсичным, чем эмбитол, и более эффективным по сравнению с пара-изомером и
эмбитолом. В связи с отсутствием рациональной лекарственной формы новэмбитол не
применяется в медицинской практике.
Хлорэтиламины на основе аминокислот, пептидов и других биологически
активных веществ. Рост опухолей связан с интенсивным синтезом белка, для
построения которого необходимы аминокислоты. В результате многочисленных
исследований с применением изотопов установлено, что ткани опухолей in vitro и in vivo активно включают различные
аминокислоты. Пептиды и аминокислоты обладают способностью проникать через
мембраны раковых клеток в 4-5 раз легче, чем через мембраны нормальных клеток,
в связи с чем в раковых клетках может происходить кумуляция этих веществ.
Использование аминокислот в синтезе хлорэтиламинов привело к созданию в
СССР препарата сарколизнна, а за рубежом - препарата мелфалана, в которых
носителем цитотоксической хлорэтиламинной группы является незаменимая
аминокислота - фенилаланин (D, L-форма для сарколизина, L-форма для мелфалана).
Результаты биологических и клинических исследований показали, что
сарколизин был первым соединением, вызывающим полное рассасывание ряда
экспериментальных опухолей, оказывающим выраженное терапевтическое действие в
отношении некоторых солидных опухолей человека. Он обладает большей
избирательностью действия по сравнению с известными β-хлорэтиламинами, но в то же время
проявляет побочное токсическое действие на кроветворение, а также
характеризуется ограниченным спектром противоопухолевой активности. В связи с
этим как в Советском Союзе, так и за рубежом были продолжены исследования по
синтезу изомеров, производных и аналогов сарколизнна. Модификация молекулы
сарколизина путем получения изомеров (мета-, орто-, D- и L-сарколизин),
введение в ароматическое ядро галогенов (хлор, бром, фтор) привели к созданию
препаратов, которые в некоторых случаях превосходили по активности сарколизин.
Введение в онкологическую практику во второй половине 50-х годов
отечественного препарата сарколизина вызвало большой интерес к
хлорэтиламино-производным других аминокислот. Были синтезированы и изучены в
эксперименте хлорэтиламинопроизводиые пролина.
Применение пролина в качестве носителя хлорэтиламиногруппы связано с его
способностью превращаться в организме в орнитин и глютаминовую кислоту.
Указанные хлорэтиламинопроизводиые пролииа обладают сравнительно малой
токсичностью. Более эффективным в эксперименте оказалось 3-бис-(β-хлорэтил)аминопроизводное (азиприн),
которое проявило выраженную противоопухолевую активность в отношении ряда
перевиваемых опухолей. Азиприн, обладающий иным спектром действия, чем
сарколизин, несколько уступает ему по активности. В клинике он не нашел
применения.
Введение хлорэтиламиногруппы в тирозии привело к получению активного
препарата - фентирина, применяемого в медицинской практике для лечения острого
лейкоза.
Синтезированы и экспериментально изучены хлорэтиламинные производные
фенилалкановых кислот на основе гистидина, являющегося, как известно, основной
аминокислотой. Гистидин входит как составная часть в ядерные нуклеопротеиды,
которые представляют собой одну из «мишеней» при действии алкилирующих веществ.
Можно было предположить, что использование в качестве «носителя»
хлорэтиламиногруппы производных гистидина отразится на направленности и
избирательности действия новых веществ.
Наибольшей антибластической активностью обладает препарат гисфен
метиловый эфир п-бис-(β-хлорэтил)аминофенацетил-L-гистидина. Он был предложен для клинического изучения при
лейкозных заболеваниях, однако оказался малоэффективным.
Таким образом, характер аминокислоты, несущей хлорэтиламиногруппу,
определяет токсичность, силу и спектр противоопухолевого действия; иа
биологическую активность влияет и положение хлорэтиламииогруппы в аминокислотном
остатке. Присоединение алкилирующей β-хлорэтил аминогруппы к аминокислоте
привело к изменению спектра и повышению избирательности противоопухолевого
действия. В связи с этим дальнейшие поиски соединений с возможно большей
избирательностью действия были направлены на использование в качестве носителей
цитотоксической группы пептидов и других биологически активных веществ.
На основе п-бис-(β-хлорэтил) аминофенилуксусной и п-бис-(β-хлорэтил)аминофенилмасляной кислот
был синтезирован ряд соединений типа амидов, близких к пептидам сарколизииа.
Соединения отличаются друг от друга природой аминокислоты, присоединенной
к п-бис-(β-хлорэтил)аминофенилуксусной кислоте (хлорфенацил), которая
обладает выраженным антибластическим действием. Присоединение к хлорфенацилу посредством
амидной связи различных аминокислот приводит к значительному снижению
токсичности полученных соединений. При замене одной аминокислоты в молекуле
амида на другую изменяются сила и спектр противоопухолевого действия
хлорэтиламинов. Предполагается, что неодинаковая противоопухолевая активность
этих соединений обусловлена различиями в использовании разными видами опухолей
тех незаменимых аминокислот, которые входят в состав указанных соединений.
Кроме того, структура аминокислоты - носителя алкилирующей группы,
очевидно, оказывает влияние на процессы распределения вещества в организме,
перенос его через мембраны и катаболический распад. При замене природной,
аминокислоты (β-фенил-а-аланина) на неприродную (β-фенил-β-аланин)антибластическое действие соединения
снижается. Это свидетельствует о том, что противоопухолевые препараты следует
создавать на основе природных соединений, которые могут играть определенную
роль в обмене веществ опухолевой ткани. Среди бис-(β-хлорэтил)аминных производных
фенилалкановых кислот наиболее эффективным оказался лофенал - п-(бис-(β-хлорэтил)аминофенацетил)-D,L-фенилаланин.
Этот препарат отличается от сарколизина спектром противоопухолевого
действия.
В связи с предположением Л. Ф. Ларионова о возможности усиления противоопухолевых
свойств хлорэтиламинов и этилениминов путем присоединения их к природным
гетероциклическим соединениям, играющим значительную роль в обмене веществ,
были синтезированы хлорэтиламинные производные пиримидина. Пиримидины являются
биологически важными соединениями, так как входят в состав нуклеиновых кислот,
некоторых витаминов и коферментов. При получении таких соединений учитывались
особенности метаболизма опухолевых клеток, заключающиеся в том, что утилизация
производных пиримидина происходит легче, а активность ферментов, разрушающих
пиримидины, в опухолевой ткани ниже, чем в нормальных тканях. Так были созданы
допан и нордопан.
Допан был синтезирован и испытан в 1952-1954 гг. и в настоящее время
применяется в медицинской практике. Нордопан, полученный одновременно с
допаном, обладает почти таким же противоопухолевым действием, как и допан. В то
же время технология получения нордопана отличается большей трудоемкостью.
Высокая противоопухолевая активность допана и нордопана, в которых носителем
цитотоксической бис-(β-хлорэтил)аминогруппы были соответственно 6-метилурацил
и урацил, побудила исследователей к использованию в качестве носителей
галогеналкиламинной группировки других производных пиримидина и различных
гетероциклических структур.
В 1957 - 1958 гг. был синтезирован фторпан, который обладал высокой
противоопухолевой активностью, но оказался в 5 раз менее токсичным, чем допан.
Фосфорилированные хлорэтиламины. Одним из направлений поиска
противоопухолевых веществ является создание препаратов с латентной активностью.
Идеальными условиями для получения таких препаратов являются наличие в раковой
клетке фермента, способного активизировать препарат, и отсутствие такового в
нормальных клетках. Кроме того, продукт активации должен обладать высокой реакционной
способностью.
До настоящего времени различия в ферментном составе нормальных и
злокачественных клеток изучены недостаточно. Тем не менее исследования по
созданию препаратов с латентной активностью развиваются, и получены уже
некоторые практические достижения. Так, дифосфат стильбэстрола, применяемый для
лечения рака предстательной железы, превращается в организме нз латентной
«транспортной» формы в активную. Эти данные стимулировали исследования по
синтезу и биологическому изучению фосфорилированных хлорэтиламинов в качестве
потенциальных противоопухолевых веществ. Первый представитель фосфорилированных
хлорэтиламииов был получен в 1954 г.
Предполагалось, что это соединение при попадании в организм под действием
фосфатазы будет подвергаться ферментативному расщеплению по Р - N-связи, причем
вследствие повышенного содержания кислой фосфатазы в злокачественных опухолях
оно должно активироваться в опухолевых клетках. В то же время исследователи
считали, что фосфорилирование хлорэтиламинов приведет не только к понижению
основности атома азота, но и к уменьшению реакционной способности хлорэтильных
групп в результате образования амидной связи, а следовательно - и к уменьшению
токсичности. В эксперименте соединение действительно оказалось малотоксичиым,
однако и малоактивным.
Наиболее активными оказались соединения, среди которых по
противоопухолевому действию был выделен препарат цнклофосфан - 2-оксо-2-бис(β-хлорэтил)аминотетрагидро-2,1,3-фосфоксазин,
который и в настоящее время применяется при лимфогранулематозе, раке яичников,
раке молочной железы, раке легкого и др.
Для соединений, к которым относится циклофосфан. характерным является
наличие N-триметиленовой группы. У гомологов указанных диамидоэфиров,
содержащих менее или более трех метиленовых групп, обнаружена меньшая
противоопухолевая активность. Кроме того, введение в одну из метиленовых групп
фосфоксазинового цикла метальных групп также приводит к снижению активности.
Циклофосфан и его производные отличаются от других фосфорилированных
хлорэтиламинов более высоким терапевтическим индексом, обладают низкой
реакционной способностью in vitro и относятся к препаратам с латентной
активностью.
Подробное изучение механизма активации циклофосфана показало, что в
противоположность первоначальной гипотезе об активации препарата под действием
фосфатаз и фосфорамидаз в действительности происходит окислительное превращение
его в микросомах печени с участием большой группы ферментов, в том числе
смешанной системы микросомных оксидаз.
Высокая противоопухолевая активность циклофосфана стимулировала развитие
исследований в области фосфорилированных хлорэтиламинов.
Осуществлен синтез аналогов циклофосфана, содержащих хлорэтиламинные
группы в цикле, - трихлорфосфамида и изоциклофосфамида, обладающих высокой
антибластической активностью в эксперименте и отличающихся от циклофосфана
спектром противоопухолевого действия. Клиническое изучение трихлорфосфамида
показало его эффективность при лимфогранулематозе, ретикулосаркоме, хронических
лимфо- и миелолейкозах, которая, однако, не превосходит активность
циклофосфана.
Изоциклофосфамид оказался активным при бронхогенной карциноме, раке
яичников и поджелудочной железы, хондросаркоме и тестикулярной тератоме, прн
наличии устойчивости опухолей к циклофосфану и лучевому воздействию.
В ряду фосфорилированных хлорэтиламинов кроме соединений циклической
структуры синтезируются и изучаются соединения линейной структуры. Проведены
исследования по синтезу и изучению свойств бис-(β-хлорэтил)амидов фосфорной кислоты,
содержащих остатки фенолов или аминов.
В отличие от хлорэтиламинов алифатического и жирно-ароматического рядов
фосфорилированные хлорэтиламины линейной структуры обладают выраженным
ингибирующим действием в отношении
солидных опухолей эпителиального и мезенхимального происхождения.
Наиболее эффективными оказались диариловые эфиры бис-(β-хлорэтил)амидофосфорной кислоты,
среди которых выявлено несколько соединений, которые в отличие от других
алкилирующих соединений почти не вызывают лейкопении, проявляй в эксперименте
высокую противоопухолевую активность.
Получены фосфорилированные хлорэтиламииы, содержащие только остатки
гетероциклических соединений.
Синтезируя соединения с большим количеством электрофильных центров,
исследователи предполагали увеличить избирательность противоопухолевого
действия. Однако в большинстве случаев полифункциональные алкилирующие
соединения отличались низкой эффективностью. Это объясняется тем, что
повышенная реакционная способность таких соединений не дает им возможности
поступать в ткань опухоли в неизмененном виде, поскольку они взаимодействуют с
клеточными субстратами вблизи места их введения в организм.
Синтезированы фосфорилированные хлорэтиламины, содержащие гидразидную
группу, - хлористоводородные соли ариловых эфироз гидразидо-N,N-бис-(β-хлорэтил)амидофосфорных кислот.
Введение гидразидной группировки в молекулу фосфорилированного
хлорэтиламина привело к уменьшению токсичности соединений (примерно в 5 раз)
при сохранении характера биологического действия. Особенностями этой группы
веществ являются отсутствие угнетения системы кроветворения и избирательность
действия. Отдельные представители ее обладают выраженной противолейкозной
активностью в эксперименте, не уступая по эффективности хлорбутину.
Синтезированы аналоги циклофосфана, содержащие перекисную группу,
продолжаются исследования по получению аналогов циклофосфана, содержащих
различные заместители в цикле.
Этим объясняется значительно более медленный гидролиз фосфорилированных
хлорэтиламинов по сравнению с гидролизом нефосфорилированных. Гидролиз
фосфорилированных хлорэтиламинов линейной структуры, в частности диариловых
эфиров бис-(β-хлорэтил)амидофосфорной кислоты, изучен достаточно подробно.
Оказалось, что при растворении дифенилового эфира бис-(β-хлорэтил)амидофосфорной кислоты в
водном растворе бикарбоната натрия (рН 8,3) сразу же образуется 43% ионного
хлора. С помощью хроматографического контроля установлено, что гидролитическое
расщепление дифенилового эфира бис-(β-хлорэтил)амидофосфорной кислоты
осуществляется сравнительно быстро и заканчивается в течение 20 мин после его
растворения с образованием натриевой соли дифенилового эфира фосфорной кислоты
и 2-хлорэтил-2-оксиэтиламина. Образование последнего, вероятно, является
результатом гидролиза бис-(β-хлорэтил)амина, который получается на
первом этапе при расщеплении амидной связи Р-N.
.2 Хлорбутин

Синонимы: Хлорамбуцил, Лейкеран, Амбохлорин, Хлораминофен, Эхлорил,
Линфолизин, Лейкоран, Ленукоран.
Синтез. В качестве исходного вещества для получения хлорбутина
применяется метиловый эфир γ-(п-аминофенил)масляной кислоты (I),
который при взаимодействии с окисью этилена (II) образует метиловый эфир γ-[п-бис(β-оксиэтил)аминофенил]-масляной кислоты
(III). При действии на III хлороксида фосфора (IV) в растворе бензола
образуется метиловый эфир γ-[п-бис(β-хлорэтил) аминофенил]масляной кислоты
(V), из которого обработкой соляной кислотой получают γ-[п-бис(β-хлорэтил)аминофенил]масляную кислоту-
хлорбутин (VI).

Фармакологическое действие. Хлорбутин является алкилируюшим (вызывающим
химические процессы в клетке, приводящие к нарушению стабильности ДНК -
дезоксирибонуклеиновой кислоты, содержащейся в основном в ядре клетки и
являющейся носителем генной информации) цитостатическим (препятствующим росту
клеток) препаратом; оказывает угнетающее действие на кроветворную ткань и
гиперплазированные (опухолевые) ткани, Препарат влияет более избирательно на
лимфоидную ткань, чем на гранулоцитарные элементы (клетки крови, относящиеся к
лейкоцитам),
Показания к применению. Назначают хлорбутин внутрь при хроническом
лимфолейкозе (раке крови, при котором источником опухолевого процесса являются
лимфобласты - клетки костного мозга, из которых развиваются форменные элементы
крови - лимфоциты), преимущественно при лейкемических формах; лимфо- и
ретикулосаркоме (форме злокачественной опухоли, возникающей из лимфоидной и
рыхлой соединительной ткани); лимфогранулематозе (раке лимфатической системы,
при котором в лимфатических узлах и внутренних органах образуются плотные
образования, состоящие из быстро растущих клеток), а также при раке яичников.
Способ применения и дозы. Применяют внутрь ежедневно в виде таблеток.
Суточная доза при хроническом лимфолейкозе составляет от 2 до 10 мг (в
зависимости от степени лейкоцитоза - увеличения содержания лейкоцитов в крови).
Курс лечения - от 3 до 6 нед. Общая доза на курс лечения - 200-400 мг. При
уменьшении количества лейкоцитов в крови до 2,5-2,0*109/л больного
переводят на поддерживающую терапию, назначая по 2-6 мг в неделю в течение года
и более.
У больных лимфогранулематозом без лейко- и тромбоцитопении (без
уменьшения числа лейкоцитов и тромбоцитов в крови) начинают с ежедневной
разовой дозы 20 мг; к концу курса снижают дозу до 10 мг. Количество препарата
на курс - 400-500 мг.
Хлорбутин применяют также в качестве иммунодепрессанта (лекарственного
средства, подавляющего защитные силы организма). Доза - 5-10 мг в сутки.
Лицам, ранее лечившимся другими цитостатическими препаратами или
подвергавшимся лучевой терапии, хлобутин назначают не ранее, чем через 1,5-2
мес. после окончания предыдущего лечения при условии отсутствия выраженной
лейкопении (пониженного содержания лейкоцитов в крови), тромбоцитопении
(уменьшения числа тромбоцитов в крови) и анемии (снижения содержания гемоглобина
в крови). Препарат назначают, начиная с небольших доз.
Во время лечения хлорбутином необходимо систематически (не менее 2-3 раз
в неделю) производить анализ крови (общий) и дифференциальный подсчет
лейкоцитов, определение количества тромбоцитов, эритоцитов, гемоглобина.
Побочное действие. В процессе лечения хлорбутином могут возникнуть
лейкопения, анемия (снижение содержания гемоглобина в крови) и тромбоцитопения,
при передозировке развивается значительная лейкопения, прогрессирующая вплоть
до панцитопении (уменьшения содержания в крови всех форменных элементов -
эритоцитов, лейкоцитов, тромбоцитов и т. д.). Уменьшение количества лейкоцитов
может продолжаться в течение 10-12 дней после отмены препарата.
При развитии резкой лейкопении прекращают прием препарата, а в случае
необходимости переливают кровь или вводят лейкоцитную и тромбоцитную массу
(форменные элементы крови - лейкоциты и тромбоциты, выделенные из цельной крови
и предназначенные для переливания больному), назначают стимуляторы кроветворения,
витамины. Переливание крови (100-125 мл 1 раз в неделю) рекомендуется
производить в течение курса лечения.
Противопоказания. Препарат противопоказан при тяжелых заболеваниях печени
и почек, острых заболеваниях желудочно-кишечного тракта, непосредственно после
применения других цитостатических препаратов и лучевой терапии, при выраженной
лейкопении, тромбоиитопении и анемии, связанных с развитием злокачественного
процесса.
Форма выпуска. Таблетки, содержащие по 0,002 и 0,005 г (2 и 5 мг)
хлорбутина,
Условия хранения. Список А. В хорошо укупоренной таре в прохладном,
защищенном от света месте
.3.
Сарколизин
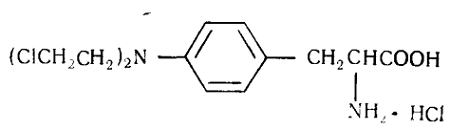
Синонимы: Рацемелфалан, Сарколорин.
Синтез. Сарколизин впервые и независимо был синтезирован советскими и
английскими исследователями. Описано несколько способов его получения.
Разработан способ, основанный на взимодействии ацетиламиномалонового
эфира с п-нитробензилбромидом в присутствии металлического натрия, последующем
восстановлении образовавшегося п-нитробензилацетиламиномалонового эфира до
п-аминобеизилацетил-аминомалонового эфира и на обработке его окисью этилена.
Образующийся при этом п-бис(β-оксиэтил)аминобензилацетиламиномалоновый
эфир при взаимодействии с хлористым тионилом превращается в п-бис(β-хлорэтил)-аминобензилацетиламиномалоновый
эфир, из которого при .кипячении с соляной кислотой получают сарколизин.
Для синтеза сарколизина в качестве исходного продукта применяют β-фенил-α-аланин, который нитруют.
Образовавшийся при этом п-нитрофенилаланин нагревают с фталевым ангидридом и
выделяют п-нитро-N-фталилфенилаланин, Из последнего при действии хлористого
водорода получают этиловый эфир п-нитро-N-фталилфенилаланина, который
восстанавливают палладием до этилового эфира п-амино-N-фталилфенилаланина, а
затем при последовательной обработке окисью этилена, хлороксидом фосфора и
соляной кислотой превращают в сарколизин.
Получают сарколизин также из 2-фенил-4-п-нитробезальоксазолона-5, который
образуется при взаимодействии п-нитробензальдегида с гиппуровой кислотой. Из
азлактона при алкоголизе синтезируют этиловый эфир α-бензоиламино-п-нитрокоричной кислоты,
который восстанавливают с палладиевым катализатором до этилового эфира
р-(п-аминофенил)-α-бензонламинопропионовой кислоты. При последовательном
окснэтилнровании, замещении оксигруппы на хлор и кислотном гидролизе этилового
эфира β-[п-(β-хлорэтил)аминофенил]-α-бензоиламннопропионовой кислоты
образуется сарколизин. Наиболее удобный способ синтеза сарколизина основан
также на применении азлактонного синтеза. Однако по этому способу в азлактонном
синтезе применяют альдегид с готовой бис-(β-хлорэтил)аминной группой. Из анилина
(I) и окиси этилена (II) или этиленхлоргидрина синтезируют бис-(β-оксиэтил)анилин (III), который при
обработке хлороксидом фосфора (IV) дает бис-(β-хлорэтил) анилин (V). При
взаимодействии V с диметилформамидом (VI) или метил (этил) форманилидом в
присутствии хлороксида фосфора образуется п-бис-(β-хлорэтил)аминобензальдегид (VII). Из
VII и гиппуровой кислоты (VIII) получают азлактон (IX), который восстанавливают
в бензоилсарколизин (X). При кислотном гидролизе X образуется сарколизин (XI).

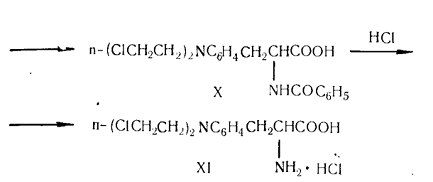
Фармакологическое действие. Сарколизин является производным бис-(β-хлорэтил)-амина и аминокислоты
фенилаланина. Подобно другим препаратам этого рода, сарколиэин обладает
алкилирующими свойствами (вызывает химические процессы в клетке, приводящие к
нарушению стабильности ДНК - дезоксирибонуклеиновой кислоты, содержащейся в
основном в ядре клетки и являющейся носителем генной информации) и подавляет
развитие гипершшэированных (разросшихся) тканей.
Показания к применению. Применяют для лечения семиномы яичка
(злокачественной опухоли, развивающейся из тканей яичек, вырабатывающих мужские
половые клетки - сперматозоиды), особенно при наличии метастазов (новых
опухолей, появившихся в результате переноса раковых клеток с кровью или лимфой
из первичного очага); ретикулосаркомы (злокачественной опухоли, возникающей из
рыхлой быстрорастущей соединительной ткани); злокачественной ангиоэндотелиомы
(рака, развивающегося из клеток внутренней оболочки кровеносного или
лимфатического сосуда); опухоли Юинга (злокачественной опухоли костной ткани);
миеломной болезни (костно-мозговой опухоли, состоящей из клеток лимфоидной
ткани различной степени зрелости).
В комбинации с колхамином сарколизин иногда применяют также при раке
пищевода и желудка.
Способ применения и дозы. Препарат назначают внутрь (после еды) и
внутривенно, вводят также в полости.
Выпускают для приема внутрь в таблетках по 0,01 г (10 мг), для инъекций -
в сухом виде в герметически закрытых флаконах, содержащих по 0,02 и 0,04 г (20
и 40 мг) препарата. Для получения раствора вводят во флакон 10-20 мл
изотонического раствора натрия хлорида; для ускорения растворения флакон
подогревают в воде до +60-+70°С. Внутрь или внутривенно назначают препарат один
раз в неделю. Разовая доза для взрослого с массой тела 60-70 кг составляет
40-50 мг; для больных с массой тела 50 кг и менее - из расчета 0,5-0,7 мг/кг. В
первые два приема дают по 50 мг, затем - по 30 мг. Детям также назначают из
расчета 0,5-0,7 мг/кг.
Курс лечения продолжается 4-7 нед. При метастазах опухолей в серозные
оболочки с выпотом в брюшную или плевральную полость (распространении опухолей
по оболочкам внутренних органов с накоплением жидкости в брюшной и грудной
полостях) можно вводить раствор сарколизина в полости. В брюшную полость вводят
от 40 до 100 мг, растворенных в 20-50 мл изотонического раствора натрия
хлорида, 1 раз в неделю; в плевральную полость (полость между оболочками
легких) - 20-80 мг, растворенных в 10-20 мл изотонического раствора натрия
хлорида, также 1 раз в неделю. До введения раствора сарколизина в полости
удаляют экссудат (богатую белком жидкость, выходящую из мелких сосудов), затем
вводят для, анестезии 60-70 мл 1% раствора новокаина, после чего, не вынимая
троакара (хирургического инструмента, предназначенного для прокола брюшной
полости) из брюшной полости или иглы из плевральной полости, придают больному положение
лежа и через 10 мин вводят свежеприготовленный раствор сарколизина, На куре
лечения - обычно 4-3 инъекций. Часто через 2-3 инъекции происходит рассасывание
или уменьшение количества экссудата: инъекции, однако, продолжают. Вводят
раствор сарколизина через иглу диаметром 0,3-0,5 мл; предварительно (за 10 мин)
вводят 100-120 мл 1% раствора новокаина. Всего на курс применяют 0,16-0,2 г
(160-200 мг) сарколизина,
Сарколиэин можно использовать и для регионарной химиотерапии (введения
препарата с использованием экстракорпорального кровообращения в кровеносные или
лимфатические сосуды вблизи опухоли при помощи искусственного кровообращения).
Применяют препарат в дозе 25-40 мг на 1 см2 тканей псрфузируемой
области.
Сарколиэин сравнительно быстро действует на опухоль: терапевтический
эффект проявляется обычно после 2-3 введений разовых доз (по 30-40 мг); если
после приема 100-150 мг эффект отсутствует, дальнейшее применение этого
препарата считают нецелесообразным из-за нечувствительности к нему данной опухоли.
Побочное действие. При применении сарколизина наблюдается угнетение
кроветворения с уменьшением количества лейкоцитов (форменных элементов крови),
особенно нейтрофилов. В процессе лечения необходимо тщательно следить за
картиной крови. Применение препарата прекращают при уменьшении числа лейкоцитов
в крови до 3,5-3,0*109/л, а также при выраженной тромбоиитопении. В
конце курса лечения необходимо определять количество лейкоцитов в крови перед
каждым введением серколиэина.
После окончания введения сарколизина количество лейкоцитов может
продолжать уменьшаться еще в течение 1-2 нед. При необходимости прибегают к
переливанию крови (1-2 раза в неделю по 100-125 мл).
При передозировке препарата и глубокой лейкопении (пониженном содержании
лейкоцитов в крови - 2,0*109/л и ниже), нейтро- и тромбоцитопении
(уменьшении числа нейтрофилов и тромбоцитов в крови) могут наблюдаться
повышение температуры тела, стоматит (воспаление слизистой оболочки полости
рта), симптомы геморрагического диатеза (повышенная кровоточивость). Для
предупреждения инфекции следует в таких случаях применять пенициллин,
переливать кровь, вводить лейкоцитную и тромбоцитную массу (форменные элементы
крови - лейкоциты, тромбоциты, выделенные из цельной крови и предназначенные
для переливания больному); при симптомах агранулоцитоза (резкого снижения числа
гранулоцитов в крови) назначают внутримышечно натрия нуклеинат, а также внутрь
пентоксил, лейкоген.
При появлении в процессе лечения повторной тошноты и рвоты назначают для
их купирования за 1 ч до введения сарколизина аминазин (0,025 г внутрь),
этаперазин или противорвотные средства.
При введении сарколизина в полости могут возникать боль, тошнота, рвота;
эти явления обычно проходят самостоятельно. Для предупреждения этих осложнений
следует до введения сарколизина производить местную анестезию плевры (или
брюшины).
Противопоказания. Сарколизин противопоказан в терминальных стадиях
заболеваний (при состоянии организма, предшествующем смерти), при кахексии
(крайней степени истощения), выраженной анемии (уменьшении числа эритроцитов в
крови), лейкопении (ниже 4,0*109/л), тромбоцитопении (ниже 150*106/л),
тяжелых поражениях печени, почек и сердечно-сосудистой системы.
Если больному производилась лучевая терапия, тоспрколизин применяют не
ранее чем через 1 мес. после окончания лечения; препарат назначают в этих
случаях в уменьшенных дозах.
Формы выпуска. Таблетки для приема внутрь по 0,01 г в упаковке по 25
штук; порошок во флаконах по 0,02 и 0,04 г для приготовления инъекционных
растворов,
Условия хранения. Список А, В хорошо укупоренной таре в сухом прохладном
месте.
2.4 Лофенал
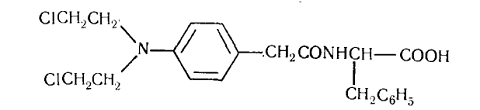
Синтез.
Способ I
По способу I вначале осуществляется конденсация п-бис-(β-хлорэтил)аминофенилуксусной кислоты
(I) с бензиловым эфиром D,L-феннлаланина (II) в присутствии
1,3-дициклогексилмочевины (III). Затем при каталитическом гидрогенолизе
бензилового эфира N-[п-бис-(β-хлорэтил)аминофенацетил]-D,L-фенилаланина (IV) образуется п-[бис-(β-хлорэтил)аминофенацетил]-D,L-феннлаланин - лофенал (V).
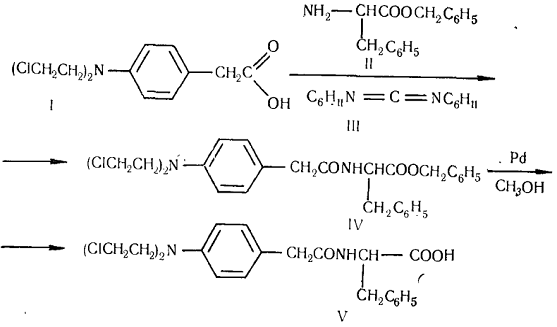
Способ II
По способу II лофенал (III) образуется ацилированнем D,L-фенилаланина (I) хлорангндрндом п-бис-(β-хлорэтил) амннофеннлуксусной кислоты
(II) в условиях реакции Шоттен-Баумана в присутствии едкого натра.

Фармакологическое действие. Обладает противоопухолевой активностью в
дозах, оказывающих слабое угнетающее действие на гемопоэз (кроветворение),
особенно на тромбоиитопоэз (процесс образования тромбоцитов в организме) после
нерадикальных операций (оперативного вмешательства, не направленного на
излечение болезни).
Показания к применению. Рак яичников (неоперабельные, не поддающиеся
хирургическому лечению, случаи); после нерадикальных операций; диссеминация по
брюшине с асцитом (рассеивание опухоли по брюшине и наличие жидкости в брюшной
полости); метастазы в легкие (появление в легких вторичных опухолей вследствие
переноса раковых клеток с кровью или лимфой из первичного очага),
сопровождающиеся гидротораксом (накоплением жидкости в грудной клетке); как
дополнительное средство при хроническом лимфолейкозе (раке крови, при котором
источником опухолевого процесса являются лимфобласты - клетки костного мозга,
из которых развиваются форменные элементы крови - лимфоциты), лимфогранулематозе
(раке лимфатической системы, при котором в лимфатических узлах и внутренних
органах образуются плотные образования, состоящие из быстро растущих клеток),
лимфо- и ретикулосаркоматозе (форме злокачественной опухоли, возникающей из
лимфоидной и рыхлой ткани).
Внутрь после еды 1-2 раза в день от 0,3 до 1,2 г в сутки. На курс лечения
- от 9 до 50 г. Для поддерживающей терапии назначают 0,3-0,6 г в неделю.
Лечение проводят под гематологическим контролем (контролем состава крови) не
реже раза в неделю в начале лечения и каждые 3-4 дня во второй половине курса.
Побочное действие. Иногда потеря аппетита, тошнота, рвота; лейкопения
(пониженное содержание лейкоцитов в крови) и тромбоцитопения (уменьшение числа
тромбоцитов в крови).
Противопоказания. Значительная анемия (снижение содержания гемоглобина в
крови), лейкопения и тромбоцитопения, терминальная стадия болезни (состояние
организма, предшествующее смерти), а также тяжелые сопутствующие заболевания.
Форма выпуска. Таблетки по 0,3 г в упаковке по 100 штук.
Условия хранения. Список Б. В защищенном от света месте при температуре
не выше +10 °С.
.5 Дегранол
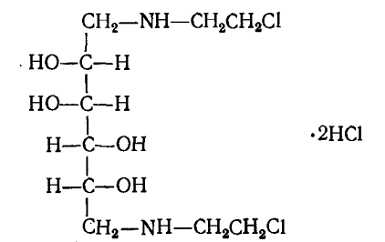
Синонимы: Манномустин.
Синтез.
Способ I.
В качестве исходного продукта применяется 1,2-5,6-диангидро-3,4-изопропилиден-D-маниитол (I), который при действии
этиленимииа (II) превращается в 1,6-D-дидезокси-1,6-ди-этиленимино-3,4-О-изопропилиден-D-маниитол (III). Из последнего при
обработке концентрированной соляной кислотой образуется дегранол (IV).
злокачественный
препарат противоопухолевый
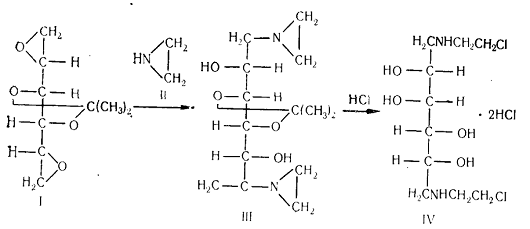
Способ II.
Для получения дегранола (VI) в качестве исходного продукта используется
2,3-4,5-ди-О-метилен-D-маннитол-1,6-ди-(п-толуолсульфонат)
(I).
При взаимодействии I с этаноламином II образуется
1,6-ди-(2'-оксиэтиламино)-2,3-4,5-ди-О-метилен-D-маннитол (III). Последний при обработке хлористым тионилом
(IV) превращается в
1,6-ди-дезокси-1,6-ди-(2'-хлорэтиламино)-2,3-4,5-ди-О-метилен-D-маннитол (V), после кипячения
которого с соляной кислотой образуется дегранол (VI).

Фармакологическое действие. Противоопухолевое средство.
Показания к применению. Хронический лимфолейкоз (рак крови, при котором
источником опухолевого процесса являются лимфобласты - клетки костного мозга,
из которых развиваются форменные элементы крови - лимфоциты); хронический
миелолейкоз (рак крови, при котором источником опухолевого процесса являются
гранулоцитарные клетки - клетки костного мозга, из которых развиваются форменные
элементы крови - лейкоциты); лимфогранулематоз (рак лимфатической системы, при
котором в лимфатических узлах и внутренних органах образуются плотные
образования, состоящие из быстро растущих клеток); лимфо- и ретикулосоркоматоз
(форма злокачественной опухоли, возникающей из лимфоидной и рыхлой
соединительной ткани); множественная мислома (заболевание костного мозга,
характеризующееся разрастанием кроветворных клеток); эритремия (болезнь,
обусловленная разрастанием клеток костного мозга и появлением в кровеносном
русле большого количества форменных элементов крови - эритроцитов, лейкоцитов и
т. д.).
Побочное действие. Лейкопения (пониженное содержание лейкоцитов в крови)
со значительной лимфопенией (уменьшением числа лимфоцитов в крови); иногда
тошнота, головокружение.
Противопоказания. Лейкопения (менее 3000 лейкоцитов в 1 мкл),
тромбоцитопения - уменьшение числа тромбоцитов в крови (менее 100 000
тромбоцитов в 1 мкл крови).
Форма выпуска. Ампулы по 0,05 г в упаковке по 5 штук.
Условия хранения. Список А. В защищенном от света месте.
2.6 Допан

Синонимы: Хлорэтиламиноурацил.
Синтез. Синтез допана состоит из трех стадий. В качестве исходного
продукта в синтезе используют 4-метил-5-аминоурацил (I), из которого при
действии окиси этилена (II) получают 4-метил-5-[бис-(2'-оксиэтил)амино]-урацил
(III). Из III при обработке хлороксидом фосфора (IV) получают
2,6-дихлор-4-метил-5-[бис-(2'-хлорэтил) амино]-пиримидин (V), который
гидролизуется соляной кислотой, и при этом образуется
4-метил-5-[бис-(2'-хлорэтил) амино]-урацил (VI).
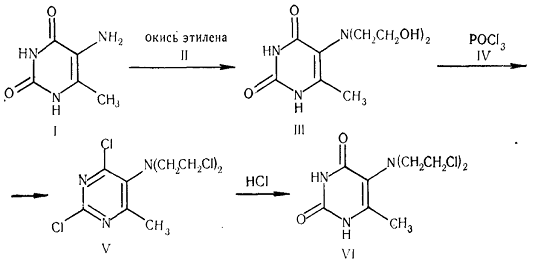
Фармакологическое действие. Обладает противоопухолевой активностью и
угнетает кроветворение (особенно костномозговое).
Показания к применению. Лимфогранулематоз (рак лимфатической системы, при
котором в лимфатических узлах и внутренних органах образуются плотные
образования, состоящие из быстро растущих клеток) II-III стадии и при
удовлетворительном общем состоянии IV стадии заболевания; сублейкемические
формы хронического миелолейкоза (рак крови, при котором в кровеносном русле
наряду с клетками - предшественниками лейкоцитов - лейкобластами - определяются
и клетки костного мозга - предшественники эритроцитов - эритробласты),
протекающие со спленомегалией (увеличением массы печени и селезенки);
хронический лимфолейкоз (рак крови, при котором источником опухолевого процесса
являются лимфобласты - клетки костного мозга, из которых развиваются форменные
элементы крови - лимфоциты).
Способ применения и дозы. Внутрь (лучше на ночь) по 0,006-0,012 г раз в
3-5-7 дней. На курс лечения - 5-7 приемов допана. Детям из расчета 0,2 мг/кг.
Побочное действие. Угнетение костномозгового кроветворения (лейкопения -
пониженное содержание лейкоцитов в крови, тромбоцитопения -уменьшение числа
тромбоцитов в крови), иногда через 8-12 ч после приема тошнота и рвота.
Противопоказания. Лимфогранулематоз при числе лейкоцитов менее 4500 и
тромбоцитов менее 150000 в 1 мкл крови, быстротекущие и острые формы этого
заболевания; острый лейкоз (общее название злокачественной опухоли крови,
возникающей из бластных клеток - клеток костного мозга, из которых образуются
лейкоциты, лимфоциты, эритроциты и т, д., и характеризующейся появлением в
кровеносном русле этих незрелых клеток) и хронический миелолейкоз (рак крови,
при котором источником опухолевого процесса являются гранулоцитарные клетки -
клетки костного мозга, из которых развиваются форменные элементы крови -
лейкоциты) с резким обострением.
Форма выпуска. Таблетки по 0,002 г в упаковке по 35 штук.
Условия хранения. Список А. В защищенном от света месте.
2.7
Циклофосфан
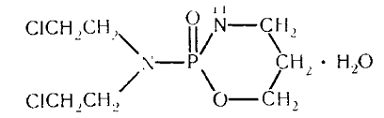
Синонимы: Циклофосфамид, Цитофорсфан, Цитоксан, Эндоксан, Эндуксан,
Геноксол, Митоксан, Процитоке, Сендоксан, СТХ, Клафен и др.
Синтез. Циклофосфаи впервые синтезирован в 1958 г. Технологичный метод
синтеза препарата разработан в Институте органического синтеза АН ЛатвССР в
1959 г. под руководством акад. АН ЛатвССР С. А. Гиллера.
Исходными продуктами при получении циклофосфана (VIII) являются
диэтаноламин, хлороксид фосфора и аминопропанол-1,3. Синтез осуществляется по
схеме
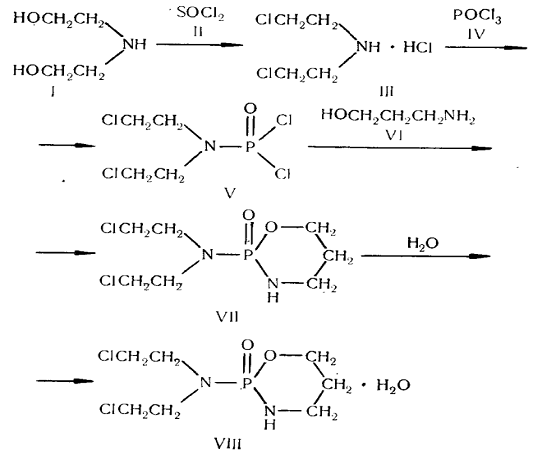
Для получения дихлорангидрида N,N-бис-(β-хлорэтил) амидофосфорной кислоты (V)
осуществляют взаимодействие III с хлороксидом фосфора (IV) в присутствии
пиридина в растворе диоксана или бензола. При конденсации V с
3-аминопропанолом-1 (VI) в присутствии триэтиламина в растворе этилового эфира
уксусной кислоты и при последующей обработке технического продукта конденсации
(VII) водой образуется моногидрат циклофосфана (VIII).
Фармакологическое действие. Является алкилируюшим (вызывающим химические
процессы в клетке, приводящие к нарушению стабильности ДНК
дезоксирибо-нуклеиновой кислоты, содержащейся в основном в ядре клетки и
являющейся носителем генной информации) иитостатическим (препятствующим росту
клеток) препаратом. Синтез препарата произведен с таким расчетом, чтобы
препарат обладал избирательной противоопухолевой активностью, т. е. чтобы он
был неактивным, находясь в крови, но при проникновении в опухолевые клетки
быстро разлагался под влиянием содержащихся в них в относительно большом
количестве фосфатаз (фосфамидаз) с освобождением бис-(β-хлорэтил)-амина.
Таким образом, препарат может рассматриваться как пролекарство с
транспортной функцией, доставляющее активное цитостатическое вещество в
опухолевые клетки.
Препарат обладает относительно широким противоопухолевым спектром
действия и оказывает более мягкое, чем другие аналогичные препараты, влияние на
тромбоцитопоэз (процесоюбразования тромбоцитов в организме).
Показания к применению. При мелкоклеточном раке легкого; раке яичников;
раке молочной железы; ретикулосаркоме (злокачественной опухоли, возникающей из
рыхлой быстрорастущей соединительной ткани); лимфосаркоме (злокачественной
опухоли, возникающей из незрелых лимфоидных клеток); хроническом лимфолейкозе
(раке крови, при котором источником опухолевого процесса являются лимфобласты -
клетки костного мозга, из которых развиваются форменные элементы крови -
лимфоциты); остром лимфобластном лейкозе (раке крови, при котором источником
опухолевого процесса являются лимфобласты - клетки костного мозга, из которых образуются
лимфоциты, которые в большом количестве выявляются в кровеносном русле);
множественной миеломе (заболевании костного мозга, характеризующемся
разрастанием кроветворных клеток); опухоли Вильмса (раке, развивающемся из
тканей почки у детей); костной ретикулосаркоме (злокачественной опухоли кости,
возникающей из рыхлой быстрорастущей соединительной ткани); опухоли Юинга
(злокачественной опухоли костной ткани); ангиосаркоме (злокачественной опухоли,
развивающейся из клеток кровеносных или .лимфатических сосудов).
Способ применения и дозы. Вводят внутривенно, внутримышечно, внутрь, а
также в полости. Растворы для инъекций готовят непосредственно перед
применением на стерильной воде для инъекций.
Используют разные схемы лечения: а) по 200 мг (3 мг/кг) ежедневно или 400
мг (6 мг/кг) через день (внутрь, внутривенно или внутримышечно); б) по 1 г (15
мг/кг) 1 раз в 5 дней внутривенно; в) 2-3 г (30- 45 мг/кг) 1 раз в 2-3 нед.
внутривенно. Курсовая доза при всех режимах лечения составляет 6-14 г.
Используют и другие схемы лечения.
При скоплении жидкости в результате ракового процесса в брюшной и
плевральной (полости между оболочками легких) полостях в дополнение к
внутривенным инъекциям вводят в полости по 0,4-1,0 г ииклофосфана (при каждой
пункции - проколе). Количество препарата, вводимого в вену, при этом
соответственно уменьшают. После окончания основного курса лечения циклофосфаном
может применяться поддерживающая терапия: 2 раза в неделю вводят внутривенно
(или внутримышечно) по 0,1-0,2 г препарата или назначают его внутрь в виде
таблеток по 0,05-0,1 г 2 роза в день, Прием препарата внутрь удобен для
длительной терапии.
Во время лечения необходимо проводить исследование крови не реже двух роз
в неделю. При снижении количества лейкоцитов до 2,5*109/л и тромбоцитов
до 100*109/л лечение прекращают. При резко выраженной лейкопении
(пониженном содержании лейкоцитов в крови) переливают кровь или лейкоцитную и
тромбоцитную массу (форменные элементы крови лейкоциты и тромбоциты, выделенные
из цельной крови и предназначенные для переливания больному), назначают
витамины, стимуляторы кроветворения. Переливание стимулирующих количеств крови
(100-125 мл 1 раз в неделю) рекомендуется производить в течение всего курса
лечения.
Побочное действие. При применении циклофосфана, особенно при
передозировке, могут наблюдаться различные побочные явления. Часто бывает
тошнота и рвота. Для уменьшения этих явлений рекомендуется введение пиридоксина
внутримышечно 0,05 г или аминазина 0,025 г внутривенно или внутримышечно через
1 ч после введения циклофосфана. Часто (до 90% случаев) через 18-20 дней после
начала применения препарата наблюдается частичная или полная алопеция (полное
или частичное выпадение волос); волосы отрастают после прекращения приема
циклофосфана. Иногда возникают головокружение, ухудшение зрения, дизурические
явления (расстройства мочеиспускания), гематурия (кровь в моче). Дизурические
явления проходят обычно через 4-5 дней. Часто больные жалуются на боль в
костях, длящуюся до 2-3 нед.
Местного раздражающего влияния циклофосфан не оказывает, однако при
внутриплевральном введении препарата может повыситься температура тела (на
2-3-й день), появиться кашель и боль в грудной клетке.
Противопоказания. Циклофосфан противопоказан при анемии (снижении
содержания гемоглобина в крови), кахексии (крайней степени истощения), тяжелых
заболеваниях сердца, печени и почек, в терминальных стадиях болезни (при
состоянии организма, предшествующем смерти).
Форма выпуска. В ампулах по 0,1 и 0,2 г препарата; таблетки, покрытые
оболочкой, по 0,05 г в упаковке по 50 штук.
Условия хранения. Список А. В сухом, защищенном от света месте при
температуре не выше +10 °С.
2.8 Эмбихин
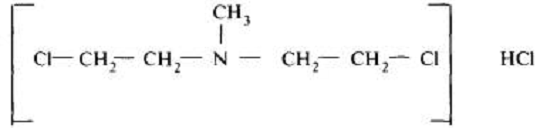
Синонимы: Хлорметин, Мустарген, Кариолизин, Хлорэтазин, Дихлорен, Димитан,
Дуамин, Эразин HZ2, Мехлорэтамин гидрохлорид, Метихлорамин, Мустин,
Нитрогранулоген, Стикстофлост, Аптимит, Азотоуперит, Хлорэтамин, Эразол, МБА,
Мебихлорамин, Митохин, Мустаарген, Мустанитроген, Нитол, Нитразин,
НитрогенМустард и др.
Фармакологическое действие. Алкилируюший (вызывающий химические процессы
в клетке, приводящие к нарушению стабильности ДНК деэоксирибонуклеиновой
кислоты, содержащейся в основном в ядре клетки и являющейся носителем генной
информации) цитостатический (препятствующий росту клеток) препарат.
Показания к применению. Препарат активен при хроническом миелоэе (потере
оболочки нервными волокнами спинного мозга); лимфо- и ретикулосаркоме (форме
злокачественной опухоли, возникающей из лимфоидной и рыхлой соединительной
ткани); лимфогранулематозе (раке лимфатической системы, при котором в
лимфатических узлах и внутренних органах образуются плотные образования,
состоящие из быстро растущих клеток); грибовидном микозе (форме рака кожи);
мелкоклеточном раке легкого. В настоящее время его применяют в основном при
лечении лимфогранулематоза в системе комплексной терапии,
Способ применения и дозы. При "ударной" методике вводят в
течение 4 дней в общей дозе 0,4 кг/кг (по 0,1 мг/кг в день) или 0,4 мг/кг
однократно. Иногда пользуются "дробнопротяжной" методикой: препарат
вводят в дозе 5-6 мг 3 раза в неделю; общая доза (за 8-20 введений) составляет
40-120 мг.
Лечение прекращают при снижении содержания лейкоцитов в крови до
2,5-3,0*109/л.
Эмбихин можно вводить также в плевральную (полость между оболочками
легких) и брюшную полости (0,2 мг/кг в 10-50 мл изотонического раствора натрия
хлорида) при наличии в них выпота (скоплении жидкости), содержащего опухолевые
клетки.
Побочное действие. Лечение эмбихином должно проводиться под тщательным
врачебным наблюдением. При применении препарата могут возникнуть осложнения и
побочные явления, связанные с его местными раздражающими свойствами и общим
токсическим действием, особенно влиянием на гемопоэз (кроветворение).
При внутривенном введении эмбихина следует тщательно следить за тем,
чтобы раствор не попал под кожу, так как возможно появление инфильтрата
(уплотнения) и некроза (омертвения ткани). В случае попадания раствора в
подкожную жировую клетчатку следует немедленно ввести в это место некоторое количество
изотонического раствора натрия хлорида. При возникновении инфильтрата применяют
компрессы.
Следует остерегаться попадания растворов препарата на слизистые оболочки
и кожу больного и медицинского, персонала. Если это произошло, необходимо сразу
же тщательно смыть препарат водой.
Для предупреждения развития у больных флебита (воспаления вен), особенно
при многократных введениях, целесообразно после инъекции вводить в вену
дополнительно 20 мл теплого раствора Рингера и не зажимать вену у места укола
после инъекции.
У части больных через 1-3 ч после введения препарата возникают тошнота и
головная боль, иногда бывает рвота. Для ослабления или устранения рвоты
назначают аминазин 0,025 г внутрь или внутримышечно через 1 ч после инъекции
эмбихина или этаперазин, Можно также вводить эмбихин вечером (после ужина), а
на ночь назначить снотворное,
В процессе лечения эмбихином необходимо следить за изменениями картины
крови. Серьезным осложнением при передозировке препарата может быть глубокое
угнетение функции костного мозга с резким подавлением гемопоэза вплоть до
явлений аплазии кроветворной ткани (состояния костного мозга,
характеризующегося резким уменьшением количества кроветворных клеток) с
летальным (смертным) исходом.
При лечении дробными дозами эмбихина влияние на лейко- и тромбоцитопоэз
(процесс образования лейкоцитов и тромбоцитов) выражено в меньшей степени,
Форма выпуска. Порошок во флаконах.
Условия хранения. Список А. В сухом, защищенном от света месте.
.9
Идентификация препаратов
По физическим свойствам препараты представляют собой белые вещества,
могут иметь желтоватый или розовый оттенок.
Производные бис-(β-хлорэтил)-амина идентифицируют и количественно
определяют методом УФ-спектрофотометрии. Производные бис-(β-хлорэтил)-амина, как правило, имеют
три максимума поглощения при 210, 260 и 300 нм.
ИК-спектрофотометрическое установление подлинности и количественное
определение основано на использовании полосы валентных колебаний (С-О)-связи
р-хлорэтиламинной группы, которая соответствует 770-760 см-1
(растворитель ацетон).
Модифицированная реакция Фудживара использована для идентификации ряда
производных бис-(β-хлорэтил)-амина. Вместо пиридина применен 20%-ный
раствор никотиновой кислоты в присутствии 20%-ного раствора гидроксида натрия.
Образовавшееся производное глютаконового альдегида сочетают с бензидином в
смеси с этанолом и уксусной кислотой до образования полиметинового красителя,
имеющего светопоглощение в области 521-527 нм. Производные бис-(β-хлорэтил)-амина дают положительные
результаты, если вместо пиридина брать его производное - γ-(4-нитробензил)-пиридин. При
нагревании в кислой среде образуются соли пиридиния, а после внесения избытка
щелочи - хиноидное соединение, окрашенное в фиолетовый цвет с максимумом
светопоглошения при 600 нм. При действии раствором реактива в ацетоне при рН 4
появляется фиолетовое или красно-фиолетовое окрашивание.
Производные бис-(β-хлорэтил)-амина разрушают с помощью дихромата калия, а
затем образующиеся летучие альдегиды открывают в парах с помощью нитропруссида
натрия, которым смачивают фильтровальную бумагу. Указанная реакция обнаружения
хлорэтилпроизводных происходит в присутствии концентрированной серной кислоты
по схеме.
Характерным свойством водных растворов производных бис-(β-хлорэтил)-амина в разведении 1:14
является способность после охлаждения превращаться в гелеобразную массу (в
более разбавленных растворах такое явление не наблюдается).
Для обнаружения производных бис-(β-хлорэтил)-амина выполняют реакцию с
реактивом Драгендорфа. Производные бис-(β-хлорэтил)-амина дают цветную реакцию
с сульфатом церия и образует окрашенные в желтый цвет продукты нитрования.
Хлорид-ион открывают реакцией с раствором нитрата серебра.
Для испытаний на подлинность и количественного определения используют
реакции на органически связанный хлор. Атом хлора можно открыть реакцией
Бейльштейна, суть которой заключается в нагревании крупинки лекарственного
вещества на медной проволоке в пламени горелки.
Пламя приобретает зеленую окраску.
В молекулах атом хлора непрочно связан с углеродом. Поэтому для
обнаружения и количественного определения органически связанного хлора
производное бис-(β-хлорэтил)-амина достаточно нагреть в водно-спиртовой
среде с раствором нитрата серебра.
Количественное аргентометрическое определение производных
бис-(Р-хлорэтил)-амина, выполняют следующим образом. Навеску нагревают в колбе
с обратным холодильником на кипящей водяной бане в течение 30-60 мин,
предварительно прибавив избыток 0,1М раствора нитрата серебра, защищая колбу от
влияния света. По истечении указанного срока холодильник промывают водой,
добавляют разведенную азотную кислоту и оттитровывают избыток нитрата серебра
0,1М раствором тиоцианата аммония (индикатор железоаммониевые квасцы).
Производные бис-(β-хлорэтил)-амина можно определить методом неводного
титрования в среде диоксана с использованием в качестве титранта 0,1М раствора
хлорной кислоты.
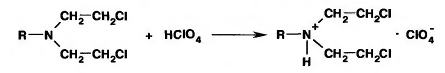
Количественный анализ в лекарственных формах выполняют
спектрофотометрическим методом при длине волны 300 нм (растворитель смесь
метанола и хлороводородной кислоты, имеющая рН = 2,3). Присутствие продуктов
гидролиза определению не мешает. Для экстракционной фотометрии в качестве
реактива используют эозинат натрия. Препараты в таблетках количественно
определяют методом ВЭЖХ по стандартному образцу в системе метанол-вода (рН
5,5). Этот же метод использован для определения препаратов и продуктов их
деструкции, в биологическом материале. Определение выполняют на хроматографе с
УФ-детектором, при длине волны 254 нм. Подвижной фазой служит смесь метанола и
фосфатного буфера (рН = 2,4).
Глава 3.
Экспериментальная часть
.1 Методика
идентификации
Таблетки циклофосфана 0,05 г, покрытые оболочкой
Состав на одну таблетку.
· Циклофосфана - 0,05 г
· Вспомогательных веществ - до получения таблетки весом 0,1 г
(без оболочки)
Подлинность. 0,4 г порошка растертых таблеток взбалтывают с 10 мл 95%
спирта и фильтруют. Прибавляют 10 мл 0,5Н раствора едкого натра и кипятят 5-10
минут. Охлаждают и подкисляют 5 мл азотной кислоты до кислой реакции. При
добавлении раствора нитрата серебра выпадает белый творожистый осадок, легко
растворимый в растворе аммиака.
,4 г порошка растертых таблеток взбалтывают с 10 мл 95% спирта и
фильтруют. Прибавляют 2 мл воды и 2 мл концентрированной серной кислоты и
кипятят; раствор становится темно-бурым. После охлаждения осторожно добавляют
25 мл 20% раствора едкого натра и снова нагревают. Влажная красная лакмусовая
бумага, помещенная в пары, окрашивается в синий цвет.
,4 г порошка растертых таблеток помещают в колбу емкостью 50 мл,
прибавляют 3 мл концентрированной азотной кислоты и 1 мл концентрированной
серной кислоты. Раствор нагревают до удаления окислов азога, после охлаждения
добавляют 10 мл воды и фильтруют. К фильтрату приоавляют 2-3 мл пергидроля и
смесь кипятят 10 минут. Раствор охлаждают до 60° и добавляют 10 мл раствора
молибдата аммония, появляется ярко-желтое окрашивание.
Определение распадаемости. 1 таблетку помещают в колбу емкостью 100 мл,
прибавляют 50 мл 0,1Н раствора соляной кислоты, имеющего температуру 37°, и
выдерживают при этой температуре 30 минут, медленно покачивая колбу 1-2 раза в
секунду. При этом таблетка не должна распадаться. Таким же образом одну
таблетку обрабатывают 0,1Н раствором гидрокарбоната натрия. Таблетка должна
распадаться в течение не более 30 минут.
Количественное определение. 5 таблеток помещают в коническую колбу
емкостью 250 мл, растворяют в 20 мл воды и 20 мл 0,5Н спиртового раствора
едкого кали. Смесь кипятят в течение 1 часа с обратным холодильником. Затем
холодильник отсоединяют, прибавляют 5 мл разведенной азотной кислоты, 5 мл пергидроля
и кипятят 10 минут, охлаждают и количественно переносят 20 мл воды в мерную
колбу емкостью 100 мл. Прибавляют 5 мл разведенной азотной кислоты, 25 мл 0,1Н
раствора нитрата серебра и доводят объем раствора водой до метки. Содержимое
колбы хорошо перемешивают и фильтруют через сухой складчатый фильтр в сухую
колбу. Первые 15-20 мл фильтрата отбрасывают. Берут 50 мл фильтрата, прибавляют
10 мл разведенной азотной кислоты, 5 мл раствора железоаммониевнх квасцов и
титруют 0,1Н раствором роданида аммония.
Параллельно проводят контрольный опыт.
мл 0,1Н раствора нитрата серебра соответствует 0,01396 г С7Н15Сl2N2O2Р*Н2О, которого в одной
таблетке должно быть 0,045-0,055 г.
Хранение. Список А. В сухом, защищенном от света месте, при температуре
не выше 10°.
3.2
Результаты идентификации
|
Название и лекарственная форма препарата
|
Показатель
|
№ п/п
|
Методика
исследования
|
Результат
|
|
Таблетки циклофосфана 0,05
г
|
Подлинность
|
1
|
0,4 г порошка растертых
таблеток взбалтывают с 10 мл 95% спирта и фильтруют. Прибавляют 10 мл 0,5Н
раствора едкого натра и кипятят 5-10 минут. Охлаждают и подкисляют 5 мл
азотной кислоты до кислой реакции. При добавлении раствора нитрата серебра
выпадает белый творожистый осадок, легко растворимый в растворе аммиака.
|
Соответствует
|
|
Таблетки циклофосфана 0,05
г
|
Подлинность
|
2
|
0,4 г порошка растертых
таблеток взбалтывают с 10 мл 95% спирта и фильтруют. Прибавляют 2 мл воды и 2
мл концентрированной серной кислоты и кипятят; раствор становится
темно-бурым. После охлаждения осторожно добавляют 25 мл 20% раствора едкого
натра и снова нагревают. Влажная красная лакмусовая бумага, помещенная в
пары, окрашивается в синий цвет.
|
Соответствует
|
|
Таблетки циклофосфана 0,05
г
|
Подлинность
|
3
|
0,4 г порошка растертых
таблеток помещают в колбу емкостью 50 мл, прибавляют 3 мл концентрированной
азотной кислоты и 1 мл концентрированной серной кислоты. Раствор нагревают до
удаления окислов азога, после охлаждения добавляют 10 мл воды и фильтруют. К
фильтрату приоавляют 2-3 мл пергидроля и смесь кипятят 10 минут. Раствор
охлаждают до 60° и добавляют 10 мл раствора молибдата аммония, появляется
ярко-желтое окрашивание.
|
Соответствует
|
|
Таблетки циклофосфана 0,05
г
|
Распадаемость
таблеток
|
4
|
1 таблетку помещают в колбу
емкостью 100 мл, прибавляют 50 мл 0,1Н раствора соляной кислоты, имеющего
температуру 37°, и выдерживают при этой температуре 30 минут, медленно
покачивая колбу 1-2 раза в секунду. При этом таблетка не должна распадаться.
Таким же образом одну таблетку обрабатывают 0,1Н раствором гидрокарбоната
натрия. Таблетка должна распадаться в течение не более 30 минут.
|
Соответствует
|
|
Таблетки циклофосфана 0,05
г
|
Количественное
определение
|
5
|
5 таблеток помещают в
коническую колбу емкостью 250 мл, растворяют в 20 мл воды и 20 мл 0,5Н
спиртового раствора едкого кали. Смесь кипятят в течение 1 часа с обратным
холодильником. Затем холодильник отсоединяют, прибавляют 5 мл разведенной
азотной кислоты, 5 мл пергидроля и кипятят 10 минут, охлаждают и
количественно переносят 20 мл воды в мерную колбу емкостью 100 мл. Прибавляют
5 мл разведенной азотной кислоты, 25 мл 0,1Н раствора нитрата серебра и
доводят объем раствора водой до метки. Содержимое колбы хорошо перемешивают и
фильтруют через сухой складчатый фильтр в сухую колбу. Первые 15-20 мл
фильтрата отбрасывают. Берут 50 мл фильтрата, прибавляют 10 мл разведенной
азотной кислоты, 5 мл раствора железоаммониевнх квасцов и титруют 0,1Н
раствором роданида аммония. Параллельно проводят контрольный опыт. 1 мл 0,1Н
раствора нитрата серебра соответствует 0,01396 г С7Н15Сl2N2O2Р*Н2О, которого в одной таблетке должно
быть 0,045-0,055 г.
|
Соответствует
|
Выводы
Фармакотерапия опухолевой патологии, наряду с лучевой терапией и
хирургией, является наиболее важной составляющей борьбы с онкологическими
заболеваниями. За последние годы она обогатилась многочисленными новыми
препаратами, повысившими ее эффективность и безопасность.
Все противоопухолевые препараты разделяют на ряд групп, исходя из их
химической структуры, механизма действия, источников получения: алкилирующие
вещества, антиметаболиты, антибиотики, агонисты и антагонисты гормонов,
алкалоиды и другие средства растительного происхождения.
Среди различных групп синтетических химических веществ, используемых для
лечения опухолевой болезни, наиболее важными продолжают оставаться алкилирующие
соединения и антиметаболиты.
Одними из первых в качестве противоопухолевых средств стали применять
вещества из группы алкилирующих соединений - производные бис-(β-хлорэтил)амина
Общая формула противоопухолевых лекарственных веществ этой группы.
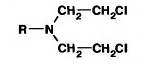
где R может быть алифатическим,
ароматическим, гетероциклическим радикалом. Свойства, большинство испытаний,
фармакологический эффект этих лекарственных веществ обусловлены наличием в
молекуле бис-(β-хлорэтил)-амина.
Производные бис-(β-хлорэтил)-амина идентифицируют и количественно
определяют методом УФ-спектрофотометрии. Производные бис-(β-хлорэтил)-амина, как правило, имеют
три максимума поглощения при 210, 260 и 300 нм.
ИК-спектрофотометрическое установление подлинности и количественное
определение основано на использовании полосы валентных колебаний (С-О)-связи
р-хлорэтиламинной группы, которая соответствует 770-760 см-1
(растворитель ацетон).
Модифицированная реакция Фудживара может быть использована для
идентификации ряда производных бис-(β-хлорэтил)-амина. При действии
раствором реактива в ацетоне при рН 4 появляется фиолетовое или
красно-фиолетовое окрашивание.
Производные бис-(β-хлорэтил)-амина разрушают с помощью дихромата калия, а
затем образующиеся летучие альдегиды открывают в парах с помощью нитропруссида
натрия, которым смачивают фильтровальную бумагу.
Для обнаружения производных бис-(β-хлорэтил)-амина выполняют реакцию с
реактивом Драгендорфа. Производные бис-(β-хлорэтил)-амина дают цветную реакцию
с сульфатом церия и образует окрашенные в желтый цвет продукты нитрования.
Хлорид-ион открывают реакцией с раствором нитрата серебра.
Для испытаний на подлинность и количественного определения используют
реакции на органически связанный хлор.
Производные бис-(β-хлорэтил)-амина можно определить методом неводного
титрования в среде диоксана с использованием в качестве титранта 0,1М раствора
хлорной кислоты.
Количественный анализ в лекарственных формах выполняют
спектрофотометрическим методом при длине волны 300 нм. Препараты в таблетках
количественно определяют методом ВЭЖХ по стандартному образцу в системе
метанол-вода (рН 5,5). Этот же метод использован для определения препаратов и
продуктов их деструкции, в биологическом материале. Определение выполняют на
хроматографе с УФ-детектором, при длине волны 254 нм. Подвижной фазой служит
смесь метанола и фосфатного буфера (рН = 2,4).
Список
использованной литературы
1. Фармацевтична хімія. Підручник для студентів вищ. фармац. начальних
закладів і фарм. фак. вищих мед. навчальних закладів III-IV рівня акредитації За заг. ред. П.О. Безуглого. - Вінниця: Нова книга,
2008. - 560 с.
2. Арзамасцев А.П. Фармакопейный анализ - М.: Медицина, 1971.
. Беликов В.Г. Фармацевтическая химия. В 2 частях. Часть 1. Общая
фармацевтическая химия: Учеб. для фармац. ин-тов и фак. мед. ин-тов. - М.:
Высш. шк., 1993. - 432 с.
. Глущенко Н. Н. Фармацевтическая химия: Учебник для студ. сред.
проф. учеб. заведений / Н. Н. Глущенко, Т. В. Плетенева, В. А. Попков; Под ред.
Т. В. Плетеневой. - М.: Издательский центр «Академия», 2004. - 384 с.
. Драго Р. Физические методы в химии - М.: Мир, 1981
. Кольтгоф И.М., Стенгер В.А. Объемный анализ В 2 томах - М.:
Государственное научно-техническое издательство химической литературы, 1950
. Коренман И.М. Фотометрический анализ - М.: Химия, 1970
8. Коростелев
П. П, Фотометрический и комплексометрический анализ в металлургии - М.:
Металлургия, 1984, 272 с.
9. Логинова Н. В., Полозов Г. И. Введение в фармацевтическую химию:
Учеб. пособие - Мн.: БГУ, 2003.-250 с.
10. Мелентьева
Г. А., Антонова Л. А. Фармацевтическая химия. - М.: Медицина, 1985.- 480 с.
11. Мискнджьян С.П. Кравченюк Л.П. Полярография лекарственных препаратов. - К.: Вища школа, 1976. 232 с
12. Фармацевтическая
химия: Учеб. пособие / Под ред. Л.П.Арзамасцева. - М.: ГЭОТАР-МЕД, 2004. - 640
с.
13. Фармацевтический
анализ лекарственных средств / Под общей редакцией В.А.Шаповаловой - Харьков:
ИМП «Рубикон», 1995
14. Фармацевтичний аналіз: Навч. посіб. для студ. вищ. фармац. навч.
закл. III - IV рівнів акредитації/П.О. Безуглий, В. О. Грудько, С. Г.
Леонова та ін.; За ред. П.О. Безуглого,- X.: Вид-во НФАУ; Золоті сторінки, 2001
- 240 с.
15. Халецкий A.M. Фармацевтическая химия - Ленинград:
Медицина, 1966
. Эшворт М.Р. Титриметрические методы анализа органических соединений
кн.1,2 - М.: Химия, 1972.