Определение содержания примесей металлов в сточных водах атомно-абсорбционным методом
Содержание
Введение
.
Выбор и обоснование способа производства
.
Методика выполнения атомно-абсорбционного анализа
.1
Устройство атомно-абсорбционного спектрометра
.2
Описание составных частей спектрометра. Оптическая схема
.3
Блок монохроматора
.4
Атомизатор
.5
Газовый блок
.
Порядок проведения исследования
.
Порядок установки спектрометра
Заключение
Приложения
Литература
Введение
В последние годы в России наблюдается некоторый
рост промышленного производства, который, с одной стороны, приводит к оживлению
производства, но, с другой сдерживается его высокой экологической опасностью.
Уменьшению экологической опасности промышленности должны служить очистные
сооружения предприятий.
В современных условиях охрана окружающей среды
стала одним из решающих факторов, определяющих дальнейшее развитие
человечества. Жидкие отходы производства обычно представляют собой
мало-концентрированные многокомпонентные органоминеральные смеси и относятся к
особо сложной для очистки группе сточных вод.
Любой водоем или водный источник связан с
окружающей его внешней средой. На него оказывают влияние условия формирования
поверхностного или подземного водного стока, разнообразные природные явления,
индустрия, промышленное и коммунальное строительство, транспорт, хозяйственная и
бытовая деятельность человека. Последствием этих влияний является привнесение в
водную среду новых, несвойственных ей веществ - загрязнителей, ухудшающих
качество воды. Загрязнения, поступающие в водную среду, классифицируют
по-разному, в зависимости от подходов, критериев и задач. Так, обычно выделяют
химическое, физическое и биологические загрязнения. Химическое загрязнение
представляет собой изменение естественных химических свойств воды за счет
увеличения содержания в ней вредных примесей (минеральные соли, кислоты,
щелочи, глинистые частицы).
Основными неорганическими (минеральными)
загрязнителями пресных и морских вод являются разнообразные химические
соединения, токсичные для обитателей водной среды. Это соединения мышьяка,
свинца, кадмия, ртути, хрома, меди, фтора. Большинство из них попадает в воду в
результате человеческой деятельности. Тяжелые металлы поглощаются
фитопланктоном, а затем передаются по пищевой цепи более высокоорганизованным
организмам. Тяжелые металлы (ртуть, свинец, кадмий, цинк, медь, мышьяк)
относятся к числу распространенных и весьма токсичных загрязняющих веществ. Они
широко применяются в металлургическом, химическом производстве, поэтому,
несмотря на очистные мероприятия, содержание соединения тяжелых металлов в
промышленных сточных водах довольно высокое. Большие массы этих соединений
поступают в океан через атмосферу. Для морских биоценозов наиболее опасными
являются ртуть, свинец и кадмий. Свинец - типичный рассеянный элемент,
содержащийся во всех компонентах окружающей среды: в горных породах, почвах,
природных водах, атмосфере, живых организмах. Наконец, свинец активно
рассеивается в окружающую среду в процессе хозяйственной деятельности человека.
Это выбросы с промышленными стоками, с дымом и пылью промышленных предприятий,
с выхлопными газами двигателей внутреннего сгорания. Миграционный поток свинца
с континента в океан идет не только с речными стоками, но и через атмосферу. С
континентальной пылью океан получает 20-30 т. свинца в год.
Высокие нормы удельного водопотребления и
большие объемы сбросов в водоемы есть результат несовершенства технологических
процессов и схем, на которых построено промышленное производство. Большое
количество отходов при современных методах промышленного производства не
является неизбежным, оно может быть сокращено путем создания новых, более
современных технологических методов. Важнейшей составной частью перестройки
технологических процессов на безотходный режим является сокращение
водопотребления, направленное, в конечном счете, на создание производства без
сброса сточных вод в водоемы.
Значительное сокращение расходов воды может быть
достигнуто за счет усовершенствования охлаждения оборудования, в первую очередь
прокатного. Одним из перспективных способов в химической промышленности,
обеспечивающих сокращение потребления воды и количества сточных вод, является
каскадная промывка металла после травления. При этом способе тракт промывки
разделяют на 3-4 отсека. Свежую или нейтрализованную оборотную воду подают в
последний по ходу металла отсек и далее перекачивают из отсека навстречу
движущемуся металлу; в результате расход промывных вод сокращается в 4-5 раз.
При рассмотрении вопроса о составе сточных вод
одним из важных понятий является концентрация загрязнений, т.е. количество
загрязнений, приходящееся на единицу объема воды и исчисляемое обычно в мг/дм3
или г/м3. Это количество зависит от нормы потребления: чем эта норма
больше, тем меньше концентрация загрязнений.
Количество примесей в сточных водах может быть
различно и зависит от характера их образования. Концентрацию загрязнений
определяют химическими анализами (взвешенные вещества, сухой остаток, азот
аммонийный, азот нитритный, азот нитратный, рН, нефтепродукты и др.),
производят определение цинка, хрома, меди, алюминия, фенолов и других химических
элементов.
Надежные данные в этом случае могут быть
получены при использовании современных методов аналитической химии, позволяющих
определить содержание тяжелых металлов на уровне фоновых концентраций.
Стоит отметить, что успехи в развитии методов
анализа позволили решить такие глобальные проблемы, как обнаружение основных
источников загрязнения. При этом тяжелые металлы были классифицированы как одни
из важнейших объектов анализа. Для осуществления такой работы химики-аналитики
должны иметь разработанные методики. Поскольку содержание металлов может
колебаться в широких пределах, то и методы их определения должны обеспечивать
решение поставленной задачи. В результате усилий ученых-аналитиков многих стран
были разработаны методы, позволяющие определять тяжелые металлы на уровне
фемтограммов (10-15 г) или в присутствии в анализируемом объеме пробы одного
(!) атома, например никеля в живой клетке.
1. Выбор и обоснование способа
производства
Раньше определяли лишь валовое содержание
тяжелого металла в воде и устанавливали распределение между взвешенной и
растворенной формами. О качестве вод, загрязненных металлами, судили на основе
сопоставления данных по их валовому содержанию с величинами ПДК. Сейчас такая
оценка считается неполной и необоснованной, так как биологическое действие
металла определяется его состоянием в водах, а это, как правило, комплексы с
различными компонентами.
В 1955 г. Австралийский ученый А. Уолш предложил
простой и практически легко осуществимый способ количественного определения
содержания элементов в растворах, распыляемых в пламя ацетилен-воздух, по
поглощению излучения атомных линий от специальных селективных ламп.
Это кажущееся сейчас простым решение, которое
лежит в основе аналитического метода атомно-абсорбционной спектрометрии,
предопределило дальнейшее стремительное развитие метода.
Атомно-абсорбционный анализ достаточно близок к
методам традиционной «мокрой» химии, поскольку определение содержания элементов
чаще всего ведется из растворов, что предусматривает во многих случаях
предварительную химическую подготовку проб. Однако, в отличие от большинства
химических методов, атомно-абсорбционная спектрометрия имеет очень высокую
селективность. Поэтому практически редко требуется отделение сопутствующих
элементов, так как их присутствие обычно не вызывает заметной систематической
погрешности определений.
По производительности работы и скорости
выполнения анализов больших партий однотипных проб атомно-абсорбционная
спектрометрия в пламенном варианте, как правило, превосходит такие как
классические химические методы, как гравиметрический, титриметрический,
спектрофотометрический, электрохимические и др. Возможно определение многих
элементов из одного и того же анализируемого раствора. Использование автоматов
для подачи пробы (автоматических дозаторов) значительно упрощает и ускоряет
выполнение массовых анализов. В современных приборах атомно-абсорбционного
анализа полностью автоматизирован процесс измерений и выдачи результатов
непосредственно в единицах концентрации элементов в реальной пробе. Метод
позволяет определять сейчас около 70 элементов.
В самом начале развития метод рассматривали как
специфический для определения только малых концентраций элементов. Но, как
показано теперь многочисленными примерами, метод атомно-абсорбционного анализа
в пламенном варианте позволяет надежно и достаточно точно определять большие
концентрации элементов в пробах нестандартного состава.
Современная техника атомно-абсорбционного
анализа, реализуя гибкость метода, позволяет устанавливать содержание элементов
в широком интервале концентраций:
· в пламени - от десятитысячных долей процента до
десятков массовых процентов;
· в электротермических атомизаторах нижняя граница
определяемых массовых долей для многих элементов составляет 10-6 -10-4%
мас., верхняя - до диапазона пламенных определений.
По воспроизводимости определений метод
атомно-абсорбционного анализа не уступает большинству классических
аналитических химических методов, за исключением гравиметрического и
кулонометрического; относительное стандартное отклонение единичного определения
в пламенном варианте метода обычно не превышает 0.005-0.03 и 0.02-0.15 при
электротермической атомизации.
Наиболее распространен в практике пламенный
вариант метода атомно-абсорбционного анализа, использующий простую, дешевую
аппаратуру и обеспечивающий самые быстрые и высокоточные измерения.
Недостатками пламенного варианта являются низкая
эффективность использования пробы при большом ее расходе, неудовлетворительные
пределы обнаружения многих элементов, фактическая невозможность определения
элементов в порошкообразных и компактных твердых пробах.
Большая часть этих недостатков отсутствует в
электротермических атомизаторах, позволивших понизить пределы обнаружения
большинства элементов по сравнению с пламенным вариантом практически на два
порядка.
Однако длительность аналитических операций при
электротермической атомизации существенно больше (в десятки раз), чем при
пламенной. Одним из наиболее существенных недостатков используемого способа
реализации метода атомно-абсорбционного анализа является необходимость
последовательного определения отдельных элементов.
Серьезным ограничением метода
атомно-абсорбционного анализа является необходимость иметь на каждый элемент
отдельный источник линейчатого излучения. Но и в этом направлении за последние
годы достигнуты серьезные успехи. Многолетние исследования аналитиков многих
стран по изучению возможности создания атомно-абсорбционных приборов без
использования селективных источников света привели к созданию конкурентоспособного
прибора, в котором применены оригинальная оптическая схема и источник со
сплошным спектром, позволяющие определять все традиционные для метода
атомно-абсорбционного анализа элементы.
Таким образом, атомно-абсорбционный анализ дает
возможность проводить универсальными приемами с высокой производительностью,
правильностью и воспроизводимостью массовое определение широкого круга
элементов в большом диапазоне концентраций. Использование электротермической
атомизации позволяет понизить на 1-2 порядка пределы обнаружения элементов по
сравнению с пламенем, сохраняя достаточно высокую воспроизводимость результатов
анализа. Решающим фактором, определяющим правильность и воспроизводимость
результатов атомно-абсорбционного анализа, является стабильность свойств поглощающего
слоя атомных паров.
Основные принципы атомно-абсорбционной
спектрометрии:
Атомно-абсорбционный анализ - метод
аналитической химии, основанный на селективном поглощении электромагнитного
излучения определенной длины волны свободными от всех молекулярных связей
нейтральными атомами определяемого элемента. Для реализации метода
атомно-абсорбционного анализа в наиболее распространенной схеме измерений
необходимо иметь:
селективный источник света изучаемого элемента
(СИС);
атомизатор (Ат) для перевода данного элемента из
реальной пробы в атомарную форму;
спектральный прибор (СП) для выделения
аналитической линии этого элемента;
электронную систему (ЭС) для детектирования,
усиления и обработки аналитического сигнала поглощения.
Определение содержания элемента в пробе проводят
с использованием градуировочного графика, так как метод атомно-абсорбционного
анализа является относительным (сравнительным).
2. Методика выполнения
атомно-абсорбционного анализа
Атомно-абсорбционный анализ (атомно-абсорбц. спектрометрия),
метод количественного элементного анализа по атомным спектрам поглощения
(абсорбции). Через слой атомных паров пробы, получаемых с помощью атомизатора,
пропускают излучение в диапазоне 190-850 нм. В результате поглощения квантов
света атомы переходят в возбужденные энергетические состояния. Этим переходам в
атомных спектрах соответствуют резонансные линии, характерные для данного
элемента. Согласно закону Бугера-Ламберта-Бера, мерой концентрации элемента
служит оптическая плотность A = lg(I0/I), где I0 и I-интенсивности излучения от
источника соответственно до и после прохождения через поглощающий слой.

Рис. 1. Принципиальная схема пламенного
атомно-абсорбционного спектрометра: 1-источник излучения; 2-пламя; 3-монохрома
гор; 4-фотоумножитель; 5-регистрирующий или показывающий прибор.
Приборы для атомно-абсорбционного анализа -
атомно-абсорбционные спектрометры - прецизионные высокоавтоматизированные
устройства, обеспечивающие воспроизводимость условий измерений, автоматическое
введение проб и регистрацию результатов измерения. В некоторые модели встроены
микро ЭВМ. В качестве примера на рис.1 приведена схема одного из спектрометров.
Источником линейчатого излучения в спектрометрах чаще всего служат одноэлементные
лампы с полым катодом, заполняемые неоном.
2.1 Устройство атомно-абсорбционного
спектрометра
Конструктивно спектрометр выполнен в
металлическом корпусе по блочной схеме на массивном основании.
Рис. 2. Схема спектрометра атомно-абсорбционного
«Квант-2»
Каркас спектрометра собран на литой
металлической плите - основании, имеющей четыре опоры для установки прибора в
устойчивом горизонтальном положении. В центральной части расположен атомизатор
с газовыми магистралями и узлом поджига, столик для проб, защитный кожух, а
также дверца с тонированным стеклом. Щиток и стекло предназначены для защиты
оператора от воздействия ультрафиолетового излучения пламени и снижения влияния
окружающих воздушных потоков на стабильность факела горелки атомизатора.
В правой части прибора находится блок
спектральных ламп, в котором расположена турель на 6 позиций, держатель
дейтериевой лампы и элементы оптической схемы. Блок спектральных ламп закрыт
крышкой, положение крышки в открытом состоянии фиксируется газовым лифтом. Под
блоком спектральных ламп находится газовый блок. Блок спереди закрыт панелью.
Подключение газовых магистралей осуществляется к штуцерам панели на задней
крышке газового блока в соответствии с выполненными обозначениями.
В левой части прибора находится блок
монохроматора, элементы оптической схемы, датчик пламени и фотоприёмник. Блок
закрыт крышкой.
Под блоком монохроматора расположен электронный
блок, закрытый панелью, на которой расположена кнопка включения прибора. На
задней крышке блока расположены: вывод заземления прибора, разъём подключения
силового кабеля, разъёмы подключения дополнительных устройств, разъём
гидрозатвора и разъём подключения интерфейсного кабеля.
Для обеспечения защиты корпуса от воздействия
паров кислот металлические детали спектрометра покрыты порошковой эмалью.
2.2 Описание составных частей
спектрометра
Оптическая схема
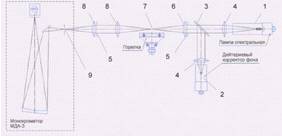
Рис. 3. Оптическая схема прибора
Оптическая схема предназначена для
формирования светового пути от источников излучения 1, 2 через область
фокусировки над газовой горелкой 7 к входной щели монохроматора 9.
Оптическая схема состоит:
Источник линейчатого спектра - лампа
спектральная с полым катодом на анализируемый элемент 1;
Светоделительная пластина 3;
Коллекторные линзы 4;
Диафрагмы 5;
Конденсорная линза 6
Линзы входного объектива монохроматора 8.
Для исключения потерь света в ультрафиолетовом
диапазоне линзы оптической схемы изготовлены из кварцевого стекла.
Светоделительное зеркало имеет покрытие,
обеспечивающее заданные значения коэффициентов отражения и пропускания в
диапазоне 190 ÷ 450 нм.
2.3 Блок монохроматора
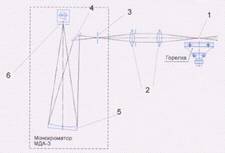
Рис. 4. Оптическая схема монохроматора
Блок монохроматора предназначен для выделения
узкого спектрального интервала, соответствующего одной из линий поглощения
(абсорбция) или испускания (эмиссия) анализируемого элемента. Блок состоит из
входного объектива и монохроматора МДА-3.
Световой поток, прошедший через пламя 1 (при
работе в режиме абсорбции) или излучённый пламенем (при работе в режиме
эмиссии) фокусируется линзами 2 объектива на входной щели 3 монохроматора.
Плоским зеркалом 4 это излучение направляется на сферическую дифракционную
решетку 5. Разложенное в спектр излучение направляется на выходную щель 6
монохроматора. Щели монохроматора расположены относительно дифракционной
решётки так, что изображение входной щели сфокусировано в плоскости выходной
щели. Длина волны, соответствующая выделяемому монохроматором спектральному
интервалу, изменяется путём поворота дифракционной решетки.
Смена ширины щелей монохроматора, поворот и
фокусировка дифракционной решётки осуществляется исполнительными механизмами,
управляемыми по командам компьютера в соответствии с методиками и
рекомендациями. Предусмотрена корректировка настройки монохроматора по длинам
волн при каждом включении спектрометра, а также точная подстройка длины волны
для элементов со сложным спектром.
Ширина щелей монохроматора выбирается исходя из
рекомендаций методики выполнения измерений.
2.4 Атомизатор
Назначение атомизатора - перевод пробы в атомный
пар с возможно большей эффективностью. Основной способ атомизации - нагревание
пробы до 2000-27000С. Для этой цели используется пламя или
электрический ток. Пламя - это низкотемпературная плазма, в которой протекают
химические реакции, поддерживающие температурный режим. В спектроскопии обычно
используется пламя горючих газов - ацетилена, реже пропана в смеси с окислителями
- воздухом или закисью азота.
В аналитической практике наиболее широкое
распространение получило пламя ацетилен-воздух. Это пламя наиболее стабильно,
имеет высокую пропускаемость в аналитическом диапазоне длин волн и слабую
собственную эмиссию, обеспечивает высокую эффективность атомизации более 30
элементов, в том числе щелочных и щелочноземельных металлов.
Использование пламени ацетилен, закись азота с
температурой до 27000С позволило определять почти все элементы.
Для получения пламени и подачи в него
исследуемой пробы используются различные конструкции горелки и распылители.
Горючее, окислитель и проба в виде аэрозоля смешиваются, как правило,
предварительно. Чувствительность атомно-абсорбционного анализа с атомизацией в
пламени ограничена происходящими в ней побочными процессами и кратким временем
пребывания в нем частиц - около 10-3 секунды. Лишь менее 5-15% атомов из
наиболее мелких аэрозольных капель атомизируются. Увеличение чувствительности
наряду с совершенствованием пламенных систем можно получить и с помощью
электротермической атомизации. Атомизация осуществляется в специально печи в
инертной атмосфере, по специальной программе, разделяющей по времени на этапы -
высушивание, озоление, атомизация, прожиг (очистка печи перед очередным анализом).
Электротермическая атомизация осуществляется обычно в графитовых трубках,
нагреваемых электрическим током большой силы. Такой способ был предложен
Львовым.
Работа атомизатора
При подготовке спектрометра к измерениям с
помощью механизма перемещения устанавливают требуемое положение горелки по
вертикали и по горизонтали. Изменение положения горелки по горизонтали
осуществляется путём вращения ручки, а регулировка положения горелки по
вертикали производится либо с помощью электропривода, управляемого по командам
ПК, либо вращением ручки.
Вращение ручки по часовой стрелке приводит к
перемещению горелки вверх, а против часовой стрелки - вниз. Параллельность щели
горелки и оптической оси прибора обеспечивается конструкцией механизма
крепления горелки. При работе спектрометра из газового блока в камеру смешения
по трубкам, подключённым к штуцерам, поступает горючий газ и газ - окислитель.
В камере эти газы смешиваются.
Анализируемый раствор через капиллярную трубку
попадает в распылитель, где превращается в мелкодисперсный аэрозоль. Через
штуцер во внутреннюю полость корпуса распылителя из газового блока поступает
сжатый газ - окислитель. Давление газа - окислителя на входе в распылитель
регулируется дросселем, а его значение контролируется по манометру.

Рис. 5. Блок пламенной атомизации
Газ проходит через зазор между иглой и сводом
капиллярного отверстия сопла.
Вследствие высокой скорости обтекания потоком
газа - окислителя иглы в капиллярном отверстии образуется область пониженного
давления (разрежения), что приводит к засасыванию раствора пробы через
капиллярную трубку. При вытекании из канала иглы раствор захватывается
потоком газа - окислителя, и дробится на капли при прохождении через сопло.
Ударяясь о шарик - импактор капли раствора со скоростью вытекающего потока
дополнительно разбиваются, образуя аэрозоль.
Крупные капли аэрозоля оседают на деталях
распылителя, внутренних стенках камеры, рассекателе и через дренажный штуцер
стекают по шлангу через гидрозатвор в сливную ёмкость. Количество аэрозоля
попадающего в пламя горелки составляет примерно 15÷25%
от
объёма потребляемой пробы, т.е. при расходе 100 мл пробы в слив уйдёт примерно
80 мл.
Временной диапазон нахождения капель аэрозоля в
пламени горелки - несколько миллисекунд. За это время происходит испарение
растворителя, сгорание материала самой пробы, атомизация. В результате всех
этих процессов в пламени образуется атомный пар анализируемого элемента.
2.5 Газовый блок
Газовый блок предназначен для коммутации
газовых потоков в атомизатор, регулировки необходимых расходов и поддержания
рабочих давлений, а также контроля для обеспечения безопасной работы с горючими
газами (рис. 6).
Газовый блок обеспечивает автоматическую подачу
заданных расходов горючего газа и газа - окислителя в камеру смешения и
горелку, газа - окислителя в распылитель, а также контроль входного давления и
наличия пламени. Управление газовым блоком осуществляется в соответствии с
программой управления по командам ПК, которые передаются исполнительным
элементам газового блока через микроконтроллер, входящий в состав электронного
блока спектрометра. Газовый блок состоит из газового модуля, датчика пламени и
гидрозатвора.
Рабочее давление в магистрали окислителей должно
быть в диапазоне 3,5 ÷ 5 атм.,
при давлении менее 3,2 атм. произвести поджиг пламени невозможно. Рабочее
давление в магистрали ацетилена должно быть в диапазоне 1,4 ÷
1,8 атм.,
при давлении менее 1,2 атм. произвести поджиг пламени ацетилен - воздух
невозможно.
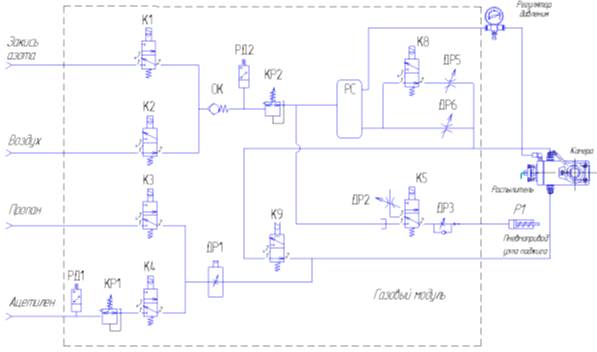
Рис. 6. Пневматическая схема газового модуля
Процесс поджига горелки разбит на три этапа: поджиг,
отжиг, стабилизация
Поджиг - при
подаче команды «поджечь» в зависимости от выбранной горючей смеси открывается
клапан К3 (пропан) или К4 (ацетилен) и газ поступает на регулятор расхода ДР1,
с выхода которого подаётся в камеру смешения. Расход газа определяется
выбранной методикой или оператором по команде управляющей программы.
Одновременно закрываются клапаны К9, К8, открывается клапан К5 и через дроссель
ДР3 воздух поступает на пневмопривод Р1, который производит поворот рычага
блока поджига для перемещения спирали к щели горелки. Спираль накаляется и
поступивший в горелку горючий газ воспламеняется. При появлении сигнала с
датчика о наличии пламени клапан К5 закрывается, и спираль отводится от
горелки.
Отжиг - в
режиме отжига в камеру смешения автоматически подаётся повышенное
количество горючего газа и воздуха. Открывается клапан К8, увеличивая расход
воздуха, также возрастает расход газа, протекающего через ДР1.
Стабилизация - в
режиме стабилизации расходы горючего газа и газа - окислителя
устанавливаются в номинальные значения необходимые для проведения анализа.
Клапан К8 закрывается и регулятором ДР1 устанавливается необходимый расход.
При подаче команды «погасить» происходит
отключение газовых магистралей (клапан К3 или К4) и включение режима,
соответствующего состоянию выключенного прибора.
Условия поджига пламени горелки.
Для обеспечения безопасности работы с горючими
газами и корректности выполнения команд газовой автоматики модуля необходимо
соблюдение следующих условий:
. Наличие давления в магистрали окислителя - не
менее 3,4 атм.;
. Наличие давления в магистрали ацетилена (при
условии работы с газовой смесью ацетилен - воздух) - не менее 1,4 атм.;
. Наличие горелки соответствующей используемой
газовой смеси;
. Наличие воды в гидрозатворе;
. Правильность установки расходов горючего газа
на этапах поджига.
При несоблюдении выше перечисленных условий
управляющая программа выводит на экран монитора соответствующее сообщение о
причине блокировки процедуры поджига пламени. Состояние сигналов с датчиков
газового модуля выводится в окне панели диагностики управляющей программы.
Газ, используемый для работы прибора
Сжатые газы, используемые в работе спектрометра,
должны соответствовать следующим техническим характеристикам:
· Ацетилен растворенный технический
марки А или В по ГОСТ 5457-75 в стальных баллонах, 40 л, ТУ 6-21-38-94 или ТУ
1412-006-00204760-2005. При работе без блока подготовки газов допускается
применение ацетилена только марки А. Номинальное давление газа в баллоне 1,9
МПа при 20ºС, остаточное
давление, при котором допускается эксплуатация, не менее 0,5 МПа;
· Пропан-бутановая смесь в
стальных баллонах (50 л, 27 л, 12 л, 5 л) бытового назначения для сжиженных
углеводородных газов типа 3-50 ГОСТ 15860 с клапанным или вентильным запорным
устройством. Номинальное давление газа в баллоне 1,1 МПа при 20ºС;
· Закись азота (сжиженный
газ) в стальных баллонах, 10 л, ГОСТ 949-73, код по квалификационной системе
ВОЗ(АТС) N01AX13. Номинальное давление газа в баллоне 5,1 МПа при 20ºС;
· Аргон газообразный,
технический, высший сорт по ГОСТ 10157 или ВЧ (ТУ 6-21-12-94) в стальных
баллонах, 40 л, ГОСТ 949-73. Номинальное давление газа в баллоне 15 МПа при 20ºС,
остаточное давление, при котором допускается эксплуатация 2 МПа.
· Сжатый воздух на
входе БПГ и установки для его получения должны удовлетворять требованиям
п.2.3.4 данного Руководства. Сжатый воздух на входе в спектрометр должен
соответствовать классу загрязненности не выше 3 по ГОСТ 17433. Изменение
давления на входе в спектрометр в процессе работы не должно превышать ±10%
относительно установленного значения.
3. Порядок проведения исследования
Метод атомно-абсорбционного анализа является относительным
(сравнительным), поэтому для установления вида градуировочной зависимости
«Абсорбция - Концентрация элемента» используют градуировочные растворы, в
которых концентрация С определяемого элемента известна. С помощью этих
растворов строят градуировочный график в данных координатах «А-С». В области
линейности число градуировочных растворов может быть минимальным - вплоть до
одного, в области нелинейности число градуировочных растворов необходимо
увеличивать. Современные приборы атомно-абсорбционного анализа позволяют с
помощью ЭВМ сразу получать математическую форму градуировочного графика с целью
последующей выдачи результатов непосредственно в концентрациях элементов. Для
этого, после измерения абсорбции элемента в пробе, по градуировочному графику
или математической форме этого графика рассчитывают концентрацию элемента в
пробе. При проведении градуирования и анализа предполагается, что коэффициент a
в уравнении поглощения одинаков для градуировочных растворов и анализируемых
проб. Качество аналитической методики или метода анализа, характеризующее
возможности определения или обнаружения элементов в области их малых
содержаний, называется чувствительностью. Простейшей численной характеристикой
чувствительности служит коэффициент чувствительности - производная
аналитического сигнала по концентрации определяемого элемента. Если
градуировочная функция является линейной, то коэффициент чувствительности - это
тангенс угла наклона градуировочной кривой. Чем выше коэффициент
чувствительности, тем меньшее содержание элемента соответствует одной и той же
величине аналитического сигнала, и тем выше (при прочих равных условиях)
чувствительность методики в целом. Если при атомно-абсорбционных измерениях
градуировочный график прямолинеен в изучаемой области концентраций, то
чувствительность определяется соотношением ΔА/ΔС
(dA/dC), т. е. изменением сигнала атомарного поглощения при изменении
концентрации определяемого элемента, что соответствует тангенсу угла наклона
градуировочного графика.
абсорбция

концентрация элемента
Рис. 7. Градуировочный график: изменение
абсорбции в зависимости от концентрации определяемого элемента
Пробоподготовка для построения
графика
Стадия пробоподготовки является очень важной в
процессе атомно-абсорбционного анализа и зачастую вносит основную погрешность в
результат анализа. Особенно это относится к твердым образцам со сложной
матрицей, которые необходимо перевести в раствор. Уровень химических помех в
значительной степени зависит от способа подготовки пробы, принятого в методике
анализа. Удачно выбранный способ разложения анализируемого материала позволяет
не только перевести в раствор определяемый элемент, но и облегчить его
отделение от сопутствующих элементов или создать благоприятные условия для
конечного определения содержания элементов без операции разделения. Разложение
проб часто является более трудоемкой операцией, чем определение. Подготовка
пробы может занимать от нескольких минут до многих часов.
Общие требования к подготовке пробы
для атомно-абсорбционных измерений включают следующие положения
1. Полное извлечение определяемого элемента из
исходной пробы в конечный раствор.
. Рациональный выбор реактивов и схемы
пробоприготовления с учетом последующей методики анализа.
. Доступность химических реактивов, посуды и
аппаратуры.
. Универсальность приемов пробоприготовления,
т.е. применимость к пробам различного состава и возможность одновременного
определения из одного раствора многих элементов.
. Быстрота, производительность и малая
трудоемкость, возможность механизации и автоматизации пробоподготовки.
. Использование минимального числа и количества
реактивов; низкая поправка контрольной (холостой) пробы, в особенности при
определении следовых содержаний элементов.
Приготовление растворов для
градуировки
Для подготовки градуировочных растворов можно
использовать коммерческие, стандартные образцы растворов металлов (обычно с
концентрацией 100 или 1000 мг/мл) или готовить головные градуировочные растворы
самостоятельно непосредственно в лаборатории с использованием стабильных при
хранении чистых металлов или устойчивых их соединений (оксиды, соли),
квалификации не хуже «ч.д.а», с известным (или специально определенным)
содержанием основного вещества. Использование высокочистых металлов и их
соединений не обязательно, так как головные растворы при доведении до рабочих
градуировочных растворов подвергаются значительному разбавлению. Но для
растворения металлов и их соединений следует применять высокочистые реагенты,
расходуемые в больших количествах. Рекомендуемые вещества для приготовления
градуировочных растворов в атомно-абсорбционном анализе представлены в табл. 1.
Приготовление головных градуировочных растворов с концентрацией 1 мг/мл (1
г/л). После растворения навески объем раствора должен доводиться до 1 литра
разбавлением водой.
Таблица
1
Приготовление градуировочных растворов
|
Элемент
|
Исходный
реагент
|
Навеска,
гр.
|
Растворение
|
|
Al
|
Al
|
1.0000
|
40мл.
конц. HCl +30% Н2О2
|
|
Al
|
1.0000
|
25мл.
конц. HCl + несколько капель конц. HNO3
|
|
Al(NO3)2·9H2O
|
11.605
|
Вода.
|
|
Сa
|
CaCl2
|
2.7692
|
Вода.
|
|
CaCO3
|
2.4973
|
25мл
1М HCl
|
|
Сl
|
NaCl
|
1.6485
|
Вода.
|
|
Сu
|
Cu(NO3)2·3H2O
|
3.8019
|
Вода.
|
|
Fe
|
Fe
|
1.0000
|
20мл.
5М HCl + 5мл. конц. HNO3
|
|
Pb
|
Pb(NO3)2
|
1.5985
|
Вода.
|
|
Zn
|
ZnO
|
1.2447
|
30мл.
5М HCl
|
В таблице 2 приведены данные для некоторых
металлов, необходимые для построения калибровочного графика.
Таблица
2
Рабочие параметры и характеристики ААС «КВАНТ-2»
|
Элемент
|
Длина
волны (нм)
|
Ширина
щели (мм)
|
Ток
ЛПК (мА)
|
Тип
пламени
|
|
Al
|
309,3
|
0,1-0,25
|
10-16
|
А-3А
|
|
Сa
|
422,7
|
0,1-025
|
7-12
|
А-3А
|
|
Сl
|
357,9
|
0,25
|
11-14
|
А-В
|
|
Сu
|
324,8
|
7-10
|
А-В
|
|
Fe
|
248,3
|
0,1
|
18-24
|
А-В
|
|
Pb
|
217,0
|
0,25-0,5
|
13-16
|
А-В
|
|
Zn
|
213,9
|
0,5
|
6-8
|
А-В
|
Порядок работы на спектрометре
Порядок работы на спектрометре определяется
структурой программного обеспечения и представляет собой выполнение
определённой последовательности действий:
. Загрузка методики измерений;
. Настройка (проверка настройки) оптической
схемы прибора;
. Построение (уточнение) калибровочной кривой;
. Проведение измерений неизвестных проб;
. Обработка и печать результатов измерения.
Загрузка листа измерений
Нажать кнопку «Измерение» панели инструментов.
Подвести курсор к нужной ячейке строки «Методики», дважды нажать левую кнопку
мыши, выбрать элемент и предварительно созданную методику в окне «Методики и
калибровки». Нажать кнопку «Выбрать». В окне «Лист измерений» нажать кнопку
«Загрузить методику» (рис. 8).

Рис. 8. Выбор методики
Если уровень сигнала лампы ЛПК менее 20%, в
закладке «ЛД, Усиление» увеличить значение усиления фотоприёмника. Добиться
уровня сигнала лампы ЛПК (жёлтый) 40÷60% (рис.
8).
Настройка оптической схемы прибора
Провести юстировку ЛПК. Настройка положения
лампы возможна в автоматическом и ручном режиме.
Автоматический режим юстировки запускается
нажатием кнопки «Юстировка лампы» в закладке «Турель».
Провести юстировку дейтериевой лампы. Если
уровень сигнала лампы D2 менее 20% в закладке «ЛД, Усиление»
увеличить значение тока лампы D2. Добиться уровня сигнала лампы D2
(бирюзовый) 40÷60%. (рис. 9)
Рис. 9. Окно ручной юстировки
Провести подстройку длины волны
монохроматора
Зайти в закладку «Монохроматор» панели
управления, нажать кнопку «Настройка».
Провести балансировку сигналов. При нажатии
кнопки «Балансировка» в закладке «ЛД, Усиление» происходит автоматическое
выравнивание сигналов ЛПК и D2, а также изменение величины до
значения 100%. Возможно провести балансировку в ручном режиме, изменяя величину
усиления фотоприёмника и тока лампы D2.
Процедуры юстировки лампы, подстройки
монохроматора (рис. 10) и балансировки сигналов управляющая программа выполняет
автоматически при загрузке методики, если произведены соответствующие настройки
в закладке главного меню «Настройки» > «Настройки программы».
Рис. 10. Автоматическая подстройка монохроматора
Проверить положение горелки относительно луча в
вертикальной плоскости. Поджечь пламя. В закладке «Газовая система» панели
управления нажать кнопку «Поджечь». На экране будут отображаться стадии
зажигания горелки. Если в течение 10 секунд после подачи команды «Поджечь»
поджига пламени не произошло, подача горючего газа автоматически прекращается и
на экран выводится сообщение «Поджечь пламя не удалось». В этом случае следует
повторно подать команду на поджиг.
Спектрометр готов к проведению измерений. Перед
построением либо уточнением калибровки рекомендуется прогреть горелку в течение
15 минут.
Порядок проведения калибровки
Для создания калибровки необходимо приготовить
серию растворов с известной концентрацией анализируемого элемента (стандартные
растворы). В их числе может использоваться раствор, не содержащий
анализируемого элемента (нулевой раствор). Для построения калибровки как правило,
используется 3-5 растворов.
Нажать кнопку «Методики» панели инструментов,
выбрать методику далее кнопку «Создание калибровки». Появится окно создания
калибровки (рис. 11). Ввести значения концентраций растворов для построения
калибровочной кривой. Выбрать режим проведения измерений (Количество измерений
на обнуление - 1). Процедура уточнения ранее созданной калибровки подробно
описана в программном обеспечении прибора.
атомный абсорбционный спектрометр
металл
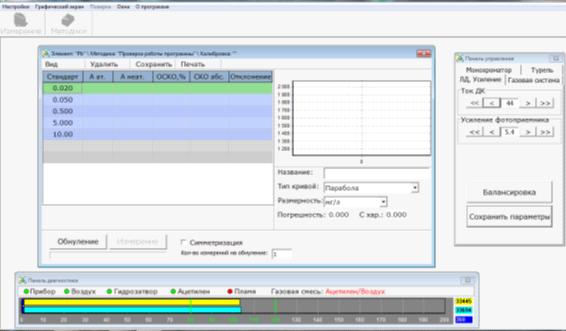
Рис. 11 Окно создания калибровки
Провести измерения аналитического сигнала для
калибровочных растворов. Для каждого из растворов рекомендуется проводить от 2
до 5 параллельных определений (замеров); анализировать калибровочные растворы
следует в порядке возрастания их концентрации (рис. 12).
Каждый из параллельных замеров состоит из двух
процедур - обнуления и измерения.
Для проведения процедуры обнуления необходимо
опустить капилляр в фоновый раствор и нажать кнопку «Обнуление».
Для проведения процедуры измерения ввести
капилляр в калибровочный стандарт, концентрация которого выделена в таблице
окна «Создание калибровки». Нажать кнопку «Измерение», либо нажать на
клавиатуре компьютера клавишу «Enter» или «Пробел».
Следуя рекомендациям программного обеспечения,
уточнить в случае необходимости, тип кривой аппроксимации, ввести название и
сохранить полученную калибровку.
На каждый металл строится свой калибровочный
график, которые остаются в памяти компьютера в разделе «Методика» (см.
приложение 1,2,3).
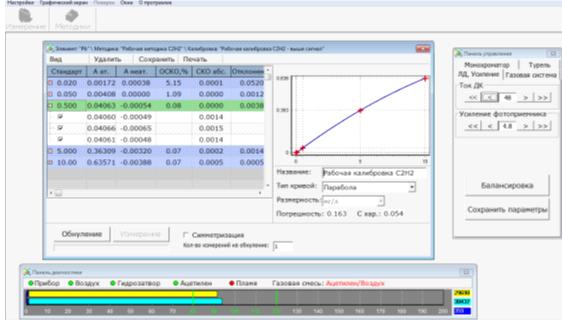
Рис. 12. Построение калибровочной кривой
Порядок проведения измерений
Подробное описание режимов измерений приведено в
Руководстве пользователя ПО (программное обеспечение). Рассмотрим проведение
измерений неизвестных проб в ручном режиме (без автомата подачи проб).
Нажать кнопку «Измерение» панели инструментов.
Появится окно «Лист измерений» рис. 11. Ввести в выбранном столбце строки
методик калибровку на анализируемый элемент (подвести курсор дважды нажать
левую кнопку мыши, выбрать в окне «Методики и калибровки» элемент, методику,
калибровку, нажать кнопку «Выбрать»).
Ввести по необходимости в столбце «Образцы»
названия образцов для анализа (подвести курсор к ячейке, дважды нажать левую
кнопку мыши, ввести название). Выбрать ячейку для измерения на пересечении
столбца с выбранной методикой и строки с анализируемым образцом. Нажать кнопку
«Загрузить методику». Выбрать режим проведения измерений (Кол-во измерений на
обнуление - 1). Провести измерения (рис. 13).
Каждый из параллельных замеров состоит из двух
процедур - обнуления и измерения.
Для проведения процедуры обнуления необходимо
опустить капилляр в фоновый раствор и нажать кнопку «Обнуление».
Рис. 13. Проведение измерений неизвестных проб
Для проведения процедуры измерения ввести
капилляр в раствор измеряемого образца. Нажать кнопку «Измерение», либо нажать
на клавиатуре компьютера клавишу «Enter» или «Пробел».
Результаты измерения заносятся в выбранную
ячейку в соответствии с заданным видом отображения результата. После проведения
измерений сохранить результат. Результаты измерений сохраняются в «Базе данных»
в виде листов и таблицы измерений (рис. 14).
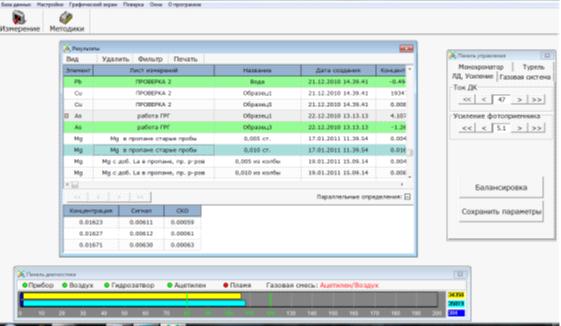
Рис. 14. База данных «Результаты»
Согласно выше изложенных инструкций проводится
измерение проб, которые поступили в лабораторию для исследования (каждой пробе
в лаборатории присваивается свой лабораторный номер, см. приложение 4).
Результатом анализа считается среднее из трех
измерений (рис.15).
Проведен анализ трех проб, по три замера в
каждой, выведена средняя концентрация.
Согласно Санитарным правилам и нормам СанПиН
2.1.7.573-96 «Гигиенические требования к использованию сточных вод и их осадков
для орошения и удобрения», (утв. постановлением Госкомсанэпиднадзора РФ от 31
октября 1996 г. №46) концентрация меди должна быть не более 3,0 мг/л.

Рис. 15. Протокол анализа
4. Порядок установки спектрометра
Требование к помещению.
· Прибор следует устанавливать в закрытом,
отапливаемом, сухом, проветриваемом помещении, вдали от нагревательных
устройств и источников резких потоков воздуха (кондиционеры, вентиляторы,
двери, форточки).
· Помещение, в котором установлен
прибор, должно иметь площадь не менее 10 м2 и объём не менее 30 м3
из расчёта на одно рабочее место.
· Помещение должно быть обеспечено
вытяжной вентиляцией. Над горелкой устанавливается металлический зонт,
подключённый к системе принудительной вытяжной вентиляции с расходом воздуха
5-10 м3/мин.
· Помещение следует выбирать, исходя
из требований минимального уровня запыленности и задымлённости, так как
взвешенные частицы пыли и дыма вызывают трудно-контролируемые неселективные
помехи, обусловленные рассеянием света. Наличие пыли в помещении приводит со
временем к ухудшению пропускания и увеличению рассеяния оптическими элементами
прибора. Городская пыль является источником загрязнений при анализе тяжёлых
металлов и элементов щелочной группы.
· Покрытие пола, потолка, стен должно
соответствовать требованиям технологической гигиены и не накапливать пыль и
влагу.
· Воздух в помещении не должен
содержать пары химически активных веществ (особенно соляной и серной кислот) и
органических растворителей в количествах, превышающих установленные санитарные
нормы.
· Не следует располагать спектрометр
вблизи оборудования, являющегося источником вибрации и электромагнитных помех.
· Желательно устанавливать спектрометр
в помещении, где располагается рабочее место химика - аналитика, оснащённое
раковиной и канализационным сливом.
Организация газоснабжения.
· К рабочему месту должны быть подведены сжатые
газы, необходимое число которых определяется используемым типом пламени.
· Для увеличения ресурса работы
спектрометра, снижения требований к техническим характеристикам сжатых газов и
газовых магистралей рекомендуется при эксплуатации спектрометра использовать
блок подготовки газов (БПГ).
· Баллоны со сжатыми газами должны
быть установлены в отдельном нерабочем помещении или за пределами здания в
закрытых металлических шкафах с соблюдением «Правил устройства и безопасной
эксплуатации сосудов, работающих под давлением» (постановление Госгортехнадзора
России №91 от 11.06.2003 г.).
· Помещения, используемые для
установки спектрометра и размещения баллонов, а также газовые магистрали,
должны быть приняты пожарной инспекцией и Госгортехнадзором.
· Баллоны с ацетиленом и сжиженными
газами (закись азота и пропан-бутановая смесь) должны устанавливаться только в
вертикальном положении.
· За пределами здания и вне
отапливаемых помещениях допускается устанавливать только ацетиленовые баллоны
или баллоны с пропан-бутановой смесью, снабженные вентильным запорным
устройством и баллонным редуктором типа «БПО 5-4».
· Компрессорная установка для
получения сжатого воздуха должна быть расположена на расстоянии не менее 5 м от
прибора. При этом компрессорную установку, являющуюся источником шума и
вибраций, желательно разместить в отдельном помещении. Компрессорная установка
должна иметь в своем составе ресивер вместимостью не менее 20 л. и регулируемый
газовый редуктор. Максимальная производительность должна быть не менее 105
л/мин, производительность при противодавлении в ресивере 0,6 МПа должна быть не
менее 45 л/мин.
· Допускается использование сжатого
воздуха компрессорных станций c давлением на выходе 0,45÷0,6
МПа.
При этом на выходе воздушной магистрали перед спектрометром должны быть
установлены запорное устройство и ресивер с устройством для удаления конденсата
и масляных паров.
· На баллонах с газами должны быть
установлены исправные редукторы давления с поверенными манометрами.
Магистрали, по которым сжатые газы поступают к
спектрометру, должны удовлетворять следующим техническим требованиям:
· Газовые магистрали, выходящие за пределы
помещения, в котором установлен спектрометр, должны быть выполнены из стальных
нержавеющих труб, или с помощью полипропиленовой трубки, проложенной для защиты
от механических и тепловых воздействий внутри стальных труб большего диаметра.
· Для подводки сжатого воздуха кроме
того могут быть использованы резиновые газовые рукава класса III, ГОСТ 9356-75,
или трубки из других полимерных материалов с техническими характеристиками,
рассчитанными на максимальное рабочее давление не менее 2 МПа.
· Внутренний диаметр труб для подводки сжатого
воздуха должен быть не менее 7 мм, а для подводки других газов 6 ÷
9 мм.
Состав растворов, вводимых в
атомизатор
Не допускается вводить в пламя растворы на
основе хлор- и фторсодержащих органических растворителей. Содержание в растворе
хлор- и фторсодержащих органических соединений, а также хлорной и серной
кислоты не должно превышать 5%. Содержание других кислот не должно превышать
20%. Общее содержание солей металлов в растворе не должно превышать 5%.
Введение в пламя “ацетилен-закись азота”
раствора, содержащего хлорную кислоту в концентрации выше 5%, может привести к
взрыву.
Превышение общего содержания в растворе солей
металлов сверх указанного выше приводит к понижению температуры пламени,
возрастанию матричных (химических) помех, что существенно ухудшает
аналитические характеристики спектрометра.
Во избежание повреждения деталей распылителя, не
допускается вводить в атомизатор растворы, содержащие плавиковую кислоту. При
вводе растворов, содержащих серную кислоту, следует соблюдать крайнюю
осторожность - по окончании работы перед выключением пламени необходимо
пропускать через пламя промывочный раствор или дистиллированную воду в течение
10 минут.
Следует избегать ввода в атомизатор растворов,
содержащих частицы размером более 200 мкм и волокна, это может привести к
засорению канала распылителя и (или) капиллярной трубки.
Заключение
В данном курсовом проекте представлено
устройство и принцип работы атомно-абсорбционного спектрометра «Квант-2А»,
описаны основные элементы и схемы прибора, проведено исследование проб сточных
вод на наличии в них меди (Cu).
В ходе измерения было определено, что концентрация меди в отобранных пробах не
превышает норм, указанных в Санитарных правилах.
Согласно проведенному исследованию стоит
отметить, что назначение атомно-абсорбционного спектрометра заключается в том,
что он может определить порядка 70 элементов (главным образом, металлов).
Этот метод имеет ряд
преимуществ:
· простота,
· высокая селективность и малое
влияние состава пробы на результаты анализа.
· высокая скорость анализа (в
автоматическом режиме работы):
пламенный спектрометр до 500 проб в час,
спектрометр с графитовой печью - до 30 проб в
час.
· высокая чувствительность ААС:
Пределы обнаружения большинства элементов в
растворах при атомизации: в пламени 1-100мкг/л, в графитовой печи 0,001- 1
мкг/л (абсолютные пределы обнаружения 0,1-100 мкг/л).
Относительное стандартное отклонение
(повторяемость и воспроизводимость) в оптимальных условиях измерений 0,2-0,5%
для пламени и 0,5-1,0% для печи.
Недостаток метода - необходимость переведения
пробы в раствор; невозможность одновременного определения нескольких элементов
при использовании линейчатых источников излучения.
Высокая скорость проведения анализа, качество
выполненного исследования, полная автоматизация процесса позволяет считать его
самым эффективным в определении концентрации металлов в растворах.
Очень широко данный метод используется для
анализа экологических объектов: природные и сточные воды, почвы, растения,
биологические ткани, жидкости, корма, продукты питания, атмосферные выбросы.
Литература
1. Прайс
В. Аналитическая атомно-абсорбционная спектроскопия, пер.с англ., М., 1976.
2. Брицке
М.Э. Атомно-абсорбционный спектрохимический анализ. // М. Химия. 1982.
. Полуэктов
Н.С. Методы анализа по фотометрии пламени // М. Химия, 1967.
. Славин
У. Атомно-абсорбционная спектроскопия.// пер. с англ., М., Химия, 1971.
. Пупышев
А.А. Практический курс атомно-абсорбционного анализа: Курс лекций.
Екатеринбург: ГОУ ВПО УГТУ-УПИ, 2003.
. Золотов
Ю.А. Основы аналитической химии. Т.2. М.
Химия, 1996, стр.199-262.
Приложение 1
Калибровочный график на медь

Приложение 2
Калибровочный график на свинец
Приложение 3
Калибровочный график на железо
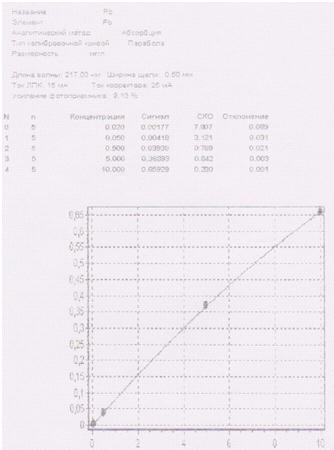
Приложение 4
Протокол исследуемых проб, поступивших в
лабораторию на определение содержания меди в сточных водах
