Термодинамика и кинетика процессов с участием твердых фаз
Контрольно-практическая
работа
Термодинамика
и кинетика процессов с участием твердых фаз
Содержание
1. Методы расчета изменений функций
состояния в процессах взаимодействия твердых фаз
. Экспериментальные методы
исследования термодинамики реакций с участием твердых фаз
. Механизмы твердофазных реакций
.1 Диффузия в твердых фазах
.2 Методы, позволяющие определить
механизм процессов с участием твердых фаз
.3 Теория твердофазного
взаимодействия
.4 Твердофазные превращения без
изменения состава
.4.1 Типы и механизмы полиморфных
превращений
.4.2 Характеристика полиморфных
превращений
.4.3 Способы управления фазовыми
превращениями без изменения состава фаз
. Кинетика реакций с участием
твердых фаз
.1 Диффузионные модели
.2 Модели процессов, лимитируемых
реакциями на границе раздела фаз
.3 Модели зародышеобразования
. Активные прекурсоры
.1 Степень активности твердых фаз и
методы ее оценки
.2 Способы активирования твердых
фаз.
.3 Влияние состава прекурсора и
условий получения на активность синтезируемой фазы
.4 Повышение активности твердых фаз
методом легирования
.5 Механохимическое активирование
твердых фаз
.6 Практика активации твердофазных
процессов
Литература
1. Методы расчета изменений функций состояния в
процессах взаимодействия твердых фаз
Из курса классической термодинамики известно,
что термодинамические уравнения связывают между собой свойства любой
равновесной системы, каждое из которых может быть измерено независимыми
методами. В частности, при постоянном давлении справедливо соотношение ∆G
= ∆H - T∆S,
а принципиальная возможность самопроизвольного протекания (при этих условиях)
любого химического процесса, в отсутствии кинетических затруднений,
определяется неравенством ∆G
< 0. По определению химический потенциал любого компонента системы может
быть определен с помощью уравнения:
μi
= μio + RTℓn
αi
где μio
- стандартный химический потенциал i-го
компонента, αi - его
активность.
Химический потенциал - термодинамический
параметр, характеризующий состояние химического и фазового равновесия в
макроскопической системе. Наряду с давлением, температурой и другими
параметрами состояния системы, химический потенциал относится к интенсивным
величинам, т.к. не зависит от массы системы. Поскольку для
избарно-изотермического потенциала системы количество молей компонентов n1,…..,nj
(j - число
компонентов в системе) является единственным экстенсивным переменным, то
справедливо соотношение:
G = ∑kiμi
(i от 1 до j)
где ki
- стехиометрический коэффициент перед i-тым
компонентом в уравнении протекающего в системе химического процесса. Отсюда
следует, что μi
по определению представляет собой парциальную молярную энергию Гиббса, т.к. для
однокомпонентной системы из 4. получаем μ1 = G/n1.
Тогда возможность протекания любого химического процесса можно оценить,
рассчитав ∆G по
уравнению:
∆G
= (∆μio)
+ RTℓn
К
где К константа равновесия, выраженная через
активности прекурсоров и продуктов реакции в начальный момент времени, или :
∆G
= (∆μio)
= -RTℓn
Кр
где Кр. константа равновесия, выраженная через
активности реагентов в состоянии равновесия.
Термодинамические задачи, возникающие при
исследовании процессов с участием твердых, можно условно разделить на четыре
основных типа: а) возможно ли протекание данного процесса при заданных
параметрах состояния; б) какой, из нескольких возможных прекурсоров, более
предпочтителен при синтезе фазы заданного состава с точки зрения термодинамики;
в) какими должны быть материалы подсыпки и тигля, которые используются при
синтезе искомой фазы (спекании керамики, формировании пленки, текстуры или
выращивании монокристалла); г) какова вероятность образования твердых растворов
между целевым и побочным продуктом реакции или с избытком прекурсора и т.д..
Рассмотрим способы решения указанных задач на
нескольких примерах.
Задача 1. Какой из прекурсоров (с
термодинамической точки зрения) более предпочтителен при синтезе фазы состава BaTiO3,
если предполагается, что он будет взаимодействовать с TiO2
(α-форма
- рутил) с образованием искомого вещества. Группу возможных прекурсоров
составляют: карбонат, оксид, нитрат и оксалат бария.
Решение поставленной задачи можно получить, если
для прекурсоров и продуктов реакции известны значения ∆Gо298,
∆Но298 и теплоемкостей (Ср) в некотором интервале температур. Имея эти
данные, можно рассчитать ∆Gо
реакции при любой температуре, относящейся к интервалу, в котором известны
значения Ср. В свою очередь, зная значения ∆Gо
можно вычислить константу равновесия, используя выражение: ∆G
= (∆μio)
= -RTℓn
Кр. Энтальпию реакции при температуре Т можно определить по уравнению:
∆НТ = ∆Но +∫ ∆СрdТ
(интегрирование в пределах от 0 до Т), где ∆Но
- гипотетическая энтальпия реакции при 0оК.
Так как ∆Ср = ∆a
+ (∆b)T
+ (∆c)T2
+ ………, тогда получаем:
∆НТ = ∆Но + (∆a)Т
+
½ (∆b)T2
+ ⅓(∆c)T3
+ ……….
Подставляя это значение ∆НТ в уравнение
Гиббса-Гельмгольца: [∂(∆G/Т)/∂(1/Т)]p
= ∆Н и, производя интегрирование, получаем:
∆G
= ∆Но - ∆aTℓnТ
- 1/2 (∆b)T2
- 1/6(∆c)T3
+ ……… + JT
где J
- константа интегрирования.
По уравнению можно рассчитать изменение
изобарного потенциала для реакции, если известны: а) зависимости теплоемкости
всех прекурсоров и продуктов реакции от температуры в интервале от 298оК до
требуемой (т.е. известны значения констант a,
b и c
для всех участников процесса); б) ∆Н реакции для какой либо температуры в
указанном интервале (это необходимо для определения значения ∆Но); в)
значение ∆G при этой же
температуре (для определения значения J).
а) Рассмотрим возможность образования титаната
бария в результате процесса: BaСO3
+ TiO2 = BaTiO3
+ СО2, для этого воспользуемся табличными данными (таб.1).
Исходя из представленных данных, находим:
∆Gо298
= - 1572,3 - 394,4 + 1137,6 + 888,6 = 59,5 кДж/моль, т.е. равновесие
рассматриваемого процесса при стандартных условиях практически полностью
смещено в сторону исходных компонентов.
Таблица 1. Данные для расчета ∆G
и константы равновесия процесса
BaСO3
+ TiO2 = BaTiO3 + СО2(г.)
(Т
= 298 - 1100оК)
|
состав
фазы
|
значения
a, b и c
|
∆Gо298
кДж/моль
|
∆Но298
кДж/моль
|
|
a
|
b
∙103
|
c
∙
106
|
|
|
|
BaСO3
|
89,96
|
46,27
|
16,13
|
-
1137,6
|
-
1216,3
|
|
TiO2 (рут.)
|
75,19
|
1,17
|
18,20
|
-
888,6
|
-
943,9
|
|
BaTiO3
|
121,46
|
8,53
|
19,16
|
-
1572,3
|
-
1659,8
|
|
СО2(г.)
|
44,22
|
8,79
|
8,62
|
-
394,4
|
-
393,4
|
∆Но298 = - 1659,8 - 393,4 + 1216,3 + 943,9
= 107 кДж/моль
∆a =
121,46 + 44,22 - 89,96 - 75,19 = 0,53
∆b
= 0,00835 + 0,00879 - 0,04627 - 0,00117 = - 0,03
∆c
= (19,16 + 8,62 - 16,13 - 18,20)∙10-6 = - 6,55∙10-6
∆Но = ∆Но298 - 0,53Т - ½
(- 0,03)T2 - ⅓(- 6,55∙10-6)T3
=
= 107000 - 0,53∙298 + 0,5∙0,03∙88804
+ ⅓∙6,55∙10-6∙26,46∙106 =
= 108231,9 Дж/моль, рассчитаем J,
используя значение ∆Но и Т = 298оК
∆Gо298
= 108231,9 - 0,53∙298 ℓnТ
+ 0,5∙0,03∙88804 + 1/6(6,55∙10-6)T3
+
+…+ JT =
108231,9 - 0,53∙298 ∙2,3∙2,474 + 1332,06 + 28,89 + …… + J∙298,
тогда: J
= - 165 и общее уравнение зависимости ∆Gо
от температуры для исследуемого процесса имеет вид:
∆Gо
= 108231,9 - 0,53TℓnТ
+ 0,015T2 + 1,09∙10-6∙T3
+ ……… - 165T, тогда: ∆Gо1000К=
- 44335,1 Дж/моль или ( - 44,335 кДж/моль), т.е при этой температуре равновесие
процесса смещено вправо и можно оценить степень превращения, вычислив константу
равновесия при 1000оК:
∆Gо1000К
= - 8,31∙1000∙2,3ℓgК
или - 44335 = - 8,31∙1000∙2,3ℓgК
ℓgК
= 2,32 → К = 208,9, т.е. выход продукта реакции, если бы при данной
температуре достигалась энергия активации рассматриваемого процесса, был бы
выше 99 мол.%.
б) Определим, как изменится, согласно
термодинамике, выход BaTiO3,
если вместо карбоната бария использовать оксид этого элемента, т.е. рассмотрим
процесс: BaO + TiO2
= BaTiO3 ( таблица 4.2).
Исходя из представленных в таб.2 данных, получим:
∆Gо298
= - 1572,3 + 525,1 + 888,6 = - 158,6 кДж/моль, т.е. равновесие рассматриваемого
процесса при с.у. смещено в сторону продукта реакции.
Таблица Данные для расчета ∆G
и константы равновесия процесса BaO
+ TiO2 = BaTiO3
(Т = 298 - 1100оК).
|
состав
фазы
|
значения
a, b и c
|
∆Gо298
кДж/моль
|
∆Но298
кДж/моль
|
|
a
|
b
∙103
|
c
∙
106
|
|
|
|
BaO
|
49,33
|
7,87
|
3,68
|
-
525,1
|
-
550,2
|
|
TiO2 (рут.)
|
75,19
|
1,17
|
18,20
|
-
888,6
|
-
943,9
|
|
BaTiO3
|
121,46
|
8,53
|
19,16
|
-
1572,3
|
-
1659,8
|
∆Но298 = - 1659,8 + 550,2 + 943,9 = -165,7
кДж/моль
∆a
= 121,46 - 49,33 - 75,19 = - 3,06
∆b
= 0,00835 - 0,00787 - 0,00117 = - 0,00051
∆c
= (19,16 - 3,68 - 18,20)∙10-6 = - 2,72∙10-6
∆Но = ∆Но298 + 3,06Т - ½
(- 0,00051)T2 - ⅓(- 2,72∙10-6)T3
=
= - 165700 + 3,06∙298 + 0,5∙0,00051∙88804
+ ⅓∙2,72∙10-6∙26,46∙106 =
= - 164741,49 Дж/моль, рассчитаем J,
используя значение ∆Но и Т = 298оК
∆Gо298
= - 164741,49 + 3,06∙298 ℓnТ
+ 0,5∙0,00051∙88804 + 1/6(2,72∙10-6)T3
+
+…+ JT =
- 164741,49 + 3,06∙298 ∙2,3∙2,474 + 22,64 + 11,99 + …… + J∙298,
тогда: J
= 3,07 и общее уравнение зависимости ∆Gо
от температуры для исследуемого процесса имеет вид:
∆Gо
= - 164741,49 + 3,06 TℓnТ
+ 0,225∙10-3T2 + 1,09∙10-6∙T3
+ ………+ + 3,07T, тогда: ∆Gо1000К=
- 139242,49(Дж/моль или ( - 139,242 кДж/моль), т.е при этой температуре
равновесие процесса остается смещенным вправо, хотя степень превращения при
этой температуре меньше, чем при с.у.(см. значение ∆Gо298
рассматриваемого процесса):
∆Gо1000К
= - 8,31∙1000∙2,3ℓgК
или - 139242,49 = - 8,31∙1000∙2,3ℓgК
ℓgК
= 7,28 → К = 19285202, т.е. равновесие практически полностью смещено
вправо и использование BaO
(с термодинамической точки зрения) при синтезе BaTiO3
более предпочтительно, по сравнению с BaСO3.
Очевидно, что при использовании Ba(NO3)2
в качестве прекурсора при высокотемпературном синтезе BaTiO3
(Т ≥ 1000оК) выход продукта реакции сравним с процессом BaO
+ TiO2 = BaTiO3.
Это объясняется тем что, несмотря на более низкие значения ∆Gо298
и ∆Но298 образования нитрата бария по сравнению с его оксидом, ∆GоТ
этого процесса быстро уменьшается с ростом температуры за счет увеличения
энтропийного фактора:
Ba(NO3)2 + TiO2 = BaTiO3 + NО2(г.)
+
NO(г.) + O2(г.)
, в чем читатель может убедиться, используя данные таблицы 3.
Таблица 3. Данные для расчета ∆G
и константы равновесия процесса Ba(NO3)2
+ TiO2 = BaTiO3
+ NО2(г.) + NO(г.)
+ O2(г.) (Т = 298 -
1100оК).
|
состав
фазы
|
значения
a, b и c
|
∆Gо298
кДж/моль
|
∆Но298
кДж/моль
|
|
a
|
b
∙103
|
c
∙
106
|
|
|
|
Ba(NO3)2
|
125,81
|
149,47
|
- 16,79
|
-
797,25
|
-
992,73
|
|
TiO2 (рут.)
|
75,19
|
1,17
|
18,20
|
-
888,6
|
-
943,9
|
|
BaTiO3
|
121,46
|
8,53
|
19,16
|
-
1572,3
|
-
1659,8
|
|
NО2(г.)
|
42,16
|
9,55
|
-
6,99
|
51,6
|
33,5
|
|
NO(г.)
|
29,43
|
3,85
|
- 0,58
|
80,64
|
90,31
|
|
O2(г.)
|
6,09
|
3,25
|
- 1,02
|
0
|
0
|
Как видно из данных таблицы 4, из потенциальных
прекурсоров наиболее низкими значениями ∆Gо298
и ∆Но298 обладает BaС2O4.
Используя данные этой таблицы и результаты расчета задачи (а), сравните ∆GоТ
процессов:
BaСO3
+ TiO2 = BaTiO3
+ СО2 и BaС2O4
+ TiO2 = BaTiO3
+ СО2 + СО при температурах ≥ 1000оК. На основании полученных Вами
данных, а также результатов приведенных выше расчетов убедитесь в
справедливости эмпирического правила, регламентирующего выбор прекурсора с
точки зрения термодинамического подхода: из нескольких возможных прекурсоров
твердофазного процесса с термодинамической точки зрения наиболее
предпочтительны фазы, имеющие максимальные значения ∆Gо298
и ∆Но298 образования и принимающие участие в процессах, сопровождающихся
значительным ростом энтропии системы.
Как будет показано ниже, термодинамическое
обоснование выбора прекурсора является необходимым, но, зачастую, недостаточным
при проведении реальных процессов синтеза твердых фаз.
Таблица 4. Данные для расчета ∆G
и константы равновесия процесса BaС2O4
+ TiO2 = BaTiO3
+ СО2(г) + СО(г.) (Т = 298 - 1100оК).
|
состав
фазы
|
значения
a, b и c
|
∆Gо298
кДж/моль
|
∆Но298
кДж/моль
|
|
a
|
b
∙103
|
c
∙
106
|
|
|
|
BaС2O4
|
85,32
|
48,66
|
12,17
|
-
1189,8
|
-
1259,7
|
|
TiO2 (рут.)
|
75,19
|
1,17
|
18,20
|
-
888,6
|
-
943,9
|
|
BaTiO3
|
121,46
|
8,53
|
19,16
|
-
1572,3
|
-
1659,8
|
|
СО2(г.)
|
44,22
|
8,79
|
8,62
|
-
394,4
|
-
393,4
|
|
СО(г.)
|
6,34
|
1,84
|
-
2,8
|
-
137,2
|
-
110,6
|
Задача Можно ли проводить спекание керамики
ниобата кальция в кварцевой трубке?
В рассматриваемой системе между фазой CaNb2O6
и фазой, образующей контейнер (SiO2)
возможно протекание процесса:
CaNb2O6
+ SiO2 = СаSiO3
+ Nb2O5
Для ответа на вопрос задачи воспользуемся
значениями μо фаз ( например
при 1500оК), являющихся прекурсорами и продуктами данного процесса:
∆Gо
= μоСаSiO3
+ μоNb2O5
- μоSiO2
- μоCaNb2O6
= (μоСаSiO3
- μоSiO2
- μоСаО)
- (μоCaNb2O6
- μоNb2O5
- μоСаО)
= ∆μоСаSiO3
- ∆μоCaNb2O6,
где ∆μоСаSiO3
и ∆μоCaNb2O6
изменение энергии Гиббса при образовании СаSiO3
и CaNb2O6
из оксидов. Как следует из приведенного уравнения, для получения последнего
выражения использован прием, который заключается во введении в расчет двух
новых слагаемых: μоСаО и (-μоСаО),
что позволило свести определение ∆Gо
к разности двух величин. Эти величины можно определить экспериментально, а их
зависимость от температуры представить в виде уравнения ∆μо
= ∆Gо = А +ВТ В
частности для процессов:
СаО + SiO2
= СаSiO3 (А = -
81,3 кДж/моль, В = 0,0005 кДж/моль∙Ко)
СаО + Nb2O5
= CaNb2O6
(А = -175,7 кДж/моль, В = 0,02259 кДж/моль∙Ко).
Тогда при 1500оК получаем: ∆μоСаSiO3
= - 81,3 + 0,0005∙1500 =- 81,3 + 0,75 = = - 80,55 кДж/моль, а ∆μоCaNb2O6
= -175,7 + 0,02259∙1500 = -175,7 + 33,88 = = - 141,82 кДж/моль и ∆Gо
= - 80,55 + 141,82 = 61,27 кДж/моль, т.е ∆Gо>>0
и равновесие смещено в сторону исходных фаз. Однако ∆Gо
- конечная величина и, следовательно, при длительном обжиге прессзаготовки
последняя содержит в своем составе некоторое количество продуктов реакции,
активность которых можно оценить, используя связь между Кравновесия и ∆Gо
реакции: ∆Gо = - RT
ℓn
α СаSiO3∙α
Nb2O5
/α CaNb2O6
∙α
SiO2 . Так как для
исходных компонентов α
CaNb2O6
≈α
SiO2 ≈ 1, то ∆Gо
≈ - RT
ℓn
α СаSiO3∙α
Nb2O5
Подставляя в последнее уравнение значение ∆Gо
= 61,27 кДж/моль, находим, что после достижения в системе равновесия при
1500оК, сформировавшаяся из порошка CaNb2O6
керамика, содержит в своем составе примесные фазы, произведение активности
которых α СаSiO3∙α
Nb2O5
= 0,0073. Полученный результат, в частности, означает, что если в системе
содержится Nb2O5
в виде самостоятельной фазы (например, из-за не полностью завершившейся реакции
синтеза), то после достижения системой состояния равновесия, активность СаSiO3,
входящего в состав твердого раствора на основе CaNb2O6,
будет составлять 7,3∙10-3. Пригодна ли такая керамика для дальнейшего
использования будет зависеть от влияния примеси на значения параметров изделия,
для изготовления которого эта керамика предназначена.
Задача 3. При каких условиях взаимодействие Ti2O3
с титаном приведет к образованию однофазного «TiO»:
Ti2O3
+ Ti = 3«TiO»,
если известно, что изменение ∆Gо
в Дж/моль для реакции 2«TiO»
+ ½О2 = Ti2O3
выражается уравнением : ∆Gо1
= (477920 + 79,96Т), а для процесса Ti
+ ½О2 = «TiO»
такая зависимость имеет вид: ∆Gо2
= (512050 + 89,2Т)?
Вычтем из второго уравнения первое: Ti
+ ½О2 + Ti2O3
= «TiO» + 2«TiO»
+ +
½О2,
что соответствует рассматриваемому процессу: Ti2O3
+ Ti = 3«TiO».
Тогда, ∆Gореакции=
∆Gо2 - ∆Gо1
= 34130 + 9,24Т , т.е ∆Gо>>0
и значения этой величины будут возрастать по мере увеличения температуры
системы. Это означает, что фаза «TiO»
метастабильна при любых температурах, а повышение температуры системы будет
способствовать ее диспропорционированию на Ti
и Ti2O3.
Задача 4. Определите равновесный состав
продуктов реакции между CaWO4
и SrO при Т = 1500оК,
если известно, что в этих условиях образуются непрерывные твердые растворы
состава: Ca1-xSrxO
и CaхSr1-хWO4.
∆Gо
обменного процесса: CaWO4
+ SrO = SrWO4
+ СаО может быть определено из уравнения ∆Gо
= ∆μоSrWO4
- ∆μоCaWO4,
где ∆μоSrWO4
и ∆μоCaWO4
- изменение энергий Гиббса при образовании соответствующих фаз из оксидов.
Согласно табличным данным и, используя уравнение:
∆μо = ∆Gо
= А +ВТ, находим:
для реакции: CaО
+ WO3 = CaWO4
∆μо
= (- 168,87 + 0,01674∙Т) кДж/моль
для реакции: SrО
+ WO3 = SrWO4
∆μо
= (- 217,10 + 0,01300∙Т) кДж/моль
тогда ∆Gо
= (- 48,23 - 3,74∙10-3∙Т) кДж/моль, а при Т = 1500оК значение ∆Gо
= - 53,84 кДж/моль, т.е. равновесие рассматриваемого процесса смещено вправо. С
учетом возможности образования в системе неограниченных твердых растворов,
активность фаз, находящихся в равновесии, будет пропорциональна молярной
концентрации этих растворов. Равновесный же состав твердых растворов при
заданной температуре можно оценить, используя уравнение ∆G
= (∆μio)
= -RTℓn
Кр. (где Кр. константа равновесия, выраженная через активности реагентов в
состоянии равновесия) и условия квазибинарности. Тогда, заменив активности
мольными долями (αi
= Ni), получим: NСаО/NSrО
= NSrWO4/NCaWO4
и, учитывая условия квазибинарности, получаем:
∆Gо
= - RT
ℓn
(NSrWO4 / NCaWO4)2,
а так как NCaWO4 + NSrWO4
= 1, то (NSrWO4 / (1 - NSrWO4))2
= exp(-∆Gо
/ RT)
Теперь, подставляя в последнее уравнение
значение ∆Gо = - 53,84
кДж/моль, находим: 2,3 ℓg
(NSrWO4 / (1 - NSrWO4))2
= 53840/8,31∙1500 = 4,32 и ℓg
(NSrWO4 / (1 - NSrWO4))2
=1,88, тогда NSrWO4 / (1 - NSrWO4)
= 8,7 и NSrWO4 = =0,897.
Следовательно, в системе при Т = 1500оК в равновесии находятся два типа твердых
растворов CaxSr1-xO
и Ca1-х SrхWO4
с х = 0,897, что предопределяется, преимущественно, большей термодинамической
стабильностью CaО по
сравнению со SrО.
Когда данные, необходимые для выполнения
описанных выше расчетов, отсутствуют в литературе их можно экспериментально
получить, использую один из представленных ниже методов.
2. Экспериментальные методы исследования
термодинамики реакций с участием твердых фаз
Ранние методы определения значений ∆G
реакций основывались либо на термохимических данных, либо на измерении констант
равновесия. Оба этих метода имеют неустранимые недостатки, связанные в первом
случае с трудностями определения энтропии процессов, а во втором - с
образованием рентгеноаморфных промежуточных фаз и установлением в системах
кажущихся или локальных равновесий. В значительной степени устранить эти
недостатки удалось после разработки метода, основанного на измерении э.д.с.
гальванического элемента, в котором роль электролита выполняет твердая
ионпроводящая фаза.
Метод электродвижущих сил. Указанный способ
определения значений ∆G
реакций, с участием твердых фаз, заключается в измерении э.д.с. гальванического
элемента, потенциалобразующая реакция которого совпадает (или тесно связана) с
исследуемым твердофазным процессом. Например, реакцию образование PbTiO3
из оксидов: PbO + TiO2
= PbTiO3 можно провести в
гальванической ячейке состоящей из оксидов свинца и титана, а также PbTiO3.
При этом в качестве электродов используют свинец, а в качестве электролита -
твердую фазу, проводящую ионы О2- (например, Ca0,15Zr0,85O1,85
или YxZr1-xO2-0,5x
с х от 0,2 до 0,3):
Pb, PbO│
O2-│ PbTiO3,
TiO2, Pb
(где │O2-│- твердый
электролит)
→ O2-
Из схемы следует, что у левого электрода
рассматриваемого элемента химический потенциал PbO,
выше чем у правого, а химический потенциал свинца в обоих полуэлементах -
одинаков. Так как химический потенциал любого вещества равен сумме химических
потенциалов составляющих его компонентов, то химический потенциал кислорода (μО)
для левого электрода μО = μPbO
- μPb, больше,
чем для правого. Следовательно для выравнивания μО
в обеих частях элемента необходим перенос ионов O2-
слева направо (показано на схеме стрелкой). Результатом такого переноса
является протекание следующих процессов:
(левый электрод) PbO
+ 2ē = Pb + O2-
(правый электрод) Pb
+ O2- + TiO2
= PbTiO3 + 2ē
∑ PbO
+ TiO2 = PbTiO3
Описанная совокупность процессов в
гальваническом элементе с замкнутой внешней цепью будет протекать
самопроизвольно, а в элементе с разомкнутой внешней цепью им будет
препятствовать разность электродных потенциалов, возникающих на границах
электрод - электролит. Если приложить к этому элементу внешнее напряжение,
равное по значению и противоположное по знаку разности потенциалов электродов,
то в системе наступит электрохимическое равновесие, а перенос ионов O2-
между электродами станет обратимым. В этих условиях для каждого компонента
системы изменение химического потенциала равно нулю: ∆μi=∆μio
+ ziEF = 0 (где F
- число Фарадея, Е - э.д.с. элемента, ziEF
- электрическая работа связанная с переносом заряда ziF,
zi -заряд иона в
единицах заряда электрона.
Для рассматриваемого элемента в состоянии
электрохимического равновесия ∆μОo
+ 2EF = 0 или ∆μОo
= - 2EF. Так как
перенос одного моля ионов кислорода от электрода к электроду сопровождается
образованием одного моля продукта реакции из оксидов, получаем:
∆Gо
реакций образования PbTiO3
из оксидов = ∆μОo
= - 2EF, тогда:
∆Ho
- T∆Sо
= - 2EF или Е = - ∆Ho/2F
+ T∆Sо/2F
Так как для многих твердофазных реакций значения
∆Ho и ∆Sо
незначительно зависят от температуры, экспериментально найденная температурная
зависимость э.д.с. элемента может быть описана уравнением:
Е = а + bT,
тогда ∆Ho = - 2аF,
а ∆Sо =2bF
Для рассматриваемого гальванического элемента
экспериментально при 900 - 1100оК было установлено, что : Е = 37,5 - 0,027Т
(мВ), тогда:
∆Gо1000К
= ∆Ho - 1000∆Sо
= - 2,03 кДж/моль
∆Ho1000К
= - 2аF = - 7,24 кДж/моль
∆Sо1000К
=2bF = - 5,2 Дж/моль∙оК
Для определения изменения функций состояния в
процессе взаимодействия оксидных кристаллических фаз могут быть использованы и
твердые электролиты не кислородного типа. Например, при расчете значений ∆Gо
реакции: CaO + SiO2
= CaSiO3, может быть
использована электрохимическая цепь:
O2, CaO
│CaF2
│ CaSiO3, SiO2,
O2 (где │CaF2│-
твердый электролит)
← F-
При μО2
= const (над обоими
электродами), с учетом (μСаО)лев.>
(μСаО)прав.
получаем : (μСа)лев.> (μСа)прав.
и, следовательно, (μF
)лев.< (μF
)прав. . Тогда, связанный с градиентом химического потенциала, перенос ионов
фтора справа налево приводит к протеканию следующих процессов:
(левый электрод) CaO
+ 2F- = CaF2
+ ½O2 + 2ē
(правый электрод) CaF2
+ ½O2 + 2ē
+ SiO2 = CaSiO3
+ 2F-
∑ CaO
+ SiO2 = CaSiO3
Следовательно, ∆Gо
рассматриваемой реакции равно (- 2ЕF),
где F - число Фарадея, Е
- э.д.с. элемента.
В указанном методе успешно применяются и
катионпроводящие твердые электролиты. Рассмотрим определение ∆Gо
следующей реакции.
½К2О + ½Nb2O5
= KNbO3
Так как К2О - фаза, которая не может
существовать в кислородной атмосфере, градиент химического потенциала по К+
зададим за счет использования смеси:
KF + NiF2
+ Ni
Ni, KF,
NiF2 │Кβ-Аl2O3│
KNbO3, Nb2O5,
O2
→ K+
(левый электрод) 2KF
+ Ni = NiF2
+ К+ + 2ē
(правый электрод) 2К++ ½O2
+ 2ē + Nb2O5
= 2KNbO3
∑ 2KF + Ni
+½O2 + Nb2O5 = NiF2 + 2KNbO3
и ∆Gо
суммарного процесса равно (- 2ЕF).
На втором этапе воспользуемся табличными значениями ∆Gо
реакций:
Ni + F2
= NiF2 (∆Gо1)
К + ½F2
= KF (∆Gо2)
К +½О2
= К2О (∆Gо3)
тогда: ∆Gо
реакции ½К2О
+ ½Nb2O5
= KNbO3 можно определить
из соотношения: ∆Gо
= - 2ЕF - ½∆Gо1
+ ∆Gо2 - ½∆Gо3.
Таким образом, метод э.д.с. позволяет изучать в
обратимых термодинамических цепях термодинамику многих твердофазных реакций.
Можно отметить, что при исследовании термодинамики интерметаллидов и некоторых
фаз внедрения в качестве электролитов успешно используются расплавы и стекла,
что расширяет область применения описанного выше метода. Метод гетерогенных
равновесий (МГР).
Метод гетерогенных равновесий может быть
использован при исследовании термодинамики любых процессов с участием твердых
фаз, в том числе и фаз переменного состава. Он использует экспериментально
полученные данные по равновесному давлению газовой фазы, находящейся над
конденсированными фазами. Обязательным условием при этом является наличие в
такой системе хотя бы одной конденсированной фазы, которая относится к
продуктам твердофазной реакции. При этом исследуемая газовая фаза может
представлять собой индивидуальный компонент или входить как составная часть в
газообразную смесь нескольких веществ. Рассмотрим несколько примеров,
позволяющих оценить возможности данного метода.
. Для определения ∆Gо
реакции: BaO + PbO2
= BaPbO3 экспериментально
определены равновесные давления (Р1 и Р2 ) кислорода в следующих процессах
термического разложения оксидных фаз:
BaPbO3 = BaO
+ ⅓ Pb3O4
+ ⅓О2 (∆Gо1)
PbO2 = ⅓ Pb3O4
+ ⅓О2 (∆Gо2)
вычитая первое уравнение из второго получаем: BaO
+ PbO2 = BaPbO3
тогда ∆Gо
= ∆Gо2 - ∆Gо1.
Пренебрегая растворимостью Pb3O4
в BaPbO3 и PbO2
и учитывая, что активность индивидуальных твердых фаз равна единице, получаем:
∆Gо1
= - RT
ℓn
(Po1)⅓ и ∆Gо2
= - RT
ℓn
(Po2)⅓
Опишем методику определения ∆Gо
для реакции: 2PbO + Nb2O5
= = Pb2Nb2O7
. Для этого рассмотрим гетерогенные равновесия:
PbO + СО = Pb
+ СО2 (∆Gо1)
Pb2Nb2O7
+ 2СО = 2Pb + 2СО2 + Nb2O5
(∆Gо2)
∆Gо1
= - RT
ℓn
(РСО2/РСО)1
∆Gо2
= - RT
ℓn
(РСО2/РСО)22
∆Gо
= 2∆Gо1 - ∆Gо2
= RT(ℓn
(РСО2/РСО)22 - ℓn
(РСО2/РСО)1).
Подставляя в последнее уравнение
экспериментально найденные парциальные давления СО и СО2 в газовых фазах,
равновесных (PbO + Pb)
, и (Pb2Nb2O7
+ Pb + + Nb2O5)
найдем ∆GоТ Pb2Nb2O7,
при ее образовании из оксидов.
Термохимические методы. В связи с низкими
значениями ∆НоТ и длительностью большинства твердофазных реакций прямое определение
их тепловых эффектов в калориметрах, независимо от конструкции последних,
проводится очень редко. Обычный путь определения ∆НоТ процессов в этом
случае заключается в исследовании термохимических циклов, включающих быстро
протекающие реакции, сопровождающиеся значительными тепловыми эффектами.
. Метод растворения.
Рассмотрим гипотетический процесс: А + АВ = А2В.
Для определения ∆НоТ этого процесса подберем растворитель, в котором с
высокой скоростью растворяются все реагенты данной системы. Тогда возможный
термохимический цикл будет иметь вид:
Тогда, в соответствии с законом Гесса, получаем:
∆Но = ∆Н1 + ∆Н2 + ∆Н4 - ∆Н3.
В качестве растворителей могут использоваться
растворы кислот, щелочей, органические растворители в случае твердых
молекулярных веществ, а также ионные расплавы для реакций с участием
тугоплавких соединений ионного или ионно-ковалентного типа.
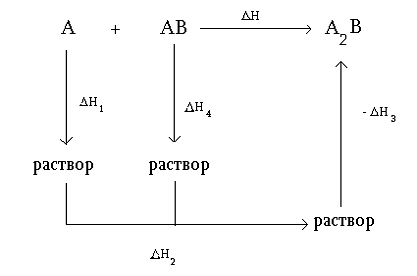
Рис 1. Cхема
определения ∆НоТ твердофазного процесса методом растворения ∆Н1, ∆Н4
и ∆Н3 энтальпии растворения веществ А, АВ и А2В, соответственно ∆Н2
-энтальпия смешения.
Метод сжигания. Этот метод основан на
непосредственном измерении теплот сгорания простых веществ с образованием
прекурсоров и продуктов реакций. Процесс окисления проводят в изолированной
системе («калориметрической бомбе»). Например, для определения ∆Но
процесса: 2ВаО + TiO2
= Вa2TiO4
необходимо экспериментально определить ∆Но реакций:
Ва + О2 = 2ВаО (∆Но1)
Ti + О2 = TiO2
(∆Но2)
Ва + Ti
+ 2О2 = Вa2TiO4
(∆Но3)
Тогда: ∆Но = ∆Но3 - ∆Но2 - ∆Но1.
Необходимо отметить, что рассматриваемый метод
дает определенную погрешность в связи с дефектностью кристаллических решеток
фаз, формирующихся в условиях горения простых веществ, а также возможностью
образования в этих условиях аморфных продуктов реакций.
. Метод окисления или восстановления продукта
твердофазного взаимодействия.
Рассмотрим использование этого метода на двух
примерах:
а) Определить ∆Но реакции: Cu2O
+ Al2O3
= 2CuAlO
Воспользуемся вариантом, основанном на
восстановлении реагентов:
Cu2O
+ Н2 = 2Cu + Н2О(Г.) (∆Но1)
CuAlO2 + Н2 = 2Cu
+ Н2О(Г.) + Al2O3
(∆Но2)
∆Но = ∆Но1 - ∆Но
б) Определить ∆Но реакции: 2SnO
+ Nb2O5
= Sn2Nb2O7.
Воспользуемся вариантом, основанном на окислении
реагентов:
SnO + О2 = 2SnO2
(∆Но1)
Sn2Nb2O7
+ О2 = 2SnO2 + Nb2O5
(∆Но2)
∆Но = ∆Но1 - ∆Но2
Заканчивая обзор калориметрических методов
целесообразно напомнить, что достоверность полученных значений ∆Но
реакций будет зависеть не только от точности измерения выделяющейся или
поглощающейся теплоты в процессе химического взаимодействия, но и от полноты
протекания процессов и степени совершенства формирующихся в системах фаз.
Помимо учета этих факторов, способных изменить значения измеряемых величин,
последние целесообразно сравнивать с данными для тех же процессов, полученными
методами э.д.с. или гетерогенных равновесий.
Оценка изменения энтропии при твердофазных
процессах. Оценку ∆SoT
процессов, протекающих между твердыми фазами, проводят на основе
фундаментального соотношения ∆GоТ
= ∆НоТ - Т∆SoT,
используя их экспериментально найденные, описанными выше методами, значения ∆GоТ
и ∆НоТ.
Другой метод оценки рассматриваемой величины
связан с использованием уравнения:
∆SoT
= RT( ∂ ℓnKравн./∂T
+ ℓnKравн. /T)
Этот метод достаточно прост, но не всегда
достаточно точен. В частности, значительные ошибки в определении значений ∆SoT
в этом случае могут быть связаны с нестандартностью состояния сосуществующих в
системе твердых фаз и с взаимной растворимостью прекурсоров и продуктов реакции
(в любом сочетании этих компонентов).
Термохимический метод определения ∆SoT,
основанный на измерении теплоемкостей прекурсоров и продуктов реакций в широком
интервале температур (вплоть до предельно низких), может быть использован
только в случае объектов, для которых справедлива тепловая теорема Нернста,
т.е. для кристаллов, приближающихся по свойствам к идеальным. Тогда:
∆ST
= ∫ Cp
dℓnT
(пределы интегрирования от 0 до Т).
3. Механизмы твердофазных реакций
Твердофазное взаимодействие, в первом
приближении, складывается из двух основных процессов: (а) формирование новой
фазы на границе контакта прекурсоров и (б) - перенос вещества к реакционной
зоне. Массоперенос в объеме твердых фаз осуществляется за счет диффузии
различных видов частиц, а их подвижность предопределяется дефектностью
структуры кристалла [11-13], следовательно, тип и концентрация дефектов в
системе должны оказывать существенное влияние на механизм и кинетику
твердофазных превращений.
.1 Диффузия в твердых фазах
Диффузия частиц в объеме твердой фазы может
протекать по различным механизмам: вакансионному, междоузельному и эстафетному.
Первый механизм заключается в перемещении атомов или ионов из регулярных
позиций в соседние вакантные узлы (рис.2в), второй - в переходе частиц из одних
междоузельных позиций в другие (рис. 2а), третий - в перемещении частиц из
одних регулярных позиций в другие через междоузлия (рис. 2б).
Кроме объемной диффузии, в кристаллах
наблюдается поверхностная диффузия, диффузия по границам зерен и блоков, а
также порам, дислокациям, разломам и т.д.. Число частиц, находящихся на
поверхности макрокристалла значительно меньше числа частиц в его объеме,
поэтому, в данном случае, доля вещества переносимого за счет поверхностной
диффузии невелика.
С уменьшением размеров кристаллов роль
поверхностной диффузии резко возрастает, чему способствует рост доли
поверхностных частиц в системе и более высокие коэффициенты поверхностной
диффузии по сравнению с объемной.
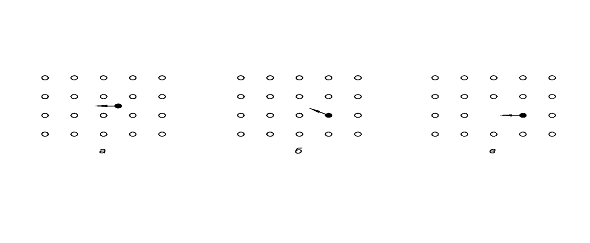
Механизмы диффузии:
а) по междоузлиям; б) эстафетный; в)
вакансионный.
Например, в металлах коэффициент диффузии по
границам зерен при фиксированной температуре выше коэффициента объемной
диффузии на три порядка, а коэффициент поверхностной диффузии превосходит
коэффициент объемной диффузии на шесть порядков. Этим, в частности, объясняется
резкое увеличение скорости твердофазных реакций при использовании наноразмерных
прекурсоров. Высокие значения коэффициентов поверхностной и пограничной
диффузии предопределяются строением поверхности кристаллов и границ зерен,
которые, даже в равновесном состоянии, имеют более высокую концентрацию
дефектов, чем в объеме кристаллов, а с учетом дислокаций и границ блоков эта
разница достигает нескольких порядков.
Процесс диффузии в определенном направлении
(например, вдоль оси х) описывается законом Фика:
f = - D
dN/dx
где f
- поток диффузии (число частиц, проходящих через единицу площади за единицу
времени; dN/dx
- градиент концентраций в направлении оси х, D
- коэффициент диффузии (м2/сек).
Повышение температуры способствует росту
значений D:
D = Dо
e‾ ∆H/RT
где ∆H
- энергия активации процесса и Dо
- константа.
D = ν l2/c
где ν - средняя
частота колебаний атомов; l
- расстояние, которое проходит диффундирующая частица в одном элементарном
акте; с - геометрический фактор, характеризующий строение рассматриваемой
решетки. Для диффузии по междоузлиям ν может
быть вычислена по уравнению:
ν = νоg
e‾ ∆G/RT
где g
- число направлений возможного перемещения частицы; ∆G
- свободная энергия перехода 1 моля частиц через потенциальный барьер,
разделяющий соседние междоузлия; νо
можно определить исходя из массы частицы m
и ее силовой константы смещения при междоузельном переходе:
νо =
1/2π √К/m
Экспериментально определить коэффициент диффузии
для твердых тел достаточно трудно. Это связано с тем, что их значения, особенно
при низких температурах, малы, т.е. расстояния, на которые перемещаются
частицы, например, в течение часа составляют от нескольких десятых до
нескольких единиц нанометров. Удовлетворительные результаты дает метод меченых
атомов, использующийся как для исследования диффузии примесных частиц, так и
для самодиффузии, а также методы, основанные на анализе газовой фазы и на
изменении электрофизических свойств образцов. Проанализируем экспериментальные
и расчетные данные для нескольких систем.
Экспериментально установлено, что диффузия
атомов водорода, гелия и лития в кристаллах германия и кремния происходит по
междоузлиям. Коэффициенты диффузии атомов водорода и гелия определены по
изменению состава газовой фазы, разделенной монокристаллической пластиной
вырезанной из кристалла кремния или германия.
Диффузию лития можно оценить по изменению
электропроводности образца (литий по отношению к основному веществу выступает
как донор электронов, т.е. его появление в системе приводит к росту ее
электропроводности). Вычисленное, с использованием уравнений (6 и 7), значение D
для рассматриваемых систем хорошо согласуется с экспериментально найденными
значениями данного коэффициента. При этом отмечено, что скорости процессов
значительны даже при низких температурах, что хорошо согласуется с качественной
теоретической оценкой диффузии, которая происходит по междоузлиям. В частности,
D для лития в
германии при 500оС составляет величину порядка 10‾6 см2/сек.
Диффузия частиц в твердых растворах замещения,
как и самодиффузия в этих системах, происходит по вакансионному механизму.
Указанным способом диффундируют в кристаллах германия и кремния атомы элементов
соседних главных подгрупп: D
для таких частиц составляет в кремнии при 1000оС 10‾13 - 10‾14см2/сек
и 10‾11 - 10‾13 см2/сек в германии при 800оС, что на несколько
порядков меньше, чем при междоузельной диффузии.
Самодиффузия в галогенидах щелочных металлов
также протекает по вакансионному механизму, что позволяет изменять ее скорость
путем введения определенных примесей, например, двухзарядных катионов
(двухзарядный катион замещает два однозарядных, что предопределяет возникновение
катионных вакансий) или анионов (вакансии в анионной подрешетке). Увеличение
числа вакансий в единице объема уменьшает расстояние между ними, что
равносильно снижению энергии активации процесса диффузии (фактор,
предопределяющий рост скорости рассматриваемого процесса). Указанный прием
позволяет повысить значения D
на несколько порядков, но только в том случае, когда имеет место вакансионный
механизм объемной диффузии.
.2 Методы, позволяющие определить механизм
процессов с участием твердых фаз.
.Метод Трубанда - Вагнера. Направление
массопереноса определяют по изменению масс прекурсоров и продуктов реакции.
Например, при взаимодействии иодида серебра с иодидом рубидия по схеме:
AgI + 2RbI
= Rb2AgI3
При контакте спрессованных таблеток прекурсоров с
таблеткой продукта реакции наблюдается изменение масс всех составных частей
системы (увеличение массы центральной таблетки, изготовленной из порошка Rb2AgI3
и практически одинаковая убыль массы образцов прекурсоров). Полученный
результат свидетельствует, что реакция протекает благодаря встречной и
практически эквивалентной диффузии ионов серебра и рубидия через слой продукта.
Метод меченых граничных поверхностей. Идея
метода заключается в том, что между реагентами помещается инертная метка
(тонкий слой металла), по положению которой по окончании процесса можно судить
о его механизме путем оценки направления и скорости миграции частиц
определенных сортов. Например, для системы МеО - ЭО2 возможны следующие
варианты:
а) метка осталась неподвижной - противодиффузия
катионов;
б) метка сместилась в таблетку состоящей из
порошка МеО - одностороннее перемещение составных частей реагента МеО;
в) метка сместилась в сторону образца реагента
ЭО2 - одностороннее перемещение составных частей данного реагента.
Следует отметить, что метод меченых поверхностей
имеет определенные недостатки, связанные, в первую очередь, с
экспериментальными трудностями определения истинного местоположения метки и
возможностью ее смещения за счет возникновения напряжения роста. К тому же при
высоких температурах метка недостаточно инертна по отношению к реагентам и
продуктам взаимодействия.
. Метод свободной поверхности. В данном случае в
качестве метки исходной границы раздела фаз используется плоскость одной из
таблеток, диаметр которой меньше, чем диаметра таблетки второго реагента.
Варианты смещение метки и способы их интерпретации в данном случае такие же,
как в предыдущем методе, однако с экспериментальной точки зрения метод
свободной поверхности более доступен и не имеет указанных выше недостатков.
. Метод микрорентгеноспектрального анализа.
Образец, представляющий собой трехслойную систему, состоящую из прекурсоров,
разделенных слоем продукта реакции, разрезается в направлении перпендикулярном
слоям, шлифуется и образовавшаяся поверхность облучается узким сканирующим
электронным пучком. Полученные данные позволяют определить распределение фаз в
зоне реакции, установить геометрию фронта реакции, характер и
последовательность превращений протекающих в системе в процессе взаимодействия
твердых фаз.
В последе время установлено, что использование
только одного из перечисленных выше методов часто приводит к неверной трактовке
механизма взаимодействия. Наиболее достоверные сведения о нем можно получить
только при комплексном исследовании, позволяющем одновременно оценивать
изменение нескольких характеристик системы, в которой протекает реакция между
твердыми фазами: фазовый состав, изменение значений функций состояния системы,
изменение массы и размеров образцов, их тепло- и электропроводности.
.3 Теория твердофазного взаимодействия
Основные положения термодинамической теории,
описывающей реакции между твердыми фазами, впервые были сформулированы Вагнером
в 1936 году и дополнены Шмальцридом в 1962 году:
. Скорость реакции между твердыми фазами лимитируется
скоростью диффузии ионов через слой формирующегося продукта.
Слой продукта реакции является компактным, и
содержащиеся в нем неравновесные дефекты (дислокации границы зерен) не вносят
определяющего вклада в подвижность ионов.
. Реакции на границе фаз протекают значительно
быстрее, чем процессы диффузии через слой продукта, и поэтому на границах фаз
устанавливается локальное термодинамическое равновесие.
. Отдельные ионы движутся в реакционном слое
независимо друг от друга.
. В любом поперечном сечении продукта
сохраняются условия электронейтральности.
На рисунке 3 показаны типичные схемы
массопереноса для реакций АО + В2О3 = АВ2О4.
В первом случае наиболее подвижными в слое
продукта являются ионы А2+ и О2- - они переносятся в равных количествах к границе
раздела АВ2О4 - В2О3 и взаимодействуют с В2О3 с образованием продукта реакции.
Во втором случае к границе АО - АВ2О4 через слой шпинели переносятся ионы В3+ и
О2-, где происходит их взаимодействие с АО. В последнем варианте наращивание
слоя продукта реакции происходит и со стороны его границы с АО и с
противоположной стороны, чему способствует противодиффузия катионов.
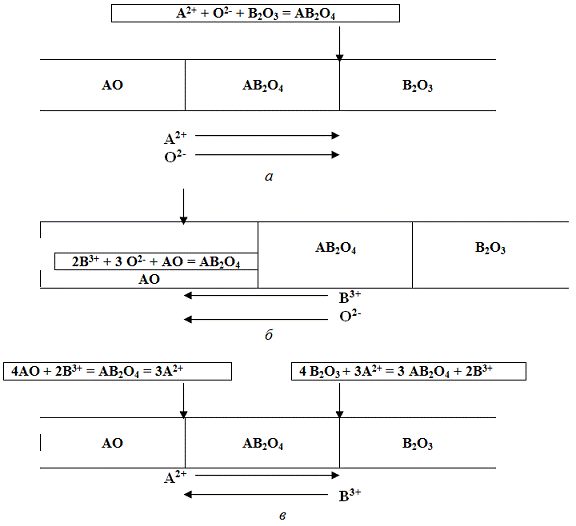
Рисунок 3. Возможные механизмы массопереноса в
процессе формирования фаз состава АВ2О4
Еще один механизм образования фаз состава АnВmОz
из оксидов (при проведении процесса на воздухе или в кислороде) заключается в
перемещении в одном направлении через слой продукта катионов с эквивалентным
потоком электронов и одновременным переносом О2 через газовую фазу к границам,
разделяющим исходные оксиды и формирующийся продукт реакции. Очевидно, что
указанный механизм может быть реализован только при высокой электронной
проводимости продукта реакции.
Необходимо помнить, что в любой реальной системе
различные механизмы массопереноса действуют одновременно, поэтому изменением
параметров состояния можно изменять характер доминирующего в системе механизма.
Например, исследование взаимодействия в системе MgO
- Fe2O3
показало, что при парциальном давлении О2, равном 105 Па, доминирующим
механизмом образования фазы со структурой типа шпинели (MgFe2O4)
является противодиффузия катионов в жесткой кислородной упаковке и скорость
процесса предопределяется подвижностью катиона Mg2+,
коэффициент диффузии которого имеет наименьшее значение . Образующийся в этих
условиях продукт реакции имеет фиксированный состав, а не вступившие в реакцию
оксиды не образуют твердых растворов с MgFe2O4.
При этом выход продукта реакции после обжига смеси исходных оксидов в течении
20 часов при 1500оК не превышает 30%. При понижении парциального давления
кислорода в системе на границе Fe2O3
- MgFe2O4
происходит частичное разложение оксида железа (III):
Fe2O3
→ ⅔Fe3O4
+ 0,167О2
Fe3O4
имеет структуру, сходную с продуктом реакции, поэтому на границе раздела MgFe2O4
- Fe3O4
формируется нестехиометрический твердый раствор. Эта фаза имеет электронную
проводимость и высокую концентрацию катионных вакансий, что способствует росту
скорости образования продукта реакции за счет увеличения скорости массопереноса
по реакции:
Fe2O3
↔ 2Fe3+ +
6ē
+ 1,5О2
При этом энергия активации процесса с
уменьшением парциального давления кислорода с 105 до 10-3 Па уменьшается в
среднем в 2 - 2,5 раза.
Если реакцию образования MgFe2O4
провести в замкнутой системе с ограниченным объемом газообразной фазы, то при
1273оК массоперенос лимитируется процессами испарения - конденсации Fe2O3
.
Таким образом, в рассмотренной системе
массоперенос одновременно осуществляется по трем различным механизмам, а
изменением условий проведения процесса можно подавить или инициировать любой из
них. Аналогичные механизмы массопереноса и способы управления ими были
обнаружены и для процессов протекающих в системе NiO
- Fe2O3:
при понижении парциального давления О2 в системе с 105 до 10-3 Па и, следовательно,
с ростом концентрации катионных вакансий в продукте реакции, энергия активации
снижается с 393 до 185 кДж/моль (при температуре синтеза 1140-1190оК).
Иная картина бала обнаружена в процессе
исследования реакций в системе ZnO
- Fe2O3:
. При одинаковых параметрах состояния системы
скорость реакции образования ZnFe2O4
оказалась выше у порошкообразных образцов шихты, которые перед обжигом не
прессовались (эффект противоположный по сравнению с ранее рассмотренными
системами).
Увеличение парциального давления О2 в системе
ведет не к снижению, а росту скорости массопереноса в динамических условиях.
. При проведении взаимодействия в среднем
вакууме скорость образования ZnFe2O4
оказалась наименьшей из трех указанных вариантов условий синтеза данной фазы.
Указанные различия связаны с тем, что в отличии
от MgFe2O4
и NiFe2O4
феррит цинка является фазой внедрения, в которой перенос катионов
осуществляется по междоузлиям. При этом в динамических условиях во всем
исследованном интервале парциальных давлений кислорода доминирующим остается
механизм односторонней диффузии ионов цинка (а не ионов Fe3+
и электронов с одновременным переносом О2 через газовую фазу). Аналогичные
изменения механизмов массопереноса и скорости формирования продукта реакции
наблюдаются и в системе LiFeO2
- Fe2O3,
в которой продукт реакции также является фазой внедрения, но в данном случае
наиболее вероятен механизм противодиффузии ионов лития и железа по междоузлиям.
Изменять энергию активации процессов
формирования сложных оксидных фаз можно и путем создания катионных или анионных
вакансий в кристаллических решетках прекурсоров. В частности, энергия активации
процессов, скорость которых лимитируется зародышеобразованием или реакциями на
границе раздела фаз, снижается с увеличением концентрации дефектов в анионной
подрешетке прекурсора.
Указанную закономерность можно проследить на
примере образования фазы со структурой перовскита, получаемую с использованием
в качестве исходного вещества фаз оксида титана с изменяющейся концентрацией
анионных вакансий:
TiO2-x + SrCO3 = SrTiO3-x +CO2
Таблица 5. Зависимость энергии активации
процесса образования SrTiO3-x
в зависимости от дефектности TiO2-x.
|
Х
|
Еак.
Кдж/моль
|
|
0,000
|
409,2
|
|
0,005
|
360,2
|
|
0,017
|
301,7
|
|
0,025
|
281,2
|
|
0,030
|
256,0
|
Подводя итог можно сделать вывод, что дефекты
нестехиометрии играют предопределяющую роль в выборе системой механизма
твердофазных реакций. В то же время, этими механизмами можно целенаправленно
управлять, если известны механизмы массопереноса и тип разупорядочения продукта
реакции. Управлять механизмами реакций можно и за счет создания прекурсоров, с
заданной концентрацией и типом дефектов.
Роль неравновесных дефектов типа дислокаций,
микроискажений и свободных поверхностей в формировании продуктов твердофазного взаимодействия
растет по мере увеличения дисперсности исходной шихты (т.е. при увеличении
размеров реакционной зоны). Если объем реакционной зоны становится соизмеримым
с объемом системы, в которой протекает твердофазное взаимодействие, то диффузия
уже не будет лимитировать скорость этого взаимодействия, т.е. концентрация
дефектов практически не влияет на скорость реакции. Кроме того, при низких
температурах роль диффузионных процессов в реакциях между твердыми фазами резко
снижается (из-за ассоциации вакансий и накопления дислокаций, возникающих в
результате исчезновения точечных дефектов) и диффузионный механизм сменяется
дислокационным механизмом массопереноса .
Роль неравновесных дефектов прекурсоров на
совершенство кристаллической решетки продукта реакции была показана на рисунке
4. Исследование продукта реакции в этом случае показало, что он состоит из двух
слоев: - плотный и совершенный по структуре, примыкающий к монокристаллу АО и -
пористый и высоко дефектный по структуре. Различие в формировании слоев
заключается в различных механизмах массопереноса из-за неодинаковой
неравновесной дефектности прекурсоров:
слой образуется в процессе противодиффузии
ионов, а второй - за счет односторонней диффузии более подвижного катиона А2+ и
электронов с одновременным переносом О2 через газовую фазу.
Особый механизм реакции был обнаружен в
системах, образованных из фаз с близкой полярностью связи Э - О. В этом случае
реакция начинается с предварительной аморфизации реагентов, которые на втором
этапе взаимодействуют между собой с образованием псевдоаморфного продукта
реакции. Это предопределяется тем, что энергия самодиффузии в этом случае
высока, а протекание реакции через аморфизацию прекурсоров позволяет снизить ее
в 5-6 раз. Указанный механизм реакции реализуется, например, при образовании BeAl2O4
из оксидов алюминия и бериллия

Рисунок 4. Механизмы образования АВ2О4 (области
1 и 2) из монокристалла АО и мелкодисперсного В2О3.
Значительны отклонения рассчитанных и
экспериментальных данных по скоростям реакций с участием твердых фаз могут
свидетельствовать об изменении механизма взаимодействия за счет возникновения
жидкой фазы в системе (плавление реагентов или образование легкоплавкой
эвтектики между продуктом реакции и исходными веществами). Например, реакции
между оксидами и галогенидами, реакции присоединения и обмена с участием
галогенидов и сульфатов элементов главных подгрупп I
и II групп и т.д.
автокаталитически ускоряются в момент появления жидких эвтектик, образованных
реагентами или реагентами и продуктами реакций. Резкое увеличение скорости
процессов в данном случае связано с ростом коэффициентов диффузии частиц при
переходе от твердых фаз к жидким.
Многостадийными являются механизмы
самораспространяющегося высокотемпературного синтеза (СВС), например, в
процессе реакции:
PbO2 + WO2
= PbWO4
последовательно происходит:
). Разложение PbO2
до PbO ( ∆Н > 0)
). Окисление WO2
до WO3 ( ∆Н<<
0)
). Перенос PbO
на поверхность зерен WO3
через газовую фазу и по механизму поверхностной диффузии через область контакта
зерен, образование продукта реакции и плавление эвтектики, кристаллизация PbWO4
из эвтектического расплава при температуре выше 1000оК.
Высокая скорость твердофазного процесса может
наблюдаться и в том случае, когда одна из реагирующих фаз относится к так
называемым «суперионникам», например галогениды и сульфиды меди(I)
и серебра(I) [33-34], которые
даже при низких температурах характеризуются аномально высокой подвижностью
катионов. Так в сульфиде серебра коэффициент диффузии ионов серебра при 500оК
достигает значения 10-2 см2/ сек., а энергия активации диффузии составляет
всего 12 кДж/моль. Поэтому, принимая во внимание, что данная фаза является
смешанным электрон-ионным проводником, очевидно, что реакции с ее участием не
будут лимитироваться скоростью диффузии ионов серебра, даже при низких
температурах системы.
.4 Твердофазные превращения без изменения
состава
.4.1 Типы и механизмы полиморфных превращений
Простые и сложные вещества фиксированного
качественного и количественного состава могут существовать в твердом состоянии
в виде нескольких модификаций, отличающихся строением кристаллических решеток -
такие модификации называются полиморфными. Каждая из них термодинамически
стабильна в строго определенном интервале значений параметров состояния системы
и способна переходить в другую при изменении этих параметров. Следует отметить,
что не все превращения без изменения состава в твердых фазах сводятся к
полиморфизму. В частности, появление новых качеств у твердого вещества
(проводимость и сверхпроводимость, магнитные превращения и оптические эффекты)
не всегда связано с изменением строения кристаллической решетки.
Рассматривая полиморфные превращения с
практической точки зрения. В первую очередь необходимо определить, какова, в
данном конкретном случае, скорость процесса и можно ли управлять этой
скоростью, тормозя или инициируя превращения, т.е. изменяя энергию активации
процесса перехода.
Учитывая структурную родственность участвующих в
процессе превращений модификаций, была предложена следующая классификация
полиморфных превращений:
. Превращения, связанные с изменением первичной
координации.
Превращения, связанные с изменением вторичной
координации.
. Превращения, обусловленные разупорядочением.
. Превращения с изменением типа химической
связи.
В процессе первого из них расположение ближайших
соседей полностью нарушается и создается новый тип кристаллической решетки. Это
сопровождается значительным изменением внутренней энергии системы, которое, в
первую очередь, обусловлено перераспределением сил притяжения и отталкивания
между соседними частицами кристаллической решетки. Указанные превращения бывают
двух видов: деформационные (с растяжением) и реконструктивные ( с перестройкой
структуры).
К деформационным превращениям относится,
например, трансформация кубической решетки хлорида цезия с КЧ=8 в структуру
типа хлорида натрия с КЧ=6, которая сопровождается вытягиванием решетки в
направлении {111}, а также характерный для металлов переход от структуры ОЦК к
структуре ГЦК с изменением КЧ от 8 до 1 В обоих случаях процессы
характеризуются низкими энергиями активации и, зачастую, протекают с высокой
скоростью. Отметим, что структура фаз с более низким КЧ имеет меньшую
плотность, более высокие значения энтропии, теплоемкости и внутренней энергии
и, следовательно, является высокотемпературной формой.
При реконструктивном превращении система
проходит через целый ряд промежуточных форм, в которых ионы имеют КЧ отличные
от значений КЧ в исходной и конечной фазах. Наблюдаемая энергия активации этих
превращений (даже с учетом образования промежуточных форм) значительна, что
предопределяет их низкую скорость. Стимулировать реконструктивный переход можно
за счет повышения давления в системе. Например, CdTiO3
при с.у. имеет термодинамически стабильную структуру типа ильменита с КЧ
каждого из катионов равное 6, а при давлении 25·108 Па и 773оК у этого
вещества наблюдается переход с образованием фазы со структурой перовскита (КЧCd2+
= 12, КЧ иона титана не изменяется). Аналогично, SiO2
при с.у. имеет кристаллическую решетку с КЧ у катиона равное 4 (кварц), которая
при р=130·108 Па и Т=1473оК трансформируется в кристаллическую решетку
типа рутила с КЧ катиона равное 6.
Превращения, связанные с изменением вторичной
координации не предусматривают изменения в расположении ближайших соседей,
структурный переход осуществляется путем перемещения - небольшого сдвига частиц
без нарушения связи, т.е. контакта с исходными соседями. Примером такого
изменения структуры может быть переход α- кварца
в β-
кварц.
В этом случае, из-за отсутствия энергетического барьера, связанного, зачастую,
с образованием промежуточных форм, превращение протекает с высокой скоростью и
лимитируется только скоростью передачи тепловой энергии, т.е. теплопроводностью
образца.
Процесс разупорядочения (третий вариант
полиморфного превращения) может быть ориентационным или позиционным. В первом
случае он осуществляется путем изменения ориентации определенных атомных групп
относительно друг друга, например путем вращения. Ориентационное
разупорядочение возможно в молекулярных (СН4), атомных (металлы) и ионных
кристаллах (галогениды аммония, некоторые виды шпинелей, с ионами меди (II)
марганца (III) в своем
составе, в оксидных ферромагнитных материалах, переходящих в парамагнитное
состояние путем изменения ориентации атомных магнитных моментов и т.д.). Позиционное
разупорядочение реализуется путем перераспределения частиц между узлами
кристаллической решетки, в то время как позиционное упорядочение приводит к
образованию сверхструктур различного типа. Например, позиционное упорядочение
вакансий и ионов Fe3+
в октаэдрической подрешетке γ-Fe2O3
способствует формированию тетрагональной решетки Fe3+[
Fe3+5/3V′1/3]
c соотношением осей
с/а=3 (упорядочение типа 1:5). Превращения в твердых фазах, связанные с
ориентационным разупорядочением протекают с высокой скоростью в отличие от
процессов позиционного разупорядочения или упорядочения, т.к. последние
осуществляются за счет диффузии ионов (атомов) и вакансий.
Превращения, сопровождающиеся разрушением одних
и образованием других химических связей выделены в самостоятельную группу т.к.
они сопровождаются не только кристаллографическими изменениями фазы, но и
изменением состояния ее электронной системы. Например, превращение алмаза
(ковалентная неполярная двухцентровая связь) в графит с межслоевой
многоцентровой делокализованной связью, а также металлической модификации олова
в полупроводниковую фору со структурой алмаза. Большинство превращений данного
типа имеют высокие энергии активации и, следовательно, протекают с низкой
скоростью.
Согласно теоретическим расчетам и
экспериментальным данным было показано [35], что «фазовое превращение на
поверхности твердого тела протекает в направлении образования новой
кристаллической решетки, находящейся в ориентационном и размерном соответствии
с кристаллической решеткой исходной поверхности». Указанное положение
справедливо только в том случае, когда энергия деформации двухмерной решетки
новой фазы (Е) меньше работы образования ее трехмерного зародыша (А), т.е. если
Е<А. В противном случае, когда Е>А, характер протекающего процесса не
зависит от структуры исходной фазы.
3.4.2 Характеристика полиморфных превращений
Наиболее общим вариантом классификации фазовых
переходов можно считать классификацию, основанную на анализе характера
изменения термодинамических функций системы в точке превращения . Согласно этим
взглядам, у переходов первого рода энергия Гиббса, как функция параметров (P,V,T)
непрерывна, тогда как ее первые производные, претерпевают разрыв:
(∂G/∂T)P = - S ; (∂G/∂P)T
= V ; [∂(G/T)/∂(1/T)]P = H
т.е. значения энтропии, энтальпии и объем
системы изменяются скачком. К превращениям этого типа относятся процессы
сопровождающиеся разрывом связей, изменением координации частиц и т.д., т.е.
характеризующиеся значительной степенью преобразования, как кристаллической
структуры, так и электронной подсистемы.
Переход второго порядка характеризуется
непрерывным изменением не только энергии Гиббса, но и ее первых производных,
тогда как значения вторых производных при температуре фазового перехода
изменяются скачком. Следовательно, в этом случае наблюдается непрерывное
изменение энтропии, энтальпии и объема системы, а скачкообразно меняются только
значения ее теплоемкости, сжимаемости и коэффициента термического расширения.
Примерами переходов второго рода могут служить процессы упорядочения и
разупорядочения сплавов, появление ферро-, антиферромагнетизма,
сегнетоэлектрических свойств, сверхпроводимости и т.д.. Аналогичным образом
определяются переходы третьего и более высоких порядков, однако они
сопровождаются небольшими изменениями в системе и поэтому их обнаружение
связано со значительными экспериментальными трудностями.
Независимо от характера структурных изменений
при фазовых переходах все они могут быть разделены на энантиотропные и
монотропные. Первый тип характеризует обратимые переходы между кристаллическими
фазами одинакового качественного и количественного состава, а второй -
необратимые переходы метастабильной (при любых параметрах состояния системы)
фазы в термодинамически стабильную. Примером монотропных переходов является превращение
γ-форм
оксидов алюминия, железа (III),
хрома (III) и т.д. в
стабильные α-модификации со
структурой корунда. В связи с метастабильностью γ-форм
при любых параметрах состояния системы (также как и для метастабильных форм
иного состава), на равновесных диаграммах состояния отсутствуют области
отвечающие этим фазам. Необходимо отметить, что температура монотропных
превращений не является константой при выбранных параметрах состояния системы и
зависит от способа получения метастабильной фазы.
Особую группу составляют превращения, которые
осуществляются путем сдвига одной или нескольких плоскостей на расстояние
меньше межатомного (межионного) - мартенситовы превращения. В связи с низким
значением энергии активации такого процесса предотвратить его методом закалки
невозможно, т.е. не удается кинетически стабилизировать высокотемпературную
модификацию.
.4.3 Способы управления фазовыми превращениями
без изменения состава фаз
Одной из задач, стоящих перед современным
материаловедением, является определение способов, с помощью которых можно было
бы управлять скоростью и механизмом полиморфных превращений. В настоящее время
известно, что достижение поставленной цели возможно как путем изменения
параметров состояния системы (температуры, давления), так и за счет введение в
систему нетепловых форм энергии - облучение, механическое (в том числе ударное)
воздействие, а также введением в исходные фазы примесей.
. Влияние температуры и скорости ее изменения в
системе на полиморфные превращения сводиться к методике стабилизации
высокотемпературных форм и появлению в ряде случаев новых метастабильных
состояний системы, которые можно рассматривать как активные комплексы,
облегчающие или затрудняющие протекание фазового перехода с кинетической точки
зрения. Если энергия активации процесса превращения достаточно высока
(реконструктивный переход, позиционное упорядочение, изменение характера
химической связи), высокотемпературное состояние достаточно легко фиксируется
методом закалки. В противном случае, как отмечалось выше, метод закалки не
эффективен (мартенситовое превращение, ориентационное упорядочение).
При медленном снижении температуры системы
последовательность формирования фаз диктует не термодинамический, а
кинетический фактор: на первом этапе в системе кристаллизуется фаза, энергия
активации зародышей которой минимальна. Последующее развитие событий уже будет
предопределяться кинетикой превращения указанной фазы в термодинамически более
стабильные, вплоть до образования фазы с наименьшим значением энергии Гиббса -
указанная последовательность превращений носит название ступенчатых переходов
Оствальда. Анализируя данную последовательность необходимо учитывать, что
скорость твердофазных реакций, имеющих диффузионный механизм, резко уменьшается
при понижении температуры системы, делая невозможным достижение равновесного
состояния. Это свидетельствует о том, что термодинамически стабильные
модификации невозможно будет получить из первоначально образовавшихся в системе
фаз в том случае, когда указанные превращения будут иметь высокие активационные
барьеры. Если же энергия активации указанных переходов низка, то в системе при
высоких температурах быстро наступает состояние равновесия, которое, например,
характеризует степень совершенства структуры при позиционном упорядочении.
Снижение температуры системы потребует все большего увеличения времени
изотермической выдержки для достижения в системе равновесия и, наконец, при
индивидуальной для каждой системы температуре, подвижность частиц в системе
станет настолько низкой, что можно будет говорить об образовании
«замороженного» состояния - состояния равновесного при какой-то высокой
температуре То и сохранившегося при с.у. за счет кинетических затруднений.
Таким образом, достигаемое реально при медленном
охлаждении образцов распределение частиц в кристаллических решетках отвечает
некоторой эффективной температуре, ниже которой подвижность ионов (атомов) и
дефектов очень мала, но величина этой подвижности отлична от нуля. Поэтому в
системе, во времени, продолжаются изменения, связанные с ее попыткой достичь
состояния равновесия. Скорость указанных процессов резко увеличивается под
влиянием, как тепловых, так и нетепловых форм вводимой в систему энергии
(термоциклирование, магнитные и электрические поля и т.д.). Указанные изменения
в образцах ведут к изменению их механических и электрофизических свойств и
называются старением.
Изменение давления в системе в процессе
твердофазного превращения оказывает кинетический и термодинамический эффект:
рост давления в равновесных условиях смещает температуру полиморфного
превращения в соответствии с уравнением Клаузиуса - Клайперона:
∆Т = (T∆V/∆H)∆Р
где ∆V
- разность молярных объемов равновесных кристаллических модификаций, ∆H
- изменение энтальпии при фазовом переходе.
Из уравнения видно, что знак изменения
температуры, при которой одна модификация превращается в другую, определяется
знаками ∆V и ∆H.
Так как, в большинстве случаев, молярный объем высокотемпературной фазы больше
молярного объема низкотемпературной фазы, а ∆H
перехода α-формы в β-
форму
всегда больше нуля, увеличение Р в равновесных условиях будет увеличивать
температуру фазового превращения.
Одновременно рост давления способствует
аннигиляции дефектов типа вакансий, внедренных частиц, дислокаций и
межкристаллитных границ, что резко снижает скорость процессов,
предопределяющихся диффузией. Это подавляет образование зародышей новой фазы в
объеме исходной и может полностью исключить возможность фазового перехода.
. Введение примесей в кристалл с низкой
дефектностью способствует образованию зародышей новой фазы. Это снижает энергию
активации фазового превращения, т.е. в присутствии примесей способность системы
сохранять метастабильное состояние снижается. Если же примесь вводится в
кристалл, характеризующийся высокой концентрацией дефектов, то наблюдается
эффект, противоположный описанному. Например, добавление к металлическому олову
серебра, свинца, висмута и ряда других металлов в микроколичествах
предотвращает его переход в α-форму
при температурах ниже 286оК. Если же ввести в эти сплавы дополнительно медь,
марганец, цинк или алюминий, то фазовый переход при указанных температурах
будет протекать с высокой скоростью. Аналогично для монотропных переходов:
оксиды марганца, кобальта и никеля стабилизируют γ-форму
оксида железа (III),
а оксид хрома (III)
ее дестабилизирует.
4. Кинетика реакций с участием твердых фаз
При планировании любого химического
эксперимента, в том числе синтеза вещества с использованием метода твердофазных
реакций, предварительно необходимо получить ответ на две группы вопросов:
а) возможно ли и при каких параметрах состояния
системы самопроизвольное превращение выбранных исходных веществ в искомый
продукт реакции - ответ на эти вопросы могут дать термодинамические расчеты;
б) при каких условиях скорость взаимодействия
будет настолько большой, чтобы процесс имел бы практическое значение, и каков
выход продукта реакции при этих условиях - ответы на эти вопросы дает
химическая кинетика и теория равновесных состояний.
При получении положительного ответа на первую группу
вопросов на втором этапе необходимо выявить факторы влияющие на скорость и
полноту рассматриваемого взаимодействия и оптимизировать условия синтеза
целевого продукта таким образом, чтобы процесс завершался за заданный период
времени и характеризовался бы заданной степенью превращения.
К сожалению, классическая теория, описывающая
скорость химических реакций, детально разработана только для гомогенных
процессов, что не позволяет без глубокой коррекции использовать ее выводы для
описания взаимодействия между твердыми фазами. Тем не менее, она все же может
служить отправной точкой для понимания факторов, определяющих скорость
твердофазных реакций, характерной особенностью которых является локализация
реакционной зоны на поверхности раздела фаз. При этом общая поверхность и
толщина реакционной зоны могут быть различны и зависят как от природы
реагентов, так и от условий осуществления процесса. Так, например, взаимное
растворение реагентов способствует увеличению ширины реакционной зоны, а
изменение степени дисперсности прекурсоров и степени их смешения может изменить
ее площадь на несколько порядков. Однако, несмотря на указанные особенности,
основные понятия классической кинетики, такие как энергия активации,
лимитирующая стадия, порядок реакции и т.д. при описании данных гетерогенных
реакций сохраняются, хотя их интерпретация несколько меняется в соответствии со
спецификой механизмов взаимодействия.
Несмотря на разнообразие процессов с участием
твердых фаз, большинство из них представляют собой совокупность последовательно
протекающих стадий:
а) «покрывание» одного из реагентов другим -
более летучим или легкоплавким;
б) активирование реагентов за счет адсорбции и
образования поверхностных промежуточных форм;
в) дезактивация поверхности;
г) активирование реагентов путем объемной
диффузии;
д) образование первоначально аморфного, а затем
и кристаллического продукта реакции;
е) отжиг дефектов кристаллической решетки
продукта .
Следует учитывать, что на различных этапах
взаимодействия лимитировать процесс в целом могут его различные стадии. Так,
например, при синтезе титанатов, цирконатов, ниобатов свинца с использованием в
качестве прекурсоров высокодисперсных порошков соответствующих оксидов, на
первом этапе происходит «покрывание» поверхности частиц оксидов d-элементов
низкоплавким и легколетучим оксидом свинца (II).
В связи с этим, на начальных этапах синтеза взаимодействие в целом лимитируется
стадией (д) - т.е. процессом формирования продукта реакции в местах контакта
реагентов. По мере образования непрерывного слоя продукта реакции
взаимодействие постепенно переходит в диффузионный режим и лимитируется
скоростью диффузии ионов свинца и кислорода через слой продукта .
Для количественной характеристики твердофазных
процессов применяют различные величины, одной из которых является степень
превращения αi:
αi
= Ni/Ni
(исх)
где Ni
и Ni (исх) - число
молей i-того реагента к
моменту времени τ и до
реакции.
Следует отметить, что αi
в отличие от концентрации не является параметром состояния системы т.к. одна и
та же степень превращения может быть реализована в различных системах
одинакового исходного состава, отличающиеся структурой реакционной зоны, а
следовательно составом и структурой промежуточных форм, а также количеством,
составом и структурой продуктов параллельных реакций.
Сложный многостадийный характер процессов с
участием твердых фаз отражается на форме кривых описывающих функции αi
= f(τ) и
∂α/∂τ
= f(α) (рис.5.).
Как видно из представленных данных, на начальных
стадиях твердофазных реакций их скорость близка к нулю (индукционный период).
На следующем этапе происходит резкое увеличение скорости процессов вплоть до
достижения максимума (k)
после чего значения скорости падают до нуля. В связи с этим зависимость αi
= f(τ) изображается
в виде сигмовидной кривой.
Индукционный период можно рассматривать как
время необходимое для формирования реакционной зоны. В этот период в системе
происходят изменения на макро и микро уровне, предшествующие началу
формирования в системе продукта реакции (переориентация частиц порошков за счет
сглаживания их поверхности, испарение и конденсация летучих компонентов,
искажение кристаллических решеток исходных фаз, диффузия частиц вдоль
поверхности и дислокаций, образование промежуточных поверхностных фаз и т.д.).
Этап быстрого роста скорости реакций зачастую связан с образованием и ростом
ядер конечного продукта в реакционной зоне.
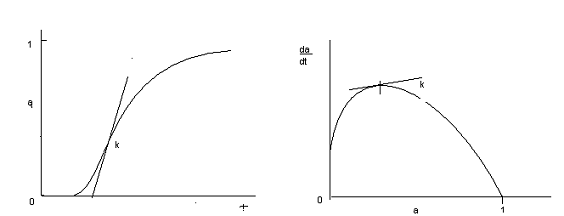
Рис.5. Кинетические кривые: степень
твердофазного превращения как функция времени αi
= f(τ) (справа)
и скорость твердофазного превращения как функция степени превращения ∂α/∂τ
= f(α) (слева)
В ряде случаев этот процесс может быть более
сложным и включать взаимное растворение реагентов с последующим распадом
твердого раствора, приводящим к образованию зародышей продукта реакции.
Снижение скорости реакции после достижения максимума объясняется образованием
сплошного слоя продукта взаимодействия за счет столкновения растущих зародышей
. Необходимо отметить, что при использовании активных прекурсоров форма кривой,
выражающей зависимость αi
= f(τ) может
не содержать участка, отражающего индукционный период взаимодействия.
Интерпретация большинства кинетических данных
для гетерогенных процессов, в большинстве случаев, базируется на двух важнейших
положениях химической кинетики связанных с понятиями «энергия активации» и
«лимитирующая стадия процесса». Первое из них заключается в том, что любая из
стадий химического взаимодействия осуществляется с участием небольшого числа
частиц (максимум трех) и этот процесс сопровождается образованием переходного
состояния («активного комплекса»), характеризующегося более высокой энергией по
сравнению со средней энергией системы. При этом из всех возможных параллельных
путей реакции один, имеющий наименьший энергетический барьер, будет предопределять
состав и строение продуктов реакции при минимальных температурах процесса (не
обязательно стабильных термодинамически при заданных параметрах состояния
рассматриваемой системы). Теория активных комплексов дает в общем виде
уравнение, описывающее скорость реакции и предлагает модель, с помощью которой
можно проводить полуэмпирические расчеты для простейших процессов. Другое
положение заключается в том, что суммарная скорость сложного процесса,
состоящего из ряда последовательных стадий, предопределяется наиболее медленной
из них. В соответствии с представлениями о переходном состоянии константа
скорости (kс ) процесса может
быть выражена в виде зависимости:
kс
= kT/h exp( -∆H/RT) exp∆S/R
где k
- постоянная Больцмана, h
- постоянная Планка, ∆H
и ∆S- энтальпия и
энтропия активации, соответственно.
Необходимо отметить, что при последовательной
схеме течения реакции равновесие достигается во всех стадиях, предшествующих
лимитирующей, тогда как для достижения равновесия у последующих стадий
необходимо использования специальных технологических приемов (изотермический
отжиг образцов, введение добавок снижающих энергию активации указанных стадий,
изменение состава и давления газовой фазы т. д.)
Как отмечалось выше, рассматриваемые процессы
протекают в пределах межфазных границ и представляют собой совокупность
нескольких стадий заключающихся в переносе реагирующих частиц к реакционной
зоне и формированию в ней продукта реакции. Зачастую эта серия стадий имеет
относительно простую кинетику, если фактическая максимальная скорость одной из
стадий намного меньше, чем фактическая максимальная скорость любой из остальных
стадий. В этом случае реализуются только три возможных типа реакций: реакции,
контролируемые: а) скоростью переноса, б) скоростью взаимодействия диффундирующих
частиц на границе раздела фаз и в) скоростью образования или роста зародышей.
.1 Диффузионные модели
Как показали проведенные исследования, кинетика
большинства процессов с участием конденсированных фаз определяется скоростью
переноса частиц к зоне реакции. Наиболее изученными являются, например, реакции
окисления металлов кислородом, скорость которых лимитируется переносом и
подчиняется параболическому закону. Если предположить, что на поверхности
металла образуется непрерывная пленка, то скорость реакции будет
предопределяться скоростью диффузии кислорода (атомарного или молекулярного в
зависимости от структуры и состава оксида). Это связано с тем, что скорость
диффузии атомов большинства металлов через слой продукта реакции на несколько
порядков меньше, чем у кислорода - т.е. наращивание слоя оксида в процессе
окисления металла происходит на границе раздела металл - оксид. Очевидно, что с
ростом толщины оксидной пленки (из-за увеличения длины пробега) суммарная
скорость переноса кислорода уменьшается. Одним из возможных способов оценки
скорости рассматриваемой реакции является зависимость изменения массы образцов
во времени. Тогда, обозначив скорость переноса Vп
г/см2 · сек получим:
Vп = dz/dτ
= D (∂c/∂x)
где z
- изменение массы образца, τ
- время, D - коэффициент
диффузии частиц, лимитирующих процесс в направлении перпендикулярном пленке
оксида, с - молярная (поверхностная) концентрация диффундирующих частиц, x
- толщина оксидной пленки.
Или, исходя из предположения о существовании
локальных термодинамических равновесий с учетом граничного условия (х = 0 при τ
= 0), получим:
x2 = kp
τ
где kp
- константа скорости параболического роста продукта.
Для того чтобы найденные уравнения могли бы быть
применены для описания процессов между порошками твердых фаз Яндер вводит ряд
ограничений для процессов, протекающих по схеме nA
+ mB = AnBm:
. Компонент А представляет собой совокупность
одинаковых микрочастиц с начальным радиусом r0.
Компонент В обладает высокой поверхностной
диффузией, за счет чего на поверхности частиц А быстро образуется непрерывный
слой продукта реакции.
. Взаимодействие лимитируется односторонней
диффузией компонента В через слой продукта к компоненту А.
. Продукт реакции не образует твердых растворов
с прекурсорами.
. Отношение объема продукта реакции к объему
прекурсоров ≈ 1.
. D,
транспортируемых частиц, не изменяется во времени, а активность реагентов на
границе реакционной зоны остается постоянной.
. Толщина слоя продукта изменяется во времени по
параболическому закону.
При выполнении указанных условий с учетом
соотношения x = r0
(1 - 3√ 1- α
), функция αi
= f(τ),
описывающая изменение степени превращения (α)
от времени принимает вид:
F1(α)
= (1 - 3√ 1- α
)2 = kя τ
(где kя - константа
Яндера)
Применимость любого кинетического уравнения к
исследуемому процессу проверяется одним из графических методов. Для этого
экспериментально полученные данные (степень превращения) обрабатываются в
координатах F1(α)
- τ
или kя - α
(рис.6).
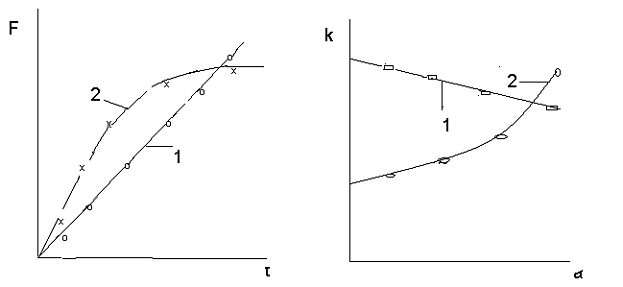
Рис.6 Графические способы проверки
экспериментальных данных (степень превращения) на соответствие модели Яндера.
- уравнение Яндера применимо; 2 - уравнение
Яндера неприменимо
Проверка соответствия экспериментальных данных
величинам α,
рассчитанных по уравнению Яндера, позволило установить, что хорошее
согласование экспериментальных и вычисленных величин наблюдается только на
начальных стадиях процесса синтеза (α
< 0,4), что связано с формированием слоя продукта реакции и постепенным
уменьшением площади поверхности, перпендикулярной диффузионному потоку. В связи
с этим Гинстлинг и Броунштейн отказались от положения о параболическом законе
роста слоя продукта, оставив представление об односторонней диффузии компонента
В и показали, что в этом случае функция αi
= f(τ)
имеет вид:
F2 (α)
= 1 - ⅔ α - (1 - α)⅔
= kГБ τ
(где kГБ
- константа Гинстлинга и Броунштейна)
Кроме указанных уравнений в настоящее время
используются и другие, основанные на представлении об односторонней диффузии, в
частности уравнение Картера -Валенси, учитывающее, что эквивалентные объемы
продукта реакции и покрывающего реагента различны, уравнение Дюнвальда -
Вагнера, основанное на законе Фика (диффузия нестационарного потока вещества из
постоянного источника в сферическое тело заданного радиуса), уравнение Журавлева
- Лесохина - Темпельмана, учитывающее изменение концентрации диффундирующего
вещества по мере протекания реакции и т.д..
Как отмечалось выше, соответствие исследуемого
процесса той или иной модели доказывается графически, однако, необходимо
отметить, что в процессе твердофазного взаимодействия происходит изменение
энергий активаций его отдельных стадий. Из этого следует, что лимитировать
реакцию на разных этапах ее протекания могут различные типы взаимодействий.
Поэтому, как правило, в целом описать кинетику твердофазной реакции одним из
приведенных выше уравнений зачастую не удается, что вызывает необходимость
использовать различные модели для отдельных интервалов степеней превращения.
По мере накопления экспериментальных данных
становилось очевидным, что обсужденные выше модели, постулирующие односторонний
характер диффузии частиц покрывающего реагента вглубь зерен покрываемого
реагента не могут описать всего разнообразия твердофазных реакций. В связи с
этим было высказано предположение (которое позднее было доказано
экспериментально), что возможны еще два механизма переноса - с направлением
диффузии от А к В (т.е. противоположному по сравнению с моделью Яндера) (рис.7)
и со встречной диффузией реагентов.
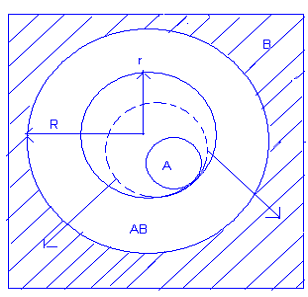
Рис.7 Схема твердофазной реакции по
антияндеровской модели (направление диффузии от А к В). Пунктиром показано
изменение объема и положения частицы А во времени.
При сохранении остальных ограничений введенных
Яндером можно получить уравнения описывающие процессы с измененным направлением
диффузии:
F11 (α)
= [(1 + α)⅓ -
1]2 = kАЯτ
(диффузионная
модель анти-Яндера)
F12 (α)
= 1 + ⅔α - (1 + α)⅔ = kАГτ
(диффузионная
модель анти-Гинстлинга) и т.д..
Как отмечено выше, основой всех рассмотренных
кинетических моделей реакций с участием твердых фаз является предположение, что
изменение α во времени обратно
пропорционально толщине образующегося слоя продукта реакции. Однако указанная
зависимость выполняется только в том случае, если в пределах слоя продукта
реакции и на границах реакционной зоны реализуется состояние локальных
термодинамических равновесий [7,10], т.е в случае порошкообразных реагентов с
низкой неравновесной дефектностью и, следовательно, постоянным значением
коэффициентов диффузии реагентов через слой формирующегося продукта. На первый
взгляд кажется, что это положение может относиться только к идеальной модели,
так как порошки прекурсоров получают, зачастую, при высоких температурах и
концентрация дефектов в них отвечает эффективной температуре, ниже которой
подвижность составляющих решетки ничтожно мала. Кроме этого все образцы
подвергаются механической обработке (перетирание, прессование) на
предварительных и конечных этапах синтеза продукта реакции, что способствует
образованию неравновесных дефектов типа дислокаций, свободной поверхности и
т.д..
С ростом концентрации неравновесной дефектности
прекурсоров, резко увеличивается диффузионная подвижность составных частей
решетки, что приводит к росту скорости процесса при фиксированной температуре.
В свою очередь термическая обработка образцов, осуществляемая для достижения
энергии активации процесса, способствует аннигиляции дефектов, уменьшая их
концентрацию и изменяя природу, что приводит к снижению скорости реакции.
Следовательно, при реальном твердофазном синтезе локальные равновесия
отсутствуют, как в объеме продукта, так и на границе раздела фаз. Учет данного
обстоятельства можно осуществить, используя уравнения предложенные Тамманом ,
Крегером и Циглером и Хальбертом и ряд других:
F3(α)
= 1 - 3√ (1- α
) = kTlnτ
F4(α)
= (1 - 3√ 1- α
)2 = kКЦ lnτ
F5 (α)
= 1 - ⅔ α - (1 - α)⅔
= kГБХ lnτ
Проверка пригодности представленных выше
уравнений для описания реальных процессов выявило ряд практически важных для
технологии закономерностей :
. Если температура, при которой осуществляется
твердофазное взаимодействие, не превышает температуру формирования решетки
прекурсоров, то последние являются достаточно стабильными даже при высокой
концентрации неравновесных дефектов. Слой продукта реакции в этом случае имеет
высокую концентрацию неравновесных дефектов, связанную с несовершенством
структуры исходных реагентов. При этом, за счет относительно низкой температуры
взаимодействия высокодефектных прекурсоров, неравновесные дефекты продукта
реакции достаточно стабильны во времени. Сформировавшуюся в этом случае
реакционную смесь можно рассматривать как систему, в которой устанавливаются
локальные равновесия, практически не зависящие от длительности процесса, и в
которой изменение толщины слоя продукта изменяется во времени по параболическому
закону. Это позволяет успешно описывать такие процессы, применяя модели Яндера,
Гинстлинга и т.д. (учитывая направления диффузионных потоков), несмотря на выше
высказанные ограничения, связанные с характером дефектности прекурсоров.
Если порошки исходных фаз были получены при
температуре более низкой, по сравнению с температурой синтеза продукта реакции,
то параллельно с процессом образования этого продукта будут протекать процессы
аннигиляции дефектов в исходных фазах и, следовательно, во времени будет
изменяться дефектность слоя продукта реакции. Так как лимитирующей стадией
большинства рассматриваемых процессов является диффузия катионов через этот
слой, то скорость реакции в данном случае будет изменяться во времени в
соответствии с уравнениями F3(α),
F4(α)
и F5(α).
. Если синтез прекурсоров осуществлялся при
температурах выше температуры синтеза продукта реакции, то в полном
соответствии с теоретическими выкладками, такие процессы (также как процессы,
описанные в пункте 1) будут хорошо описываться уравнениями, основанными на
моделях параболического роста продукта реакции.
Необходимо напомнить, что все рассмотренные выше
уравнения диффузионной кинетики выведены в предположении об одинаковом размере
сферических частиц, образующих реакционную смесь. В реальных условиях это
положение трудно выполнимо и такие смеси, получаемые специальными методами,
имеют только теоретическое значение. Используемые же реальной технологией
порошки твердых фаз полидисперсны. Тогда скорость реакций, протекающих в таких
системах, максимальна на начальных стадиях процесса (максимальная площадь
реакционной зоны за счет большого числа мелких частиц) и затем снижается (по
мере расходования наиболее мелких фракций).
 τ/τ0,5
τ/τ0,5
Рис.8. Экспериментальные данные (точки) и
теоретическая кривая, рассчитанная по модели Картера-Валенси для процесса PbO
+ ZrO2 = PbZrO3
(полидисперсные порошки исходных фаз), α
- степень превращения, τ0,5 - время
полупревращения.
С другой стороны, совместная механическая
обработка порошков на начальной стадии процесса синтеза конечного продукта
приводит к их механохимическому взаимодействию до начала обжига смеси. Учитывая
выше сказанное, удивительным представляется тот факт, что уравнения, выведенные
в представлении о монодисперсности исходной шихты, оказались пригодными для
описания кинетики процессов с участием полидисперсных порошков. Предполагается
, что это связано с взаимной компенсацией влияния гранулометрического состава и
неоднородного развития реакционной поверхности. На рисунке 8 представлена
кривая, полученная на основе модели Картера-Валенси для реакции PbO
+ ZrO2 = PbZrO3,
на которую в виде точек наложены экспериментальные данные .
Как видно из рисунка согласование между теорией
и экспериментом практически идеальное, что свидетельствует о правильности
выбора кинетической модели.
.2 Модели процессов, лимитируемых реакциями на
границе раздела фаз
При высокой скорости диффузии частиц исходных
веществ через слой продукта локальные равновесия на границах раздела фаз
отсутствуют, и скорость процесса в целом лимитируется скоростью стадии
взаимодействия на границе раздела фаз. Эта же стадия лимитирует процесс и в том
случае, когда фаза продукта реакции не образует непрерывного слоя . В этом
случае скорость реакции определяется величиной доступной межфазной границы, и
процесс будет относиться к топохимическим. Вывод кинетических уравнений
обсуждаемых процессов основывается на следующих положениях: а) скорость
процесса лимитирует стадия взаимодействия на границе раздела фаз; б) скорость
реакции прямопропорциональна площади поверхности реагента, не вступившего в
реакцию; в) скорость зародышеобразования высока, поэтому поверхность каждой
частицы покрыта непрерывным слоем продукта реакции. Тогда при сферическом и дискообразном
(цилиндрическом) строении взаимодействующих частиц соответственно получаем:
F6 (α)
= 1 - (1 - α)⅓ = kсτ
; F7 (α)
= 1 - (1 - α)0,5 = kдτ
.3 Модели зародышеобразования
Как отмечалось выше, в данном случае
лимитирующей стадией взаимодействия в системе является образование или рост
зародышей продукта реакции на активных центрах, в роли которых могут выступать
поверхностные дефекты, выходы дислокаций на поверхность кристаллов, точечные
дефекты ассоциаты и кластеры. Так как мольные объемы прекурсоров и продуктов
реакций различны, образование ядер продукта реакции на поверхности прекурсора
приводит к деформации кристаллической решетки последнего, т.е. скорость в
данном случае предопределяется не только химическими, но и кристаллохимическими
факторами. Наиболее часто для описания обсуждаемых процессов используется
уравнение предложенное Багдасарьяном :
ln ln (1/ 1- α) = ln k - n ln
τ
где n
- параметр, предопределяющийся механизмом реакции, скоростью
зародышеобразования и геометрией зародышей (значение этого параметра для
различных случаев можно найти в справочниках и монографиях). Приведенное
уравнение может быть использовано для описания кинетики любых топохимических
процессов.
Анализ уравнений формальной изотермической
кинетики, применяющихся для описания скорости процессов взаимодействия между
твердофазными реагентами, показал, что большинство из них можно выразить одним
соотношением :
∂α/∂τz
= k (1 - βα)m
где k
- константа, значения которой предопределяются природой прекурсоров; β
- безразмерный фактор, близкий к единице; z
- величина, определяемая механизмом взаимодействия; m
- величина, зависящая как от механизма процесса, так и от формы
взаимодействующих частиц, она формально может рассматриваться как порядок
реакции и называется индексом реакции.
Анализ экспериментальных данных с использованием
указанного уравнения заключается в их проверке на сходимость при заданных
значениях z, β
и m (таблица 6). Для
этого экспериментальные значения α
сравниваются с теоретическими значениями этой величины, полученными с помощью
универсального уравнение с учетом возможных комбинаций значений z,
β и m,
представленных в таблице 6, что позволяет установить лимитирующую стадию
изучаемого процесса. На втором этапе, с учетом установленной кинетической
модели, используется одно из уравнений, описывающее конкретную лимитирующую
стадию (функции типа F(α)
приведенные выше).
Таблица 6. Соответствие значений z,
β и m
различным кинетическим моделям твердофазных реакций и формам частиц порошков
|
z
|
β
|
m
|
лимитирующий
процесс и формы частиц
|
|
1
|
1
|
2/3
|
на
границе фаз, сферы
|
|
1
|
1
|
1/2
|
на
границе фаз, иглы
|
|
1
|
1
|
0
|
на
границе фаз, тонкие диски
|
|
1/2
|
1
|
0
0,29 2/3 0,43
|
диффузия
|
|
> 1
|
1
|
1
|
зародышеобразование
|
Необходимо отметить, что все эти уравнения
относятся к изотермической кинетике, тогда как в ряде случаев (с практической
точки зрения) информацию о степени превращения в данный момент времени проще
получить, используя метод непрерывного нагрева. В частности это относится к
реакциям между твердыми фазами, при которых, наряду с целевым твердым
продуктом, происходит образование побочных газообразных веществ, например, BaCO3
+ TiO2 = BaTiO3
+ CO2↑ , для
которой степень превращения, с высокой точностью, можно оценить на основе
изменения массы образцов в процессе их нагрева. Для получения кинетической
информации в данном случае применяется метод ТГА и уравнения неизотермической
кинетики, например:
lg
AE/qR
= lg G(α)
- lg P(α)
где А - предэкспотенциальный множитель, Е -
энергия активации процесса, q
= ∂Т/∂τ, R
- газовая постоянная, G(α)
и P(α)
- функции, значения которых для наиболее распространенных кинетических
уравнений можно найти в работе (G(α))
или оценить методом подбора (P(α))
.
5. Активные прекурсоры
Анализ рассмотренных выше кинетических моделей
позволяют наметить пути совершенствования процессов синтеза твердых фаз и
решения ряд проблем в рамках основной задачи стоящей перед материаловедением -
создания твердотельных материалов с заданными свойствами. Среди этих проблем
можно назвать высокие температуры синтеза большинства перспективных (с
практической точки зрения) твердых фаз, что не только предопределяет высокую
энергоемкость этих процессов, но и приводит к нарушению состава продуктов
реакций за счет испарения из систем легколетучих компонентов, нежелательного разложения
прекурсоров и, как следствие, неконтролируемой дефектности образующихся фаз.
Очевидно, что возможны два способа решения указанных проблем: а) снижение
энергии активации лимитирующих стадий процессов твердофазного синтеза и б)
разработка новых способов формирования кристаллических фаз при низких
температурах.
Первый из указанных способов связан с изменением
состояния исходных фаз за счет варьирования их химической и термической
предыстории, т.е. целенаправленного создания прекурсоров, характеризующихся
заданными типом и концентрацией неравновесных дефектов, а также стабильностью
неравновесного состояния до заданной температуры. Рассмотрим перспективы
указанного пути решения, введя понятие нормального и активного состояния
твердых фаз. Нормальным называется состояние твердой фазы, если ее дефектность
равновесна, т.е. является однозначной функцией параметров состояния системы.
Активным называется состояние твердых фаз, характеризующееся наличием
неравновесных дефектов. Эти дефекты могут быть не одинаковы по своей природе и,
следовательно, различно влиять на энергию активации процессов с участием
дефектных фаз.
С термодинамической точки зрения мерой
активности твердой фазы может является присущий ей избыток энергии Гиббса по
сравнению с фазой того же состава и строения, находящейся в нормальном
состоянии:
∆Gизб.
= G*Т - GТ
где G*Т
и GТ энергия Гиббса
фазы в активном и нормальном состоянии соответственно.
Очевидно, что численное значение ∆Gизб
не может служить единственным критерием активности реагента в любом химическом
процессе, т.к. механизмы этих процессов (лимитирующие стадии) и условия, при
которых взаимодействуют реагенты, могут быть различны. Кроме этого, избыточная
энергия фаз может быть обусловлена наличием в них дефектов, имеющих различную
природу (свободная поверхность, статические искажения кристаллической решетки,
микронапряжения, дислокации и макроскопические дефекты типа межблочных границ,
точечные дефекты и их ассоциаты, протяженные дефекты и т.д.) и, следовательно,
характеризующихся различной термической стабильностью.
Необходимо также учитывать, что в
кристаллической решетке реальных фаз все виды неравновесных дефектов
присутствуют одновременно, влияя, как на свойства друг друга, так и на
химические свойства взаимодействующих реагентов в целом. Поэтому, как
отмечалось выше, для эффективного использования активных прекурсоров необходимо
разработать методы их получения, позволяющие синтезировать фазы с необходимым
доминирующим типом неравновесных дефектов при сохранении максимально высокой избыточной
энергии фаз вплоть до температур, при которых достигается энергия активации
проводимого процесса. При этом в ряде случаев, можно не разделять этапы
формирования активных фаз и процесс синтеза целевого продукта, т.е. можно
проводить активацию исходных фаз в процессе их взаимодействия.
Для объяснения механизма формирования активных
фаз и природы их относительной стабильности можно использовать теорию
пересыщения Рогинского и принцип ориентационного и размерного соответствия
Данкова-Конобеевского. В основу последнего принципа были положены
экспериментальные данные, полученные при исследовании топохимических процессов,
и показавшие, что твердая фаза, формирующаяся в результате протекания таких
реакции, испытывает влияние прекурсора. Это явление (получившее название
«память материи»), заключающееся в том, что при образовании новой фазы,
формирующие ее частицы, кристаллографически закономерно, располагаются по
отношению к частицам, образующим кристаллическую решетку исходной фазы. При
этом понижение свободной энергии системы за счет такой взаимной ориентации
будет максимальным, при минимальном различии в строении соприкасающихся граней
кристаллов новой и исходной фаз. Основываясь на последнем положении, условие
ориентированной кристаллизации можно выразить неравенством:
A + E
- Z  A1
A1
где А и Е - соответственно работа образования и
энергия двумерного зародыша новой фазы при ориентированной кристаллизации, Z
- работа сил адгезии (деформационная составляющая), A1
- работа образования трехмерного зародыша при неориентированной кристаллизации.
Расчеты показывают, что ориентированная
кристаллизация в таких системах возможна, если параметры элементарных ячеек
сопрягающихся решеток отличаются не более чем на 18%. Очевидно, что за счет
ориентированной кристаллизации кристаллическая решетка продукта реакции
искажается и эти искажения, приводящие к росту свободной энергии системы,
максимальны в случае максимального различия в строении соприкасающихся фаз.
Согласно теории Рогинского активность твердой
фазы предопределяется условиями ее получения: G*Т
фазы тем выше, чем дальше от состояния равновесия находилась система в момент
формирования ее кристаллической решетки, т.е чем выше степень пересыщения
(отклонение от равновесного значения) одного или нескольких параметров
состояния системы. На основании данного определения можно выделить несколько
источников пересыщения:
. Фазовые пересыщения системы, в состав которой
входят термодинамически метастабильные полиморфные или аморфные модификации
прекурсоров. Например, смесь γ-Al2O3
c CaCO3
имеет фазовое пересыщение за счет наличия в ее составе метастабильной формы
оксида алюминия, которая при нагревании способна необратимо превращаться в
стабильную кристаллическую форму - корунд.
Пересыщения связанные со структурным
несовершенством кристаллов, с высокой скоростью формирующихся в процессе
распада метастабильной фазы, например, в процесс резкого охлаждения паров или
при кристаллизации из пересыщенного раствора.
. Пересыщения обусловленные изменением
химического потенциала одного из компонентов фазы в приделах области ее
гомогенности, например, TiO2-x
со структурой рутила, образующийся в процессе разложения титанилоксалата
взаимодействует с карбонатами щелочноземельных элементов с большей скоростью,
чем TiO2-x
полученный разложением титанилнитрата в связи с различной концентрацией
дефектов в указанных фазах.
. Пересыщения вызываемые увеличением суммарной
площади поверхности (поверхностной энергии) фазы по мере уменьшения объема
образующих ее частиц порошка (при неизменном количестве вещества в системе).
Количественной характеристикой этого типа пересыщения является избыточная
энергия образца заданной степени дисперсности по сравнению с равным по массе
монокристаллом того же состава и структуры.
Анализ источников пересыщения позволяет сделать
вывод, что за счет изменений условий синтеза фазы можно регулировать вид и
степень ее пересыщения и, следовательно, создавать прекурсоры с заданным
набором физико-химических и гранулометрических характеристик. С другой стороны
необходимо учитывать, что синтезируемая фаза в свою очередь сохраняет некоторые
характеристики фаз, с использованием которых она получена (см. раздел
«Диффузионные модели»), т.е. влияние предыстории на активность фаз может
проявляться не только в первом, но и во втором «поколении».
.1 Степень активности твердых фаз и методы ее
оценки
Как отмечалось выше, условной мерой активности
твердой фазы может быть присущее ей значение избыточной энергии Гиббса. С
другой стороны в большинстве случаев более наглядной является информация,
показывающая во сколько раз (с термодинамической точки зрения) данная фаза
более активна по сравнению с аналогичной фазой, находящейся в нормальном
состоянии. Ответить на данный вопрос позволяет величина, получившая название
«степень активности» (γ), являющаяся
отношением ∆Gобразования
активной фазы к ∆Gобразования
«нормальной» фазы: γ = ∆Gоб.ак
/∆Gоб.н.. Очевидно,
что уменьшение значения (γ) означает
рост активности фазы заданного состава. Оценку данной величины можно провести
методом, основанным на экспериментальном определении избыточной энтальпии
растворения активных и «нормальных» форм:
А* + растворитель → раствор + ∆Н1
А + растворитель → раствор + ∆Н2
А* → А + ∆Низб. (∆Низб = ∆Н1
- ∆Н2 ),
где А* и А активная и «нормальная» фазы,
соответственно.
Значение ∆Низб, конечно, нельзя
рассматривать как полную характеристику активного состояния фазы, т.к. ∆Gизб.=
∆Низб - Т∆Sизб
и значение ∆Sизб в ряде
случаев может быть значительной величиной . Однако, в большинстве случаев, ориентировочную
оценку активности фазы можно сделать, используя найденные значения ∆Низб.
Также для термодинамической оценки активности
твердых оксидных фаз используют значения избыточной энергии Гиббса кислорода ∆Gизб.о2,
которую определяют, измеряя э.д.с. гальванических ячеек типа:
Pt, активная
оксидная фаза │ZrO2(CaO)│O2
(Po2 = 2∙104
Па), Pt
Pt, «нормал.»
оксидная фаза │ZrO2(CaO)│O2
(Po2 = 2∙104
Па), Pt, тогда: ∆Gизб.о2
= 4F(Е2 - Е1), где Е2 и
Е1 - э.д.с. гальванической ячейки с «нормальной» и активной оксидными фазами,
соответственно. Метод э.д.с. можно применять для оценки активности любой
твердой фазы, в том числе и безкислородной, но в каждом конкретном случае
необходимо использовать твердый электролит, проводимость которого обусловлена
переносом ионов, входящих в состав исследуемых твердых фаз.
Метод контактной вольтамперометрии основан на
исследовании зависимостей электродных превращений оксидных фаз (находящихся в
контакте с твердым электродом или жидким электролитом) от их структуры и
степени дефектности . Анализ вольтамперных кривых восстановления и окисления
ряда оксидов d-элементов позволил
сделать вывод, что в условиях проведения опытов химические процессы
осуществляются непосредственно в твердой фазе и их характер предопределяется
наличием в решетке вакансий по генерируемым в процессе окисления
(восстановления) ионам. В частности, зарегистрированные в работе различия в
электрохимическом поведении γ- и
α-форм
Fe2O3
являются следствием различной концентрации дефектов в исследованных фазах, что,
после построения градуировочной кривой, позволяет с помощью указанного метода
количественно оценивать содержания γ- и
α-форм
в образцах Fe2O3.
Положение о связи определенного вида дефектов с характером
окислительно-восстановительных процессов в твердой фазе можно проиллюстрировать
и данными работы , в которой были исследованы процессы электровосстановления
фаз, содержащих ионы железа в различных степенях окисления: магнетита, вюстита,
γ-,
α-форм
Fe2O3
и гексаферрита бария. Согласно вольтамперным кривым в случае α-
Fe2O3
и гексаферрита бария, входящие в их состав ионы железа в процессе электролиза
восстанавливались до металла, тогда как во всех остальных фазах этого не
наблюдалось. Различие в строении указанных групп фаз заключается в том, что в α-
Fe2O3
и гексаферрите бария основной тип дефектов - междоузельные ионы, которые и
восстанавливаются до металла, а в магнетите, вюстите и γ-
Fe2O3
- вакансии, и, следовательно, в данном случае процесс восстановления ионов
железа невозможен. Аналогичные результаты были получены при исследовании
различных форм диоксида титана и фаз со структурой перовскита - в частности,
если анатаз легко восстанавливается до металла в электрохимическом процессе с
участием твердой фазы, то рутил в указанных условиях до металла не восстанавливается
[33]. Таким образом, вольтамперный метод позволяет определять тип дефектов в
кристаллической решетке и их относительную концентрацию.
В связи с тем, что понятие активности фазы
связано с повышенной концентрацией неравновесных дефектов, а сами дефекты при
достаточно высокой температуре ионизированы, очевидно, что любая активная фаза
по сравнению со стабильной характеризуется повышенной концентрацией электронов
или дырок. Оценить избыточную концентрацию элементарных носителей можно путем
измерения термо- э.д.с. образцов во времени в процессе изо- или
политермического обжига, что позволяет получить информацию о скорости
аннигиляции наиболее подвижных дефектов и времени перехода активной фазы в
стабильную в условиях поведения исследования. Обработка экспериментальных
данных ведется путем построения кривых в координатах х - τ
(для
изотермического режима) или х - Т (для политеримического режима), где х = (αо
- α1)/(αо
- αк)
и αо,
α1
- термо-э.д.с.
в начальный момент времени и в момент времени τ1, αк-
термо-э.д.с. стабильной фазы, полученной длительным обжигом исходной фазы при
высокой температуре. Например, анализ результатов исследования термо-э.д.с
образцов гематита, полученных при термическом разложении сульфата и нитрата
железа (III) (рис.9),
показывает, что степень аннигиляции неравновесных дефектов в образцах, имеющих
одинаковую химическую и термическую предысторию, зависит, как от температуры,
так и от времени термообработки (рис. 9а), а для образцов, синтезированных при
сходных условиях термообработки с использованием различных прекурсоров - от
природы прекурсора (рис. 9б).
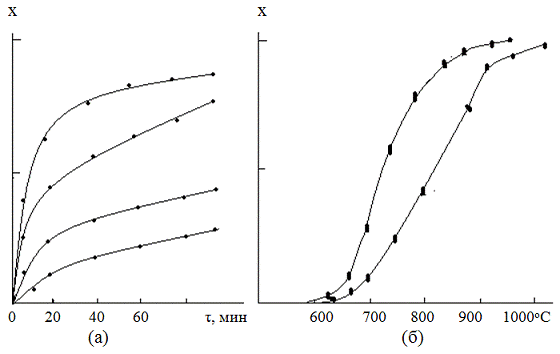
Рис. 9 Кривые изотермического (а) и
политермического (б) отжига неравновесных дефектов в гематите, полученном (а)
при разложении нитрата железа (III)
- (температуры отжига для кривых (а) снизу вверх: 680, 710,740,770оС ), а также
при разложении нитрата (левая кривая на рис.б ) и сульфата (правая кривая на
рис.б ) железа (III).
Отмеченные на рисунках значения Х составляют 0,5 и 1.
Еще одним свойством, которое в определенной
степени связано с концентрацией неравновесных атомных и электронных дефектов в
кристаллической решетке, является каталитическая активность твердых фаз.
Согласно современной теории гетерогенного катализа каталитическая активность
фазы предопределяется наличием на ее поверхности активных центров - областей с
повышенной напряженностью электрического и магнитного полей. В свою очередь,
одной из причин формирования указанных центров, является нарушение
кристаллохимического строения поверхности твердой фазы. Это нарушение может
быть вызвано, как изменением координационных чисел поверхностных атомов или
ионов, так и наличием в поверхностном слое вакансий, дислокаций, ступеней роста
и т.д..
В связи с тем, что каталитические реакции, связанные
с переносом электронов, подразделяются на донорные и акцепторные влияние
дефектов на каталитические свойства твердых фаз может быть различным: а)
варьирование концентрации и природы дефектов изменяет электронное состояние
системы (положение уровня Ферми - т.е. термодинамический потенциал электрона на
поверхности кристалла) - коллективный эффект, б) дефекты могут принимать
участие в сорбционном и каталитическом акте т.е. выступать в роли активного
центра - локальный эффект. В реальных системах одновременно реализуются оба
указанных эффекта, один из которых доминирует над другим, поэтому положение
уровня Ферми практически не определяет каталитическую активность фазы, если в
системе доминирует локальный эффект. В частности, это наблюдается при значительных
дислокационных нарушениях структуры (локализация электронов поверхностной зоны
в местах выхода дислокаций на поверхность т. е. образование активного центра).
Таким образом, можно сделать вывод, что рост каталитической активности фазы
свидетельствует об увеличении ее дефектности, однако определить роль каждого из
возможных типов дефектов в изменении свойств изучаемой фазы только на основе
данных по ее каталитической активности в большинстве случаев невозможно. В
качестве модельных реакций для оценки степени дефектности оксидных фаз
предложены: окисление СО кислородом, термическое разложение N2O,
одноатомных спиртов, KClO3
и ряд других.
К прямым методам определения, как точечных, так
и протяженных дефектов различного типа относится метод рентгеноструктурного
анализа (РСА), основанный на обсчете физического уширения дифракционных
максимумов, который осуществляется методами аппроксимации, моментов или
гармонического анализа формы максимумов на дифрактограммах (ГАФРЛ). В
частности, при использовании наиболее распространенного метода ГАФРЛ,
дифрактограмму рассматривают как проекцию сечения обратной решетки, которая по
отношению к кристаллу является Фурье-пространством, преобразование в котором
распределения интенсивности позволяет судить о нарушениях кристаллической
структуры, вызвавших размытие этих узлов. Использование указанного метода
позволяет рассчитать изменение среднестатистических смещений в отдельных
подрешетках и, следовательно, установить тип доминирующих дефектов в
исследуемых фазах, определить размер областей когерентного рассеивания (размер
кристаллических блоков), вероятность образования двойниковых и деформационных
дефектов упаковки, а также размер микронапряжений.
К относительно простым (с экспериментальной
точки зрения) методам оценки активности твердых фаз можно отнести методы
измерения истинной теплоемкости, усадки прессзаготовок при непрерывной
термообработке и свободной поверхности порошков. В первом из них сравниваются
кривые экспериментальных зависимостей Cp
= f(T)
двух твердых фаз, имеющих одинаковый качественный и количественный состав, одна
из которых находится в нормальном, а другая в активном состоянии. В этом случае
сопоставление кривых первичного нагрева с последующими этапами термообработки
дает возможность установить область температур, в которой происходит
аннигиляция дефектов, измерить тепловой эффект отжига дефектов и их
относительную стабильность. Например, у металлов, подвергнутых механической
деформации, при нагревании в калориметре наблюдаются два максимума на кривой
тепловыделения, один из которых соответствует отжигу дислокаций, а другой -
аннигиляции точечных дефектов. Так как энергии активации этих процессов
различны, они реализуются в различных температурных интервалах . Однако этот
метод не позволяет установить тип дефектов, предопределяющих различие значений Cp
для активной и нормальной фаз. В связи с этим правильная интерпретация
результатов исследований возможна только с учетом данных по типу доминирующих
дефектов, полученных, например, методом РСА.
Экспериментальные результаты по усадке образцов
прессзаготовок математически обрабатываются путем построения зависимостей Ω
= ∂(∆L/L)/∂T
(рис.10), где Ω - скорость усадки
прессзаготовки , L и ∆L
- исходный линейный размер и его изменение при достижении системой заданной
температуры.
Анализируя представленные данные, можно сделать
вывод, что скорость усадки прессзаготовок NiO
является функцией трех параметров - температуры системы, скорости нагрева, а
также химической и термической предыстории оксидной фазы. Так увеличение скорости
нагрева способствует быстрой аннигиляции термически нестабильных дефектов, что
приводит к снижению скорости диффузии и, следовательно, к минимальной усадке
образцов при достижении фиксированной температуры (рис. 10а). С ростом
термической стабильности неравновесных дефектов удается сохранить их высокую
концентрацию при температурах, отвечающих интенсивному спеканию, и в
значительной степени нивелировать роль скорости нагрева (рис. 10б).
В то же время термическая стабильность
неравновесных дефектов зависит то состава фазы, подвергающейся разложению, и от
условий проведения этого процесса (рис. 10 а и б). С практической точки зрения
полученные результаты свидетельствуют о том, что максимальной плотностью будет
характеризоваться керамика, изготовленная из порошкообразной фазы, имеющей
максимальную концентрацию термически стабильных неравновесных дефектов, т.е.
полученной разложением термически стабильного прекурсора при температуре
близкой к температуре спекания продукта разложения. В частности, плотность керамики,
изготовленной при одинаковых температурах спекания из «сульфатного» NiO
в среднем на 10% выше, чем из «оксалатного» NiO.
Рис. 10. Скорость усадки прессзаготовок оксида
никеля полученного разложением (а) - оксалата никеля при 300оС (время
изотермической выдержки 8 часов) с дополнительной термообработкой при 900оС (2
часа) и (б) - сульфата никеля при 900оС (8 часов). Скорость нагрева для кривых
слева направо: 3; 20; 100 оС/мин.
В связи с тем, что любая поверхность твердой
фазы является высокодефектной, в случае порошков, характеризующихся высокой
степенью дисперсности, наблюдается изменение многих физико-химических свойств
твердых фаз, в том числе температуры плавления (таблица 7) , давления паров и
растворимости
Таблица 7. Зависимость температуры плавления
порошкообразных металлов от размеров частиц порошка
|
r, нм
|
Тпл.
оС
|
|
К
(Тпл.= 63 оС)
|
Ag (Тпл.= 960 оС)
|
Pt (Тпл.= 1673 оС)
|
|
20
|
-
223
|
320
|
634
|
|
30
|
-
129
|
527
|
953
|
|
40
|
-
80
|
637
|
1158
|
|
50
|
-
51
|
700
|
1233
|
|
100
|
6
|
837
|
1528
|
Изменение указанных свойств также можно
использовать для характеристики активности высокодисперсных фаз.
.2 Способы активирования твердых фаз
Реакционную способность твердых фаз можно в
широких пределах варьировать за счет изменения качественного и количественного
состава, а также строения прекурсоров, используемых при синтезе искомого
продукта реакции. В связи с тем что, реакционная способность фаз
предопределяется концентрацией в них неравновесных дефектов, на ее величину оказывают
влияние так же: а) условия проведения процесса синтеза твердых фаз, б)
концентрация и вид микропримесей, в) механическое, радиационное или химического
воздействие и т.д..
Все эти приемы позволяют получать активные фазы,
характеризующиеся различной природой и концентрацией неравновесных дефектов,
которые (наряду с равновесными дефектами) определяют поведение фаз в любых
твердофазных процессах (взаимодействие нескольких твердых фаз, спекание,
рекристаллизация, разложение и т.д.). В связи с тем, что эти превращения могут
лимитироваться различными стадиями, приемы активирования фаз не могут быть
универсальными. Это означает, что значительное снижение энергии активации одной
группы процессов за счет использования той или иной активной фазы не
гарантирует, что эта же фаза будет способствовать снижению энергии активации
других процессов с ее участием. Использование активной фазы будет эффективным
только в том случае, если в ней в процессе синтеза сформировались неравновесные
дефекты заданного типа и их концентрация в системе достаточна для обеспечения
высокой скорости процесса получения целевого продукта. Следовательно, приступая
к выбору приема активации фазы, необходимо предварительно получить ответ на три
основных вопроса:
а) какая стадия лимитирует процесс?;
б) каков ее механизм и, следовательно,
изменением концентрации дефектов какого типа можно регулировать ее скорость?;
в) как получить активную фазу с достаточной
концентрацией неравновесных дефектов заданного типа, устойчивых при температуре
проведения основного процесса?
Подход к решению двух первых вопросов был
рассмотрен выше, остановимся на способах активации фаз и их возможностях в
плане влияния этих способов на природу и концентрацию неравновесных дефектов.
.3 Влияние состава прекурсора и условий получения
на активность синтезируемой фазы
Модельные процессы, используемые для оценки
активности твердой фазы, условно можно разделить на три группы:
а) процессы протекающие на поверхности твердой
фазы и предопределяющиеся, преимущественно, площадью этой поверхности, а также
степенью ее несовершенства ( катализ, сорбция и т.д.);
б) процессы, сопровождающиеся полным разрушением
структуры фазы (реакции окисления, восстановления, соединения и разложения без
изменения степеней окисления атомов или ионов, входящих в состав фазы,
растворение фаз в растворах кислот или щелочей и т.д.);
в) процессы, в результате которых не происходит
существенного изменение качественного или количественного состава фазы, но
наблюдается упорядочение ее структуры (спекание, первичная и вторичная
рекристаллизация).
Высокая скорость процессов первой группы может
быть обеспечена только при сочетании большой суммарной площади поверхности
образца с высокой степенью кристаллохимического несовершенства его свободной
поверхности. Используя представления о влиянии температуры на процесс
аннигиляции дефектов, а также теорию пересыщения Рогинского, можно сделать
вывод, что указанным условиям будут отвечать фазы, синтезированные при
минимальных температурах в условиях высокого пересыщения по любому параметру
системы (высокая скорость нагрева, разлагающегося прекурсора; выделение искомой
фазы из пересыщенного раствора; синтез в условиях значительного градиента
температуры, давления или концентрации и т.д.). При этом время воздействия на
систему фактора, способного снижать концентрацию неравновесных дефектов, должно
быть минимальным (например, изотермическая выдержка при температуре разложения
исходного реагента). Определенное значение при получении активной фазы в
процессе термического разложения прекурсора должен иметь и температурный
интервал, в котором протекает реакция разложения - чем он меньше, тем более
вероятно образование фазы с высокой концентрацией неравновесных дефектов за
счет высокой скорости ее кристаллизации.
Еще одним условием высокой активности фазы в
процессах первого типа является температура, при которой осуществляется катализ
или сорбция - она должна быть существенно ниже, чем температура, при которой
происходило формирование активной фазы. В противном случае активность фазы быстро
снижается во времени за счет аннигиляции неравновесных дефектов. В качестве
примеров рассмотрим свойства оксидных фаз, синтезированных в различных условиях
из нескольких видов прекурсоров. Так Fe2O3
можно получить термическим разложением солей кислородных кислот или гидроксида:
Fe(NO3)3∙9H2O
→
плавление и частичный гидролиз при Т< 100oC
→
удаление из системы воды в интервале температур 100-150оС → формирование
аморфного Fe2O3
при 150-240оС → образование кристаллического α-
Fe2O3
в интервале температур 250-350оС;
FeC2O4∙2H2O + O2 = γ- Fe2O3 +
2CO2 + 2H2O (T = 200-350oC)
γ-
Fe2O3 → α-
Fe2O3 (T = 450-540 oC);O3 ∙х
H2O →2α-FeOOH + (x-1) H2O
(T = 50-140 oC)
α-FeOOH → α-
Fe2O3 + H2O (T = 290-380 oC)
(в интервале температур T
= 290-350 oC в образцах
одновременно содержатся и α- и
γ-
формы
Fe2O3).
Анализ представленных экспериментальных данных
показывает, что формирование кристаллического Fe2O3
в первой системе протекает из образующейся на первом этапе разложения аморфной
формы в условиях низкой степени пересыщения в широком интервале температур. Это
способствует кристаллизации α-Fe2O3,
характеризующейся низкой дефектностью. Одновременно, окислительная атмосфера,
создаваемая газообразными продуктами разложения, исключает высокую дефектность
продукта реакции по анионной подрешетке. Поэтому, несмотря на высокую удельную
поверхность синтезированной фазы, ее каталитическая и адсорбционная активность,
а также способность к спеканию невелики.
Аналогичные свойства имеет и Fe2O3,
полученный по способу (в): термические условия образования фаз в системах (а) и
(в) сходны, однако в последней системе отсутствует окислительная атмосфера. Это
способствует формированию в системе (в) на первом этапе γ-Fe2O3
с высокой дефектностью анионной подрешетки, а ее превращение в α-Fe2O3
протекает в узком температурном интервале, что способствует сохранению
достаточно высокой дефектности и у конечного продукта реакции. В связи с этим α-Fe2O3
«гидроксильный» при сравнимой степени дисперсности имеет несколько большую
сорбционную и каталитическую активность и лучшую спекаемость, чем «нитратный» α-Fe2O3.
И, наконец, в системе (б) Fe2O3
формируется при сравнимых с системами (а) и (в) температурных условиях, но в
восстановительной атмосфере. Это предопределяет высокую концентрацию
неравновесных дефектов сначала у γ-
Fe2O3
(который по этой причине сохраняется в системе вплоть до 540оС), а при более
высоких температурах синтеза и у α-Fe2O3,
образующегося из промежуточного продукта в процессе монотропного перехода.
Поэтому, несмотря на бóльший размер
частиц порошка «оксалатного» Fe2O3
(следствие высокой скорости формирования этой фазы в условиях средней степени
пересыщения), эта форма (из рассматриваемых форм) оказывается наиболее активным
для модельных процессов первого типа.
Эффект скорости нагревания и его
продолжительности можно рассмотреть на том же примере α-Fe2O3,
образующегося при разложении сульфатов и оксосульфатов железа (II
и III. Исследования этих
фаз показали, что максимальными каталитическими и сорбционными свойствами
обладают образцы α-Fe2O3
полученные в результате разложения соли Мора в условиях резкого роста
температуры обжига и минимальной конечной изотермической выдержке. По этим
свойствам им не уступают и продукты непрерывного горения смеси частично
обезвоженного сульфата и нитрата железа (III)
с глюкозой. Обращает на себя внимание, что наряду с кратковременностью
температурного воздействия на систему, в рассматриваемых примерах процесс
формирования фаз протекал в присутствии восстановителей (в первой системе -
аммиака, а во второй - углерода и СО), что также способствовало формированию
кристаллической решетки α-Fe2O3
с высокой концентрацией неравновесных дефектов.
Скорость процессов второй группы также
предопределяется суммарной площадью свободной поверхности фазы, обратно
пропорциональной размерам ее телесных комплексов, и, в отличие от процессов
первой группы, объемной концентрацией неравновесных дефектов в отдельных
частицах.
Из данных экспериментов по свойствам Fe2O3
и TiO2 следует, что
понятие «активная фаза» (с учетом сформулированных выше условий) для
конкретного процесса предопределяется еще двумя факторами: а)температура
процесса с участием активной фазы не должна превышать температуру ее синтеза
более, чем на 100-150оС; б) сама активная фаза должна быть синтезирована при минимальной
температуре, отвечающей разложению, окислению или восстановлению прекурсора.
Например, образцы TiO2
были получены разложением ряда прекурсоров с фиксированной скоростью подъема
температуры в системе (50оС/час):
а) TiOSO4∙2Н2О→
TiO2 + SO2
+ ½O2 + 2Н2О - согласно
данным ДТА и ТГА процесс протекает в несколько стадий (дегидратация, гидролиз,
испарение газообразных продуктов реакции) в интервале температур 70 - 865оС в
окислительной атмосфере (основная форма рутил с примесью анатаза);
б) H2[TiO(C2O4)2]∙2Н2О
→ TiO2 + 3Н2О + CO2
+ CO - процесс также
протекает в несколько стадий, но в более узком температурном интервале (50-
470оС) - основная форма - анатаз;
в) TiO2∙хН2О
→ TiO2 + хН2О -
гидроксид титана был осажден из 0,5М (в пересчете на TiO2)
нитратного раствора, путем добавления последнего в 10% раствор аммиака, т.е. в
условиях высокого пересыщения. Аморфная форма TiO2
в этих условиях образуется при 150-200оС (конечная температура дегидратации
зависит от скорости нагрева). Эта форма с высокой скоростью кристаллизуется при
280- 290оС (инициирует кристаллизацию процесс разложения нитрата аммония,
который является побочным продуктом разложения гидроксида титана).
Кристаллизация TiO2
и разложение NH4NO3
являются экзотермическими процессами, что приводит резкому и кратковременному
(в пределах нескольких минут) подъему температуры в системе до 650-800оС и
образованию смеси двух полиморфных модификаций TiO2
(анатаз с примесью рутила).
По данным РСА и электронной микроскопии
минимальный размер частиц TiO2
в порошках, синтезированных по реакциям (б) и (в): ОКР от 22 до 50 нм,
агломераты до 340 нм, тогда как частицы «сульфатного» TiO2
имеют размер порядка нескольких мкм.
«Оксалатный» TiO2
имеет высокую активность в реакциях, протекающих при низких температурах - он
характеризуется наибольшей растворимостью в концентрированной серной кислоте
при 60-90оС, максимальной скоростью взаимодействия с PbO,
Bi2O3,
карбонатами элементов главной подгруппы первой группы вплоть до температур
изотермического обжига порядка 600оС. В то же время, его реакции с карбонатами
кальция, стронция и бария, для активации которых требуется температура порядка
1000оС, полностью не завершаются даже после 20 часового изотермического обжига
при 1000оС, что свидетельствует о значительной степени аннигиляции
неравновесных дефектов в интервале температур 600 - 1000оС.
«Гидроксидный» TiO2,
формирующийся фактически в процессе «теплового удара», также имеет высокую
концентрацию неравновесных дефектов, вызванных высокой скоростью процесса
кристаллизации. Однако, в отличие от предыдущего случая, когда образование
кристаллической решетки фазы происходит в восстановительной атмосфере
содержащей СО, кристаллический TiO2
в рассматриваемой системе формируется в присутствии окислителей (N2O
и О2 ), что исключает высокую дефектность анионной подрешетки продукта реакции.
В связи с этим природа, а следовательно, и термическая стабильность
неравновесных дефектов у двух рассматриваемых типов образцов TiO2,
будет различной: неравновесные дефекты вызванные частичным восстановлением фазы
«оксалатного» TiO2 будут
быстро исчезать при нагревании образцов на воздухе (скорость аннигиляции
предопределяется скоростью диффузии сорбирующегося кислорода в объем
кристалла), тогда как неравновесная дефектность «гидроксидного» TiO2,
предопределяющаяся (помимо свободной поверхности, которая у двух типов образцов
примерно одинакова) статическими искажениями решетки, микронапряжениями,
дислокациями и макроскопической дефектностью типа блочных границ, имеет
значительно большую энергию активации процесса аннигиляции и, следовательно,
сохраняется при более высоких температурах.
В связи с этим, как следует из экспериментальных
данных: а) растворимость «гидроксидного» TiO2
в концентрированной серной кислоте ниже, чем у «оксалатного» TiO2;
б) скорости взаимодействия обоих типов образцов с PbO,
Bi2O3,
карбонатами элементов главной подгруппы первой группы при 600оС приблизительно
одинаковы, а при 750оС скорость этих же процессов с участием «гидроксидного» TiO2
в 1,8- 4,3 раза выше, чем для «оксалатной» формы. Успешно «гидроксидный» TiO2
был использован и для синтеза титанатов кальция и стронция ( 1,8 и 3,5 часа
обжига при 1030оС соответственно). При этом было отмечено, что если температура
синтеза поднималась выше оптимальной, скорость образования продукта реакции
резко снижалась. По-видимому, именно аннигиляция дефектов является причиной
низкой эффективности «гидроксидного» TiO2
при синтезе титаната бария: установлено , что при 1030оС скорость реакции
образования титаната бария в этой системе низкая за счет высокой
термодинамической стабильности ВаСО3, а увеличение температуры обжига
значительно снижает реакционную способность TiO
Наибольшая скорость реакции TiO2
+ ВаСО3 = ВаTiО3 + СО2
зафиксирована при использовании «сульфатного» TiO2
(время обжига 3 часа при 1180оС). Важно отметить, что увеличение температуры
системы в этом случае вплоть до 1300оС практически не изменяет скорость
процесса т.к. рост скорости диффузии за счет увеличении температуры
компенсируется снижением активности оксида титана в связи с аннигиляцией
неравновесных дефектов. Очевидно, что высокая термическая стабильность этих
дефектов обусловлена способом синтеза «сульфатного» TiO2:
максимальный из рассмотренных типов TiO2
температурный интервал формирования фазы и самая высокая конечная температура
обжига.
Для высоких скоростей процессов спекания и
рекристаллизации необходима высокая скорость как объемной, так и поверхностной
диффузии, т.е. исходные порошки должны характеризоваться значительной объемной
и поверхностной концентрацией неравновесных дефектов и, следовательно, их
формирование должно протекать в предельно неравновесных условиях. Из
рассмотренных выше образцов в наибольшей степени таким условиям удовлетворяет
«гидроксидный» TiO2,
с использованием которого при Т = 1250оС (τобжига=
2 часа) можно получить крупнозернистую керамику TiO2
с плотностью ≈ 85% от теоретически возможной. Установлено, что увеличение
температуры обжига или его продолжительности не позволяет получить керамику бóльшей
плотности при использовании указанной шихты. Для изготовления мелкозернистой
керамики с плотностью ≥ 93% наиболее эффективным является «сульфатный» TiO2,
частицы которого имеют максимальную кажущуюся плотность и минимальную объемную
дефектность, однако использование такой шихты повышает температуру спекания на
150-200оС и время обжига в 2-3 раза.
Аналогичные зависимости скоростей низко- и
высокотемпературных процессов с участием образцов Fe2O3,
синтезированных с использованием различных прекурсоров, была установлена при
изучении кинетики их растворения в кислотах, а также кинетики процессов
формирования фаз со структурой шпинели.
.4 Повышение активности твердых фаз методом
легирования
Прием, позволяющий изменить активность твердой
фазы путем введения в систему микродобавок, называется легированием или
модифицированием. Роль добавки в системе предопределяется ее качественным и
количественным составом, кристаллохимическим строением, концентрацией,
характером взаимодействия с основной фазой (растворение или поверхностные
процессы) и равномерностью распределения в объеме или на поверхности телесных
комплексов матрицы. Причины, приводящие к изменению свойств фаз в процессе их
легирования, могут быть связаны:
а) с изменением объемной концентрации точечных и
протяженных дефектов (фактор позволяющий варьировать скорости диффузионных
потоков в объеме взаимодействующих частиц и, следовательно, предопределяющий
скорость процессов, сопровождающихся полным разрушением исходной фазы, скорость
полиморфных превращений и т.д.);
б) с ростом концентрации примесных атомов или
ионов в поверхностном слое основной фазы (фактор, влияющий на подвижность
дислокаций и структуру реакционной зоны, а, следовательно, на скорость
начальных стадий твердофазного взаимодействия, усадку при спекании,
каталитические, сорбционные и другие свойства).
Кроме этого сорбированные частицы способны
препятствовать рекристаллизации материалов (за счет блокирования диффузионных
потоков от мелких к крупным кристаллам) или наоборот усиливать ее за счет
образования высокодефектных промежуточных продуктов в поверхностных слоях.
Таким образом, в проблеме определения роли добавки в процессе формирования фазы
можно выделить термодинамический и кинетический аспекты: способна ли добавка
растворятся в основной фазе и если способна, то с какой скоростью.
Обсуждая роль легирующего компонента по
отношению к фазе определенного состава и кристаллохимического строения,
необходимо учитывать особенности технологических приемов, используемых при
легировании, от которых зависит равномерность распределения добавки в объеме
системы. Из известных в настоящее время методов, обеспечивающих равномерное
распределение малорастворимых добавок по поверхности частиц легируемой фазы
можно назвать: а) пропитку порошков растворами солей, при термическом
разложении которых, на поверхности частиц порошка формируется заданное
количество вводимой в систему добавки; б) обработку порошков газообразными
реагентами, с последующим гидролизом и термолизом продуктов сорбции; в)
использование для этих же целей аэрозолей и лиозолей (как самой легирующей
фазы, так и ее прекурсоров). Эти же методы эффективны и в случае объемного
легирования, если скорость растворения добавки в исходной фазе достаточно
велика. Высокую степень гомогенности конечной системы, практически независимо
от скорости растворения фаз друг в друге, можно достичь, используя методы
соосаждения из пересыщенных растворов, совместной кристаллизации,
криохимическим методом и т.д..
Прямых способов, позволяющих оценить степень
однородности распределения добавки в системе в настоящее время неизвестно.
Однако, если исследуемое вещество имеет несколько полиморфных модификаций, а
фазовый переход приводит к исчезновению или появлению каких либо свойств,
поддающихся прямому измерению, то однородность состава телесных комплексов фазы
можно оценить по температурному интервалу фазового перехода - чем больше этот
интервал тем выше степень флуктуации состава отдельных частиц системы и тем
ниже эффективность использованного при легировании технологического приема.
Влияние способа введения микродобавок на скорость взаимодействия в конкретной
системе предопределяется природой лимитирующей стадии процесса. Например,
скорость реакции BaCO3
+ TiO2 = BaTiO3
+ СО2 можно значительно увеличить путем формирования на поверхности частиц TiO2
высокодефектных слоев, за счет легирования поверхности этих частиц оксидами
кобальта или марганца, тогда как введение того же количества добавки в объем
частиц, практически не изменяет скорость указанного взаимодействии при
фиксированных режимах обжига . Кинетика реакций подобного типа лучше всего
описывается моделью зародышеобразования , при этом за счет увеличения
концентрации внедренных ионов по мере роста степени нестехиометрии рутила,
вызванной растворением в исходной фазе добавок, подвижность ионов Ва2+
снижается, при одновременном уменьшении энергии активации процесса в целом,
что, суммарно, мало изменяет скорость рассматриваемого взаимодействия при
объемном легировании. В свою очередь, поверхностное легирование способствует
протеканию рассмотренных выше процессов формирования реакционной зоны и
возникновению промежуточных продуктов с высокой дефектностью, что увеличивает
скорость диффузионных потоков в приграничных областях. Судя по
экспериментальным данным, именно эти стадии являются лимитирующими для
рассматриваемого процесса (по крайней мере, на начальных этапах синтеза), что и
способствует увеличению его средней скорости при поверхностном легировании.
Если лимитирующая стадия массопереноса связана с сопряженной диффузией ионов
(например, сопряженная диффузия Рb2+
и О2- по вакансионному механизму для реакций РbО
+ ЭO2 = PbЭO3
где Э = Ti, Zr)
объемное легирование TiO2
способствует формированию продукта с повышенной концентрацией вакансий, как в
катионной, так и в анионной подрешетках, за счет чего средняя скорость процесса
увеличивается. При этом внедренные ионы в оксиде титана не оказывают
существенного влияния на скорости диффузионных потоков, так как они
осуществляются по вакансионному, а не по междоузельному или эстафетному
механизму. Роль поверхностного легирования TiO2
при синтезе рассмотренных типов соединений бария и свинца примерно одинакова.
Понимание роли легирования в процессе активации
твердых фаз позволило установить еще одну причину, связывающую активность фазы
с ее предысторией. Очевидно, что влияние прекурсора, продуктом топохимического
превращения которого является рассматриваемая фаза, не ограничивается только
его ориентационным воздействием при формировании кристаллической решетки этой
фазы. Подавляющее большинство реакций с участием твердых фаз, в априори, протекает,
как минимум, в микронеравновесных условиях (различный размер и дефектность
телесных комплексов фазы, градиент температур и парциальных давлений, как
газообразных прекурсоров, так и продуктов реакций, длительность и
многостадийность процессов, полиморфизм и т.д.). В связи с этим в продукте
реакции остаются группировки, входившие в состав исходной фазы, концентрация
которых зависит от условий проведения процесса. Не исключено, что данное
явление предопределяется не только кинетическими, но и термодинамическими
факторами - смешанная фаза, фактически представляющая собой твердый раствор,
может оказаться более стабильной по сравнению с индивидуальной при высоких
температурах, когда состояние системы в значительной степени предопределяется
энтропийным фактором.
Кристаллохимическая роль легирования фазы за
счет собственных прекурсоров может заключаться в термодинамической и
кинетической стабилизации некоторых полиморфных модификаций (γ-Al2O3,
анатаза, α-SbSI
и т.д.), а также способствовать изменению ее объемной и поверхностной
активности. С кристаллографической точки зрения механизмы реализации указанных
явлений могут быть различны: так «сульфатный» γ-Al2O3
содержит ионы серы в тетраэдрических узлах кубической упаковки типа шпинели, а
в α-SbSI
макромолекулы (SbSI)n
связываются между собой через мостики из атомов серы.
Независимо от того вводится ли добавка
специально или легирование фазы происходит за счет исходных веществ, в процессе
синтеза, роль добавки (помимо ее природы) предопределяется способом распределения
в системе. Если добавка полностью растворяется в основной фазе, то ее влияние
обусловлено, преимущественно, образованием атомных и электронных дефектов.
Особенно эффективно введение в ионно-ковалентные фазы ионов, имеющих больший
или меньший заряды по сравнению с ионами, формирующими основную решетку - этот
прием носит название «гетеровалентное замещение».
Для достижения определенного эффекта, вызванного
легированием, необходимо знать механизм исследуемого процесса и природу
прекурсоров. Известно, например, что кристаллический оксид цинка содержит
избыток цинка в форме внедренных атомов, концентрация которых предопределяет
химическую активность данной фазы. Тогда для увеличения концентрации внедренных
частиц необходимо провести замещение ионов цинка ионами, имеющими более низкий
заряд (для выполнения кристаллографических условий процесса замещающий ион
должен иметь радиус близкий к радиусу Zn2+),
например, ионами лития. Так как Li+
имеет более низкий заряд по сравнению с Zn2+,
то для сохранения электронейтральности кристалла один из двух замещаемых ионов
цинка, поглощая электроны, должен перейти в междоузельное пространство в виде
атома или иона с меньшим зарядом: ZnО
↔ Zn* +
ē + ½О2 (где Zn*
внедренный однозарядный ион цинка), тогда при растворении Li2О
в ZnО происходит
процесс:
Li2О + ½О2
+ 2ē
↔ 2Li+Zn
+ 2О2-о
(где - Li+Zn
ионы лития в регулярных позициях ионов цинка, О2-о ионы кислорода в регулярных
позициях кристаллической решетки ZnО).
Как видно из приведенных схем, протекание
последнего процесса смещает равновесие первого вправо, т.е. способствует росту
концентрации внедренных частиц, а, следовательно, и активности формирующейся
фазы.
Аналогично можно показать, что замена Zn2+
на катионы с большим зарядом приведет к необходимости перехода части внедренных
частиц цинка в регулярные позиции, что снизит активность рассматриваемой фазы.
Влияние растворимых примесей на процесс спекания
может быть различен, как с точки зрения скорости, так и степени
рекристаллизации получаемого керамического материала. В связи с тем, что
скорость спекания лимитируется объемной диффузией, можно предположить, что при
«гетеро- и изовалентном замещении» скорости процессов спекания легированных фаз
будут различны, т.к. при изменении состава добавки, изменяется природа дефектов.
Например, при растворении МО в Э2О3 состав образующейся фазы можно выразить в
виде Э1-хМхО1,5-х, что свидетельствует об увеличении дефектности в анионной
подрешетке по мере роста концентрации добавки в системе. Если же состав добавки
М2О3, то состав легированной фазы Э1-хМхО1,5, т.е. изменение дефектности по
сравнение с основной фазой может быть вызвана только различием в значениях
равновесных концентраций дефектов образующих систему фаз или условиями
формирования твердого раствора. Если же состав добавки МО2, то четыре иона Э3+
должны замещаться тремя ионами М4+, т.е. легированная фаза будет иметь
повышенную концентрацию дефектов к катионной подрешетке. Однако, в отличие от
процессов химического взаимодействия твердых фаз, сопровождающихся разрушением кристаллических
решеток прекурсоров, процесс спекания на макроуровне можно рассматривать как
взаимодействие между близкими по составу телесными комплексами, приводящее к
уменьшению пористости прессзаготовки. Причиной возникновения диффузионных
потоков в таких системах является разница между значениями
изобарно-изотермического потенциала границы контакта двух зерен и значениями
этой энергии, характерными для свободной поверхности. Процесс диффузии
равноценен миграции вакансий (дефектов различной природы) решетки в
противоположном направлении, при этом вакансии могут мигрировать и разряжаться
не только в месте контакта, но и на поверхности зерен, что, в свою очередь,
равноценно переносу вещества с поверхности частиц к поверхности контакта .
Очевидно, что разряжение неравновесных вакансий способствует уменьшению
объемной и поверхностной дефектности кристаллической решетки телесных
комплексов фазы - в этом заключается микроскопический эффект спекания. В связи
с тем, что скорость рассматриваемого процесса зависит от коэффициентов
диффузии, как катионов, так и анионов, природа легирующей добавки при
«гетеровалентном» замещении сказывается только в пределах различия этих
коэффициентов для отдельных частиц. Поэтому активировать процессы усадки
приблизительно в равной степени можно за счет введения в исходные фазы
различных по составу и структуре веществ, обеспечивающих примерно равную
дефектность легированной фазы, при условии, что термическая и термодинамическая
устойчивость применяющихся добавок близки. Последнее положение связано с
представлением о возможности изменения равновесной дефектности твердой фазы при
фиксированной температуре за счет образования твердого раствора, т.к. значения
термодинамических функций легированной и исходной фазы различны. Нивелированию
роли природы растворяемой добавки в процессах спекания, по сравнению с ее
концентрацией в системе и способом синтеза легированной фазы, способствует и
значительная роль поверхностных дефектов частиц порошка, как в возникновении
самих диффузионных потоков, так и при формировании реакционной зоны.
При выборе легирующего компонента необходим учет
еще нескольких факторов, способных повлиять на активность модифицированной
фазы: а) возникающие в процессе легирования дополнительные дефекты могут
образовывать ассоциаты, которые способны в значительной степени изменить
свойство твердой фазы; б) упорядочение ионов добавки в основной фазе, как
правило, приводит к образованию структур сдвига; в) вводимые добавки способны
аннигилировать неравновесные дефекты, характерные для исходной фазы, т.е.
нивелировать различия фаз одного и того же состава и строения, связанные с их
предысторией.
Легирующие добавки, растворимость которых в
основной фазе мала, по механизму воздействия на процесс спекания можно
разделить на две группы: а) способные к поверхностному взаимодействию с
образованием при температуре спекания жидких продуктов, в которых растворимость
основной фазы незначительна; б) легкоплавкие фазы, способные заметно растворять
вещество матрицы.
Роль добавок первой группы сводится к подавлению
процессов вторичной рекристаллизации за счет формирования протяженной
реакционной зоны, характеризующейся относительно низкими коэффициентами
диффузии частиц, входящих в состав спекаемой фазы. Тогда в процессе спекания
концентрация дефектов в отдельных зернах приближается к равновесной, а продукт
взаимодействия заполняет межзеренное пространство. Этот прием позволяет
получать мелкозернистую керамику с минимальной пористостью, но только при
высоких температурах и длительном обжиге. В качестве примера можно привести
получение керамики корунда с использованием в качестве легирующей добавки
оксида магния, который при взаимодействии с Al2O3
образует фазу состава MgAl2O4,
которая в виде тонкой пленки концентрируется на поверхности частиц корунда. При
оптимальном способе распределения легирующего компонента в системе указанный
выше эффект достигается при содержании MgО
в системе менее 1 мол.%.
Добавки второй группы также образуют протяженную
реакционную зону, характеризующуюся (в отличии от добавок первой группы)
высокой скоростью массопереноса спекаемого вещества, что способствует
значительному увеличению скоростей спекания, но, одновременно, приводит к
значительной рекристаллизации образцов. Использование добавок этой группы
позволяет снизить температуру и длительность процесса спекания и предотвратить
коробление изделий, но получаемая в этом случае керамика является
крупнозернистой, кристаллическая решетка этих зерен, характеризуется повышенной
концентрацией неравновесных дефектов, за счет формирования кристаллов в
условиях среднего и высокого пересыщения, а сами изделия достаточно высокой
пористостью. Характеристики керамики в этом случае можно улучшить, если добавка
второго типа способна, при высокой температуре, испарятся, что вызывает
значительную усадку образцов, увеличивает их плотность и уменьшает пористость.
5.5 Механохимическое активирование твердых фаз
Если энергия механического воздействия на
систему сравнима или превышает энергию химических связей в телесных комплексах
фазы, то в процессе этого воздействия наблюдаются значительные изменения макро-
и микроструктуры отдельных частиц фазы. При механическом воздействии твердые
частицы до тех пор претерпевают упругую и пластическую деформацию, пока в
каком-либо сечении напряжение не превысит предел прочности объекта и не
произойдет его разрушение вдоль этого сечения с образованием нескольких
осколков, удаляющихся от места разрушения с определенной скоростью. Таким
образом, механическая энергия в описанном процессе может расходоваться на упругую
и пластическую деформацию кристаллической решетки фаз, образование новой
поверхности (разрыв химических связей) и превращаться в кинетическую энергию
осколков. В связи с тем, что и при деформации и при разрушении частиц
происходит перегруппировка или разрыв химических связей указанные процессы
должны быть отнесены к химическим. Раздел химии, изучающий химические изменения
и превращения, происходящие во время или после завершения механического
воздействия, получил название механохимии. Увеличение значений свободной
энергии Гиббса в процессе механической обработки образцов объясняется не только
увеличением суммарной площади их поверхности, изменением строения и
энергетического состояния поверхностных слоев, но и образованием дислокаций,
несовершенств решетки, связанных с формированием плоскостей сдвига или
скольжения, метастабильных (при стандартных условиях) полиморфных модификаций и
другими кристаллохимическими изменениями кристаллической структуры исходных
фаз. Разрушение твердых тел также сопровождается возникновением электрических
зарядов на поверхности осколков и эмиссией электронов. Заряды, возникающие в
результате трения частиц друг об друга, разряжаются с той или иной скоростью,
при этом в ряде случаев разряд сопровождается холодным излучением (триболюминесценция)
и всегда химическими изменениями в поверхностном слое или в объеме частиц.
Схема механохимического процесса в рамках модели плазмы представлена на рис.11.
Согласно этой модели, на границе сталкивающихся тел образуется область высокого
беспорядка, в пределах которого беспрерывно формируются дислокации,
концентрирующиеся в предповерхностном слое.
Рис.11. Схема механохимических процессов по
Тиссену.
Как видно из рисунка, две сильно деформированные
поверхности разделены слоем плазмы, образующейся в результате концентрации
механических напряжений в зоне контакта. Плазма испускает электроны (эффект
Крамера) и световые кванты (триболюминесценция). Состояние веществ в области
плазмы характеризуется аномально высокими значениями функций состояния и
энергией, достаточной для преодоления активационного барьера любого химического
процесса - т.е. в данной области возможно протекание с высокой скоростью
различных химических процессов и перенос вещества между частицами, находящимися
в контакте.
Как отмечалось выше, механическое воздействие на
твердые фазы может вызывать в них изменения различной природы: а) образование
дефектов, процесс аннигиляции которых характеризуется относительно высокими
значениями энергии активации, (т.е. изменения, предопределяющие
термодинамическую метастабильность фазы, но устойчивые во времени); б)
короткоживущие дефекты, возникающие и исчезающие в процессе механического
воздействия на систему. В связи с этим различают механохимическое активирование
прекурсоров, для которых характерно наличие дефектов первого типа (роль этих
дефектов для процессов с участием твердых фаз была рассмотрена выше) и
активирование химического процесса в целом за счет подвода к системе
механической энергии - механохимические реакции.
Очевидно, что в свою очередь механохимические
реакции могут быть разделены на две группы: а) подвод механической энергии к
образцу обеспечивает достижение энергии активации процесса способного
развиваться самопроизвольно при данных параметрах состояния системы (например,
механохимическое активирования процессов окисления и разложения взрывчатых и
горючих смесей, снижение температуры восстановления оксидов р и d-элементов
углеродом и т.д..); б) реакции имеющие значения ∆G
>> 0 и осуществляемые за счет совершения работы над системой и возможные
только в том случае, когда подводимая механическая энергия ∆Gомех.
больше ∆Gо298
рассматриваемого химического процесса, например процессы:
4Cu + CO2 = 2Cu2O + C+ 2H2 = Si +
CH4
можно провести при повышении давления в системе
до 700 - 900 ат или за счет механического воздействия на систему, при условии
достижения соотношения ∆Gо
= ∆Gо298 - ∆Gомех
< 0.
Механохимическая реакция может быть изображена в
виде схемы, представленной на рис.12.
Период (а) отвечает стационарному течению
процесса в отсутствии механохимического активирования. Период (б) соответствует
увеличению активности реагентов в процессе роста воздействия на систему
механической энергии (индукционный период), после чего скорость процесса при
выбранных условиях обработки стабилизируется (этап (в)), а при прекращении
механического воздействия происходит аннигиляция нестабильных неравновесных
дефектов, что вызывает снижение скорости процесса (г).
Длительность периодов (б) и (г) зависит от
энергии активации проводимого процесса, скорости изменения и типа механической
нагрузки, а также от относительной стабильности неравновесных дефектов,
генерируемых в рассматриваемом процессе.

Рис.12 Изменение скорости механохимических
реакций во времени: - (а)- стационарное течение реакции без механического
воздействия на систему, (б) - течение реакции на начальной стадии механического
воздействия, (в)- стационарный этап течения процесса при выбранных условиях
обработки, (г) этап прекращения механического воздействия на систему; - вариант
механического воздействия типа удара или взрыва(τн
и
τк
- время начала и конца механического воздействия на систему).
В пределе, когда энергия активации процесса
мала, а скорость нарастания механического воздействия на систему очень велика
(ударная, взрывная волна), время индукционного периода стремится к нулю. То же
происходит с длительностью периода (г) при крайне низкой стабильности дефектов,
возникающих в процессе механической активации материала (рис.12).
Химические изменения в индивидуальной фазе,
подвергающейся механическому воздействию, могут иметь различную природу:
а) полиморфные превращения, например, переход
кальцита в арагонит, связанный с накоплением дислокаций, приводящих к фазовому
переходу благодаря скольжению атомных слоев относительно друг друга ;
б) аморфизация поверхностного слоя частиц,
например, образование на поверхности частиц кварца аморфного слоя, обладающего
высокой химической и каталитической активностью;
в) разложение веществ, с образованием различных
продуктов реакции в зависимости от условий проведения процесса. В этом плане
показателен пример, приведенный в работе: при механическом измельчении FeCO3 в
вакууме при комнатной температуре происходит разложение этой фазы на FeО и СО
Образующийся при этих условиях FeО настолько активен, что способен при с.у. с
высокой скоростью окисляться кислородом воздуха с образованием Fe2О3 или
взаимодействовать с водяным паром по реакции 2FeО +(1+х) Н2О = Fe2О3•хН2О + Н В
результате этого механическая обработка данной фазы на воздухе приводит к
образованию соединений Fe(III). Аналогично протекает механохимическое
разложение и других карбонатов. При этом с ростом поляризующего действия
катиона напряженность механического поля, необходимого для разложения фазы
снижается. В процессе механического воздействия происходит разложение солей и
других кислородных кислот. Так нитраты, в состав которых входят
низкополяризующие катионы, разлагаются с образованием нитрита и кислорода, а
содержащие катионы, характеризующиеся средним ПД, - на оксид металла, кислород
и соединения азота (N2, NO, NO2) - (при низкой термодинамической стабильности
оксида он, в свою очередь, распадается на металл и кислород ), а оксалаты - с
образованием оксида металла, СО и СО При этом, если в условиях проведения
процесса одновременно разрушается и оксид металла, то процесс развивается со
скоростью взрыва, за счет резкого увеличения температуры системы, вызванной
окислением СО, например, Ag2C2O4 →
Ag2O + CO + CO2, одновременно:
2Ag2O → 4Ag + O2 и 2СО + О2 = 2СО2 или суммарно:
Ag2C2O4 → 2Ag + 2CO2 с ∆Н <<0; Сульфиты разлагаются на
сульфаты и сульфиты (реакция диспропорционирования), а соли одноосновных
органических кислот - на оксид металла (реже карбид или карбонат) и, в зависимости
от состава аниона, на СО, СО2 и низкомолекулярные органические соединения.
Приведенные примеры показывают, что воздействие механической и тепловой энергии
на процесс разложения солей во многом схожи, что послужило толчком для
создания, альтернативной модели плазмы, теории механохимических процессов,
которая связывает наблюдающиеся в системе изменения с локальным ростом
температуры в отдельных точках системы. Согласно этой теории в процессе
столкновения и трения частиц механическая энергия превращается в тепловую, что
локально повышает температуру контактов до температуры плавления или даже
кипения (возгонки), что импульсно повышает давление в микрообъеме. Эти факторы
и предопределяют возможность протекания описанных выше процессов, а также
способствуют формированию фаз, характеризующихся высокой концентрацией
неравновесных дефектов.
Таким образом, активность фазы после ее
механического активирования предопределяется остаточной концентрацией
неравновесных дефектов, образующихся в результате увеличения суммарной площади
поверхности частиц порошка при его измельчении. При механическом воздействии на
систему происходит формирование «замороженных состояний», возникающих в
условиях резкого изменения значений параметров состояния в ее локальных
объемах. Например, за счет локального повышения температуры в данной области
системы, концентрация точечных дефектов в ней резко возрастает, даже если не
происходит плавление сталкивающихся частиц. Так как это воздействие
кратковременно, быстрый рост температуры сменяется быстрым ее снижением, что
«замораживает» процесс упорядочения кристаллической решетки или вызывает
кристаллизацию при крайне неравновесных условиях. При механической активации
смеси фаз («мокрый» или «сухой» помол зачастую предшествует процессу твердофазного
синтеза) в ряде случаев наблюдается протекание между фазами химических
процессов, которые не всегда желательны, так как механосинтез является
неравновесным процессом и, зачастую, приводит к образованию не основной, а
промежуточных фаз, энергия активации образования которых в условиях проведения
взаимодействия минимальна. Например, при «сухом», а в случае использования
активных форм прекурсоров и при «мокром» помоле смесей PbO с Nb2O5 с мольным
соотношением реагентов 1:1, в системе наблюдается образование не фазы PbNb2O6,
а фаз со структурой пирохлора. Стабильность этих фаз достаточно высока и они,
совместно с избытком оксида ниобия сохраняются в системе вплоть до 1200оС, что
резко снижает скорость формирования целевого продукта реакции.
Еще одним фактором, влияющим на эффективность
мехобработки исходных фаз, перед использованием их в качестве прекурсоров
синтеза или спекания, является их природа и предыстория. Например, при
одинаковых условиях механического воздействия на стабильные при с.у.
модификации TiO2 и ZrO2,
приводящего к получению шихты примерно одинаковой дисперсности, активность ZrO2
возрастает в большей степени, чем активность TiO2,
что связано с большей энергией кристаллической решетки ZrO2
и, следовательно, с большей стабильностью неравновесных дефектов, образующихся
в результате указанного воздействия. Аналогичная обработка образцов,
представляющих собой две модификации TiO2
(рутил и анатаз), увеличивая реакционную способность фазы первого типа,
приводит к снижению активности второй или практически ее не изменяет. Это
связано с тем, что α-форма
(рутил) является термодинамически стабильной при с.у. и имеет относительно
низкую концентрацию неравновесных дефектов, и, следовательно, мехобработка, в
соответствии с ранее рассмотренным механизмом, будет способствовать росту этой
концентрации. Анатаз же - метастабильная форма, характеризующаяся повышенной
концентрацией неравновесных дефектов, поэтому введение механической энергии в
систему будет способствовать не только возникновению новых дефектов, но и
аннигиляции дефектов, уже присутствовавших в исходной фазе. Влияние
механического взаимодействия на фазы одинаковой структуры и состава полученные
с использованием различных прекурсоров также не одинаково: образцы α-форма
TiO2, полученные с
использованием сульфатного прекурсора изменяют свою активность в большей
степени, чем аналогичные образцы, синтезированные в нитратных или оксалатных
системах [50]. Очевидно, что это связано с более высокой исходной дефектностью
фаз второго и третьего типа (в нитратных системах TiO2
формируется при относительно низких температурах, а в оксалатных - в
восстановительной атмосфере).
Одним из приемов механического воздействия на
систему является прокатка, которая может использоваться для изменения
дефектности, как простых веществ так и соединений различных типов. Установлено,
что плотность дислокаций в поверхностном слое металлов при прокатке возрастает
до 1012 на 1см2, при этом поверхностная энергия, за счет образования дислокаций
увеличивается, на 50 Дж/см В свою очередь рост концентрации неравновесных
дефектов резко увеличивает реакционную способность, как компактных образцов,
так и порошков металлов. Например, значительный рост скорости химической
коррозии механически активированного порошка железа (взаимодействие с кислотами,
с газообразными окислителями и растворенным в воде кислородом ), увеличение его
каталитической активность в процессе синтезы аммиака, а также скорости
взаимодействия с простыми веществами различных типов. Аналогичные изменения в
структуре поверхностных слоев происходят и в процессе прокатки смесей оксидов,
что приводит к резкому увеличению скорости взаимодействия между реагентами,
образующими смесь, например, при прокатке смеси оксидов железа и молибдена с
карбонатом кальция реакция развивается со скоростью взрыва.
Необходимо еще раз подчеркнуть, что любое
механическое воздействие приводит не только к образованию дислокаций в
поверхностном слое, но и способно изменять внутреннюю структуру фазы. Это
связано с тем, что поглощаемая образцом механическая энергия приводит к
деформации решетки (Ес -энергия сжатия решетки, направлена на преодоление сил
межатомного отталкивания и предопределяющаяся эластичностью материалов),
возбуждает тепловые колебания составных частей решетки (Ет) и изменяет энергию
электронов системы (Еē - энергия
возбуждения электронов): ЕМ = Ес + Ет + Еē . Очевидно,
что указанные факторы способны изменять концентрацию как точечных, так и
протяженных дефектов в объеме кристаллов, а в ряде случаев способствовать
формированию сверхструктуры или протеканию полиморфного превращения. При этом
влияние механического воздействия на внутреннюю структуру частиц фазы
пропорционально напряженности механического воздействия (количеству
механической энергии в единицу времени). Отсюда можно сделать вывод, что
максимальные изменения в системе должна происходить под воздействием ударной
волны или использовании пресс-форм типа «наковальня», что позволяет
синтезировать термодинамически нестабильные фазы, например, со структурой
перовскита.
Большое влияние оказывает механическое
воздействие и на процесс спекания порошков. Как отмечалось ранее, при
диспергировании образцов за счет резкого увеличения суммарной площади
поверхности частиц многократно увеличивается протяженность реакционной зоны при
одновременном уменьшении слоя продукта. Влияние этих факторов способно
увеличивать скорость любых процессов с участием твердых фаз, в том числе и
процесса спекания. Кроме этого, вклад движения дислокаций в процессах спекания
и истинной рекристаллизации является доминирующим, что также увеличивает
скорость этих процессов с участием механически активированной шихты.
(Необходимо напомнить, что концентрация неравновесных дефектов типа дислокаций
увеличивается с ростом суммарной поверхности частиц порошка твердой фазы и
интенсивностью механического воздействия на систему, а истинная
рекристаллизация представляет собой процесс образования кристаллов,
характеризующихся равновесной дефектностью, из деформированных исходных
частиц). Отсюда можно сделать практический вывод, связанный с макроструктурой
изготавливаемой керамики: чем выше давление прессования порошка (выше
деформационные изменения кристаллической решетки отдельных частиц), тем ниже
температуры, при которых происходит рекристаллизация материала, выше
концентрация центров рекристаллизации и, следовательно, меньше размер зерен,
формирующейся в процессе спекания керамики.
Помимо описанных выше методов активации твердых
фаз необходимо отметить способы образования дефектов в процессе фотолиза или
радиолиза (частичное разрушение фаз при их взаимодействии с электромагнитными
квантами различной энергии, электронами, α-частицами
и т.д.).
.6 Практика активации твердофазных процессов
На практике основным способом активации
процессов, протекающих между твердыми фазами, остается повышение температуры
системы, которое инициирует процессы, имеющие диффузионную природу. Однако, как
было показано выше, рост температуры системы приводит к увеличению скорости
процессов аннигиляции неравновесных дефектов во взаимодействующих фазах и,
следовательно, к снижению их активности. Кроме этого при повышенных
температурах становится невозможным эстафетный механизм диффузии, за счет
которого скорость массопереноса максимальна. Кроме этого при нагревании
возможно протекание и необратимых процессов, не позволяющих получить продукт
реакции заданного состава или строения, а, следовательно изготовить материал, с
заданными электрофизическими и механическими параметрами. Это может быть
связано:
а) с низкой термической стабильностью
прекурсоров (например, разложение оксидов некоторых p-
и d- элементов - Sb2O5,
V2O5,
PbO2, MnO2,
Fe2O3,
CuO и т.д. при
нагревании );
б) с недостаточной термической стабильностью
продукта реакции: например: PbZrO3
↔ ZrO2 + PbO↑;
в) с испарением из шихты легколетучих фаз (PbO,
Bi2O3,
MoO3 и т.д.).
г) с неконтролируемой вторичной
рекристаллизацией, приводящей к ухудшению механических характеристик керамики в
связи с различной геометрией и размером ее зерен;
д) с эффектом разбухания, связанного с
перетеканием газов из мелких пор в крупные за счет разности давлений
(разбухание прессзаготовок смесей исходных фаз при синтезе материалов типе
ЦТС);
е) с неконтролируемой дефектностью продуктов
твердофазного синтеза за счет формирования в процессе охлаждения «замороженных»
состояний.
В настоящее время предложены более или менее
эффективные пути решения некоторых из указанных проблем. В частности, для
уменьшения влияния температуры на процесс аннигиляции неравновесных дефектов
было предложено при обжиге образцов повышать температуру системы с высокой скоростью.
Предполагалось, что увеличение скорости подъема температуры системы не позволит
завершиться процессу аннигиляции неравновесных дефектов к началу спекания - это
даст возможность провести спекание и рекристаллизацию за сокращенный период
времени и повысить эффективность указанных процессов (бóльшая
плотность керамики в сочетании с ее мелкозернистостью). Очевидно, что указанный
прием не является универсальным, так как энергия активации процесса аннигиляции
может изменяться в широких пределах и зависит, как от состава и строения фазы,
так и от ее термической и химической предыстории. Обозначив скорость
относительной линейной усадки через (ΰ), можно
показать:
ln ΰ = ln
(BN) - Eус./(RT)
- где В - константа для данного образца, зависящая от химической и термической
предыстории образца, N
- общая концентрация дефектов кристаллической решетки фазы, Eус.
- энергия активации процесса усадки.
Тогда, при неизменном значении N
во всем температурном интервале проводимых процессов, зависимость ln
ΰ = f(1/T)
прямолинейна, если же N
за счет аннигиляции снижается, значения ln
ΰ будут
уменьшаться (участки bc
и cd кривой 2 -
рис.13).

Рис.13. Зависимость ln
ΰ от
1/T : 1- при
постоянном значении N, 2 - при
изменяющимся N в процессе
спекания гематита.
Приведенные данные свидетельствуют, что эффект
быстрого увеличения температуры в системе, наблюдается всегда, независимо от
энергии активации процесса аннигиляции дефектов (участок ab
кривой 2 - рис.13), при этом его роль в процессе тем значительнее, чем выше
кинетическая стабильность неравновесных дефектов. Например, для гематита,
полученного с использованием сульфатного прекурсора и имеющего высокое значение
Еак. аннигиляции, данный прием позволяет значительно повысить скорость спекания
и получить керамику высокой плотности. В то же время для Fe2O3,
полученного при низкой температуре (например, при разложении нитрата Fe(III)),
этот эффект наблюдается только на начальной стадии процесса и имеет
кратковременный характер. С учетом приведенных выше исследований, а также
данных работ можно сделать вывод, что для увеличения плотности керамики методом
быстрого подъема температуры системы необходимо использование шихты, частицы
которой характеризуются наличием кинетически стабильных дефектов (при
максимально возможной концентрации последних).
Другой технологический прием, позволяющий
повысить скорость спекания, заключается в циклическом изменении температуры в
системе в процессе обжига образцов . Использование этого приема может быть
эффективным, если спекаемая фаза в пределах термоцикла испытывает полиморфное
превращение, т.е. периодически активируется за счет перестройки структуры, а
также при резком изменении температуры на этапе охлаждения (нагрева). В
последнем случае в кристаллической решетке создаются напряжения (на этапе
охлаждения), которые сохраняются вплоть до высоких температур за счет высокой
скорости нагревания. Очевидно, что возможности данного приема ограничены
коэффициентами термического расширения спекающегося материала и стабильностью
«замороженных» состояний. Это связано с тем, что при резком охлаждении или
нагревании возможно образование микротрещин в изделии, которые увеличиваются
при повторных циклах, что приводит к разрушению образца. Так же при высоких
скоростях диффузии атомов или ионов, образующих кристалл, прием
термоциклирования может оказаться не эффективным в связи с быстрым снятием напряжения,
вызванного термоударом. Вариант же полиморфного превращения, температура
которого находится в пределах ∆Т цикла, более перспективен. В частности,
использование термоциклирования в пределах 700 - 1050оС при спекании керамики Bi4Ti3O12
и Na0,5Bi4,5Ti4O15,
фазы которой имеют точку Кюри порядка 750оС, удалось увеличить скорость
спекания практически в два раза (при снижении максимальной температуры обжига в
среднем на 100оС) и получить образцы с размером зерна порядка 2-3 мкм.
Размер зерна керамики является ее важнейшей
характеристикой, в связи с тем, что она, зачастую, непосредственно
предопределяет такие свойства образцов, как плотность, упругая податливость,
коэффициент Пуассона, модуль Юнга и так далие. Так как скорость изменения
температуры системы влияет на характер первичной и вторичной рекристаллизации,
то в условиях быстрого изменения температуры (высокого пересыщения по одному из
параметров состояния) количество центров кристаллизации резко возрастает и,
следовательно, указанный фактор позволяет в широких пределах варьировать
гранулометрический состав конечных изделий. Например, при нагревании
прессзаготовок материала типа ЦТС (марка ПКР- 1М) до 1240оС со скоростью 900,
400 и 100оС/ч была получена керамика со средним размером зерна 2, 5 и 22 мкм, соответственно
(средний размер частиц исходной шихты 0,7 мкм). Аналогичные результаты были
получены и при спекании порошка состава Ni0.4Zn0.6Fe2O4
(Тобжига = 1300оС, скорость подъема температуры 800, 300 и 50оС/ч, размер зерен
1, 5 и 18 мкм, соответственно).
В ряде случаев активировать реакционную смесь
твердых фаз или индивидуальную фазу можно за счет варьирования состава газовой
фазы, в объеме которой происходит твердофазный синтез или спекание. Указанный
прием эффективен при спекании оксидных фаз, в состав которых входят катионы p-
и d-элементов, которые
способны при изменении параметров состояния системы изменять свою степень
окисления. Это связано с тем, что при изменении окислительного потенциала
газовой фазы, указанные твердые вещества могут изменять свой состав. Так в
случае восстановительной атмосферы уменьшение средней степени окисления
катионов вызывает появление вакансий в анионной подрешетке (TiO2-x)
или дефектов типа внедренных атомов или ионов в катионной (Zn1+xO).
В присутствии газообразных окислителей возможен рост средней степени окисления
катионов, что вызовет появление вакансий в катионной подрешетке (Ni1-xO,
U1-xO2).
Каким бы ни был механизм дефектообразования, варьирование состава фазы в
пределах области гомогенности, приводит к перестройке ее кристаллической
структуры тем в большей степени, чем шире эта область.
Поэтому, во-первых, изменение окислительного
потенциала во времени позволяет поддерживать высокую активность твердой фазы
вплоть до завершения процесса. Во-вторых, этот прием малоэффективен в случае
узкой области гомогенности фазы, высокой энергии активации процесса ее
окисления (восстановления), а также в случае термодинамических затруднений
протекания окислительно-восстановительных процессов при выбранных параметрах
состояния системы. И, в третьих, необходимо помнить, что процесс спекания в
этом случае происходит в условиях значительного и переменного пересыщения, т.е
в условиях способствующих увеличению скорости процесса и получению
мелкозернистой керамики, но кристаллическая решетка формирующихся телесных
комплексов будет характеризоваться высокой концентрацией неравновесных
дефектов, что хорошо для прекурсоров, но недопустимо для конечного продукта,
если этот продукт - функциональный материал. Если в состав прекурсоров входят ионы
кислорода, то эффект состава газовой фазы может быть достигнут и при замене
воздуха инертным газом. Это связано с тем, что изменение парциального давления
кислорода способствует смещению равновесий дефектообразования: независимо от
типа дефектов в этих условиях фаза теряет кислород, что изменяет дефектность
анионной или катионной подрешеток (в первую очередь в поверхностном слое) и
способствует росту скоростей спекания и рекристаллизации. Однако, используя
данный прием, необходимо учитывать, что некоторые примеси, содержащиеся в
газовой фазе, могут сорбироваться поверхностью порошков, резко снижая ее
поверхностную энергию и, следовательно, уменьшая скорость твердофазного
взаимодействия. Очевидно, что данное явление аналогично «отравлению» твердого
катализатора при гетерогенном катализе (блокирование активных центров), что не
удивительно, учитывая роль поверхностных дефектов в обоих этих процессах. Для
устранения описанных затруднений можно использовать добавки, способствующие
десорбции нежелательных примесей, что особенно актуально при получении
бескислородной керамики. Это связано с тем, что гелий или аргон, применяющиеся
при синтезе и спекании боридов, карбидов, силицидов и т.д., всегда содержат
некоторое количество кислорода, который, взаимодействуя с основной фазой,
образует на ее поверхности слой оксидов, которые блокируют протекание всех
типов взаимодействий с участием данных фаз. Восстановить поверхность фаз можно
путем введения в образцы различных восстановителей (например, металлов триады
железа при спекании карбидов и боридов титана, циркония, ниобия), способных
связывать кислород с образованием летучих, в условиях обжига, оксидов. Другой
прием предусматривает введение в газовую фазу восстановителей (углеводородов,
водорода или смеси этих веществ), уменьшающих парциальное давление кислорода в
системе. Однако использование газообразных восстановителей ограничено
температурами синтеза, т.к. с ростом температуры возрастают окислительные
свойства воды и оксидов углерода, являющихся продуктами окисления добавок.
Повысить скорость процесса спекания и добиться
высокой плотности образцов, близкой к рентгенографической можно, используя
метод горячего прессования, который заключается в одновременном воздействии на
образец высокой температуры и высокого объемного или одноосного давления
статического или импульсного типа. Увеличение скорости спекания в этих условиях
связано с тем, что при высоких температурах возрастает пластичность материала,
а рост давления в системе может приводить к появлению в точках контакта частиц
(играющих роль концентраторов энергии) жидкой фазы. Эти факторы способствуют:
а) росту протяженности реакционной зоны за счет увеличения суммарной площади
контакта между спекающимися частицами; б) миграции пор к поверхности образцов,
связанной с интенсификацией процессов первичной и вторичной рекристаллизации;
в) образованию большого числа зародышей (т.к. процесс развивается в условиях
высокого пересыщения), что приводит к формированию мелкозернистой керамики.
Очевидно, что изменить энергию системы и
повлиять на скорость процессов с участием твердых фаз можно и другими
способами, однако всегда необходимо учитывать, что эффективность того или иного
воздействия предопределяется не только видом вводимой энергии, но и способом ее
введения в систему. Например, данные о возможности активации рассматриваемых
процессов с помощью ультразвуковых колебаний крайне противоречивы в связи с
тем, что в большинстве случаев исследователями учитывается только мощность
акустического воздействия и не учитывается геометрия звукового поля, геометрия
объекта, подвергающегося воздействию, и способ его контакта с источником
ультразвуковых колебаний, а также тип данного источника.
Рассмотрим возможные изменения, возникающие в
системе под действием ультразвука. В качестве первого примера возьмем
металлическую (для определенности, медную) пластинку, с жестко соединенным с
ней пьезоэлементом. Поместим изделие в воду, контактирующую с воздухом. За счет
растворенного в воде кислорода поверхность пластинки окисляется, и слой
гидроксида блокирует поверхность металла - процесс переходит в диффузионный
режим и его скорость контролируется коэффициентом диффузии кислорода через слой
продукта, толщиной этого слоя и его сплошностью. Если подать на пьезоэлемент
напряжение то он начнет деформироваться, в том числе и в направлении
параллельном плоскости пластины. Так как пластина и пьезоэлемент представляют
собой единое целое, то деформация пьезоэлемента вызовет деформацию медной
пластины типа растяжение - сжатие. При достижении критических значений
амплитуды колебаний пленка оксида будет разрушена и свободная поверхность с
высокой скоростью будет окисляться. При постоянной работе пьезоэлемента
скорость окисления металла будет предопределяться скоростью растворения
кислорода в воде или скоростью его диффузии в жидкой фазе (в зависимости от
величины парциального давления О2 над водой). Замена медной пластинки на
алюминиевую принципиально не изменит влияние излучателя на характер процесса,
за исключением, того, что алюминий, имеющий низкое значение электродного
потенциала, может окисляться не только растворенным в воде кислородом, но и
водой, а продукты его окисления имеют более высокую энергию адгезии к металлу.
Это приводит к тому, что резкий рост скорости коррозии алюминия будет
наблюдается при большей амплитуде колебаний, но при ее достижении скорость
окисления алюминия в несколько раз будет превосходить скорость окисления меди в
тех же условиях. Другой пример - воздействие внешнего ультразвукового
излучателя на порошки твердых фаз в процессе их помола. Ультразвук в данном
случае может способствовать развитию микротрещин, т.е. увеличивать
эффективность помола. При этом его влияние на дисперсность частиц (при
фиксированной напряженности звукового поля) будет предопределяться отношением
плотностей материала излучателя и дисперсионной среды: чем оно ближе к единице,
тем выше эффективность ультразвукового воздействия (при переходе через границу
раздела фаз напряженность акустического поля уменьшается пропорционально
значению указанного отношения). Поэтому влияние ультразвука на дисперсность
порошка при «мокром» помоле значительно выше, чем при «воздушном». Этим же
объясняется различное влияние ультразвука на твердофазные процессы,
проводящиеся при непосредственном контакте прессзаготовки с излучателем и в условиях,
когда излучатель и прессзаготовка пространственно разделены, например, слоем
воздуха. В процессе твердофазного взаимодействия ультразвук может увеличивать
скорости массопереноса, способствуя формированию тонкоблочной структуры,
уменьшать расстояния между источниками и стоками вакансий, а также разрушать
слой продукта, образующегося на границе раздела между частицами. Однако энергия
активации этих процессов достаточно велика, поэтому воздействие ультразвука на
их скорость процессов при низкой температуре не может быть значительным:
положительный эффект указанного воздействия может наблюдаться только при
высоких температурах при условии высокой дефектности поверхностных слоев
взаимодействующих частиц.
Влияние электрического поля на скорость реакций
с участием твердых фаз можно разделить на три группы:
а) поверхностные реакции окисления -
восстановления, ∆G которых предопределяется значением электродных
потенциалов. В этом случае, создавая внешнюю э.д.с., можно управлять
термодинамикой и кинетикой указанных процессов, например, подавлять химическую
и электрохимическую коррозию металлов или, наоборот, увеличивать скорость их
окисления, изменять адсорбционную и каталитическую активность твердых фаз, а
также любых процессов, скорость которых предопределяется состоянием
поверхности;
б) изменение концентрации неравновесных дефектов
различных типов и возбуждение электронной подсистемы, связанные со смещением
ионов из регулярных позиций (процесс поляризации) - фактор способствующий не
только увеличению искажения решетки, но и снижающий энергию активации всех
механизмов дефектообразования и, следовательно, способный изменять, как
скорость твердофазных процессов, так и их термодинамику, например, смещать
температуру полиморфных превращений, если они сопровождаются переходом от
полупроводниковой проводимости к металлической;
в) локальное увеличение температуры в местах
контакта частиц при прохождении тока через спрессованный порошок (за счет
разрыва сплошности контакта в образцах и возникновения контактной разности потенциалов
на границах зерен, электрическое сопротивление этих участков системы
максимально), что повышает дефектность поверхностных слоев и скорости
процессов, зависящих от данного параметра, например, процесса спекания.
В реальных системах по действием электрического
поля в той или иной степени происходят все указанные изменения и, зачастую,
выделить предопределяющий фактор способствующий протеканию рассматриваемого
процесса не представляется возможным. Кроме этого роль данных факторов помимо
их природы будет предопределяться технологическими параметрами формирования
образцов, подвергающихся воздействию электрического поля. В качестве примера
можно привести разложение соли Ag2C2O4
в поле напряженностью 3 - 4 В/см (Ag2C2O4=
2Ag + 2СО2), которое
может быть вызвано как фактором (б), при низкой проводимости образцов,
(электрохимическое восстановление Ag+
ведущее к разрушению кристаллической решетки со скоростью взрыва), так и
фактором (в), при достаточной проводимости поверхностных слоев частиц фазы,
например, за счет сорбированной воды.
твердый
фаза диффузия легирование
Литература
1. Ардашникова
Е.И.: Сборник задач по неорганической химии. - М.: Академия, 2010
2. Балецкая
Л.Г.: Неорганическая химия. - Ростов н/Д: Феникс, 2010
. Радушев
А.В.: Гидразиды и 1,2-диацилгидразины. - Екатеринбург: УрО РАН, 2010
. Федотов
М.А.: Ядерный магнитный резонанс в неорганической и координационной химии. -
М.: ФИЗМАТЛИТ, 2009
. Ардашникова
Е.И.: Сборник задач по неорганической химии. - М.: Академия, 2008
. Балятинская
Л.Н.: Химия элементов и их соединений. - Белгород: БелГУ, 2008
. Гольбрайх
З.Е.: Практикум по неорганической химии. - М.: Альянс, 2008
. Саенко,
О.Е.: Химия для колледжей. - Ростов н/Д: Феникс, 2008
. Субботина
Н.А.: Демонстрационные опыты по неорганической химии. - М.: Академия, 2008
. Федеральное
агентство по образованию, БелГУ ; Авт.-сост.: Л.А. Дейнека и др.; Рец.: И.В.
Тикунова, Г.Е. Лунина: Основы химии. - Белгород: БелГУ, 2008
. БелГУ
; авт.-сост.: Л.А. Дейнека и др. ; рец.: И.В. Тикунова, Г.Е. Лунина: Основы
химии. - Белгород: БелГУ, 2007
. каф.
общей, неорганической и аналитической химии БелГУ ; Л.Н. Балятинская и др. ;
рец.: В.И. Павленко, В.В. Ядыкина: Методические указания к выполнению
лабораторного практикума по дисциплине "Неорганическая химия". -
Белгород: БелГУ, 2007
. Киселев
Ю.М.: Химия координационных соединений. - М.: Академия, 2007
. Олейникова
И.И.: Лабораторикум. - Белгород: БелГУ, 2007
. Балятинская
Л.Н.: Задания для самоподготовки по дисциплине "Неорганическая
химия". - Белгород: БелГУ, 2006
. Олейникова
И.И.: Химия. - Белгород: БелГУ, 2006
. Раков
Э.Г.: Нанотрубки и фуллерены. - М.: Университетская книга; Логос, 2006
. Ахметов
Н.С.: Общая и неорганическая химия. - М.: Высшая школа, 2005
. Балятинская
Л.Н.: Задания для самоподготовки по дисциплине "Неорганическая
химия". - Белгород: БелГУ, 2005