Синтез и модификация биологически активных макрогетероциклических соединений
Министерство образования и науки РФ
Федеральное государственное бюджетное
образовательное учреждение
высшего образования
Ивановский государственный
химико-технологический университет
МАГИСТЕРСКАЯ ДИССЕРТАЦИЯ
на тему:
Синтез и модификация биологически
активных макрогетероциклических соединений
Автор: Петрова Д.В.
Научный руководитель: д.х.н. проф. Семейкин А.С.
Руководитель магистерской программы: д.х.н. проф. Макаров
С.В.
Иваново, 2016г.
Автореферат
магистерской диссертации Петровой Д.В.
на тему «Синтез и модификация биологически активных
макрогетероциклических соединений»
Актуальность темы. Синтетические аналоги порфиринов находят своё применение как
катализаторы, жидкие кристаллы, фотосенсебилизаторы для фотодинамической
терапии раковых опухолей и фотодезактивации патогенной микрофлоры. Поэтому
разработка синтеза таких соединений представляет научный интерес.
Цель работы: синтез фенилзамещённых порфиринов, изомерных им порфиценов,
аналогичных корролов, а также изучение спектральных и координационных свойств
этих соединений.
Практическая значимость. С целью повышения выхода модифицирован синтез
2,7,12,17-тетрафенилпорфицена. Разработаны методики синтеза фенилзамещенных
пирролов для синтеза β-фенилзамещенных порфиринов и порфиценов.
Апробация работы. Отдельные разделы диссертации были представлены на 6
конференциях различного уровня.
Публикации. Материалы, изложенные в диссертации, нашли свое отражение в
8 печатных работах (направлена в печать в журнал ВАК - 1 статья, опубликованы:
1 статья в журнале, 6 тезисов).
Структура и объем работы. Диссертация состоит из введения, аналитического
обзора литературы, практической части с обсуждением результатов, выводов и
списка использованных источников, насчитывающего 99 наименований.
Материал диссертации изложен на 60 страницах, содержит 23 схемы, 6
рисунков, и 2 таблицы.
Abstract
’s thesis by Petrova D.V.
«Synthesis and modification of macroheterocyclic biologically
active compounds»
Actuality of the subject. Synthetic analogues of porphyrins
find their application as catalysts, liquid crystals, photosensitizers for
photodynamic therapy of tumors and photo dezactivation of pathogenic bacterial
flora. Therefore, the development of synthesis of these compounds is
interesting scientific direction.
The purposes of the research are synthesis of
fenilsubstituted porphyrin, isomers them porphycenes, similar corroles, and the
study of the spectral and coordination properties of these compounds.
Practical significance. Modification of synthesis of
2,7,12,17-tetrafenilporphycen was performed to increase the yield. Synthesis
techniques of the phenyl-substituted pyrroles for the synthesis of β-phenyl-substituted porphyrins and
porphycenes were developed.
Approval of the work. Certain sections of the master's
thesis were presented at 6 conferences of different levels.
Publications. The materials contained in the
thesis are reflected in the 8 printed works, among them: 1 article in press (in
the magazines of SCADT), 1 magazine article, 6 theses.
Structure and wordage of work. The master's thesis
consists of introduction, analytical review of literature, the experimental
part and the discussion of results, conclusion and list of references, which
includes 99 titles.material is presented at 60 pages, contains 23 circuits, 6
figures and 2 tables.
Содержание
Введение
. Теоретическая часть
.1Виды изомеров и аналогов порфиринов
.1.1 Виды изомеров порфиринов
.1.2 Виды аналогов порфиринов
.2 Методы синтеза макрогетероциклических соединений
.2.1 Методы синтеза порфиценов
.2.1.1 Метод синтеза исходных α-незамещённых пирролов методом
Бартона-Зарда
.2.2 Получение (мезо-)5,10,15,20-тетразамещённыхпорфиринов
.2.3 Методы синтеза корролов
.2.3.1 Тетрамеризация 2-замещенных пирролов
.2.3.2 Конденсация пирролов с альдегидами
.2.3.3 Синтез из бипиррольных соединений
.3 Синтез металлокомплексов макрогетероциклов
1.3.1 Металлопорфирины
.3.2 Металлокомплексы порфиценов
1.3.3 Металлокомплексы корролов
. Экспериментальная часть и обсуждение результатов
.1 Синтез 2,7,12,17-тетрафенилпорфицена
.2 Синтез 5,10,15,20-тетрафенилпорфина
.3 Синтез 5,10,15-трифенилкоррола
.4 Синтез металлокомплексов тетрафенилпорфина,
тетрафенилпорфицена, трифенилкоррола
.4.1 Металлокомплексы 5,10,15,20-тетрафенилпорфина
.4.2 Металлокомплексы 2,7,12,17-тетрафенипорфицена
.4.3 Металлокомплексы 5,10,15-трифенилкоррола
.5 Попытки синтеза β-замещенных порфиринов
.5.1 Попытки синтеза 3,7,13,17-тетрафенилпорфирицена
.5.2 Попытки синтеза смеси изомерных β-тетрафенилпор-фиринов
.6 Попытки синтеза β-замещенных порфиценов
.6.1 Синтез эфиров изоцианоуксусной кислоты
.6.2 Эфиры муравьиной кислоты
.6.3 Синтез 2,4-дикарбэтокси-3-фенилпиррола
.6.4 Синтез пирролов методом Бартона-Зарда
.7 Электронные и 1Н ЯМР-спектры мезо-тетрафенилпорфирина,
тетрафенилпорфицена, трифенилкоррола и их металлокомплексов
Выводы по работе
Список литературы
Введение
Порфирины и их металлокомплексы большой класс соединений широко
распространеных в природе, к ним относятся производные гема - гемоглобин,
отвечающий за перенос кислорода в крови и многочисленные
окислительно-восстановительные ферменты - цитохромы. Магниевые комплексы
гидрированных порфиринов - хлорофиллы, отвечают за утилизацию солнечной энергии
водорослями и высшими растениями.
Родоначальником всех порфиринов является простейший макроцикл - порфин.
Синтезировано большое количество искусственных производных порфина (1) -
наиболее известные и доступные из них β-октаэтилпорфин (2) и мезо-тетрафенилпорфин
(3).
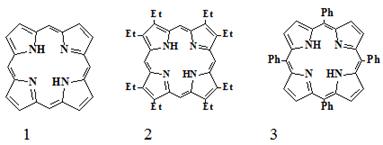
Характерной особенностью порфиринов является их многообразие, которое
вместе с уникальной молекулярной структурой определяет их биологические функции
в природе, способность выступать в качестве катализаторов, фотосенсибилизаторов,
разнообразных красителей, органических полуприводников. Порфирины являются
макроциклами, которые из-за особой многоконтурной ароматической системы,
способности образовывать сверхпрочные внутрикомплексные соединения практически
со всеми металлами периодической системы находят практическое применение во
многих областях науки.
В связи с этим возрос интерес не только к порфириноам но и к не известным
в природе их аналогам и изомерам. Известно большое количество таких соединений
относящихся к классу порфириноидов: - изомеров и аналогов порфиринов, имеющих
искаженное строение реакционного центра таких, например, как порфицены (4)
и корролы (5).
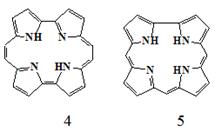
Таким образом, целью данной дипломной работы стал синтез фенилзамещённых
порфиринов, изомерных им порфиценов, аналогичных корролов, а также изучение
спектральных и координационных свойств этих соединений.
1. Теоретическая часть
1.1 Виды изомеров и аналогов порфиринов
синтез порфирин макрогетероциклический металлокомплекс
1.1.1 Виды изомеров порфиринов
Порфирины, производные порфина C20H14N4 (1),
являются самыми распространенными и важными среди природных макроциклов. Долгое
время синтетические изомерные порфириноподобные системы, так называемые
порфириноиды, как отдельный от порфиринов класс соединений, не рассматривались,
до исследований Вогеля [1-2]. Он сообщил о синтезе первого ароматического
изомера порфирина, порфицене (4). Эта разновидность порфириноидов
демонстрировала спектроскопические и координационные свойства, которые
отличались от таковых для порфиринов, а их исследование как потенциальных PDT фотосенсибилизаторов служили
«исходной точкой» для последующего синтеза нескольких других порфириноидов,
имеющих внутрициклические атомы азота, изомерных порфину. Эти последние
разновидности включают корфицен (6), полученный независимо Сесслером [3]
и Вогелем [4], гемипорфицен (7), полученный Калло [5], а также
изопорфицен (8), синтезированный Вогелем [6]. Кроме приведённых изомеров
существуют также общее описания синтеза алкил-производного диметина (9)
[7]. Эти соединения являются, с одной стороны изомерами порфиринов, с другой
стороны их аналогами, имеющими искаженное строение внутреннего реакционного
центра, в отличие от порфиринов, реакционный центр которых квадратный.
Данные изомеры, равно как и порфицен являются ароматическими, что
доказывается наличием полосы Соре и полос Q-типа в видимых спектрах поглощения.
Кроме того ЯМР-спектры этих производных показывают сигналы характерные для
внутренних протонов, которые смещены в сильное поле, что доказывает наличие
кольцевого тока в системе, и их ароматичность.

К приведенным изомерам примыкают также сравнительно недавно
синтезированные производные «извернутых» порфиринов в которых один из атомов
азота находится снаружи макроцикла (10) [8,9]. Данные полученные
группами Латос-Гражинского и Фуруты, подтверждают богатство химии таких
порфириноидов. Обобщённая формула C20H14N4 может включать в себя такие
порфириноидные структуры как (11), которые включают замещение пиррола на
пиридин, а также, например, (12).
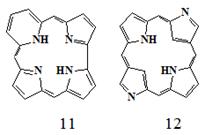
Одними из наиболее важных и доступных изомеров порфина является так
называемый класс порфиценов, которые являются одновременно как аналогами, так и
изомерами порфиринов. Порфицен является структурно новым соединением и состоит
из двух 2,2’-биспирролов соединённых двумя этиленовыми мостиками (4). В
результате это плоская, ароматичная макроциклическая система официально
известная как порфирин-(2.0.2.0.) [10]. Название порфицен было предложено
потому, что данное соединение обладает структурными особенностями отличными
таковых для порфиринов. Порфицен не единственный первый синтезированный изомер
порфирина, хотя он является наиболее упоминаемым, по крайней мере, среди систем
с N-4 центральным ядром (т.е. состоит из 4-х молекул пиррола). Следовательно,
его химия изучается, особенно в качестве лиганда для синтеза металлокомплексов.
Эти комплексы потенциально могут найти своё применение, как катализаторы,
аналоги белка, и материальной химии [11-13]. Они представляют значительный
интерес, обладают уникальными оптическими свойствами. Порфицены имеют сильное
поглощение в дальней красной области видимого спектра (в зависимости от
структуры порфицена, мксимум поглощения находиться в области 620-760 нм.) Эти
спектральные особенности позволяют использовать порфицены в фотодинамической
терапии. Другое биомедицинское применение, которое возникло недавно, включает
использование порфиценов для фотоинактивации вирусов и бактерий [14]. Однако, в
дополнение к потенциальному практическому применению, порфицены остаются
интересными обьектам для теоретического изучения электронной структуры,
ароматичности и замены протона из-за их родства с порфиринами.
1.1.2 Виды аналогов порфиринов
Аналоги порфиринов могут быть определены как пирролсодержащие макроциклы,
которые в природе не встречаются. Это общее определение включает в себя два
подкласса соединений: первая группа включает в себя системы, которые содержат
три или более пиррольных колец и некоторую степень сопряжения связей, вторая -
системы обладающими только четырьмя пиррольными кольцами, но не имеют
сопряжения связей. Традиционно, первая из этих двух групп была представлена
аналогами порфирина, которые заужены, расширены или изомерны относительно
порфирина. Наиболее известны: коррол (5), сапфирин (13) и
порфицен (6).
13
Происхождение химии аналогов порфиринов может быть прослежено из ранних
изучений структуры витамина В12.
История изучения корролов начинается с 1960 годов, как результат
исследования Джонсона, посвященного развитию синтетических методов получения
витамина B12. Это связано с тем, что в составе витамина присутствует
кобальтовое производное коррина (14), стабильный октадегидро аналог которого
- коррол. Идея Джонсона состояла в том, чтобы использовать коррол как
предшественник корринового цикла [15]; но этот подход был неудачен, и в течение
длительного времени, этот макроцикл практически не изучался. Однако химия
корролов представляет научный интерес из-за некоторых интересных и
специфических свойств лиганда этого макроцикла, например, способность к
стабилизации более высоких окислительных состояний металлов
комплексообразователей [1,2]. Кроме того, специфическое поведение корролов может
быть интересным для их применения в качестве материалов в химических датчиках
[16].
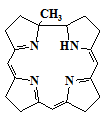
14
Корролы ароматические соединения - видимые спектры
показывают интенсивное поглощение около 400 нм и более слабое поглощение в области
500-600 нм. Это поглощение может быть связано с B и Q-полосами порфиринов и
указывают на присутствие ароматической системы. Корролы имеют интенсивные
полосы люминесценции около 600 нм, с продолжительностью жизни в около
наносекунды и очень малый Стоксов сдвиг. Кроме того, диамагнитный кольцевой ток
также присутствует в спектрах ЯМР корролов, и все сигналы показывают
значительные сдвиги, подобные, тем которые наблюдаются в аналогах порфиринов.
Одна из интереснейших характеристик корролов - это
наличие трех протонов во внутренней полости, по этой причине корролы существуют
как трианионные лиганды, в отличие от корринов и порфиринов, которые,
соответственно, являются моноанионными и дианионными лигандами.
Исследования витамина B12 также являются исходной точкой
для изучения расширенных порфиринов. В этом случае ключевое открытие было
сделано Вудвардом и его группой. В ходе работ, посвященных синтезу коррола, эти
исследователи случайно выделили пентапиррольный макроцикл. Это соединение,
содержащее 22p-электрона в
его ароматической периферии, является тёмно синим в твердом состоянии и
интенсивно зеленым в органическом растворе. Вудвард назвал этот класс
макроциклов «сапфиринами».
В результате исследований этого класса соединений,
выполненных Ibers [17], стало очевидно, что
протонированные формы сапфирина являются потенциальными анион-связывающими
агентами. Эта анион-связывающая способность, изучена с помощью широких
исследований фазового распределения, хроматографии и фазового переноса. В ходе
исследований выяснилось, что анион-связывающие свойства сапфирина связаны с
тем, что сапфирины, обладая большей полостью, более легко протонируются чем
порфирины и пиррольные NH
остатки, если не депротонируются (например, не связанные с центральным
металлом), являются хорошими донорами водородной связи для таких классических
основных анионов Льюиса как фторид, хлорид и фосфат [15].
В то время как анион-связывающий потенциал сапфиринов
интересен с позиции молекулярного распознавания, их светопоглощающие свойства
позволяют рассмотреть сапфирины, как потенциальные фотосенсибилизаторы для
использования в фотодинамической терапии. Несколько металлокомплексов сапфирина
были описаны в литературе, однако пока не существует ни одного примера, где
сапфирин действует как пятиугольный плоский лиганд.
1.2 Методы синтеза макрогетероциклических соединений
1.2.1 Методы синтеза порфиценов
Ключевой стадией в синтезе порфиценов, использующей реакцию МакМурри,
является восстановительная димеризации 5,5’-диформил-2,2’-биспирролов в
присутствии низковалентного титана активированного медью, с последущим
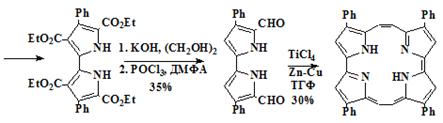
самопроизвольным окислением атмосферным кислородом (Схема 1).
Это единственный на настоящее время метод получения порфиценов. Исходя из
этого, основной стратегией синтеза, является получение биспиррола необходимого
строения. Поэтому стратегии синтеза могут быть различными.
Схема 1

Впервые порфицены были синтезированы Вогелем в 1986 году. Метод синтеза
приведённый в его работах [18] позволял получать не только сам порфицен (24),
но и тетразамещенные порфицены содержащие в качестве заместителей метильные,
этильные, трет-бутильные, н-пропильные и β-метоксиэтильные группы [19-20]
(схема 2).
Схема 2
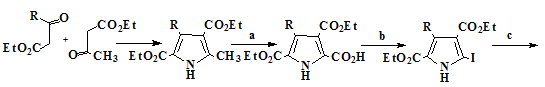
15 16 17 18 19
20 21 22

23 24
R=-CH3, -C2H5,-С(СH3)3, -н- С3H5, - С2Н4ОСH3
Условия реакций и выходы продуктов: a) Br2, SO2Cl2, AcOH/HCOOH, 0°C, 4 часа, (50-58%). b) KI/I2, EtOH/H2O, 75°C,
(75-77%). c) Cu, ДМФА, 20°С, 17 часов, (67-71%). d) NaOH, EtOH/H2O, 15 часов (96%).
e) Сублимация при 230°С, (89%). f) POCl3/ДМФА, NaOAc/H2O, (86%). g) TiCl4/Zn/пиридин, ТГФ, 0,5 часа, (10%)
Исходными соединениями для этого синтеза являются кетоэфиры (15, 16).
Из них по реакции Кнорра проводится синтез 2-метилпиррола соответствующего
строения (17), окисление которого позволяет получить пиррол
монокарбоксилат (18). Дальнейшей стадией синтеза является реакция
электрофильного ипсо-замещения карбоксильной группы монокарбоксилата
пиррола (19) на атом иода, с получением иодпиррола (20),
Конденсацией иодпиррола (20) по Ульману получают биспиррол тетраэфир (21),
который далее через стадии декарбоксилирования и формилирования по Вильсмаеру
[21] приводит к диформилбиспирролу (23) нужной структуры. Для синтеза
порфицена (24) использовался метод восстановительной димеризацией по
Мак-Мурри диформил-2,2’-биспиррола (23) в присутствии низковалентного
титана активированного медью, с последующим самопроизвольным окислением
атмосферным кислородом. Димеризация по Мак-Мурри даёт различные выхода
порфиценов (24) в зависимости от замещающих групп. По литературным данным
[22] выход порфиценов, имеющих различные заместители варьируется от 10 до 25%,
тогда как выход незамещённого порфицена всего 2%.
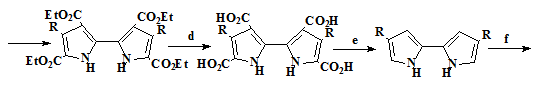
Метод синтеза, разработанный Вогелем, также используется и при получении
порфиценов, несущих ароматические заместители в своём составе. Разработкой
синтеза 2,7,12,17-тетрафенилпорфицена, занимались, не зависимо друг от друга,
две группы учёных - Вогель [18] и Нонелл [23]. Методы синтеза, применяемые
этими учёными, имеют некоторые различия. В своём методе Нонелл из биспирролтетраэфира
вида (20 R = Ph), сразу получает 2,2’-биспиррол (22)
минуя промежуточную стадию тетракарбоновой кислоты (21). Это достигается
путём обработки тэтраэфира щёлочью в этиленгликоле, что позволяет избежать
сублимации продукта, которая неизбежна в синтезе по методу Вогеля и упрощает
синтез. В последствии Нонелл [23] разработал новую стратегию получения
замещенных биспирролов (схема 3). Она основана на использовании катализируемой
палладием реакции Судзуки. В этом методе исходный тиобиспиррол (25)
имеющий блокированные 3,3’-положения легко электрофильно бромируется и, после
защиты триметилсилильными группами атомов азота, вступает в реакцию Судзуки.
Дальнейшая десульфуризация никелем по Ренею и десилилирование тетра-н-бутиламмонийфторидом
приводит с высоким выходом к требуемому биспирролу (27).
Схема 3
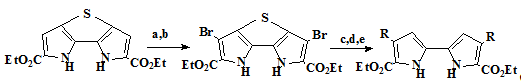
25 26 27
Условия проведения реакций и выходы продуктов: a) Br2, SO2 Cl2, AcOH/AcOEt (89%). b) SEMCl, NaH, ТГФ, (94%). с) RB(OH)2, Pd(PPH3)4, Na2CO3, 1,4-диоксан, R=Ph (100%). d) Ni, EtOH, R=Ph (100%), e) тетра-n-бутиламмоний
фторид, 1,4-диоксан, R=Ph (93%).
Хотя реакция Mак-Мурри является
устоявшимся эффективным методом получения различных порфиценов, незамещенный
порфицен по данной реакциеи получается с малым выходом. На основе
экспериментальные наблюдений, был сделан вывод, что отсутствие растворимости
исходного биспиррола в значительной степени ответственно за низкий выход [22].
Основывась на этом предположении, Waluk и его коллегии [24], синтезировали незамещенный порфицен по
усовершенствованному методу, в котором можно осуществлять временное растворение
биспирролов содержащих трет-бутильные заместители. Таким образом,
тозилирование и функционализация пиррола (28), дает исходный пиррол (30),
окислительная конденсация которого обеспечивает получение биспиррола (31),
детозилирование и формилирование его по Вильсмайеру приводит к формилбиспирролу
(32) превращенного реакцией Мак-Мурри в трет-бутилзамещенный
порфицен (33), трет-бутильные заместители в котором удалялись с
65%-ным выходом, путем нагревания в H2SO4. Другим отличием этого синтеза
является то, что осуществлена замена реакции Ульмана, при получении биспиррола
(31) на окислительную конденсацию. (Схема 4)
Схема 4

28 29 30 31 32
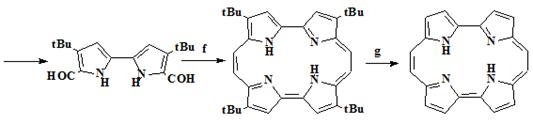
33 34 35
Условия проведения реакций: a) TSCl, NaOH. b) t-BuCl, AlCl3, CH2Cl2. c) n-BuLi, ТМП, CuCl2, ТГФ. d) KOH, CH2OH. e) POCl3/ДМФА, CHCl3.
f) TiCl4/Zn/Cu, ТГФ, 0,5 часа. g) H2SO4
Сесслер и Sanchez - Garcia
[25] упростили классический синтез порфиценов (34), использованием
нового подхода для синтеза иодпирролов (19) из альдегидов (36)
конденсацией их этилизоцианоацетатом с образованием α-незамещенных пирролов (32) и
последующим электрофильным иодированием (схема 5). Этот метод дает как
алкильные, так и ароматические пирролы общей структуры (19) с хорошими
выходами (50-71%). Новый метод позволяет сократить количество стадий, по
сравнению с классическим методом, и дает возможность получения порфиценов
имеющих различные как алкильные, так и арильные заместители. В подтверждение
этого, были синтезированы новые порфицены, имеющие в позициях 2,7,12,17
заместители R = н-C7H15 и R = п-CH3C6H4.
Схема 5

36 37 19 20

22 23
Условие протекания реакций: a) этил изоцианоацетат, 1.8-диазабицикло[5.4.0]-ундец-7-ен, 50°С. b) I2/NaI, дихлорэтан, 100°С. с) (BOC)2O, дихлорметан, 23°С; Cu, ДМФА, 110°С. d) NaOH, осушенный этиленгликоль, 180°С. е)
POCl3, ДМФА, 60°С; NaOAc, 100°C.
1.2.1.1 Метод синтеза исходных α-незамещённых пирролов методом
Бартона-Зарда
Кроме широко используемого на практике синтеза Кнорра, для синтеза
исходных пирролов используется метод Бартона-Зарда, (Схема 6)
зарекомендовавшего себя как способ получения α-незамещённых пирролов с высокими
выходами, конденсацией активированных электроноакцепторными группами алкенов с
изоцианоацетатами в присутствии сильных ненуклеофильных оснований [26-27],
например DBU.
Схема 6

Метод Бартона-Зарда наиболее широко применяется для синтеза α-незамещённых пирролов, имеющих как
алкильные заместители: метильные, этильные, а так же карбэтоксиметильные группы
так, и арильные заместители такие как: метоксифенил, триметилфенил, метилфенил,
моно- и дихлорфенил, а также нафтильные заместители. [28-29]
Данный метод синтеза пирролов очень интересен, поскольку полученный α-незамещённый пиррол может быть в
дальнейшем служить исходным соединением для синтеза не только порфиценов
имеющих в β-положениях два разных заместителя (37), но и
аналогичных изомерных порфиринов (38).
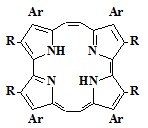
37
Ar= C6H5, C6H4OCH3= Me, Et
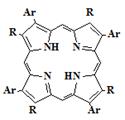
38
Ar = C6H5, C6H5CH3, C6H3(CH3)3, C6H5OCH3, C6H4(OCH3)2, C6H5Cl,
C6H3Cl3
R= Me
1.2.2 Получение (мезо-) 5,10,15,20-тетразамещенных
порфиринов
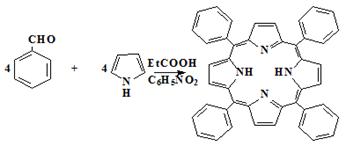
Изучение синтезов порфиринов из a-незамещенных пирролов и альдегидов началось в 1935 г., когда
Ротмунд обнаружил [30], что при взаимодействии пиррола с альдегидами образуются
мезо-замещенные порфирины (1b) (схема 7)
Схема 7

b
Реакция ацетальдегида с пирролом приводит с низким выходом к мезо-тетраметилпорфину
( R = СН3), а пиррола с формальдегидом - к
порфину ( R = H) с ничтожным выходом 0,03% [30].
В последующих работах Ротмунда [31-32] выход порфиринов был существенно
улучшен за счет проведения реакции в пиридине в запаянной ампуле при высокой
температуре (140-240оС). Конденсацией пиррола с бензальдегидом был получен мезо-тетрафенилпорфин
(R = C6H5, H2ТФП) [32].
Этот порфирин образуется с достаточно высоким выходом и легко выделяется из
реакционной смеси [31].
Позднее в работах [33-34], было показано, что выход Н2ТФП
повышается при добавлении в реакционную смесь ацетата цинка. Получающийся в
этом случае цинковый комплекс мезо-тетрафенилпорфина (ZnТФП) далее переводится действием минеральной кислоты в
свободный порфирин. Однако, выход Н2ТФП даже в этих условиях, оптимальных
для реакции конденсации в пиридине, не превышает 18%. В настоящее время в
качестве реакционной среды кроме пиридина используется также
2,4,6-триметилпиридин (коллидин) [35-36] и хинолин, которые кипят при более
высокой температуре, чем пиридин (171°С и 237°С против 115°С соответственно), что позволяет
проводить реакцию конденсации при атмосферном давлении и в присутствии
кислорода воздуха в качестве окислителя.
По методу Ротмунда осуществляют реакцию при высоких концентрациях
реагирующих веществ, но низкий выход ограничивает его применение. Методы
высокотемпературного синтеза в настоящее время модифицированы, что позволяет
полностью избежать применения растворителя. Так реакция смеси пиррола и
бензальдегида в присутствии соли металла (в отсутствии растворителя) в
запаянной ампуле при 150-2500C дает Н2ТФП или мезо-тетра(1-нафтил)порфин
с выходами превышающими 50%. Обработка смесь пиррола, бензальдегида и
силикагеля или цеолита в микроволновой печи приводит после хроматографической
очистки к H2TФП с выходом, достигающим 9% [39-37].
Следует отметить, что метод синтеза мезо-замещенных порфиринов в
основных средах незаслуженно забытый в последнее время в связи с открытием
методов кислотного катализа имеет свои специфические области применения, такие,
например, как синтез порфиринов с лабильными в кислой среде группами или
содержащими в мезо-положениях некоторые гетероциклические остатки
(например, фурановые или пиррольные).
В дальнейшем, на основании работ [31,40], их авторы получили с небольшим
выходом Н2ТФП при длительном кипячении смеси пиррола и бензальдегида в
среде метанол-пиридин, Адлер [41], исследуя влияние различных растворителей,
обнаружил, что реакция конденсации лучше катализируется кислотами, чем
основаниями. Эти выводы были подтверждены в работе Трайбса [42]. Адлер [41]
определил, что при проведении реакции в кипящих, содержащих кислоту
органических растворителях, при доступе воздуха, выход Н2ТФП достигает
40% по спектрофотометрической оценке.
В настоящее время проведение реакции конденсации пиррола с альдегидами
(схема 8) в средах, содержащих кислоту, в присутствии воздуха является одним из
основных методов получения мезо-замещенных порфиринов (метод Адлера). В
качестве кислотных растворителей в основном применяются: уксусная кислота
[41,43], пропионовая кислота [44-45]; смешанные растворители: пиридин-уксусная
кислота [42], бензол-хлоруксусная кислота [41], ксилол-хлоруксусная кислота
[46-48] и некоторые другие. Реакция обычно проводится при температуре кипения
растворителя, иногда при пропускании через реакционную смесь воздуха [42].
Установлено [49], что выход порфиринов зависит от температуры реакционной среды
и оптимальный выход реализуется при температурах близких к 1400С, При более
низких температурах мала скорость образования порфиринов, а при более высоких
велика скорость окисления полученных порфиринов. Оптимальной концентрацией
реагентов в реакционной смеси является величина порядка 0,2-0,4 моль/л.
Схема 8

b
Наконец, существуют достаточно новые модификации метода Адлера для
синтеза мезо-замещеных порфиринов: конденсация пиррола с альдегидами в
среде диметилформамид (диметилсульфоксид)-трихлорид алюминия (выход Н2ТФП
30%) [50], или в пропионовой кислоте при микроволновом облучении (выход мезо-тетраарилпорфиринов
20-43%) [51].
Следует отметить, что в условиях реакции кислотной конденсации также
образуются хлорины, а в некоторых случаях они становятся основными продуктами
реакции, однако их легко можно перевести в соответствующие порфирины обработкой
производными бензохинона: пара-хлоранилом (п-XA) или
2,3-дихлор-5,6-дицианбензохиноном-1,4 (DDQ) [52-54].
Сравнительно недавно появился новый метод синтеза мезо-замещенных
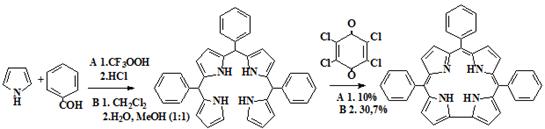
порфиринов в мягких условиях [55-64], заключающийся в конденсации пиррола с
альдегидами в хлороформе или хлористом метилене в присутствии трифторуксусной
кислоты, или эфирата трифторида бора в инертной атмосфере при комнатной
температуре до соответствующего порфириногена 39 с последующим
окислением реакционной смеси стехиометрическим количеством DDQ или п-XA (метод Линдсея) (схема 9). Иногда в
качестве окислителя используется перекись водорода в уксусной кислоте [57]
Схема 9
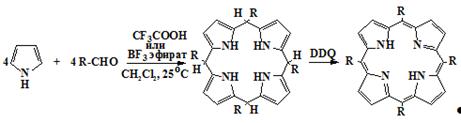
1b
Реакция, как было найдено [55], весьма чувствительна к
концентрации реагентов. Самые высокие выхода H2ТФП (35-40%) получаются при реакции по
10 ммоль бензальдегида и пиррола, и выход уменьшается примерно в два раза при
концентрации реагентов по 100 ммоль и по 1 ммоль. Снижение выхода при более
высокой концентрации реагентов можно частично уменьшить увеличением количеств
кислотного катализатора. Например, при 100 ммоль бензальдегида и пиррола, выход
H2ТФП был 23% с 1 ммоль BF3 эфирата, 30% с
3,2 ммоль и 29% с 10 м моль BF3 эфирата. Эти значения все же меньше чем
35-40%-ный выход, полученный при концентрации реагентов по 10 ммоль в CH2C12 с
BF3 эфиратом или ТФУК в качестве катализатора [58]. Реакция также чувствительна
к концентрации кислотного катализатора: для концентрации реагентов по 10 ммоль,
BF3 эфират эффективен при концентрации 1 ммоль, в то время как ТФУК требуется в
более высокой концентрации 20-50 ммоль [55].
На примере тетра(мезитил)порфина (R = 2,4,6-триметилфенил; Н2ТМП)
показано [59-61], что для синтеза тетрафенилпорфиринов содержащих в обоих
орто-положениях фенильных колец донорные заместители в стадии конденсации
требуется использовать в качестве катализатора BF3-эфират в присутствии
сокатализатора - 0,75% этанола. Этот метод был улучшен, для получения граммовых
количеств H2TMП [62-63]. Сокатализаторами в реакции синтеза H2TMП
и подобных ему порфиринов кроме этанола могут служить этиленгликоль,
2-метоксиэтанол [61] или метанол [64]. Проведение синтезов тетрафенилпорфиринов
содержащих в обоих орто-положениях фенильных колец акцепторные заместители не
требует применения сокатализатора.
Мягкие условия проведения реакции на стадиях
конденсации и окисления позволяют использовать метод Линдсея для широкого
спектра альдегидов и пирролов. Этот метод имеет особое значение для синтеза
пространственно затрудненных порфиринов [60,63,65] и мезо-алкилпорфиринов,
которые другими методами получаются с низкими выходами. Выход порфиринов может
достигать 50% в зависимости от альдегида. В настоящее время это наиболее
применяемый метод синтеза мезо-замещенных порфиринов.
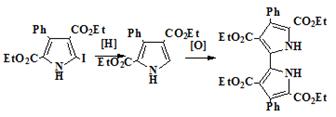
1.2.3 Методы синтеза корролов
Существует несколько различных методов синтеза корролов:
· тетрамеризация 2-замещённых пирролов;
· конденсация пирролов с альдегидами;
· синтез корролов из биспирольных предшественников;
· циклизация биладиенов.
В данной работе будут рассмотрены первые три метода. Наибольшую
практическую значимость для данной работы имеет второй метод.
.2.3.1 Тетрамеризация 2-замещенных пирролов
Тетрамеризация 2-замещенных пирролов и конденсация пирролов с альдегидами
являются успешными путями синтеза порфиринов, однако в случае корролов, данные
методы не использовались, частично потому что, в ходе данных реакций побочным
процессом является образование порфиринов. Однако, некоторые примеры,
сообщенные в литературе [66-71], показывают возможность получения корролов из
монопирролов.
Первый пример синтеза корролов с использованием тетрамеризации
2-замещенных пирролов включает получение октаметилтрифенилкоррола
кобальт-трифенилфосфинового комплекса (OMTФКор)CoPPh3 40 из 2-(α-гидроксибензил)пиррола 41
(Схема 10) [66]. Пиррол 41 сначала реагирует в кислом этиловом спирте,
после чего в реакционную смесь был введён ацетат кобальта(II) в присутствии PPh3, с получением трифенилфосфинового
комплекса кобальта 40 с выходом 25%. Присутствие ионов кобальта было
необходимо для получения кольца коррола. Заслуживает внимания то, что 40
- почти плоский [66], в отличие от аналогичных додеказамещенных порфиринов,
которые показывают значительные отклонения от плоскости из-за пространственных
взаимодействий между соседними мезо-фенильными и β-алкильными заместителями [73-77].
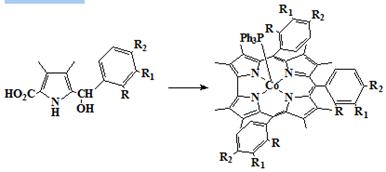
Исходя из пирролов 44-50 содержащих замещенные фенильные кольца, можно
получить корролы 51-57 имеющие различные групппы в орто-, мета- и
пара-положениях мезо-фенильных групп (Схема 11) [68].
Схема 10
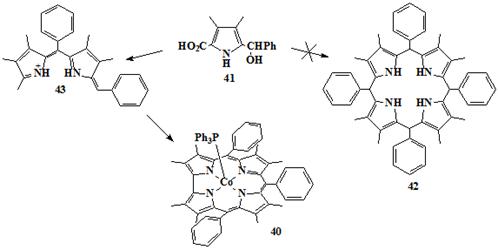
Схема 11
|
44
|
R=H R1=H R2=OCH3
|
51
|
R=H R1=H R2=OCH3
|
|
45
|
R=H R1=H R2=CH3
|
52
|
R=H R1=H R2=CH3
|
|
46
|
R=H R1=H R2=Cl
|
53
|
R=H R1=H R2=Cl
|
|
47
|
R=H R1=Cl R2=H
|
54
|
R=H R1=Cl R2=H
|
|
48
|
R=Cl R1=H R2=H
|
55
|
R=Cl R1=H R2=H
|
|
49
|
R=H R1=F R2=CH
|
56
|
R=H R1=F R2=CH
|
|
50
|
R=F R1=H R2=H
|
57
|
R=F R1=H R2=H
|
Синтез корролов из 2-замещенных пирролов был
осуществлён при использовании 2-формилпирролов в тех же самых условиях реакции,
что описаны для 41 [69]. В этом случая в присутствии ионов кобальта(II)
образуются корролы, в то время как присутствие ионов никеля(II) и родия(III)
приводит к порфиринам, а в присутствии солей железа(III) не образуется никаких
макроциклических продуктов. Реакция 2-формилпиррола в присутствии ионов
кобальта(II) дает смеси коррола и порфирина. Механизм реакции с исходным
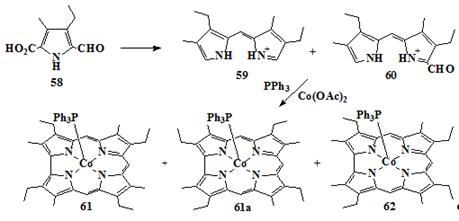
пирролом 58 был подробно изучен, в этом случае тетрамеризация дает
четыре различных продукта [70]. Кроме кобальт этиопорфирина-I образуется смесь трех комплексов
коррола (Схема 12) [69]. Два из комплексов были диастереомерами 61 и 61a,
а третий изомер 27. На основе электронных спектров реакционной смеси,
формирование этих изомеров было приписано присутствию промежуточных
дипирролилметенов 59 и 60. Эти соединения самоконденсируются, давая
конечные корроловые или порфириновые кольца.
Схема 12
1.2.3.2 Конденсация пиррола с
альдегидами
Известны пути [72] получения мезо-замещенных порфиринов
конденсацией пирролов с алкил- или арилальдегидами. Реакцию проводили при
различных условиях, в ходе работ были получены различные производные H2TФП [72]. В 1994 и 1995 годах, было сообщено, что кроме H2TФП в этой реакции образуются другие продукты, такие как
сапфирин 63 [78] и «перевернутый» порфирин 64 [79,9]. Эти примеры
доказывают возможность получения мезо-трифенилкоррола при конденсации
пиррола и бензальдегида. Два примера этого маршрута были сообщены в литературе
[70-71]; каждый включает образование мезо-трифенилкоррола 65 в
качестве побочного продукта при реакции пиррола и 2,6-динитро-4-трет-бутилбензальдегида
[70]. Дальнейшие детали этой реакции не были сообщены, и это соединение было
охарактеризовано только спектром ЯМР.
Второй пример был сообщен Лоймом с сотр. [71], которые
показали, что 5,10,15-трицимантренилкоррол можно синтезировать реакцией пиррола
и цимантренкарбоксальдегида в уксусной кислоте. Для формирования кольца коррола
реакция проводили в жестких условиях из-за конкурентного образования
5,10,15,20-тетрацимантренилпорфирина. При проведении данного синтеза в условиях
метода Линдсея [80] происходит преимущественное образование
5,10,15,20-тетрацимантренилпорфирина. Эти примеры показывают возможность
легкого с синтетической точки зрения метода получения мезо-замещенных
корролов, из доступных исходных соединений.

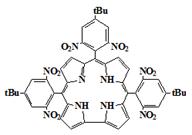
63 64 65
1.2.3.3 Синтез из бипиррольных соединений
Синтез порфиринов из бипиррольных соединений называется [2+2] методом
[72] и в случае корролов этот [2+2], подход должен включать конденсацию
дипирролилметанов и бипирролов. Успешный синтез коррола с использованием этого
метода был сообщен Джонсоном и коллегами [81]; в этом случае, бипиррол 66
и дипирролилметан 67 сначала конденсировали в кислых условиях, получая
красный осадок, который после реакции с ацетатом кобальта(II) и
трифенилфосфином в метаноле, дает комплекс коррола 70. Тот же самый
продукт было получен реакцией 68 и 69 (Схема 13). Присутствие
кобальта необходимо для образования 70; без металла реакция была
неудачна, и кобальт необходим, как было предположено, для стабилизации
предполагаемого тетрапиррольного промежуточного продукта и для гарантии
правильной геометрии в реакции циклизации.
Схема 13
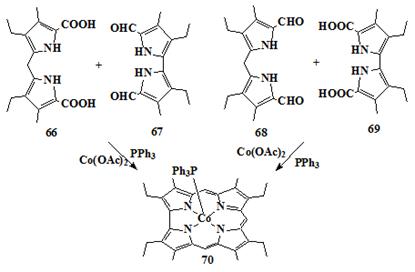
.3 Синтез металлокомплексов макрогетероциклов
.3.1 Металлопорфирины
Металлопорфирины, к числу которых относятся важнейшие природные
биокомплексы: хлорофилл, гем крови и многие ферменты, а также их синтетические мезо-азапроизводные,
из которых наиболее известны фталоцианины, имеющие промышленное значение в
качестве стойких пигментов и катализаторов различных процессов, составляют
обширную и своеобразную группу внутрикомплексных соединений.
Центральный атом металла, вытесняя из порфиринового лиганда два протона,
оказывается практически в симметричном электростатическом поле четырех атомов
азота, с которыми он может образовывать четыре эквивалентные координационные
связи донорно-акцепторного типа. Если взаимодействие металла с анионом
порфирина ограничивается преимущественно электростатическим взаимодействием, то
образуются лабильные комплексы ионного типа. Если же электростатическое
взаимодействие сопровождается заполнением вакантных орбиталей центрального
атома металла электронами донорных N-атомов
лиганда, то образуются стабильные комплексы порфиринов ковалентного или
преимущественно ковалентного типа.
Особенности металлопорфиринов как внутрикомплексных соединений
определяются не только их тетрадентантностью но и относительной жесткостью
планарного макрогетероцикла. Благодаря высокой жесткости порфириновый лиганд
предъявляет особые требования к геометрическим параметрам иона металла,
разделяя их на те которые образуют стабильные или лабильные комплексы, а также
определяя их валентное состояние.
Способы синтеза комплексных соединений порфиринов и их аналогов
базируются на использовании трех основных реакций:
· синтез макрогетероциклов на ионах металлов, как на матрице
при использовании непорфириновых фрагментов (темплатный синтез);
· введение иона металла в готовый макрогетероцикл
(комплексообразование);
· переметаллирование лабильных металлокомплексов.
В нашем случае наибольший интерес представляет использование второго
метода синтеза с использованием готового макрогетероциклического лиганда [82].
Метод комплексообразования лигандов порфиринов с донорами металлов
является наиболее общим и позволяет получать металлокомплексы которые с трудом
или с низкими выходами получаются при использовании первого метода, особенно
это относится к синтезу металлокомплексов собственно порфиринов различной
структуры, а также комплексов их аналогов с высокозарядными металлами.
Реакция комплексообразования, как правило, проводится в растворителях и
описывается основным уравнением:
H2P + MXn → MXn-2P + 2HX
Поскольку комплексообразование протекает в растворах, реакцию следует
отнести к процессам замещения лигандов в первой координационной сфере катиона
металла, поэтому роль растворителя очень велика.
Реакции комплексообразования с порфиринами относятся к медленным
процессам и характеризуются значительными энергиями активации и отрицательными
значениями величины энтропии активации.
Механизм реакции комплексообразования порфиринов с двухзарядными
катионами металлов детально изучен, чего нельзя сказать о реакциях порфиринов с
многозарядными катионами. Как правило, последние идут в более жестких условиях.
Однако в настоящее время получены многочисленные экспериментальные данные
по синтезу металлопорфиринов с катионами различных степеней окисления, которые
позволяют сделать некоторые обобщения имеющие практическое значение.
Растворитель для проведения реакции комплексообразования должен обладать
весьма противоречивыми свойствами. С одной стороны он должен обладать высокой
растворяющей способностью по отношению как к липофильному порфирину, так и к гидрофильным
солям металлов. С другой стороны он не должен слишком хорошо координироваться с
солями и, кроме того, должен иметь достаточно высокую температуру кипения для
обеспечения более быстрого прохождения реакции комплексообразования.
В качестве таких растворителей для получения металлокомплексов с II и III зарядными металлами используются в основном
карбоновые кислоты, пиридин, ДМФА. Однако, очевидно, что карбоновые кислоты не
годятся для синтеза лабильных металлопорфиринов. Для синтеза металлокомплексов
с высокозарядными металлами, кроме этого, используются более высококипящие
растворители - бензонитрил, хинолин, имидазол, а также фенол.
В настоящее время в качестве доноров катионов стали использоваться
достаточно доступные органорастворимые ацетилацетонаты, феноляты и карбонилы
металлов, что позволяет использовать в качестве реакционной среды неполярные и
малополярные растворители, такие как ароматические и полихлорированные
ароматические углеводороды.
В случае использования фенола в качестве растворителя многие исходные
доноры катионов переходят в активные в реакции комплексообразования феноляты,
что позволяет использовать в качестве доноров катионов даже оксиды некоторых
высокозарядных металлов.
В таблице 1 представлены основные доноры катиона металла и растворители.
используемые для проведения реакции комплексообразования с порфиринами и их
аналогами.
Таблица 1
|
Доноры катионов металла
|
Растворители
|
|
Галогениды перхлораты MXn ацетаты
|
карбоновые кислоты (AcOH,
EtCO2H) азотистые гетероциклы (пиридин, хинолин, имидазол)
спирты, фенол, кетоны, простые эфиры ДМФА, бензонитрил, сульфолан
|
|
алкоголяты M(OR)n феноляты
|
азотистые гетероциклы
(пиридин, хинолин, имидазол) спирты, фенол
|
|
Оксиды MOn/2
|
Фенол
|
|
ацетилацетонаты M(AcAc)n
|
бескислотные растворители
|
|
органометал. соед. MRn
|
Углеводороды (бензол,
толуол, тетралин, декалин)
|
|
карбонилы Mn(CO)m
|
Углеводороды (бензол,
толуол, тетралин, декалин)
|
Галогениды, ацетаты или ацетилацетонаты большинства двухзарядных катионов
тяжелых и переходных металлов образуют комплексы с порфиринами во многих
органических растворителях [83]. Высокие скорости образования металлопорфиринов
наблюдаются в карбоновых кислотах (уксусная, пропионовая), спиртах и кетонах,
т.е. в таких растворителях в которых достаточно хорошо растворяются как свободные
лиганды, так и соли металлов. Однако карбоновые кислоты не пригодны для
получения лабильных металлопорфиринов, например, с катионами магния, кадмия,
ртути и т. д.
Достаточно универсальной средой для проведения реакции
комплексообразования служит ДМФА - хороший растворитель для большинства
порфиринов и солей металлов.
Следует отметить, что большое значение имеет соответствие ионного радиуса
катиона металла порфириновой полости, что наряду с другими факторами в
некоторых случаях приводит к образованию металлопорфиринов с необычными
степенями окисления металла и соответственно прохождению окислительно
восстановительных процессов при комплексообразовании. Эти факторы в некоторых
случаях определяют и относительную стабильность металлопорфиринов, кроме того
стабильность металлопорфиринов определяется и структурой порфиринового лиганда,
например его искажением. Искажение порфиринового цикла может приводить и к
изменению состояния катиона металла по сравнению с обычными плоскими
порфиринами.
Как уже говорилось ранее, элементы главных подгрупп первой и второй
группы образуют крайне лабильные ионные комплексы, которые крайне неустойчивы и
разлагаются даже следами воды. Их получают, в основном, реакцией лигандов
порфиринов с алкоголятами металлов в пиридине или безводных спиртах. Несколько
в стороне находятся порфиринаты бериллия и магния. Образование бериллиевых
комплексов порфиринов до сих пор полностью не доказано, а сравнительно
устойчивые магниевые комплексы можно получить при использовании магнийорганических
соединений или перхлората магния (ангидрона) в пиидине.
Порфириновые комплексы переходных металлов легко получаются при
взаимодействии хлоридов или ацетатов двухвалентных металлов в уксусной кислоте
или ДМФА. Причем в процессе синтеза в присутствии воздуха происходит окисление
катионов хрома, марганца и железа в металлокомплексах до трехвалентного
состояния. В уксусной кислоте проходит реакция комплексообразования порфиринов
с галогенидами (с добавкой ацетата натрия) или ацетатами Ag(I), Au(III), Ga(III), In(III), Tl(III), Sn(II). Причем в
процессе реакции порфириновый комплекс серебра(I) диспропорционирует до комплекса серебра(II) и металлического серебра, а олово(II) окисляется до порфиринового
комплекса олово(IV).
Катионы Cd(II), Hg(II), Pb(II) имеют большой
ионный радиус, поэтому соответствующие порфириновые металлокомплексы лабильны и
их получают из ацетатов в пиридине.
Наиболее легко алюминий(III)
вводится в порфирины при использовании триалкилалюминия в инертном растворителе
(углеводороды), однако при этом требуется отсутствие кислорода и влаги, что
достаточно сложно практически. Другой метод заключается во взаимодействии
порфирина лиганда с безводным галоенидом алюминия в пиридине или
ацетилацетонатом алюминия в феноле при кипении. Алюминий(III) порфирины относятся к
сверхстабильным комплексам, они не разрушаются даже под действием
концентрированной серной кислоты.
Комплексы порфиринов со Sc(III), Y(III), La(III) и редкоземельными элементами обычно получают из
соответствующих галогенидов или ацетилацетонатов в кипящем имидазоле или (для
ацетилацетонатов) в трихлорбензоле. Металлокомплексы La(III), Сe(III), Pr(III) и Nd(III) крайне неустойчивы.
Ti(IV), Ge(IV), Zr(IV), Hf(IV) V(IV) Th(IV) комплексы порфиринов получают реакцией порфирина с
соответствующими галогенидами в пиридине или ацетилацетонатами в феноле или
трихлорбензоле [84]. Все эти металлокомплексы имеют экстралиганды по одну
сторону порфиринового цикла, причем титановый и ванадиевый комплексы существуют
в «иловой» форме.
Мышьяк, сурьма, висмут вводятся в порфириновый цикл взаимодействием их
тригалогенидов с порфирином в пиридине. Полученные комплексы имеют
тетраэдрическую структуру, причем мышьяк и сурьма переходят в пятивалентное
состояние и имеют один из лигандов во внешней сфере.
Порфириновые комплексы ниобия и тантала можно получить из порфирина
лиганда и соответствующих пентагалогенидов реакцией в бензонитриле или феноле,
а комплексы молибдена, вольфрама и рения из высших оксидов в феноле при этом
идет окислительно-восстановительный процесс в котором фенол является
восстановителем. Аналогичным образом взаимодействием порфирина и пятиокиси
ванадия можно получить ванадиловый комплекс порфирина. Молибденовый,
вольфрамовый и рениевый комплексы можно также достаточно легко получить
взаимодействием карбонилов соответстующих металлов с порфирином в
углеводородном растворителе с высокой температурой кипения.
Пятивалентные порфириновые комплексы ниобия, тантала, молибдена, рения и
вольфрама зачастую имеют одинаковый состав, но существенно разное строение.
Так, если комплексы ниобия и тантала гептакоординационны и имеют заместители по
одну сторону порфиринового цикла, то комплексы молибдена, вольфрама и рения
октаэдрические.
Палладий(II) и платина(II) порфирины получают взаимодействием
соответствующих дигалогенидов с порфирином в бензонитриле, палладиевый комплекс
можно проще получить в диметилформамиде. Рутениевый(II), осмиевый(II), а
также родиевый(III) и иридиевый(III) комплексы лучше всего получать из
соответствующих карбонилов в высококипящих углеводородах.
В порфириновую полость, кроме того, могут встраиваться атомы типичных
неметаллов: бора, кремния, фосфора, теллура.
1.3.2 Металлокомплексы порфиценов
В литературе имеется очень мало сведений о синтезах металлокомплексов
порфиценов однако существуют методики получения некоторых комплексов порфицена,
а именно, комплекса порфицена с никелем [10], с медью [11], и с цинком [11].
При этом, процессы комплексообразования проводятся в более жестких условиях,
чем для подобных металлокомплексов порфиринов. Для получения медного и
никелевого комплекса ипользуется метод кипячения тетрафенилпорфицена с
ацетатами соответствующих металлов в уксусной кислоте, следует отметить, что в
методиках [11] получения никелевого комплекса реакцию проводят в атмосфере
азота. При получении цинкового комплекса, тетрафенилпорфицен растворяли в ТГФ,
и при кипении вводили в метанольный раствор ацетата цинка. Решающую роль в
способности к комплексообразованию играет строение и симметрия координационного
центра порфицена. По сравнению с порфиринами, порфицены имеют меньший размер
координационного ядра, и, как следствие, меньшую координационную активность.
1.3.3 Металлокомплексы корролов
Корролы обладают превосходными хелатирующими
свойствами, и по крайней мере 18 ионов металлов пока были введены в макроцикл.
Хотя координационная химия корролов далеко не так развита как химия порфиринов,
не надо полагать, что число металлов, координированных с корролами может быть
значительно расширено в будущем. Имея три амино- и один имино-подобные атомы
азота во внутренней полости, полностью депротонированная форма коррола
действует как трианионный лиганд, отличный от порфиринов и порфиценов, которые,
являются дианионными лигандами. Кроме того, способность корролов
стабилизировать более высокие состояния окисления металла по сравнению с
порфиринами делает его координационную химию особенно интересной [87].
Существуют два метода получения металлокорролов.
Первый метод - циклизация a,c-биладиенов
в присутствии соли металла - наиболее прямой подход, однако он не всегда
применим, так как некоторые ионы металлов способны катализировать циклизацию
линейных тетрапирролов без координации, продуктом реакции в этом случае
является соответствующее свободное основание коррола. В этом случае существует
второй подход - использование готового макроцикла с соответствующим
переносчиком металла, этот метод позволяет получить более высокие выхода
соответствующих металлокорролов.
В таблице 2 представлены основные доноры катиона металла и растворители,
а так же методы используемые для проведения реакции комплексообразования с
корролами.
Таблица 2
|
Источник металла
|
Растворитель
|
Предшественник
|
Вводимый металл
|
|
Галогениды и перхлоратыMXn
|
MeOH
|
a,c-биладиены
|
Mn, Fe, Co, Ni,
Cu, Rh, Sn, Ge, In
|
|
Ацетат Co(OAc)2
|
ДМФА
|
a,c-биладиены
|
Co
|
|
Пиридин
|
|
|
|
M(OAc)2
|
MeOH
|
Свободное основание коррола
|
Co, Ni
|
|
MCl5
|
PhCN
|
|
Nb, Re
|
|
MXn
|
Пиридин
|
|
Co, Cu, Pd, Zn,
P
|
|
MXn
|
ДМФА
|
|
Cr, Co, Ni, Cu,
Rh, In, Zn
|
|
MoCl5
|
Декалин
|
|
Mo
|
|
Ацетилацетонаты OM(АcАc)2
|
Фенол
|
|
Ti, V
|
|
Карбонилы MmCln(CO)p
|
Толуол
|
|
Cr, Mo, Mn, Fe,
Rh
|
Не смотря на то, что в данной таблице в качестве
вводимого металла представлен и цинк, единственным охарактеризованным цинковым
комплексом коррола является продукт полученный реакцией (OMCor)H3 с Zn(OAc)2 при
кипячении в пиридине [88]. Был выделен металлокомплекс [(OMCor)Zn]-pyH+, однако соответствующий нейтральный Zn коррол слишком неустойчив,
охарактеризовать его не удалось.
Никелевые и медные комплексы были среди первых
металлокорролов, о которых сообщено в литературе [89]. Их строение, в течение
длительного времени было ошибочно интерпретировано, и только недавно состав
этих комплексов был до конца изучен.
Никель корролы были получены как циклизацией a,c-биладиенов так и металлированием готового коррола.
Спектральные свойства этих комплексов дали неожиданные
результаты [89,90,91]: никель корролы - парамагнитные соединения, и их
оптические спектры показывали отсутствие полосы Соре. На основе этих данных,
комплексы были идентифицированы как нейтральные Ni(II) производные, где коррол существовал как дианионный
лиганд с остаточным водородным атомом, помещенным в макроцикл, разрушая тем
самым, ароматическую систему (Схема 14). Без более точной информации, этот
водородный атом был произвольно расположен в 10-положении; отсутствие полосы Соре,
объясненное разрушением ароматической системы, причина парамагнетизма никель
корролов осталась без обьяснения и была приписана частичной потере остаточного
водородного атома, давая металлокоррольный радикал [92]. Эта гипотеза была
поддержана данными полученными при проведении реакции с основаниями, которые
интерпретировались, как отрыв протона от исходного нейтрального никель коррола,
давая соответствующий ароматический анионный никель коррольный комплекс.
Медь корролы были получены, используя методы, подобные
тем, что использовались для производных никеля [89,91]. Эти комплексы, однако,
имели полосу Соре в оптических спектрах, и по этой причине они были
представлены как 71 (Схема 14).
Способность корролов стабилизировать более высокие
степени окисления координированных металлов дало возможность Vogel с коллегами выдвинуть гипотезу, что
никель и медь корролы могут рассматриваться как металл(III) нейтральные
комплексы 72 (Схема 14). Рентгеноструктурные исследования подтверждали
эту гипотезу и демонстрировали, что оба комплекса почти плоские без присутствия
остаточного водородного атома.
Схема 14
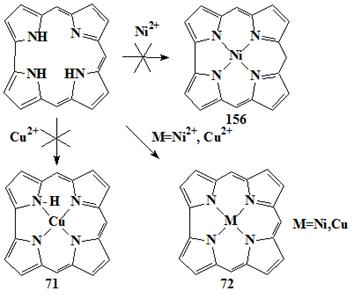
Реакция никель и меди корролов с основаниями, ранее
рассмотренная как процесс отрыва протона, должна теперь рассматриваться как
одно-електронное восстановление, ведущее к коррол-анионному комплексу, что
подтверждено электрохимическими изучениями. Возможность получения этих
соединений натриевым пленочным восстановлением соответствующих нейтральных
комплексов меди или никеля в ТГФ ранее была сообщена в литературе даже при том,
что эти данные не интерпретировалось таким образом.
2. Экспериментальная часть и
обсуждение результатов
Электронные спектры поглощения (ЭСП) снимали на сканирующем спектрометре
СПЕК ССП-715 в хлороформе, инфракрасные спектры (ИК) снимали на спектрометре Avatar 360 FT-IR в таблетках KBr, спектры протонного магнитного
резонанса (1Н ЯМР) снимали на спектрофотометре Bruker 500 (внутренний стандарт ТМС) в центре коллективного
пользования научным оборудованием «Верхневолжский региональный центр
физико-химических исследований» ФГБУН Институт химии растворов им. Г. А.
Крестова РАН. Масс - спектры снимали на времяпролетном масс-спектрометре Shimadzu Axima Confidence (MALDI -TOF).
Тонкослойную хроматографию (ТСХ) осуществляли на пластинах силуфола.
2.1 Синтез 2,7,12,17-тетрафенилпорфицена
Для изучения особенностей поведения в различных реакциях как
внутрисферного комплексообразования и протонирования, так и электрофильного
замещения на периферии макроцикла нами был синтезирован
2,7,12,17-тетрафенилпорфицен (78), изомер широко известного
5,10,15,20-тетрафенилпорфина (1b).
Методика получения 2,7,12,17-тетрафенилпорфицена описана Вогелем, который
также успешно разработал методики синтеза и для других порфиценов имеющих алкильные
боковые цепями, например: 2,7,12,17-тетраметил-, 2,7,12,17-тетраэтил- и
2,7,12,17-тетрапропилпорфицен. Основываясь на выводах сделанным Вогелем, Нонелл
и его коллеги провели усовершенствованный постадийный синтез
тетрафенилпорфицена (схема 15). При проверке синтезов в методики, приведённые в
литературе, [93] в некоторых случаях были внесены изменения.
Схема 15
73 74 75
76 77
78
В этом синтезе исходным соединением служит пиррол 73, который
радикальным замещением α-метильной группы с последующим
гидролизом превращают в карбоксипиррол 74, из которого электрофильным ипсо-замещением
получают иодпиррол 75, а далее самоконденсацией по Ульману превращают в
тетраэфир биспиррола 76 далее декарбоксилированием и формилированием по
Вильсмайеру превращаемому в диформил биспиррол 77 и востановительной
конденсацией по Мак-Мури превращаемого в требуемый порфицен 78.
Рассмотрим каждую стадию синтеза с самого начала, а также внесённые на
этих стадиях в методики изменения.
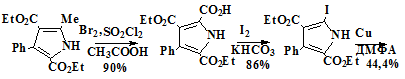
,4-диэтоксикарбонил-3-фенил-5-метилпиррол (73)
В работе Нонела [93] (схема 15), отсутствует стадия получения исходного
2,4-диэтоксикарбонил-3-фенил-5-метилпиррола (73). Поэтому данную
методику нам пришлось разрабатывать самим исходя из общих указаний
представленных в статьe
[94]. Синтез осуществлялся нитрозированием бензоилуксусного эфира 79
нитритом натрия в уксусной к-те с последующей восстановительной конденсацией
полученного гидроксиламинобензоилуксусного эфира 80 с ацетоуксусным
эфиром 81 по Кнорру до пиррольного предшественника 73 (схема 16).
Схема 16

79 80,
81 73
При перемешивании и охлаждении (< 25°С) к раствору 104,1 мл (0,6 моль)
бензоилуксусного эфира в 700 мл уксусной к-ты постепенно прибавляли раствор
41,5 г (0,6 моль) нитрита натрия в 60 мл воды, затем смесь перемешивали при
комнатной температуре ночь, добавляли 80 мл (0,63 моль) ацетоуксусного эфира и
к перемешиваемому раствору постепенно порциями добавляли 128,0 г (2,0 моль)
цинковой пыли так чтобы реакционная смесь слабо кипела. Смесь перемешивали при
кипении 2 часа и выливали в 5 л воды, осадок отфильтровывали, промывали водой и
высушивали. Пиррол растворяли в хлористом метилене, отфильтровывали
неорганические соли, раствор упаривали и остаток высушивали на воздухе при
комнатной температуре. Выход 130,7 г (72,3%). Для очистки пиррол
перекристаллизовывали из 150 мл метанола. Продукт синтеза имел температуру
плавления 112-114°С, что ниже значений приведенных в литературе [93]. Структура
полученного вещества доказана. 1H ЯМР
спектром.
H ЯМР
(вн. ст. ТМС)
δ, м.д.: 9,72bs (1H, NH); 7,35-7,28m (5H, H-Ph); 4,12q, 4,05q (4H, CH2-Et); 2,61s (3H, CH3); 1,05t, 1,00t (6H, CH3-Et) (CDCl3)
-карбокси-3,5 дикарбэтокси-4-фенилпиррол (74)
Замещение метильной группы на карбоксильную в полученном пирроле
проводили радикальным хлорированием последнего с последующим гидролизом
трихлорметильной группы.
г (66 ммоль) 2,4-диэтоксикарбонил-3-фенил-5-метилпиррола растворяли в 100
мл уксусной кислоты и охлаждали до 15°С. Добавляли в реакционную смесь 3,4 мл
(65,4 ммоль) брома в одну порцию, затем 16,0 мл (0,2 моль) сульфуридхлорида по
каплям. Реакционная смесь перемешивалась при температуре 0-5°С, 6 часов. После
этого в реакционную смесь добавляли 25 мл, воды и кипятили реакционную смесь
при температуре 65°С 25 минут. Затем смесь охлаждали, выпавший осадок
отфильтровывали, промывали водой и высушивали на воздухе при комнатной
температуре. Осадок размешивали с 50 мл диэтилового эфиром и в полученную смесь
при перемешивании приливали раствор 5,5 г (52 ммоль) бикарбоната натрия в 200
мл воды. Не растворившийся 2-формил-3,5-дикарбоэтокси-4-фенилпиррол - побочный
продукт образующийся в ходе реакции, отфильтровывали. Фильтрат подкисляли
раствором соляной кислоты (1:5) образующийся осадок отфильтровали, промывали
водой, высушивали на воздухе. Выход 19,6 г (90%). Продукт представляет собой
кристаллы белого цвета. Температура плавления 164,5-165,5°С.
ИК (KBr)
ν (см-1): 3414,9;
3229,9; 2629,9; 1737,7; 1699,1; 1621,8; 1509,7; 785,6; 705,1.
В качестве побочного продукта на этой стадии получили 2,1 г (8%)
2-формил-3,5-дикарбэтокси-4-фенилпиррол температура плавления 123-125°С.
ИК (KBr)
ν (см-1): 3426,1;
3243,2; 1699,9; 1615,4; 1510,5; 866,8; 787,2; 706,5.
Мы попытались видоизменить синтез и вместо рекомендуемой методикой
уксусной кислоты в качестве растворителя использовали хлористый метилен. В
данном случае выход продукта оказался ниже и составил 16,6 г (74,6%).
Тот же синтез был проведён нами без предварительного бромирования
пиррола, роль которого не совсем понятна.
,0 г (72,5 ммоль) 2,4-диэтоксикарбонил-3-фенил-5-метилпиррол растворяли в
100 мл уксусной кислоты, охлаждали раствор при перемешивании до температуры
менее 15°С затем по каплям вводили 17,8 мл (0,22моль) сульфурилхлорида, далее
как и в предыдущей методики проводился гидролиз. Однако при этом выход
конечного продукта составил лишь 12,7 г ( 52,9 %).
Схема 17

2-иод-3,5-дикарбэтокси-4-фенилпиррол (75)
Иодпирол получали электрофильным ипсо-замещения карбоксильной
группы иодом в присутствии бикарбоната натрия.
,0 г (27,2 ммоля) 2-карбокси-3,5-дикарбэтокси-4-фенилпиррол растворяли в
30 мл метанола (в оригинальной методике [93] для этой цели используют этанол),
к реакционной смеси при нагревании и перемешивании добавляли раствор 7,0г (66
ммоль) бикарбоната натрия в 100 мл воды, затем смесь нагревали до 75°С и при
этой температуре по каплям добавляли раствор 7,6 г (29,9 ммоль) иода 12,0 г
(72, 3 ммоль) иодида калия в 50 мл воды. Реакционную смесь фильтровали, осадок
иодпиррола промывали водой и высушивали на воздухе при комнатной температуре.
Выход 11,3 г (96%). Кристаллический порошок белого цвета. Температура плавления
продукта 177-179°С.
ИК ν (см-1): 3228; 1672,7; 1549,7; 1501; 791,1; 696,8. (KBr)
Н ЯМР (вн. ст. ТМС) δ, м.д.: 10,06bs (1H, NH); 7,34m (3H, m,p-H-Ph); 7,26m (2H, o-H-Ph); 4,16q, 4,08q (2 x 2H, OCH2); 1,06t,
0,99t (2 x 2H, CH3) (CDCl3)
3,3’,5,5’-тетраэтоксикарбонил -4,4’-дифенил-2,2’-биспиррол (76)
Вещество получали проведением конденсации полученного на предыдущей
стадии продукта по Ульману в присутствии меди.
,9 г (40,8 ммоль) 2-иод-3,5-дикарбэтокси-4-фенилпиррол, растворяли в 100
мл ДМФА, добавляли 17,4 г (0,265 моль) меди и смесь кипятили при перемешивании
9 часов, после чего оставляли на ночь при комнатной температуре. Смесь для
отделения от меди фильтровали, а осадок промывали кипящим хлористым метиленом.
Фильтрат и промывные жидкости сливали в делительную воронку, промывали сначала
раствором соляной кислоты (1:5), затем водой. Нижний слой хлористого метилена,
сливали, и высушивали сульфатом натрия, из фильтрата отгоняли хлористый метилен
до минимального объема. Биспирол высаживали из раствора метанолом, осадок
фильтровали и промывали метанолом. Выход продукта 5,3 г (44,7 %). Температура
плавления продукта 174-176°С.
ИК
ν: 3065,5; 2985,7; 1715,7; 762,2; 698,3 cm-1. (KBr)
Малый выход продукта на данной стадии, при котором теряется почти
половина иодпиррола, побудило нас искать методы решения данной проблемы, путём
усовершенствования методик. Нами были предприняты попытки заменить
рекомендованный методикой диметилформамид (ДМФА) (температура кипения 153°С)
более высококипящим растворителем N,N-диметилацетамидом
(ДМАА) (температура кипения 165,5°С), что не привело к желаемому результату,
так как выход 76 уменьшился до 33,7%.
Далее мы попытались увеличить выход 76 заменой реакции Ульмана на
окислительную конденсацию с использованием α-незамещенного пиррола 80 с его
получением восстановлением иодпиррола 75 (схема 18). Однако все наши
попытки восстановить иодпиррол (двухлористым оловом в метаноле или водородом
при катализе палладием) не увенчались успехом. В обоих случаях по спектральным
данным и данным элементного анализа получался лишь неизменный иодпиррол.
Схема 18
75 80 76
5,5’-диформил-4,4’-дифенил-2,2’-биспиррол (77)
Синтез осуществлялся в одну стадию гидролизом и одновременным
декарбоксилированием биспиррола 76 в присутствии гидроксида калия в
кипящем этиленгликоле до α-незамещенного биспиррола.
Раствор 3,0 г (5,24 ммоль) 3,3’,5,5’-тетраэтоксикарбонил-4,4’-дифенил-2,2’-биспиррола
и 3,0 г (53,5 ммоль) гидроксида калия в 30 мл этиленгликоля кипятили 2 часа,
смесь вылили в 200 мл холодной воды, отфильтровывали выпавший осадок, промывали
его водой, высушивали на воздухе при комнатной температуре и далее использовали
без дальнейшей очистки ввиду его малой устойчивости. Выход 1,45 г (97%).
Температура плавления более 350°С.
ИК
ν: 3254,9; 3103,9; 1627,5; 1344,6; 765,9; 668,9 cm-1. (KBr)
Вторая стадия - формилирование дифенилпирола комплексом хлорокиси фосфора
в ДМФА по Вильсмаеру до формилбиспиррола (77)
К раствору 1,4 г (4,92 ммоль) не очищенного 4,4'-дифенил-2,2'-биспиррола
в 24 мл осушенного ДМФА прибавляли 2,6 мл (28,4 ммоль) хлорокиси фосфора и
нагревали смесь на кипящей водяной бане два часа, затем раствор охлаждали,
выливали в 600 мл воды, нейтрализовали раствором 10,0 г (0,18 ммоль) гидроксида
калия в 100 мл воды. Осадок отфильтровывали, промывали водой, высушивали на
воздухе при комнатной температуре. Выход 1,59 г (95%). Из за плохой
растворимости продукта 1Н ЯМР спектр снять не удалось. Выход продукта реакции
на данных стадиях оказался выше заявленного в литературе [93] 97 и 95% вместо
35% суммарного выхода двух стадий.
2,7,12,17-тетрафенилпорфицен (78)
К суспензии 3,1 г (48,0 ммоль) активированной цинковой пыли, (промытой
5%-ной соляной кислотой, водой, метанолом, диэтиловым эфиром и высушенной) 0,48
г (2,4 ммоль) хлорида меди (ІІ) в 150 мл ТГФ при перемешивании и комнатной
температуре прибавляли 2,6 мл (23,7 ммоль) хлорида титана(ІV) и смесь кипятили
три часа, затем постепенно прибавляли раствор 0,5 г (1,47 ммоль)
5,5’-диформил-4,4’-дифенилбиспиррол-2,2’ в 50 мл ТГФ, и перемешивали при
кипении ещё три минуты. После охлаждения реакционную смесь нейтрализовывали
раствором 8,0 г (57,9 ммоль) поташа в 72 мл воды (10%-ный раствор). Очистка
продукта проводилась хромотографией на силикагеле хлористым метиленом. Отделяли
тёмно-синюю зону, которая по колонке сходит в первую очередь, элюат упаривали,
оставляли высушиваться на воздухе. Выход 0,16 г (12%).
Не смотря на многие попытки, и неоднократное проведение синтеза, 30%-ного
выхода указанного в литературе [93] нам достичь, не удалось.
2,7,12,17-Тетрафенилпорфицен представляет собой темно-синий порошок, обладает
хорошей растворимостью в органических растворителях (хлористом метилене,
бензоле, хлороформе, ацетоне, ДМФА а также в ТГФ).
1Н ЯМР (вн. ст. ТМС) δ, м.д.: 9,95s (4H, ms-H);
9,72s (4H, β-H); 8,36d (8H, J = 7,5 Hz,
2’,6’-H-Ph); 7,85t (8H, J = 7,5 Hz, 3’,5’-H-Ph); 7,71t (4H, J =
7,5 Hz, 4’-H-Ph); 3,77bs (2H, NH) (CDCl3)
ЭСП lmax,нм (e·10-3); 657 (45,6); 626
(42,4); 584 (32,7); 376 (119,6) (хлороформ).
ИК ν (см-1):3640,9; 2957,3; 1625,8; 1435,5;
1154,4; 769; 706,1. (KBr)
Rf (силуфол): 0,83 (бензол).TOF (m/z) найдено: 614,3 [М]+ ; вычислено: 614,8.
Таким образом, нам удалось получить 2,7,12,17-тетрафенилпорфицен 78
используя метод представленный в работе [93] с ведением некоторых упрощающих
процедуру изменений, а также разработать метод синтеза исходного пиррола 73.
.2 Синтез 5,10,15,20-тетрафенилпорфина
Для сравнения свойств порфиринов, порфиценов и корролов нами был
синтезирован 5,10,15,20-тетрафенилпорфин (1b) одностадийной окислитель-ной конденсацией пиррола с
бензальдегидом по методу Альдера (схема 19).
Схема 19
b
К раствору 1,0 мл (10,0 ммоль) бензальдегида в 15 мл нитробензола и 35 мл
пропионовой кислоты при кипении прибавляли 0,7 мл (10,0 ммоль) пиррола и смесь
кипятили 2 часа. После охлаждения осадок отфильтровывали, промывали метанолом
до полного обесцвечивания вытека и высушивали на воздухе при 70ºС. Выход - 0,6 г (39,0%) (фиолетовый
крупнокристаллический продукт). Для дальнейшей очистки порфирин растворяли в
хлороформе и хроматографировали на оксиде алюминия II степени активности по Брокману. Элюат упаривали, порфирин
осаждали метанолом и высушивали на воздухе при 70°C.
Rf
(силуфол): 0,36 (бензол - гексан, 1 : 1)
ЭСП λmax,
нм (ε·10-3):
647 (6,6); 590 (7,2); 550 (10,8); 515 (18,7); 418 (471,6) (хлороформ)
Н ЯМР (вн. ст. ТМС) δ, м. д.: 8,70s (8H, β-H); 8,07m (8H, o-H-Ph); 7,60m (12H, m,p-H-Ph); -2,81bs (2H, NH) (CDCl3)
MALDI-TOF (m/z) найдено: 614,4 [М]+ ; вычислено: 614,7.
2.3 Синтез 5,10,15-трифенилкоррола
Для сравнения свойств корролов с аналогичными порфиринами и порфиценами,
нами реакцией конденсации пиррола и бензальдегида, был синтезирован
5,10,15-трифенилкоррол (82).
Данный синтез проводился нами двумя методами: в первом случае
конденсацией пиррола с бензальдегидом в присутствии трифторуксусной кислоты в
хлористом метилене с последующим окислением продукта конденсации (81)
без выделения, хлоранилом (A),
данный метод аналогичен методу Линдсея для получения тетраарилпорфиринов. Во
втором методе при конденсации использовалась водно-метанольная смесь в
соотношении 1:1, подкисленная соляной кислотой с последующим выделением
промежуточного продукта (81) и его последующим окислением [96] (В)
(схема 20)
Схема 20
81 82
5,10,15-трифенилкоррол
(A) К перемешиваемому раствору 5,0
(72,2 ммоль) пиррола и 0,83мл (8,3 ммоль) бензальдегида в 30 мл хлористого
метилена в инертной атмосфере (под аргоном) прибавляли 0,02 мл трифторуксусной
кислоты, Реакционную смесь в инерной атмосфере перемешивали два часа. Далее
добавляли 1,57 г (6,4 ммоль) п-хлоранила и перемешивали смесь еще 0,5
часа. Оставляли реакционную смесь на ночь. Растворитель отгоняли на ротационном
испарителе, избыток пиррола отгоняли с водяным паром, грязный продукт отфильтровывали
и высушивали на воздухе. Высушенный коррол очищали с помощью колоночной
хроматографии на силикагеле, элюируя хлористым метиленом. Выход продукта 146 мг
(10%)
(B) 0,5 мл (5 ммоль) бензальдегида и
0,7 мл (10 ммоль) пиррола растворяли в смеси 200 мл метанола и 200 мл воды, при
пропускании через смесь аргона. Затем в смесь разом приливали 4,25 мл
концентрированной соляной кислоты, и перемешивали при комнатной температуре в
инертной атмосфере три часа. полученный в колбе осадок мы отфильтровали, растворяли
в хлороформе, промывали водой полученный раствор, высушивали сульфатом натрия,
раствор доводили хлороформом до объема 300 мл и кипятили с 1,23 г (5 ммоль) п-хлоранила
в течении 1 часа. Хлороформ отгоняли, остаток промывали 5%-ным раствором
гидроксида калия, водой, высушивали на воздухе при 70оС, растворяли в хлористом
метилене и хроматографировали на сикагеле элюируя хлористым метиленом. Элюат
упаривали, коррол высаживали метанолом, отфильтровывали, промывали метанолом и
высушивали на воздухе. Выход 267 мг (30%)
Rf =
0,35 (бензол)
MALDI-TOF (m/z) найдено: 526,6 [М]+ ; вычислено: 526,4
ЭСП lmax,нм (e·10-3); 646,7 (11,5); 614,6 (14,4); 575,7 (17,6); 415 (132,7) (хлороформ).
Н ЯМР (вн. ст. ТМС) δ, м. д. 8,94s, 8,90s, 8,60s,
8,59s (4x2H, β-H); 8,38d (4H, J = 6,0Hz, 2,6-
H-Ph); 8,19d (2H, 1J = 3,9Hz, 2,6-H-Ph); 7,70-7,80m (8H); 7,80-7,89m (9H,
3,4,5-H-Ph); -1,98bs (3H, NH) (CDCl3)
При осуществлении метода (В) нами были внесены изменения по сравнению с
методикой представленной литературе. В оригинальной методике [96] на стадии
выделения линейного промежуточного продукта, проводится экстрагирование его
хлороформом. Затем органический слой промывается водой, высушивается сульфатом
натрия. Растворитель отгоняется до объема 300мл. В нашем случае промежуточный
продукт в виде осадка, был отфильтрован, и затем растворён в хлороформе, что на
практике оказалась не менее эффективно чем экстракция, так как выход,
заявленный в литературе сошелся с выходом, полученным практически.
Так же стоит отметить, что метод (B) позволяет получить коррол с большим выходом (в три раза)
чем, метод (А), предположительно это связано присутствующей в методе (В)
стадией выделения промежуточного продукта. Благодаря этому последующая стадия
окисления п-хлоранилом проходит более направленно чем в методе(А), давая
больший выход целевого продукта.
2.4 Синтез металлокомплексов тетрафенилпорфина,
тетрафенилпорфицена и трифенилкоррола
2.4.1 Металлокомплексы 5,10,15,20-тетрафенилпорфина
Нами были синтезированы металлокомплексы порфирина 1b взаимодействием его с ацетатами меди
или никеля в уксусной к-те и ацетилацетонатом цинка в хлороформе. Таким
образом, были опробованы наиболее известные методы комплексообразования
порфиринов с двухвалентными металлами.
Медный(II) комплекс
5,10,15,20-тетрафенилпорфина
,3 г (0,49 ммоль) 5,10,15,20-тетрафенилпорфина при кипении смывали в
раствор 0,2 г (0,98 ммоль) гидрата ацетата меди(II) в 15 мл уксусной к-ты, дополнительно кипятили 1 ч и
охлаждали. Осадок медного комплекса отфильтровывали, промывали водой и
высушивали на воздухе при 70°C.
Выход - 0,32 г (96,6%). Для очистки растворяли в хлороформе и
хроматографировали на оксиде алюминия II степени активности по Брокману. Элюат упаривали, комплекс осаждали метанолом
и высушивали на воздухе при 70°C.
Rf
(силуфол): 0,68 (хлороформ-гептан, 3:1)
ЭСП λmax,
нм (ε·10-3):
539 (21,8); 415 (506,9) (хлороформ)
Никелевый(II) комплекс
5,10,15,20-тетрафенилпорфина
Получали аналогично медному комплексу с использованием тетрагидрата
ацетата никеля. Выход 0,29 г (91,3%).
Rf
(силуфол): 0,68 (хлороформ-гептан, 3:1)
ЭСП λmax,
нм (ε·10-3):
529 (16,4); 416 (241,0) (хлороформ)
Цинковый(II) комплекс
5,10,15,20-тетрафенилпорфина
Кипятили 1 ч раствор 0,5 г (0,81 ммоль) 5,10,15,20-тетрафенилпорфина и
0,5 г (1,9 ммоль) ацетилацетоната цинка в 100 мл хлороформа, затем полученный
раствор хроматографировали на оксиде алюминия II степени активности по Брокману. Элюат упаривали, комплекс
осаждали метанолом и высушивали на воздухе при 70°C. Выход - 0,46 г (83,4%)
Rf
(силуфол): 0,20 (хлороформ - гептан, 3 : 1)
ЭСП λmax,
нм (ε·10-3):
593 (6,2); 550 (18,1); 421(415,7) (хлороформ)
2.4.2 Металлокомплексы 2,712,17-тетрафенилпорфицена
В рамках изучения реакционной способности внутрисферного
комплексообразования порфицена, были получены его металлокомплексы с цинком и
медью.
Цинковый(II) комплекс
2,712,17-тетрафенилпорфицена
,0 мг (0,00814 ммоль) тетрафенилпорфицена растворяли в 5 мл ДМФА, в
полученный раствор добавляли 0,1 г (0,38 ммоль) ацетилацетоната цинка(II). Смесь кипятили 10 часов. В
охлаждённную реакционную смесь добавляли воду, выпавший осадок отфильтровывали.
Очистку комплекса проводилось хроматографией на силикагеле в хлористом
метилене, в данном случае первая сходящая по колонке зона - цинковый комплекс
(судя по ЭСП). Элюат цинкового комплекса упаривали до минимального объема и
остаток высушивали на воздухе при комнатной температуре. Вещество представляет
собой кристаллический порошок сине-фиолетового цвета. Выход 4,4 мг (79,8%)
ЭСП lmax,нм (e·10-3); 658 (78,06), 397,3 (89,39)
(хлороформ).
MALDI-TOF (m/z) найдено: 676,3
[М-2H]+ ; вычислено: 676,1
Медный комплекс(II)
2,7,12,17-тетрафенилпорфицена.
,0 мг (0,00814 ммоль) тетрафенилпорфицена растворяли в 5 мл ДМФА, в
полученный раствор добавляли 0,1 г (0,55 ммоль) ацетата меди гидрата,
реакционную смесь кипятили три часа. Прореагировавший раствор выливали в 10 мл
воды, добавляли ацетат натрия в небольших количествах на кончике шпателя, и
отфильтровывали выпавший осадок. Очистка цинкового комплекса проводилась
хроматографией хлористым метиленом на силикагеле, элюат упаривали до
минимального объема, остаток высушивали на воздухе при комнатной температуре.
Выход вещества, представляющего собой мелкие тёмно-синие кристаллы составил 5,4
мг (98,9%).
ЭСП lmax,нм (e·10-3); 638(82,04), 395(102,4)
(хлороформ)
MALDI-TOF (m/z) найдено: 675,3
[М-H]+ ; вычислено: 675,28
Наличие полосы Соре на границе видимой области свидетельствует об
ароматичности системы. Спектр порфицена является трёх полосным, а спектры его
металлокомплексов однополосны.
Попытка синтезировать другие металлокомплексы
2,7,12,17-тетрафенилпорфицена
Нами были предприняты попытки синтеза металлокомплексов порфицена и с
другими металлами, такими как никель(II),марганец(III), кадмий(II), а также с серебром(I).
Попытки получить комплекс тетрафенилпорфицена с никелем предпринимались
дважды в различных растворителях. В первом случае 5 мг (0,00814 ммоль)
тетрафенилпорфицена растворяли в 5мл ДМФА, к раствору добавляли 0,1 (0,40
ммоль) ацетат никеля(II) гидрат, после этого реакционную смесь кипятили, каждые
2 часа снимая электронный спектр. Однако даже после 10 часов реакции никаких
признаков введения металла не было.
Во втором случае 5 мг (0,00814 ммоль) тетрафенилпорфицена растворяли в 5
мл уксусной кислоты, в реакционную смесь вводили 0,1 г (0,40 ммоль) ацетата
никеля(II) гидрата, а также 50 мг ацетата натрия, смесь кипятили около 10
часов. Согласно ЭСП реакционной смеси никелевый комплекс в ней присутствовал в
следовых количествах.
В случае попыток получения марганцевого комплекса порфицена в ДМФА, имел
место неожиданный факт. Смесь обесцветилась и выпал желтый осадок, однако при
охлаждении восстановилась синяя окраска раствора, характерная для порфицена,
что и было подтверждено его ЭСП.
В случае реакции порфицена с ацетатом серебра(I) в ДМФА в процессе длительного кипячения происходит
выпадение осадка металлического серебра, предположительно, из-за взаимодействия
ацетата серебра с ДМФА.
Таким образом, нами было установлено, что порфицен обладает гораздо
меньшей способностью к координации, нежели чем тетрафенил-порфин.
Рассмотрим на примере цинковых комплексов: комплекс тетрафе-нилпорфина с
цинком образуется за час, в среде низкокипящего растворителя, такого как
хлороформ, при этом выход продукта составил 83%, в то время как выход цинкового
комплекса тетрафенилпорфицена 79%, при этом использовался высококипящий
растворитель - ДМФА, и время синтеза составило 10 часов.
Тетрафенилпорфицен намного легче, чем с цинком, образует комплекс с
медью, причем выход близок к 100%. Возможно это происходит из-за того что
атомный радиус иона Cu(ІІ), меньше,
чем у атома Zn(ІІ) и, кроме того, координационное
число меди 4, в отличии от 6 для цинка.
Таким образом на основании литературных и экспериментальных данных
следует вывод о том, что тетрафенилпорфицен обладает меньшей
комплексообразующей способностью. Решающую роль здесь играет строение и
симметрия координационных центров этих соединений.
2.4.3 Металлокомплексы 5,10,15-трифенилкоррола
Медный(II) комплекс 5,10,15трифенилкоррола
20 мг (0,038 ммоль) 5,10,15-трифенилкоррола и 0,1г (0,55 ммоль) ацетата
меди в ТГФ перемешивали при комнатной температуре в течении 10-15 мин.
Прохождение реакции контролировали по Rf. Прореагировавшую смесь выливал на чашку Петри и высушивали на воздухе.
Осадок растворяли в бензоле и хроматографировали на силикагеле элюируя
бензолом. Бензол отгоняли, остаток высаживали метанолом, полученный осадок
отфильтровывали и высушивали на воздухе. Выход 14 мг (63,2%)
Rf =
0,86 (бензол)
MALDI-TOF (m/z) найдено: 586,36
[М-Н]+ ; вычислено: 586,16
ЭСП lmax, нм (e·10-3); 622,5 (6,1); 541,4 (9,0); 409,9 (119,9) (хлоро-форм).
Попытки синтезировать другие металлокомплексы 5,10,15-трифенилкоррола
Также нами были предприняты попытки получения металлокомплексов
5,10,15-трифенилкоррола с трёхвалентными металлами: индием и галлием.
Для получения металлокомплекса 5,10,15-трифенилкоррола с индием смесь 50
мг (0,095 ммоль) трифенилкоррола и 0,2 г (0,9 ммоль) хлорида индия(III), кипятили в 10 мл пиридина с
обратным холодильником. Прохождение реакции контролировали по ЭСП. Однако, даже
после 7 часов кипячения смеси, существенных изменений по ЭСП замечено не было.
Для получения металлокомплекса 5,10,15- трифенилкоррола с галлием смесь
50 мг (0,095 ммоль) трифенилкоррола и 0,2 г (0,089 ммоль) ацетата галлия,
кипятили в 10 мл пиридина, в течении 3 часов. Прохождение реакции
контролировали по ЭСП. Далее полученный осадок отфильтровывали и высушивали на
воздухе. Осадок растворялив хлороформе и хроматографировали на силикагеле
элюируя хлороформом. Затем растворитель отгоняли и высаживали метанолом.
Выпавший осадок отфильтровывали, промывали метанолом и высушивали на воздухе.
Однако полученный осадок по данным масс (MALDI-TOF) и
1Н ЯМР спектров не являлся галлиевым комплексом 5,10,15-трифенилкоррола. Таким
образом, нам не удалось пока получить металлокомплексы 5,10,15-трифенилкоррола
с трёхвалентными металлами.
.5 Попытки синтеза β-фенилзамещенных порфиринов
.5.1 Попытки синтеза 3,7,13,17-тетрафенилпорфина
Таким образом, получив порфицен, заданной структуры, дальнейшей задачей
было получить аналогичные ему изомерные порфирины. Кроме
5,10,15,20-тетрафенилпорфина, возможен вариант получения более близкого по
структуре 3,7,13,17-тетрафенилпорфина (85). Основной задачей синтеза,
как и в осуществлении синтеза тетрафенилпорфицена, оставалась получение пиррола
заданной конфигурации, а затем и соответствующего дипирролилметана или
дипирролилметена, с целью дальнейшего превращения данного соединения в
3,7,13,17-тетрафенилпорфин. Учитывая огромные возможности превращения пирролов,
а именно в данном случае 5-метилпиррол-2-карбоновых эфиров, нами было выбрано
два пути синтеза (схема 21).
Схема 21
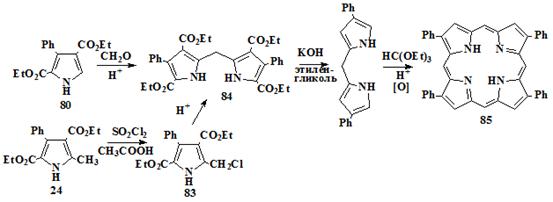
Первый путь для осуществления синтеза был связан с попыткой
восстановления 2-иод-3,5-дикарбэтокси-4-фенилпиррола до
3,5-дикарбэтокси-4-фенилпиррола (80), с последующей конденсацией
полученного продукта с формальдегидом до дипирролилметана (84)
Нам не удалось осуществить этот метод т. к. не удалось получить α-незамещенный пиррол (см. выше стр.
32)
Дальнейшие попытки были связаны с получением
2,2'-диметил-4,4'-дифенил-3,3',5,5'-тетракарбэтоксидипирролилметана (84)
из пиррола (24) его радикальным хлорированием сульфурил хлоридом в
уксусной к-те до хлорметилпиррола (83).
2-хлорметил-3,5-дикарбэтокси-4-фенилпиррол (83)
3,0 г (9,9 ммоль) 2-метил-4-фенил-3,5-дикарбэтоксипиррола растворяли в 20
мл ледяной уксусной кислоты. Далее при перемешивании в реакционную смесь
вводили по каплям 1,5 мл (9,9 ммоль) сульфурилхлорида, так, чтобы темпертура
реакционной смеси не подымалась выше 50-60°С. После добавления сульфурил
хлорида, смесь нагревали до 70°С, и перемешивали при этой температуре 30 мин.
Прореагировавшую смесь медленно охлаждали до 20°С. Выпавший в процессе
охлаждения осадок отфильтровывали, несколько раз промывали осадок маточным
раствором, затем водой и высушивали при комнатной температуре на воздухе.
Продукт представляет собой белое кристаллическое вещество с температурой
плавления 118-120°С. Выход продукта 3,0 г (89,8%.)
1H ЯМР (вн. ст. ТМС) δ, м.д.: 9,91bs (1H, NH); 7,32-7,38m (3H, m,p-H-Ph); 7,26-7,30m (2H, o-H-Ph);
5,06s (2H, CH2Cl); 4,16q (2H, J = 7,1Hz, CH2-Et); 4,07q (2H, J =
7,1Hz, CH2-Et); 1,07t (6H, J = 7,1Hz, CH3-Et); 1,01t (6H, J =
7,1Hz, CH3-Et) (CDCl3)
3,5,3’,5’-Тетракарбэтокси-4,4’-дифенилпиррол (84)
,8 г (8,3 ммоль) 2-хлорметил-3,5-дикарбэтокси-4-фенилпиррола растворяли в
3 мл ледяной уксусной кислоты, смесь нагревали до кипения, после чего, добавляли
3 мл воды. Полученную смесь кипятили 1,5 часа. Из охлаждённой прореагировавшей
смеси, содержащую, воду и вязкое вещество, декантацией сливали воду, а вещество
растворяли в бензоле и хроматографировали на силикагеле, элюируя хлористым
метиленом. Элюат упаривали до полного удаления растворителя, при этом получился
желтоватый вязкий не кристаллизующийся жидкий продукт, изучение которого
ведется в настоящее время.
2.5.2 Попытки синтеза смеси изомерных β-тетрафенилпорфиринов
Другим предполагаемым путем получения изомерного порфицену порфирина
может быть синтез его через 3-фенилпиррол (86) полученный
декарбоксилированием пиррола (74) (схема 22)
Схема 22
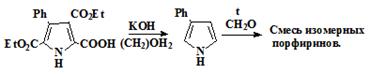
74 86
В ходе данной цепочки превращений теоретически должна получиться смесь
изомерных порфиринов, с различным взаимным положением фенильных групп
(рандомеров), однако, по стерическим причинам наиболее вероятна конфигурация
2,7,12,17-тетрафенилпорфина (87) с наиболее пространственно удаленными
фенильными кольцами:
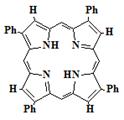
3-фенилпиррол (86)
,0 г (9,1 ммоль) 3,5-диэтоксикарбонил-4-фенил-2-карбоксипиррола
растворяли в 50 мл этиленгликоля, добавляли 3,0 г (53,6 ммоль) гидроксида калия
и немного гидразин сульфата и кипятили реакционную смесь 1,5 часа, затем
реакционную смесь выливали в воду и пиррол отгоняли с водяным паром, далее
проводили экстракцию дистиллята хлористым метиленом. К сожалению, выбранный
метод оказался тупиковым, т. к. выход продукта ничтожно мал.
2.6
Попытки синтеза окта-β-замещенных порфиценов
Как было рассмотрено ранее, все существующие стратегии синтеза порфиценов
отличаются друг от друга только стадиями подготовки исходных пирролов и
биспирролов. Ключевой стадией к синтезу порфицена является получение пиррола
заданной структуры, который и будет определять строение и положение
заместителей в конечном макроцикле.
Получение окта-β-замещенных порфиценов в первую очередь было связано с
попытками заменить синтез исходного пиррола по Кнору с последующим его
иодированием (Схемы 15, 16) на Синтез Бартона-Зарда зарекомендовавшего себя как
способ получения α-незамещённых пирролов с высокими выходами,
конденсацией активированных электроноакцепторными группами алкенов с
изоцианоацетатами в присутствии сильных ненуклеофильных оснований [26-27]
(Схема 22), а так же на, весьма схожий с ним метод синтеза пирроллов
взаимодействием бензальдегида с эфирамии изоцианоуксусной кислоты, в
присутствии DBU. (Схема 23)
Схема 22
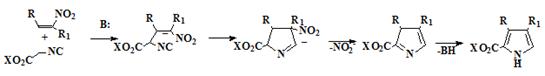
Схема 23
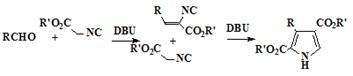
Предполагалась что синтез исходных пирролов по методу Бартона -Зарда,
позволит не только сократить количество стадий синтеза, но и повысить выхода
пирролов необходимого строения.
Предполагалось так же, что этими методами можно будет получать пирролы из
которых впоследствии возможно получить порфицены содержащие несколько
заместителей в β-положениях, одни из которых будут арильными а другие
алкильными.
 =C6H5, C6H4OCH3= Me, Et
=C6H5, C6H4OCH3= Me, Et
Однако при осуществлении данной задачи мы столкнулись с некоторыми
осложнениями, которые в основном связаны с использованием в качестве реагентов,
так называемых прекурсоров и необходимостью синтеза некоторых дорогостоящих
соединений.
2.6.1 Синтез эфиров изоцианоуксусной кислоты
Для получения пирролов из бензальдегида и методом Бартона-Зарда, были
проведены синтезы двух дорогостоящих эфиров изоцианоуксусной кислоты: этилового
и метилового.
Исходными соединениями для данного синтеза стала аминокислота-глицин.
Взаимодействием глицина и этилового или метилового спирта в присутствии
тионилхлорида синтезировали соответствующий эфир гидрохлорида глицина, которым
амидировали этил- или метилформиат в присутствии триэтиламина в качестве
основания. Полученный эфир N-формилглицина
дегидратировали хлорокисью фосфора в присутствии триэтиламина до требуемого
этил или метил изоцианоацетата. На схеме 24 представлена цепочка синтезов для
этилового эфира изоцианоуксусной кислоты.
Схема 24

88
89 90
Этилового эфира глицина хлоргидрата (88)
В суспензию 150 г, (2 моль) глицина и 130 мл, (2,23 моль) этилового
спирта при перемешивании добавляли 216 мл ( 2,9 моль ) тионил хлорида по каплям
до полного растворения, полученную смесь кипятили 2 часа. Далее колбу с
реакционной смесью охлаждали в холодной воде, выпавший осадок отфильтровывали.
Из полученного фильтрата отгоняли растворитель (спирт). Выпавший осадок
отфильтровывали и промывали диэтиловым эфиром, сушили на воздухе, затем в
сушильном шкафу. Выход продукта составил 258,6 г (93%).
Этиловый эфир N-формилглицина
(89)
140 г (1,0 моль) хлоргидрата этилового эфира глицина, 100 мг моногидрата п-толуолсульфокислоты,
в качестве катализатора реакции, 500 мл (6,2 моль) этилового эфира муравьиной
кислоты, нагревали до кипения, в кипящую смесь добавляли по каплям 154 мл, (1,1
моль) триэтиламина. Полученную смесь кипятили 20 ч, охлаждали до 20°С, выпавший
осадок отфильтровывали. Из фильтрата отгоняли на ротационном испарителе
этилформиат, выпавший дополнительно осадок отфильтровывали. Фильтрат перегоняли
под вакуумом, собирая фракцию, выкипающую при 114-116°С. Выход 71,1 г (40%).
Н ЯМР (вн. ст. ТМС) δ, м.д.:8,01s (1H, CHO); 6,83bs (1H, NH); 4,06q (2H, J=7,2 Hz, CH2O); 3,85d (2H, 1J = 5,4Hz, CH2N); 1,19t (3H, J = 7,2Hz, CH3) (CCl4)
Этиловый эфир изоцианоуксусной кислоты (90)
К раствору 16,4 г (0,13 моль) этилового эфира N-формилглицина и 44 мл (0,31 моль) триэтиламина в 100
мл осушенного хлористого метилена прикапывали при температуре 0°С 12 мл (0,13
моль) хлорокиси фосфора, затем смесь перемешивали при этой температуре 1 час.
Далее в смесь медленно добавляли при энергичном перемешивании раствор 25 г
безводного бикарбоната натрия в 100 мл воды, так, чтобы температура реакционной
смеси не подымалась выше 20-25°С. Полученную смесь перемешивали 30 мин при
комнатной температуре, после разделения фаз водный слой отделяли, разбавляли до
300 мл, экстрагировали хлористым метиленом. Органический слой объединяли,
промывали насыщенным раствором NaCl,
высушивали сульфатом натрия, отгоняли растворитель и остаток перегоняли под
вакуумом. Выход продукта 8,2 г (55,8%).
Н ЯМР (вн. ст. ТМС) δ, м.д.: 4,20q (2H, J=7,2Hz, CH2CO); 4,18s (2H, CH2CO); 1,28t (3H, J=7,2Hz, CH3) (CCl4)
Была попытка заменить в данной методике триэтиламин (в связи с его
дефицитом) на 30 мл (0,37 моль) пиридина. Однако это не дало положительных
результатов.
.6.2 Эфиры муравьиной кислоты
Из-за катастрофического недостатка реактивов, для осуществления данного
метода, были синтезированы метиловый и этиловый эфиры муравьиной кислоты.
Этилформиат [97]
В круглодонной колбе смешивают 50 г безводного CaCl2, 500 мл (13,0 моль), 96% муравьиной кислоты, 660 мл
(14,2 моль) этилового спирта, нагревали с дефлегматором, соединённым с прямым
холодильником. Отогнанный этилформиат высушивали K2CO3 и перегоняли с
дефлегматором, собирая фракцию 55С. Выход 510 г (80%).
Метилформиат
Метилформиат получали аналогичным предыдущему методом, только вместо этилового
спирта использовали метанол. Выход 503 г (72%).
2.6.3 Синтез 2,4-дикарбэтокси-3-фенилпиррола [26]
В смесь 2,0 г (0,02 ммоль) этилизоцианоацетата и 3,0 г (0,02 ммоль) DBU в 30 мл ТГФ добавляли 1,06 г (0,01
ммоль) бензальдегида в 10 мл ТГФ, при температуре 45-50°С, за период 15 мин,
при перемешивании. После перемешивали в течение 5 часов при этой температуре,
смесь нейтрализовали небольшим количеством уксусной кислоты и отгоняли
растворитель на ротационном испарителе. К остатку добавляют воду, водный слой
отделяли, и экстрагировали хлористым метиленом. Органический слой объединяли и
промывали соляной кислотой, водой, и после высушивания сульфатом натрия
растворитель отгоняли. Остаток перекристаллизовывали из водно-этанольной смеси.
Синтез также проводился нами большое количество раз, однако выход продукта
оказывался ничтожно мал.
Н ЯМР (вн. ст. ТМС) δ, м.д.: 9,56bs (1H, NH); 7,61d (1H, J
= 3,4Hz, 5-H); 7,31-7,40m (5H, H-Ph); 4,15q, 4,13q (2x2H, 1J = 7,1Hz,
CH2-Et); 1,14t, 1,09t (2x3H, 1J = 7,1Hz, CH3-Et) (CDCl3)
.6.4 Синтез пирролов методом Бартона-Зарда [27]
Исходными веществами для синтеза арилпирролов этим методом являются
арилзамещённые нитропропены, которые можно получить, конденсацией по
Кневенагелю бензальдегидов с нитроэтаном в присутствии в качестве
дегидратирующего агента этилортоформиата и катализатора ацетата калия.
Схема 25
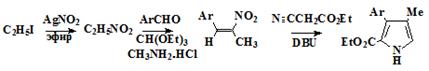
Нитроэтан
Невозможность (из-за недостатка денежных средств и законов РФ) приобрести
нитроэтан, заставила нас синтезировать его самостоятельно. Молекулярное
серебро, растворяли в азотной кислоте, с получением нитрата серебра, далее
полученный нитрат переводили в нитрит, взаимодействием с нитритом натрия. Сам
нитроэтан получали из нитрита серебра и иодэтана.
В колбу с обратным холодильником помещали 106,3 г (0,69 моль) нитрита
серебра, 150 мл диэтиловогоэфира,охложденног до 0˚С, к смеси медленно
порциями, через холодильник приливали 44,5 мл (0,552 моль) иодистого этила, так
чтобы реакционная смесь слабо кипела, далее полученную смесь нагревали на
водяной бане 40 мин, охлаждали до комнатной температуры, отфильтровывали осадок
и промывали хлористым метиленом. Растворитель из фильтрата отгоняли на
ротационном испарителе. Выход продукта составил 3,4 г (8,2%).
Синтез 1-арил-2-нитропропенов
1-(4-метоксифенил)-2-нитропропен [28]
Смесь 3.4 г (45 ммоль) нитроэтана, 7.0 г (42 ммоль) анисового альдегида,
2,4 г (40 ммоль) CH3CO2K, 3,4 г (35 ммоль) гидрохлорида метиламина, 10 мл
ортоформиата и 30 мл метанола, перемешивали при комнатной температуре 7 часов.
После этого реакционную смесь выливали в воду, отфильтровывали осадок. Продукт
перекристаллизовывали из метанола. Выход продукта реакции 3,5 г (37%).
1H ЯМР (вн. ст. ТМС) δ, м.д.: 8,08s (1H, 1-H); 7,43d (2H, J =8,8Hz, 2,6-H-Ar); 6,99d (2H, J
=8,8Hz, 3,5-H-Ar); 3,87s (3H, CH3O); 2,48s (3H, CH3) (CDCl3)
Тпл = 40-41˚С
-(4-нитрофенил)-2-нитропропен
Синтез проводился по той же схеме, только вместо анисового альдегида был
взят 4-нитробензальдегид, в количестве 5,0 г (49 ммоль).
Выход вещества составил 3,0 г (31%)
1H ЯМР (вн. ст. ТМС) δ, м.д.: 8,32d (2H, J =8,8Hz, 3,5-H-Ar);8,10s (1H, 1-H); 7,61d (2H, J
=8,8Hz, 2,6-H-Ar); 2,47s (3H, CH3) (CDCl3)
Тпл = 109-110˚С
-фенил-2-нитропропен [29]
Смесь 7 мл (69,5 ммоль) бензальдегида, 10 мл (66,6 ммоль) нитроэтана, 0,5
мл н-бутиламина, и 7 мл метанола нагревали в течение 8 часов с обратным
холодильником. Прореагировавшую смесь охлаждали, выпавший осадок
отфильтровывали, сушили на воздухе. Фильтрат, подкисляли уксусной кислотой
(0,5мл) охлаждали, выпавший осадок отфильтровывали. Выход 4,9 г (43,5%).
H ЯМР (вн. ст. ТМС) δ (м.д.): 8,75s (1H, 1-H); 8,11-8,20m (5H, H-Ph); 3,20s (3H, CH3) (CCl4)
Тпл = 65 - 66 ˚С
-этоксикарбонил-3-метоксифенил-4-метилпиррол [28]
К охлаждённому раствору 2,0 г (12,35 ммоль) 1-(4-метоксифенил)-2-нитропропена,
и 1,6 г (15,73 ммоль) этилизоцианоацетата в 15 мл ТГФ, через капельную воронку
медленно добавляли 2,35мл (15,73 ммоль) DBU. Реакционную смесь при перемешивании выдерживали при
комнатной температуре 12 ч. Реакционную смесь выливали в воду, осадок
отфильтровывали и перекристаллизовывали из метанола. Выход 1,4г. (50%)
H ЯМР
(вн. ст. ТМС)
δ, м.д.: 8,96bs (1H, NH); 7,30d (2H, J = 8,7Hz, 2,6-H-Ar); 6,95d (2H, J
= 8,7Hz, 3,5-H-Ar); 6,79d (1H, 1J = 2,7Hz, 5-H); 4,20q (2H, 2J =
7,1Hz, CH2-Et); 3,87s (3H, OCH3); 2,03s (3H, CH3); 1,20t (3H, CH3-Et); (CDCl3)
Синтез пирролов из изоцианоуксусной кислоты может быть перспективным,
поскольку сокращает количество стадии, однако в нашем случае данный метод
оказался более долгим и сложно с синтетической точки зрения осуществимым по
сравнению с синтезом Кнорра. Другой проблемой, являются малые выхода пирролов,
возможно, это связано с не достаточно хорошим качеством эфира изоцианоуксусной
кислоты, которые являются нестабильными соединениями, и подвержены окислению и
разложению при хранении.
2.7 Электронные и 1Н ЯМР-спектры мезо-тетрафенилпорфина,
тетрафенилпорфицена, трифенилкоррола и их металлокомплексов
В электронных спектрах поглощения порфиринов (рис.1) в видимой области
присутствуют 4 полосы поглощения, соответствующие энергетическим переходам,
которые относятся к числу чисто электронных переходов, а также к
электронно-колебательным переходам. Интенсивная полоса в ближайшем
ультрафиолете - полоса Соре. Ею обладают спектры всех тетрапиррольных ароматических
макроциклов. Все полосы в электронных спектрах порфирина вызваны π-π*
переходами. Полоса Соре
обусловлена электронным переходом на наиболее высокую по энергии вакантную π
орбиталь. Полосы видимой
части спектра І и ІІІ относятся к квазизапрещённым электронным переходам, а
полосы ІІ и ІV имеют колебательное происхождение, являются колебательными
спутниками полос І и ІІІ [94].
При образовании комплексов порфиринов, полоса І которая в спектрах
является наименьшей, становится намного интенсивней. Полоса І является наиболее
чувствительной к изменениям в структуре порфирина.
Спектр порфицена (рис.2) является трёх полосным, а спектры его
металлокомплексов однополосными. Наличие полосы Соре на границе видимой области
свидетельствует об ароматичности системы порфиценов. При этом самым высоким
пиком является пик I находящийся как
и у порфинов около 650 нм. Наиболее чувствительным к изменению структуры
является пик ІІ, при образовании металлокомплексов пик ІІ наиболее интенсивен,
тогда как пики І и ІІІ пропадают. Металлокомплексы тетрафенилпорфицена имеют
лишь полосу ІІ, при этом полоса Соре практически не изменяется. В некоторых
случаях, (образование металлокомплекса тетрафенилпорфицена с медью) пик ІІІ
присутствует в спектре в виде своеобразной «ступени».
ЭСП трифенилкоррола (рис.3) является также трёхполосным. Наличие полосы
Соре на границе видимой области свидетельствует об ароматичности структуры
трифенилкоррола. В отличие от порфиценов и порфиринов самым интенсивным пиком
является пик III находящийся около 580 нм, напротив
пик I, находящийся около 650 нм, является
у корролов наименее интенсивным. При образовании медного комплекса три узких
пика переходят в две широкие полосы, при этом полоса Соре не изменяется.
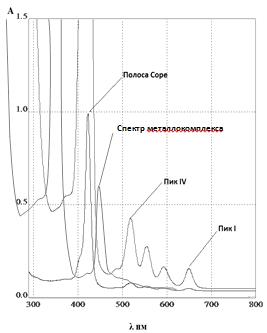
Рис. 1. ЭСП тетрафенилпорфина и его медного комплекса

Рис. 2. ЭСП тетрафенилпорфицена и его медного комплекса

Рис. 3. ЭСП трифенилкоррола и его медного комплекса
Слабопольный сдвиг сигналов мезо- и β-протонов и сильнопольный сдвиг
сигналов NH-протонов в его1Н ЯМР-спектре
указывает на наличие кольцевого тока и ароматичность полученного
тетрафенилпорфицена.(рис 5) Вместе с тем сигналы NH-протонов координационного центра молекулы сдвинуты в слабое
поле более чем на 6,5 м.д., что, от части, вызвано образованием прочных
внутримолекулярных Н-связей. Для порфиринов такое явление не характерно, они не
могут образовывать прочных водородных связей внутри цикла. Препятствием к их
образованию является не только большее расстояние N(3) и NH(2),
но и низкая протонодонорность NH и
низкая протоноакцепторность N.
Вследствии того что иминные атомы водорода в порфиринах расположены на одной
прямой в плоскости N4 возникают не
обычные условия для NH связей, они
становятся короче чем обычные связи NH. Подобные условия препятствуют образованию водородных связей. В
порфиценах из за их прямоугольного строения реакционного центра, расстояние
между N и NH намного меньше чем в молекуле порфирина, поэтому образование
водородной связи становится возможным, это и показывает нам 1Н ЯМР спектр.
У 5,10,15-трифенилкоррола, как и у тетрафенилпорфина и
тетрафенилпорфицена, имеет место сильнопольный сдвиг сигналов NH- протонов в 1Н ЯМР-спектре в сильное
поле (три протона около -2 м.д.), и сдвиг сигналов β-протонов макроцикла и H-протонов фенильных колец в слабое
поле, что свидетельствует о наличии кольцевого тока. (рис.6)
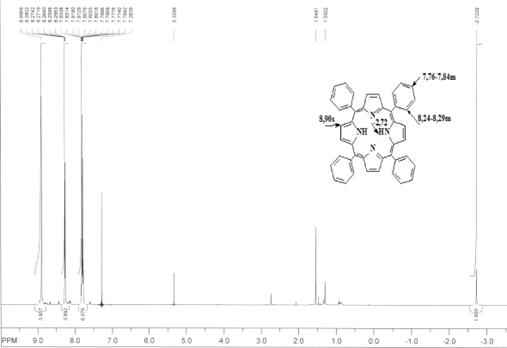
Рис.4. Спектр 1Н ЯМР 5,10,15,20-тетрафенилпорфицена в CDCl3, 298 К
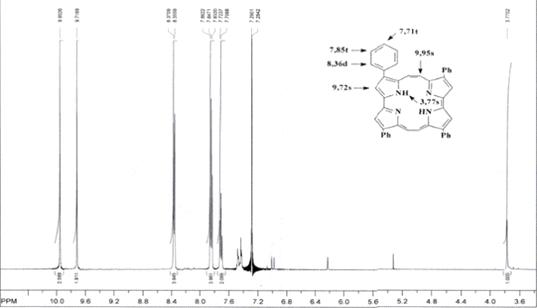
Рис. 5. Спектр 1Н ЯМР 2,7,12,17-тетрафенилпорфицена в CDCl3, 298 К
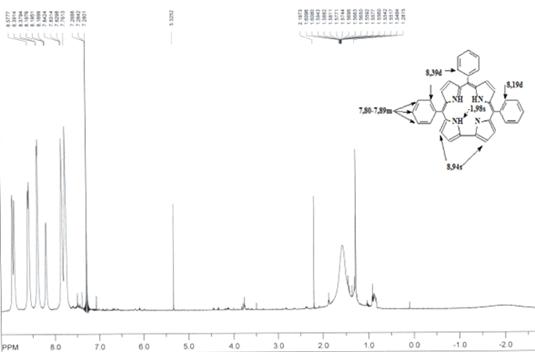
Рис.6. Спектр 1Н ЯМР 5,10,15-трифенилкоррола в CDCl3, 298 К
Выводы по работе
1. По модифицированной методике, включающей синтез фенилзамещённого
пиррола, его превращение в биспиррольное производное и циклизацию, получен и
спектрально охарактеризован (ЭСП, ИК, 1Н ЯМР, Масс-(MALDI-TOF))
2,7,12,17-тетрафенилпорфицен.
2. Синтезированы металлокомплексы 2,7,12,17-тетрафенилпорфицена с Zn(II), и Cu(II), данные соединения также
охарактеризованы спектрально (ЭСП, Масс-(MALDI-TOF))
. По методикам, разработанным в нашей лаборатории получен
изомерный 2,7,12,17-тетрафенилпорфицену, 5,10,15,20-тетрафенилпорфин, который
охарактеризован спектрально (ЭСП, 1Н ЯМР, Масс-(MALDI-TOF)).
Синтезированы металлокомплексы 5,10,15,20-тетрафенилпор-фина с цинком, медью, и
никелем. Данные вещества охарактиризованны спектрально. (ЭСП).
. Разработаны методики для начальных стадий получения
3,7,13,17-тетрафенилпорфина. Доказана структура впервые полученного
2-хлорметил-3,5-дикарбэтокси-4-фенилпиррола.
. Двумя методами получен несимметричный аналог 5,10,15,20-тетрафенилпорфина
и 2,7,12,17-тетрафенилпорфицена 5,10,15-трифенилкоррол, который охарактеризован
спектрально (ЭСП, 1Н ЯМР, Масс-(MALDI-TOF)).
. Синтезирован металлокомплекс 5,10,15-трифенилкоррола с Cu(II), данное соединение так же охарактеризовано спектрально
(ЭСП, Масс-(MALDI-TOF))
. Для синтеза α-незамещенных пирролов методом
Бартона-Зарда, освоены и отработаны методики получения исходных нитропропенов и
этилового эфира изоцианоуксусной кислоты.
Список литературы
1. Vogel E., Kocher M., Schmickler H., Lex J. A
Porfisen a Novel Porphin Isomer. // Angew. Chem., Int. Ed. Engl. 1986, № 25, Р 257;
2. Vogel E. Novel porphyrinoids.
// Pure Appl. Chem. 1990.-V. 62. -N 3. - P. 557-564.
3. Sessler J.L., Brucker E.A.,
Weghorn S.J., Kisters M., Schafer M., Lex J., Vogel E. Synthesis and crystal structure of a
novel tripyrrane-containing porphyrinogen-like macrocycle.//Angew.Chem. Int. Ed. Engl. 1994. V.
33. P. 2308.
4. Vogel E., Ko¨cher
M., Schmickler H., Lex J. Porphycen - ein neuartiges Porphin-Isomer. // Angew Chem.,Int. Ed.
Engl., 1986, N
25, Р. 257.
5. Callot H.J., Rohrer A.,
Tschamber T. New. J. Ring expansions and contractions of
metalloporphyrins.//Chem. 1995. V. 19. P. 155.
6. Vogel E., Broring M., Scholz
P., Deponte R., and all. Α
pyrrolophanediene-porphycene redox system.// Angew Chem. Int. Ed. Engl. 1991. V. 36. P.
1651.
7. Jonathan L. Sessler. New Porphyrin Isomers.
//, Angew. Chem., Int. Ed. Engl. 1994.
8. Chmielewski P.,
Latos-Grazynski L., Rachlewicz K., Glowiak T. Monoinvertiertes Tetra-p-tolylpor-phyrin:
ein neues Porphyrinisomer // Angew. Chem. Int. Ed. Engl. 1994. V. 33. N 7. P.
779-781
. Furuta H., Asano T., Ogawa
T. “N-Confused porphyrin”: a new isomer of tetraphenylporphyrin. / H. Furuta,
// J. Amer. Chem. Soc. 1994. V. 116. N 2. P. 767-768.
10. Gosmann M., Franck B. Novel synthesis of
5,10,15,20-tetraaryl-porphyrins using high-valent transition metal salts.// Angew. Chem., Int. Ed. Engl.
1986. N 25. Р. 1100.
11. Rachlewicz K., Latos-Grazynski L., Vogel E.,
Ciunik Z., Jerzykiewicz L. B. Novel reactions of iron(III) tetraphenylporphyrin
π-cation radicals with pyridine.//Inorg. Chem. 2002. N
41. Р. 1979.
. Hayashi T., Okazaki K., Urakawa N.,
Shimakoshi H., Sessler J. L, Vogel E. Hisaeda Y. Dynamic molecular recognition
in a multifunctional porphyrin and a ubiquinone analogue.//Organometallics,
2001. N 20. Р. 3074.
. Saoiabi А., Maroufi N., Laghzizil A., Barbe J. M., Guilard R., Maro J. Alkyl and
aryl substituted corroles.// Chim. Heterocycl. 2005. N 4. P. 65.
. MacDonald I., Dougherty T. J. Basic
principles of photodynamic therapy. //J. Porphyrins Phthalocyanines. 2001.V. 5.
P. 105-129.
15. Paolesse R. Corrole: the
little big porphyrinoid. //Synlett. 2008. N 15. P. 2215-2230.
. Vogel E., Koch P., Hou
X.-L., Lex J., Lausmann M., Kisters M., and all. New porphycene ligands: octaethyl-
and etioporphycene (OEPc and EtioPc) - tetra- and pentacoordinated zinc
complexes of OEPc.// Angew.Chem. 1993. N 105. P. 1670.
17. Paolesse R., Sagone F.,
Macagnano A., Boschi T., Prodi L., Montalti M., Zaccheroni N., Bolletta F.,
Smith K. M. Synthesis and functionalization of meso-aryl-substituted
corroles.// J. Porphyrins and Phthalocyanines 1999. N 3. P. 364.
18. Vogel E., Broring M., Erben
C., Demuth R., Lex J., Nendel M., Houk K.N. Porphycen - ein neuartiges
Porphin-Isomer.//Angew. Chem. Int. Ed. Engl. 1997. V. 36. P. 353с
19. Vogel E., Balci M., Pramod K., Koch P., Lex J.,
Ermer O. Reihe 2,7,12,17-Tetrapropylporphycen - Pendant des Octaethylporphyrins
in der Porphycen./ E. Vogel, // Angew. Chem., Int. Ed. Engl., 1987. N 26. P. 928.
. Richert C., Wessels J. M., MuЁ ller M.,
Kisters M., Benninghaus T.,. Goetz A. E. Preparation, mass spectrometry and
electrochemical studies of metal connected porphyrin oligomers.//J. Med. Chem. 1994. N 37. P. 2797
21. Bauer V. J., Clive D. L. J.,
Dolphin D., Paine J. B., Harris F. L., King M. M., Loder J., Wang S.-W. C.,
Woodward R. B. Sapphyrins: novel aromatic pentapyrrolic macrocycles// J. Amer. Chem. Soc.1983. V. 105. N 21.
P.6429-6436.
22. Sanches-Garcia D., Sessler
J.L. Porphycenes: synthesis and derivatives. // Chem. Soc. Rev. 2008. V 37. P.
215-232.
. Nonell S., Bou N., Borell
JI., Texido J., Villanueva A., Juarranz A., Canete M. Syntyesis of
2,7,12.17-tetraphenylporficene. First Aril-substituted porficene for the
potodinamic theropy of tumors. // Tetrahedron Lett. 1995. V.36. P. 3405-3408.
24. Urbanska N., Pietraszkiewicz M. Waluk J. Материалы 4-й Международной конференции по порфирина и
фталоцианинов, Рим, Италия, июль 2-7, 2006.
25. Sessler J. L. Sanchez-Garcia D. Материалы на 4-й Международной конференции по порфиринам и
фталоцианинам, Рим, Италия, июль 2-7, 2006.
26. Suzuki M., Miyashi M., Matsumoto K. Α
convenient synthesis of 3-substituted pyrrole-2,4-dicarboxylic acid esters.// Tetrahedron. 1990. V 3.
P. 3057-3059.
. Barton D. H. R., Zard S. Z. A new synthesis
of pyrroles from nitroalkenes. // J. Chem. Soc., Chem. Commun. 1985. P.
1098-1100.
. Ono N., Kawamura H., Bougauchi M., Maruyama
K. Porphyrin synthesis from nitrocompounds.// Tetrahedron 1990. V. 46. P.
7483-7496.
. Ono N., Katayamu H.,Nisyiyamu S.,Ogawa T.
Regioselective Synthesis of 5-unsubstituted benzyl pyrrole-2-carboxylates from
benzyl isocyanoacetate//J. Heterocyclic Chem. 1994 V.31 P.707.
30. Rothemund P. Formation of
porphyrins from pyrrole and aldehydes. // J. Amer. Chem. Soc. 1935. V. 57. P.
2010-2011.
. Rothemund P., Menotti A. R.
Porphyrin studies. IV. The synthesis of α,β,γ,δ-tetraphenylporphine. //J.
Amer. Chem. Soc. 1941. V. 63. N 1. P. 267-270.
. Rothemund P. Porphyrin
studies. III. The Structure of the porphine ring system. // J. Amer. Chem. Soc.
1939. V. 61. N 10. P. 2912-2915.
. Ball R. H., Dorough G. D.,
Calvin M. A further study of the porphine-like products of the reaction of
benzaldehyde and pyrrole. // J. Amer. Chem. Soc. 1946. V. 68. N 11. P.
2278-2281.
. Priesthoff J. H., Banks C.
V. A new method of purifying α,β,γ,δ-tetraphenylporphine. //
J. Amer. Chem. Soc. 1954. V. 76. N 3. P. 937-938.
35. Williamson M. M.,
Prosser-McCartha C. M., Mukundan S., Jr., Hill C. L. Isolation and
characterization of the principal kinetic products in the Rothemund synthesis
of sterically hindered tetraarylporphyrins. Crystal and molecular structures of
[tetrakis(2,6-dichlorophenyl)-porphinato]zinc(II) and bis[(meso-2,6-dichlorophenyl)-5-(o,o'-dichlorobenzyl)dipyrromethene]-zinc(II)
complexes. // Inorg. Chem. 1988. V. 27. N 6. P. 1061-1068.
. Bortolini O., Ricci M.,
Mennier B. Frant P., Ascone I., Goulon J. Isolation, cyaracterization
andstructural investigation by exafs/xanes of high valent manganese porhyrin
complexes as active species in the NaOCl/Mn (porpyerin)x oxygenation system.//
Nouv. J. Chem. 1986. V. 10. N 1. P. 39-49.
. Petit A., Loupy A., Maillard
Ph., Momenteau M. Microwave irradiation in dry media: a new and easy method for
synthesis of tetrapyrrolic compounds. // Synth. Commun. 1992. V. 22. N 8. P.
1137-1142.
. Onaka M., Shinoda T., Izumi
Y., Nolen E. Porphyrin synthesis in clay nanospaces. // Chem. Lett. 1993. N 1.
P. 117-120.
. Laszlo P., Luchetti J.
Porphyrin synthesis using clays. Taking advantage of statistical product
distributions. // Chem. Lett. 1993. N 3. P. 449-452.
. Thomas D. W., Martell A. E.
Absorption spectra of para-substituted tetraphenylporphines. //J. Amer. Chem.
Soc. 1956. V. 78. N 7. P. 1338-1343.
. Adler A. D., Longo F. R.,
Shergalis W. Mechanistic investigations of porphyrin syntheses. I. Preliminary
studies on ms-tetraphenylporphin. // J. Amer. Chem. Soc. 1964. V. 86. N
15. P. 3145-3149.
42. Treibs A. Haberle H.
Darstellung von-porhiren und porhyrinen. // J. Liebigs Ann. Chem. 1968. B. 718.
P. 183-207.
43. Dolphin D. J. Porphyrinogens
and Porphodimethenes, intermediates in the Synthesis of
meso-Tetraphenylporphins from Pyrroles fnl Benzaldehyde. // J. Heterocycl.
Chem. 1970. N 2.P. 275-283.
. Adler A. D., Longo F. R.
Finarelli J. D., Goldmacher J., Assour J., Korsakoff L. A Simplified Synthesis
fjr meso- Tetraphenylporhin. // J. Org. Chem. 1967. V. 32. P. 476.
. Kim J. B., Leonard J. J.,
Longo F. R. A Mechanistic Studu of the Syntyesis and Spectral Propeties of
Meso-Tetraarylporhyrins. // J. Amer. Chem. Soc. 1972. V. 94. N 11. P. 3986-3992.
46. Семейкин А. С., Койфман О. И.,
Березин Б. Д. Улучшенный метод синтеза замещенных тетрафенилпорфинов.// Химия гетероцикл. соедин.
1986. № 6. С. 798-801.
47. Kagan N. E., Mauzerall D.,
Merrifield R. B. strati-Bisporphyrins. A novel cyclophane system. // J.
Amer. Chem. Soc. 1977. V. 99. N 16. P. 5484-5486.
. Banfi S., Montanari F.,
Penso M., Sosnovskikh V., Vigano P. NaOCl Olefin epjxidations catalyzed by Mn-porhyrins
under two-phase conditions. Influence of structural factors on the stability,
catalytic activity and selectivity of porphyrins. // Gazz. Chim. Ital. 1987. N
11. V. 117. P. 689-693.
. Chauhan S. M. S., Sahoo B.
B., Srinivas K. A. Microwave-assisted synthesis of
5,10,15,20-tetraarylporphyrins. // Synth. Commun. 2001. V. 31. N 1. P. 33-37
. Barnett G. H., Hudson M. F.,
Smith K. M. Concerning meso-Tttraphenylporphyrin purification. // J. Chem.
Soc., Perkin Trans. 1975. Pt. 1. N 14. P. 1401-1403.
. Barnett G. H., Hudson M. F.,
Smith K. M. Concerning meso-tetraphenylporphyrin purification. // J.
Chem. Soc., Perkin Trans. I. 1975. P. 1401-1403.
. Rousseau K., Dolphin D. A
Purification of meso-Tetraphenylporphirhyrin.// Tetrahedron Lett. 1974.
N 48. P. 4251-4254.
. Lindsey J. S., Scheriman I.
C., Hsu H. C., Kearney P. C., Marguerettaz A. M. Rothemund and Adler-Longo reactions
revisited: synthesis of tetraphenylporphyrins under equilibrium conditions. // J. Org. Chem. 1987.
V.52. N 5. P. 827-836.
. Lindsey J. S., Hsu H. C.,
Schreiman I. C. Synthesis of tetraphenylporphyrins under very mild conditions. //Tetrahedron Lett. 1986.
V. 27. N 41. P. 4969-4970.
. Rocha Gonsalves A. M. d'A.,
Pereira M. M., Serra A. C., Johnstone R. A. W., Nunes M. L. P. G.
5,10,15,20-Tetrakisaryl- and
2,3,7,8,12,13,17,18-octahalogeno-5,10,15,20-tetrakis-arylporphyrins and their
metal complexes as catalysts in hypochlorite epoxidations. // J. Chem. Soc.,
Perkin Trans. 1. 1994. P. 2053-2057.
. Lindsey J. S., MacCrum K.
A., Tyhonas J. S., Chuang Y.-Y. Investigation of a synthesis of meso-porphyrins
employing high concentration conditions and an electron transport chain for
aerobic oxidation. // J. Org. Chem. 1994. V. 59. N 3. P. 579-587.
. Lindsey J. S., Wagner R. W.
Investigation of the synthesis of ortho-substituted tetraphenylporphyrins. //
J. Org. Chem. 1989. V.54. N 4. P. 828-836.
. Wagner R. W., Lawrence D.
S., Lindsey J. S. An improved synthesis of tetramesitylporphyrin. //
Tetrahedron Lett. 1987. V. 28. N 27. P. 3069-3070
. Wagner R. W., Li F., Du H.,
Lindsey J. S. Investigation of cocatalysis conditions using an automated
microscale multireactor workstation: synthesis of meso-tetramesitylporphyrin.
// Org. Proc. Res. Devel. 1999. V. 3. N 1. P. 28-37.
. Safo M. K., Gupta G. P.,
Walker F. A., Scheidt W. R. Models of the cytochromes b. Control of axial
ligand orientation with a hindered porphyrin system. // J. Amer. Chem. Soc.
1991. V. 113. N 15. P. 5497-5510.
. Van der Made A. W.,
Hoppenbrauwer E. J. H., Nolte R. J. M., Drenth W. An improved of
tetraarilporphirins.// Rec. Trav. Chim. Pays-Bas. 1988. V. 107. N 1. P. 15-16.
. Kihm-Botulinski M., Meunier
B. Synthesis of tetramesitylporphyrin. // Inorg. Chem. 1988. V. 27. N 1. P.
209-210.
63. Семейкин А. С., Кузьмин Н. Г.,
Койфман О. И. Изучение условий конденсации пиррола с альдегидами в порфирины.
// Журн. прикл. химии. 1988. Т. 61. № 6. С. 1426-1429.
64. Guo C.-C., He X.-T., Zhon
G.-Y. A new synthesis method for tetraphenylporphyrin and its derivatives. //
Chin. J. Org. Chem. 1991. V. 11. N 4. P. 416-419.
. Banfi S., Montanari F.,
Quici S. New Manganese Tetrakis (halogenoaryl) porherins Featuring Sterically
Hindering Electronegative Substituens: Sinthesis of Highly Stable Catalysts in
Olefin Epoxidation.// J. Org. Chem. 1987. V. 53. N 12. P. 2863-2866.
. Paolesse R., Licoccia S.,
Bandoli G., Dolmella A., Boschi T. 5,10,15-Triphenylcorrole: a product from a
modified Rothemund reaction. // Inorg. Chem. 1994. N 33. P. 1171.
. Licoccia S., Tassoni E.,
Paolesse R., Boschi T. Tetracoordinated manganese(III) alkylcorrolates. Spectroscopic studies and
the crystal and molecular structure of
(7,13-Dimethyl-2,3,8,12,17,18-hexaethylcorrolato)manganese(III). // Inorg.
Chim. Acta. 1995. N 235. P. 15.
68. Adamian V. A., D'Souza F.,
Licoccia S., Di Vona M. L., Tassoni E., Paolesse R., Boschi T., Smith K. M.
Synthesis, characterization, and electrochemical behavior of
(5,10,15-tri-X-phenyl-2,3,7,8,12,13,17,18-octamethylcorrolato)cobalt(III)
phenylphosphine complexes, where X = p-OCH3, p-CH3, p-Cl, m-Cl,
o-Cl, m-F, o-F, or H. // Inorg. Chem. 1995. N 34. P. 532.
. Paolesse R., Tassoni E.,
Licoccia S., Paci M., Boschi T. Vilsmeier formylation of
5,10,15-triphenylcorrole: expected and unusual products. Inorg. Chim.
Acta 1996. N 241. P. 55.
. Rose E., Kossanyi A.;
Quelquejeu M., Soleilhavoup M., Duwavran F., Bernard N., Lecas A. Synthesis of
biomimetic heme precursors: the “double picket fence”
5,10,15,20-tetrakis(2,6-dinitro-4-tert-butylphenyl)porphyrin. // J. Am.
Chem, Soc. 1996. N 118. P. 1567.
. Loim N. M., Grishko E. V.,
Pyshnograeva N. I., Vorontsov E. V., Sokolov V I.
5,10,15,20-Tetracymantrenylporphyrin and 5,10,15-tricymantrenylcorrol. // Izv.
Akad. Nauk. Ser. Khim. 1994. N 5. P. 925
. Dolphin D. Sheldon R.
Reversible cobalt-nitrogen alkyl and acyl group migration in cobalt
porphyrins.// Metalloporphyrins in Catalytic Oxidations;, Ed.; Marcel Dekker
Inc.: New York, 1994, p. 193.
. Scheidt W. R., Lee Y.
Metalloporphyrin mixed-valence π-cation radicals:
solid-state structures. // Struct. Bonding. 1987. N 64. P. 1.
. Barkjgia K M., Chantranupong
L., Smith K. M., Fajer J. Structural and theoretical models of photosynthetic
chromophores. Implication for redox, light absorption properties and vectorial
electron flow. // J. Am. Chem Soc. 1988. N 110. P. 7566.
. Sparks L. D., Medforth C.
J., Park M.-S., Chamberlain J. R., Ondrias M. R., Senge M. O., Smith K M.,
Shelnutt J. A. Metal dependence of the nonplanar distortion of
octaalkyltetraphenylporphyrins. // J. Am Chem. Soc. 1993. N 115. P. 581.
. Ravikanth M., Chandrashekar
T. K. π-Cation radicals of copper(II)
derivatives
of short chain basket handle porphyrins. // Struct. Bonding. 1995. N 82.
P. 105.
. Nurco D J., Medforth C. J.,
Forsyth T. P., Olmstead M. M., Smith K. M. J. Conformational flexibility
in dodecasubstituted porphyrins. // Am. Chem. Soc. 1996. N 118. P. 10918.
78. Chmielewski P. J.,
Latos-Grazynski L., Rachlewicz K. 5,10,15,20-Tetraphenylsapphyrin - identification of a
pentapyrrolic expanded porphyrin in the Rothemund synthesis.// Chem. Eur J. 1995. N 1. P. 68.
. Chmielewski P. J.,
Latos-Grazynski L., Rachlewicz K., Glowiak T. Monoinvertiertes Tetra-p-tolylpor-phyrin:
ein neues Porphyrinisomer.// Angew. Chem., Int. Ed. Engl. 1994. N 33. P. 779.
. Lindsey J. S., Schreiman I.
C., Hsu H. S., Kearney P. C., Marguerettaz A. M. Rothemund and Adler-Longo Reactions
revisited: synthesis of tetraphenylporphyrins under equilibrium conditions.// J Org. Chem. 1987. N 52. P. 827.
. Conlon M., Johnson A. W.,
Overend W. R., Rajapaksa D. Synthesis of bilenes-b. Some side reactions.
// J. Chem. Soc., Perkin Trans. I 1973. P. 2281.
. Fanning J. C., Datta-Gupta
N. Metalloporphyrin synthesis. // Ann. Rep. Inorg. General Synth. 1974. P.
297-312.
83. Buchler J. W., Eikelmann G.,
Puppe L., Rohbock K., Scheehage H. H., Weck D. Metallkomplexe mit
Tetrapyrrol-Liganden. III. Darstellung von Metallokomplexen des
Octaathylporphins aus Metaii-acetonaten. // Liebigs Ann. Chem. 1971. B. 745. S. 135-151.
84. Gouterman M., Hanson L. K.,
Khalil G.-E., Buchler J. W., Rohbock K., Dolphin D. Porphyrins. XXXI. Chemical properties
and electronic spectra of d0 transition-metal complexes. // J. Amer. Chem. Soc.
1975. V. 97. N 11. P. 3142-3149.
. E. Vogel, P. Koch, X-L, Hon,
J. Lex, M. Lausmann, M. Kisters, M.A. Aukauloo, P. Richard, R. Guilard. Angew.
New porficene Ligands: Octaetil- and Etioporficen- tetra and peatacoordinated
zinc cjmhlexes jf OEPc. // Chem. Int. Ed. Engl. 1993. V.32. P.1600-1603.
86. M. W. Renner, A. Forman, W.
Wu, C. K. Chang, J. Fajer. Electrochemical, Theoretical, and ESR Characterizations of
Porphycenes. The 7r Anion Radical of Nickel(II) Porphycene // J. Am. Chem. Soc.
1989. V.1 1 1. P. 8618-8621.
. Sessler J. L., Weghorn S. J.
5,15,25-tris-nor-hexapyrrin: the first structurally characterized linear
hexapyrrin.// Expanded, Contacted and Isomeric Porphyrins. Tetrahedron Org.
Chem. Series, Vol. 15; Pergamon: Oxford, 1997, p. 11.
. Paolesse R., Licoccia S.,
Boschi T. Synthesis and functionalization of meso-aryl-substituted
corroles. // Inorg. Chim. Acta. 1990. P. 178.
. Johson A. W., Kay I. T.
Corroles. Part I. Synthesis. // J.Chem.Soc. 1965. P. 1620.
90. Will S., Lex J., Vogel E., Schmickler
H., Gisselbrecht J.-P., Haubtmann C., Bernard M., Gross M. Synthesis of sterically
hindered corroles with catalase-like activity. Angew. Chem., Int. Ed. Engl. 1997. N
36. P. 357.
. Grigg R., Johnson A. W.,
Shelton G. Struktur und Umlagerung alkylierter Nickel-corrole. // Liebigs Ann.
Chem. 1971. N 746. P. 32.
. Murakami Y., Matsuda Y.,
Sakata K., Yamada S., Tanaka Y., Aoyama Y. Transition metal complexes of
pyrrole pigments. 1V. Electronic and vibrational spectra of cobalt(II),
nickel(II), and copper(II) complexes of some substituted dipyrromethenes. //
Bull. Chem. Soc. Jpn. 1981. N 54. P. 163.
. Grigg R. Dolphin. D. N-Methylation
and electrophilic substitution reactions of octa-alkylporphins,
octaethylchlorin, and metalloporphins.- Academic Press; New York. 1978. Vol.
II, p. 327.
. Nonell S., Bou N.,.Borell
JI, Texido J., Villanueva A., Juarranz A., Canete M. Syntyesis of
2,7,12.17-tetraphenylporficene. First Aril-substituted porficene for the
potodinamic theropy of tumors. // Tetrahedron Lett. 1995. V.36. P. 3405-3408.
. Edith JU-Hwa Chu and Т. С. Chu. Decarboxylation and formulation of certain pyrrole derivatives.
//J. Org. Chem. 1954. P. 266-269.
. Korszara B., Gryko D.
Efficient synthesis of meso-substituted corroles in a H2O-MeOH mixture.//J Org.
Chem. 2006. V.71. P. 3707-3717.
. Corwin A. H., Bailey W. A.,
Viohl P. Structural investigations upon a substituted dipyrrylmethane. An
unusual melting point-symmetry relationship. // J. Amer. Chem. Soc. 1942. V.
64. N 6. P. 1267-1273.
98. Агрономов А.Е.
<http://padabum.com/search.php?author=%D0%90%D0%B3%D1%80%D0%BE%D0%BD%D0%BE%D0%BC%D0%BE%D0%B2%20%D0%90.%D0%95.>
Шабаров Ю.С.
<http://padabum.com/search.php?author=%D0%A8%D0%B0%D0%B1%D0%B0%D1%80%D0%BE%D0%B2%20%D0%AE.%D0%A1.>
Лабораторные работы в органическом практикуме. М: Химия, 1974. 188 с.
99. Порфирины: структура,
свойства, синтез /К.А. Аскаров, Б.Д. Березин, Р.П. Евстигнеева и др. М.:Наука,
1985. 333с.