Синдром Элерса-Данло
Синдром
Элерса-Данло
Синдром Элерса-Данло
- гетерогенная наследственная болезнь соединительной ткани с разными типами
наследования
Этиология
Генетика синдрома Элерса-Данло должна
рассматриваться раздельно для каждого из 10 типов. Из них аутосомно-доминантно
наследуются 1-4, 7 и 8-й, аутосомно-рецессивно - 6-й, Х-сцепленно - 5-й и 9-й
типы. Для 10-го типа закономерность наследования не установлена, поскольку он
встречается крайне редко.
Синдром Элерса-Данло - типичный пример
разнолокусной гетерогенности. Все локусы, мутации в которых вызывают синдром,
имеют отношение к синтезу белков волокнистых элементов соединительной ткани
(главным образом коллагена). Коллагеновые волокна имеют неправильную форму и
расположены неупорядоченно.
Клиническая картина
Основные клинические характеристики синдрома
Элерса-Данло обусловлены врождённой гиперрастяжимостью соединительной ткани в
связи с нарушениями синтеза коллагена, обусловленными мутациями в разных генах
коллагена. Идентифицировано (клинически, биохимически, молекулярно-генетически)
11 типов синдрома Элерса-Данло, которые с клинико-генетической точки зрения
должны считаться самостоятельными нозологическими формами.
Основные симптомы синдрома Элерса-Данло:
. Кожа: сверхрастяжимость (щёки, под наружными
концами ключиц, локти, колени), бархатистость, хрупкость, кровоточивость,
тёмно-коричневые веснушки (более 20), рубцы (множественные, типа папиросной
бумаги, келоидные), стрии в области поясницы, просвечивающие вены, расхождение
послеоперационных швов.
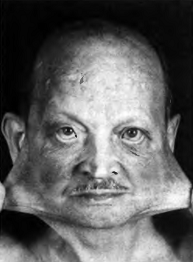
Повышенная растяжимость кожи
лица, рубцы на лбу
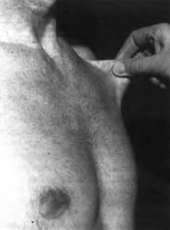
Повышенная растяжимость кожи
под ключицей
. Суставы: пассивное разгибание мизинца на 90° и
более, приведение большого пальца кисти к предплечью, переразгибание локтевого
сустава на 10° и более, переразгибание коленного сустава на 10° и более,
свободное касание ладонями пола при несогнутых коленях, переразгибание
межфаланговых, запястных, голеностопных и других суставов, привычный вывих суставов,
плоскостопие.
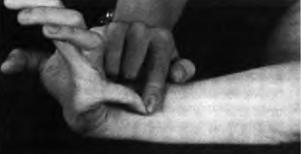
Переразгибание 5 пальца
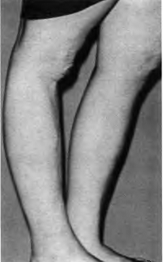
Переразгибание коленного сустава. Рубцы в
области коленных суставов
. Глаза: птоз, периорбитальная полнота, отслойка
сетчатки, остатки эпиканта, разрыв глазного яблока.
. Уши: сверхрастяжимость.
. Грудная клетка: сколиоз, кифоз, лордоз,
плоская спина, вдавление грудины.
. Живот: грыжи (пупочная, белой линии, паховая,
диафрагмальная), спонтанная перфорация кишечника.
. Конечности: варикозные вены, подкожные
подвижные узелки на голенях.
. Сердце: пролапс митрального клапана, аритмии,
вегетососудистая дистония.
. Внутренние органы: птоз желудка, почек и
матки.
. Мозг: аневризма сосудов мозга,
субарахноидальное кровоизлияния.
. Стремительные роды.
Таким образом, при синдроме Элерса-Данло имеются
нарушения (первичные или вторичные) во всех системах организма. Наиболее
важными диагностическими признаками являются гиперэластичность кожи; подкожные
узелки (сферулы), легче прощупываемые на передней поверхности голени;
переразгибание суставов; повышенная ранимость тканей; симптомы геморрагического
диатеза; пролапс митрального клапана.
В настоящее время выделяют 11 типов болезни,
различающихся между собой по первичному дефекту гена и типу наследования,
характеру и тяжести клинической симптоматики. Молекулярные механизмы патологии
соединительной ткани выявлены только при отдельных типах синдрома. Для
большинства типов характерны разболтанность (гиперподвижность) суставов,
перерастяжимость кожи без вялости и дряблости с замедленным заживлением ран и
формированием тонкого (пергаментного) рубца, склонность к кровотечениям и
кровоизлияниям. Поражение сердечно-сосудистой системы наблюдается
преимущественно при первых трех типах, которые составляют примерно 90% всех
случаев синдрома. Однако, высокая частота сердечно-сосудистых нарушений в
известных типах синдрома Элерса-Данлоса вынуждает проводить обследования во
всех неклассифицируемых формах синдрома.
Синдром
Элерса-Данло I типа
Тип наследования синдрома аутосомно-доминантный.
Молекулярный механизм патолгии неизвестен. У некоторых больных определяется
нарушение межмолекулярных поперечных связей и необычное строение коллагеновых
фибрилл.
Дети рождаются недоношенными из-за
преждевременного разрыва плодных оболочек. Течение болезни тяжелое. Характерны
генерализованная разболтанность суставов, склонность к вывихам, выраженная
гиперрастяжимость кожи, ранимость ее повышена, на месте травмы образуются
пергаментные рубцы. В местах наибольшего давления (коленные и локтевые суставы)
часто образуются псевдоопухоли, состоящие из конгломератов деструктивных
элементов соединительной ткани и организующихся гематом.
Сердечно-сосудистая система. Патология сердца по
данным аускультации и ЭКГ выявляется у всех больных, однако лишь в 30% случаев
сопровождается тяжелыми нарушениями кровообращения и сходна с таковой при
синдроме Марфана. Наиболее характерно возникновение пролапса митрального
клапана, который у большинства больных протекает бессимптомно и лишь в ряде
случаев обуславливает возникновение митральной недостаточности. Обычно пролапс
митрального клапана является аускультативной или эхокардиографической находкой.
Некоторые дети имеют жалобы на боли в грудной клетке или перебои в работе
сердца. Аускультативно чаще выслушивается сочетание щелков с
позднесистолическим шумом. На ЭКГ регистрируются признаки повышения
электрической активности левого желудочка, неспецифические изменения процесса
реполяризации, усиливающиеся при проведении ортостатической пробы или
физической нагрузки. Реже наблюдаются атриовентрикулярные блокады и блокады
ножек пучка Гиса. Эхокардиография выявляет голосистолическое и реже
позднесистолическое прогибание створок, наряду с этим может определяться
умеренная дилатация корня аорты (90-95 процентиль нормального распределения
показателя). Если дилатация аорты не выявляется в подростковом возрасте, то
она, в отличие от синдрома Марфана, не развивается и у взрослых. Митральная
недостаточность возникает на поздних стадиях заболевания и связана с
деформацией створок клапана и дилатацией атриовентрикулярного кольца. Известны
единичные наблюдения расслаивающей аневризмы аорты.
Врожденные пороки сердца наблюдаются у 5-10%
больных. Наиболее часто наблюдаются дефект межпредсердной перегородки и дефект
межжелудочковой перегородки, реже - декстрокардия, коарктация аорты,
периферические стенозы легочной артерии, двухстворчатый трикуспидальный клапан.
При морфологическом исследовании эндокард,
миокард, и перикард обычно интактны, описан случай идиопатической
концентрической гипертрофии миокарда желудочков.
Лечение.
Дети с синдромом Элерса-Данло I типа должны находиться под регулярным
наблюдением кардиолога. Проводится динамичный эхокардиографический контроль за
состоянием митрального клапана и размером аорты. При нарушении процесса
реполяризации в миокарде рекомендуется проведение курсов кардиотрофической
терапии. Санация очагов инфекции и оперативные вмешательства должны проводиться
на фоне антибактериальной терапии с целью профилактики инфекционного
эндокардита.
генетический гиперрастяжимость
соединительный ткань
Синдром
Элерса-Данло II
типа (классический тип)
Разделён на два типа: тип I (тяжёлый) и тип II
(умеренный). Поражает приблизительно от 2 до 5 человек на 100 000, и является
вторым по распространённости. Поражает коллаген V и I типа. Тип наследования:
аутосомно-доминантный.
Важные симптомы затрагивают кожу и суставы.
Больные как правило проявляют:
· уродливые или необычайно обширные
шрамы, особенно на лбу, коленях, локтях и подбородке
· гиперподвижные суставы имеют
тенденцию к вывихам, растяжению связок и подвывихам (обычно в коленной чашечке,
в плече, в пястно-фаланговом суставе en:Metacarpophalangeal joint, и в
височно-челюстном суставеen:Temporomandibular joint
· Поражение сердца протекает
субклинически, как и при I типе характеризуется пролапсом митрального клапана
редко осложняющегося митральной регургитацией. Аневризмы магистральных сосудов
наблюдаются казуистически редко.
· Для данного типа более характерно
развитие варикоза вен нижних конечностей, наблюдаемого уже в подростковом
возрасте.
Из-за сниженного мышечного тонуса, у младенцев
может быть нарушено развитие моторных навыков
Дети могут иметь склонность к развитию грыжи или
смешению любого внутреннего органа.
В настоящее время не существует определённого
теста для диагностики этого типа синдрома. И ДНК анализ и биохимические
исследования используются для выявления пораженных болезнью. В некоторых
случаях биопсия кожи была признана полезной при постановке диагноза. К
сожалению эти тесты не достаточно надёжны, чтобы выявить всех больных. Если в
семье есть несколько больных членов, то можно провести внутриутробную ДНК
диагностику.
Лечение.
Как и при I типе синдрома.
Синдром
Элерса-Данло Ш типа (гипермобильный)
Тип наследования: аутосомно-доминантный.
Заболевание имеет доброкачественное течение.
Имеет место выраженная гиперподвижность крупных суставов, кожные изменения
минимальны.
Сердечно-сосудистая система. Наиболее часто
патология сердца проявляется пролапсом митрального клапана, возможно развитие
эктопических аритмий, блокад атриовентрикулярного проведения.
Лечение.
Как и при I типе синдрома.
Синдром Элерса-Данло
IV
типа (сосудистый тип)
Поражает приблизительно 1 человека на 100 000,
вызван аутосомным доминантным дефектом в синтезе коллагена типа III. Этот тип
является самой опасной разновидностью синдрома. Проведенные исследования
определяют ожидаемую продолжительность жизни примерно в 48 лет. Тем не менее,
эта цифра вероятно искажена и основана на факте, что этот тип (как все
остальные типы синдрома) значительно не выявлен, и высокая пропорция
смертельных случаев вызвана посмертным диагностированием. Повышение
осведомлённости среди врачей и населения может помочь сделать эту цифру более
точной, и сократить число преждевременных смертей.
Признаки и симптомы:
· Гиперподвижность, наиболее очевидна на пальцах
рук и ног
· Хрупкие стенки сосудов оболочек
органов и нежной кожи, имеют склонность к разрыву или образованию аневризмы
· Больные как правило имеют тонкую,
бледную и прозрачную кожу (можно видеть вены на груди)
· Артериальная/кишечная/маточная
хрупкость или трещины, разрывы
· Некоторые пациенты выражают характерные черты
лица (большие глаза, маленький подбородок, тонкий нос и губы, мягкие уши) и
имеют маленький рост
В результате возможности маточного разрыва,
беременность может оказаться опасной для жизни. Доступно лабораторное тестирование.
Кожная биопсия может служить доказательством аномальной структуры коллагена.
Этот биомеханический анализ выявляет более 95 % случаев. Лабораторное
тестирование рекомендуется лицам имеющим два или более значительных симптома.
ДНК анализ может выявить изменения в пределах гена COL3A1. Эта информация может
помочь при предродовой генетической консультации, когда один из родителей был
диагностирован и известна его генетическая мутация или был продемонстрирован
биомеханический дефект.
Синдром Элерса-Данло
V типа
Тип наследования: рецессивный, сцепленный с
Х-хромосомой. В культуре фибробластов обнаруживается недостаточность
лизилоксидазы.
Клиническая симтоматика идентична признакам II
типа синдрома Элерса-Данло, ведущими из которых являются гиперподвижность
суставов и большая растяжимость кожи.
Сердечно-сосудистая система. В большинстве
описанных случаев синдрома больные имели тяжелую митральную регургитацию, с
ранним развитием застойной сердечной недостаточности. Аускультативно
определяется грубый систолический шум на верхушке с проведением в подмышечную
область, акцент II тона на легочной артерии. При эхокардиографии створки
митрального клапана и фиброзное кольцо увеличены в размерах, створки имеют
большое прогибание в полость левого предсердия.
Лечение.
При развитии кардиомегалии, легочной гипертензии (не более 2 стадии),
мерцания-трепетания предсердий показано протезирование клапана. В остальных
случаях проводится консервативная терапия сердечными гликозидами, диуретиками,
вазодилататорами. Всем детям должна проводиться антибактериальная профилактика
инфекционного эндокардита.
Синдром
Элерса-Данло VI
типа (кифосколиоз)
Он очень редок, обнаружено немногим более 60
случаев, передаётся аутосомно-рецессивным механизмом. Основным симптом является
общая нестабильность (непрочность) суставов. У младенцев наблюдается слабый
мышечный тонус, задержка в развитии моторных навыков, прогрессирующее в течение
жизни ненормальное искривление позвоночника сколиоз, при котором обычно больные
не могут ходить к 20 годам. Легко ранимые глаза и кожа, также вероятна
уязвимость кровеносных сосудов. Наблюдаются: спонтанная отслойка сетчатки,
кровоизлияния в стекловидное тело, разрывы глазного яблока и роговицы, склер.
Также у костей может быть снижена плотность. У большинства больных как и при
I-II типах синдрома определяются пролапс митрального клапана, митральная
регургитация, аневризмы аорты. У ряда больных, как и при IV типе синдрома
наблюдаются разрывы артерий, аневризмы головного мозга, артериовенозные
фистулы. Существует четыре основных медицинских критерия при диагностике этого
типа. Это гиперподвижные суставы, слабый мышечный тонус у новорождённых,
прогрессирующий с рождения сколиоз и хрупкие (уязвимые) глаза, что может
придать голубоватый оттенок склерам или вызвать разрыв глаза. Вызван
изменениями в хромосоме 1 гена PLOD, который кодирует фермент Лизин Гидролаза.
Возможен лабораторный тест, в нём измеряется содержание в моче hydroxylysyl
pryridinoline'а. Он крайне чувствителен и специфичен для данного типа синдрома,
рекомендуется младенцам с тремя и более основными симптомами. Внутриутробный
анализ применяется, если известно о существующем риске: диагностированы больные
члены семьи, имеющие положительные результаты тестирования. Вышеупомянутый
амниоцентез может быть выполнен, если зародышевые клетки берутся из
амниотической жидкости и измерена активность фермента.
Лечение
см. синдром Элерса-Данлоса I типа.
Синдром Элерса-Данло VII
типа (артроклазийная мультикомплексная миотония)
Больные с этой формой болезни маленького роста,
у них наблюдается высокая гибкость суставов и дислокации. Изменения на коже
также различны. Наследуется по аутосомно-доминантному и аутосомно-рецессивному
типу. Для диагностики заболевания проводится биопсия кожи.
Синдром
Элерса-Данло VIII
типа (тип дермоспараксис)
У больных с данной формой синдрома очень вялая,
мягкая, одутловатая кожа, которая обвисает. Диагностика этой формы синдрома
проводится при помощи биопсии кожи.
Синдром Элерса-Данло X типа
Тип наследования: аутосомно-рецессивный. Предполагается
дефект в молекуле фибронектина.
Характерны разболтанность суставов, повышенная
растяжимость кожи, поражение слизистых, кровоподтеки, петехии.
Сердечно-сосудистая система. У большинства
больных обнаруживаются пролапс митрального клапана, умеренная дилатация аорты в
области синусов Вальсальвы без развития аневризм. На ЭКГ может определяться
атриовентрикулярная блокада I степени.
Клиническая картина разных типов синдрома
Элерса-Данло «перекрывается» почти по всем симптомам. Для дифференциальной
диагностики используют критерии, основанные на сочетаниях выраженности
симптомов и осложнений. Для этого нужен опыт лечения больных с врождёнными
нарушениями соединительной ткани. 5-10-й типы встречаются очень редко. Наиболее
«тяжёлыми» типами являются 1-й и 4-й. «Лёгкий» 2-й тип трудно отличить от
врождённой дисплазии соединительной ткани.
Лечение. Сидром
лечится в соответствие с теми проявлениями болезни, которые наблюдаются у
каждого пациента в отдельности.
Также следует избегать повреждений суставов.
Время от времени, необходимо фиксировать суставы. Необходимо выполнять
упражнения, которые укрепляют мышцы и поддерживают суставы, чтобы снизить
повреждение суставов. Необходимо избегать занятий контактным спортом и
деятельности, при которой возможно повреждение суставов.
Синдром Элерса-Данло мало отражается на
репродуктивной функции, хотя у больных частично снижено число потомков. Имеются
изоляты с выраженным эффектом родоначальника на протяжении нескольких
поколений, в которых больные с синдромом 1-го типа составляют 10% всего
населения.