Определение ионов хлорида цинка, хлорида железа, хлорида натрия
Федеральное
агентство по образованию
Государственное
образовательное учреждение высшего профессионального образования
Алтайский
государственный университет
Химический
факультет
Кафедра
аналитической химии
Курсовая
работа
Определение
ионов ZnCl2, FeCl3, NaCl
Барнаул -
2011
Содержание
Введение
Литературный
обзор
. Методы
определения ионов цинка (Zn2+)
1.1 Титрование цинка (II)
раствором этилендиаминтетрауксусной кислоты (ЭДТА) с индикатором эриохромовым
черным Т
.2 Совместное обнаружение ионов Cu2+, Cd2+, Zn2+, Mn2+
.3 Определение ионов Cd2+ И Zn2+
.4 Определение ионов Zn2+
1.5
Разделение и определение меди и цинка
2. Методы определения Fe3+-ионов
.1 Роданильный метод. Люминесцентный анализ
.2 Определение С сульфосалициловой кислотой. Фотометрический метод
.3 Определение С 1,10-фенантролином (О-фенантролином). Фотометрический
метод
.4 Определение высокочастотным титрованием
.5 Определение железа в растворе
Потенциометрическое титрование
. Методы определения хлора
.1 Нефелометрическое определение хлорид-ионов в электролите никелирования
.2 Кондуктометрическое титрование хлороводородной и уксусной кислот
.3 Потенциометрическое определение иодид- и хлорид-ионов в их смеси
.4 Фотометрическое определение по окраске роданоферрата (III)
.5 Фотометрическое определение по окраске роданоферрата (III)
. Методы определения натрия
4.1
Пламенно-фотометрический метод определения окисей натрия и калия
.2
Пламенно-фотометрический метод определения окисей натрия и калия (в огнеупорных
материалах и изделиях с массовой долей двуокиси циркония до 65%, кроме
бадделеитовых)
.3 Метод
определения натрия и калия в водной вытяжке
.4
Определение содержания натрия и калия, растворимых в разбавленной соляной
кислоте, методом экстракции
.5 Метод
определения концентрации обменных катионов натрия и калия
5. Экспериментальная часть: определение ионов Zn2+
Заключение
Список
литературы
Введение
Современная аналитическая химия испытывает сильное влияние
экспериментальной физики и физической химии. Прогресс этих наук, разнообразие и
точность их методов изучения материи в значительной степени изменяют основное
направление развития аналитической химии. Все большее значение в аналитической
химии приобретают физические и физико-химические (инструментальные) методы
анализа.
Инструментальные методы применяют для решения задач химического анализа,
они составляют неотъемлемую часть аналитической химии. Несмотря на прогресс
инструментальных методов анализа, позволяющих решать химико-аналитические
задачи, неразрешимые обычными методами гравиметрического и титриметрического
анализа, классические методы по-прежнему играют доминирующую роль и являются
основой аналитической химии - науки о методах анализа. Не учитывая характер
исследуемого объекта, его назначение, агрегатное состояние, концентрацию,
наличие примесей, цели анализа, требуемую точность определения, длительность
выполнения анализа и многие другие факторы, нельзя отдать предпочтение тому или
иному методу анализа. Лишь овладев самыми разнообразными методами анализа и
сочетая химические, физические и физико-химические методы анализа с методами
разделения и концентрирования и использования ЭВМ, химик-аналитик сможет
успешно разрешить любую поставленную перед ним химико-аналитическую задачу.
Физические методы анализа. Определение состава и структуры самых
разнообразных веществ можно осуществлять, не прибегая к химическим или
электрохимическим реакциям. Такого рода методы определения основываются на
изучении с помощью различных прецизионных приборов физических свойств
исследуемого вещества. Физические методы обладают существенными достоинствами,
например многие из них характеризуются высокой чувствительностью и
экспрессностью. Применение этих методов дает возможность автоматизировать
измерения и решать задачи, которые нельзя разрешить методами химического
анализа. К недостаткам физических методов относятся сравнительно слабая
избирательность их и необходимость использования стандартных образцов
(эталонов) - материалов с точно установленным химическим составом по ряду
элементов.
Физико-химические методы анализа. Для анализа веществ широко используются
химические реакции, которые сопровождаются изменением физических свойств
анализируемой системы. Все методы такого рода объединяют под общим названием
«физико-химические методы». Другими словами, сущность физико-химических методов
анализа сводится к изучению соотношений между составом и свойствами исследуемых
систем. Различают прямые и косвенные физико-химические методы. В прямых методах
анализа данное свойство является критерием содержания определяемого вещества,
эти методы основаны на изучении диаграмм «состав-свойство». В косвенных методах
определенное свойство служит указателем (подобно индикатору) конца реакции,
т.е. в косвенных методах используется данное свойство определяемого вещества для
фиксирования конца процесса взаимодействия определяемого вещества с реактивом
точно известной концентрации.
Физико-химические методы, отличаясь высокой чувствительностью и
экспрессностью выполнения, дают возможность автоматизировать
химико-аналитические определения и являются незаменимыми при анализе малых и
ультрамалых количеств неорганических и органических веществ. Физико-химические
методам принадлежит ведущая роль в аналитическом контроле производства на
больших предприятиях химической промышленности, и особенно в контроле
производств, использующих в технологических процессах высокие температуры и
давления, огнеопасные, ядовитые, взрывчатые и радиоактивные вещества.
Недостатками физико-химических методов является относительно невысокая
точность многих из них и неуниверсальность. Многие из них целесообразно
применять лишь для массовых анализов.
Предмет работы: определение ионов цинка, железа, натрия, хлора.
Цель работы: отбор и изучение методик определения ионов Zn3+, Fe3+, Na+, Cl-.
Для достижения поставленной цели следует решить ряд задач:
1. Поиск литературы по теме
2. Сбор и обобщение материала по теме
. Выделение интересующих моментов
Поставленные задачи были решены с помощью методов
научного исследования: изучения литературы по теме.
1. Методы определения ионов Zn2+
.1 Титрование цинка(II) раствором этилендиаминтетрауксусной
кислоты (ЭДТА) с индикатором эриохромовым черным Т
Метод основан на индикаторном фотометрическом
титровании ионов цинка раствором ЭДТА с индикатором эриохромовым черным Т.
Zn2++H2Y2- → ZnY2- +2H+.
Взаимодействие ионов цинка с ЭДТА приводит к
образованию устойчивого хелата:
H2C - CH2
−OOCCH2 - N N - CH2COO−2CCOO - Zn - OOCCH2
Устойчивость образующегося комплексоната может быть
определена через условную константу устойчивости
βустZnY2- = β ZnY2-/ αZn(NH ) αY(H)
где αZn(NH ), αY(H) - коэффициенты побочных реакций ионов
металла и ЭДТА.
Эта константа учитывает влияние рН раствора и побочные
реакции ионов металла с ЭДТА, из которых наибольшее значение имеют реакции
образования аммиакатов ионами цинка и протонирование реагента. Титрование
проводят в среде аммиачного буферного раствора при рН = 9, так как в этих
условиях достигается наибольшая устойчивость комплексоната цинка, что
иллюстрируется данными рис. 1.30.
В качестве металлоиндикатора на ионы цинка используют
эриохромовый черный Т, проявляющий кислотно-основные свойства, за счет
ионизации комплексообразующих групп.
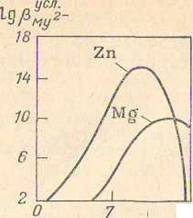
Рис. 1.30 Зависимость условных констант устойчивости
комплексонатов цинка и магния от рН раствора
Взаимодействие ионов цинка с эриохромовым черным Т
происходит по о-окси-о'-оксиазогруппировке и приводит к образованию комплекса,
менее устойчивого, чем комплексонат цинка, что подтверждается приведенным ниже
расчетом условных констант устойчивости комплексоната цинка и комплекса цинка с
металлоиндикатором:
O - Zn - O
−O3S N = N2
lgβZnY2- = lgKZnY2-
- lgαZn(NHs)n= 16,5-4,2 = 12,3
lgβZnInd= lgKZnInd
- lgαZn(NH3)n= 12,9-4,2= 8,7
Ионное состояние эриохромового черного Т сильно подвержено
влиянию рН раствора. Значительное различие в условных константах устойчивости
комплексов цинка с ЭДТА и с эриохромовым черным Т, а также достаточная
контрастность цветной реакции позволяют использовать ее для фотометрической
индикации конечной точки титрования.
Учитывая сложность равновесий в растворе, оптимальной
для фотометрического титрования ионов цинка будет среда, содержащая 0,1 М NH3 (pH =
9). Фотометрическому титрованию ионов цинка раствором ЭДТА не мешают ионы Ba(II), Ca(II), Mg(II), Sr(II), так как у них lgβMY<10. Если различие в устойчивости
комплексов оказывается недостаточным, то для обеспечения избирательности
применяют маскирование и разделение.
Приборы и реактивы:
Абсорбциомер ЛМФ-69, ЛМФ-72 с оранжевым светофильтром.
Рабочий раствор сульфата цинка ZnSO4· 7H2O хч, с концентрацией цинка 6,54
мг/мл, 0,05 М.
Раствор ЭДТА чда, 0,05 М.
Раствор хлорида аммиака NH4C1 хч
0,1 М.
Раствор аммиака хч, 0,1 М
Раствор эриохромового черного Т, 0,1%-ный.
Установка титра ЭДТА по цинку. В стакан для фотометрического
титрования помещают 1 мл стандартного раствора ZnSO4 7H2O, 5 мл раствора NH4C1, 25 мл раствора аммиака, 0,5 мл раствора эриохромового
черного Т и 10 мл дистиллированной воды. Стакан с приготовленным
раствором помещают в прибор и титруют раствором ЭДТА, снимая показания прибора
после прибавления каждой 0,1 мл титранта (вблизи точки эквивалентности по 0,05
мл). По результатам титрования строят график T=f(V). Титруют не менее трех проб,
находят среднее значение объема титранта и рассчитывают титр ЭДТА:
ТЭДТА/Zn = TZn (VZn/VЭДТА)
Выполнение работы
К анализируемому раствору добавляют 5 мл раствора NH4C1, 25 мл раствора аммиака, 0,5 мл эриохромового черного Т, 10
мл дистиллированной воды и титруют раствором ЭДТА (не менее трех титрований).
По результатам строят кривые титрования и находят Vэдта. По среднему значению рассчитывают
содержание цинка в растворе. Методом наименьших квадратов находят доверительный
интервал и стандартное отклонение.
1.2 Совместное обнаружение ионов Cu2+, Cd2+, Zn2+, Mn2+
На фоне аммиачного буферного раствора
полярографические волны ионов, образующих аммиакаты, достаточно четко выражены
и E½
существенно различаются.
Приборы:
Полярограф любой марки
Электролизер
Ртутный капающий электрод
Насыщенный каломельный электрод
Очищенный азот
Градуированные пипетки вместимостью 10 мл
Кристаллический сульфат натрия
Фоновый электролит - аммиачный буферный раствор
Стандартные растворы сульфатов кадмия, марганца, меди,
цинка, 10~3 М
Анализируемый раствор сульфатов солей, 10~3
М
Выполнение работы
Регистрируют полярограмму фонового электролита. Для
этого в электролизер вносят 10 мл аммиачного буферного раствора, несколько
кристаллов Na2SO3, перемешивают, опускают электроды, ячейку подключают к
полярографу и регистрируют полярограмму при потенциалах: -0,2 − -1,8В.
Регистрируют полярограмму стандартных растворов: в
электролизер вносят 10 мл аммиачного буферного раствора, 0,5 мл стандартного
раствора CdSO4 и полярографируют, как указано выше. Аналогично
полярографируют растворы MnSO4, CuSO4, ZnSO4.
Исследуемые растворы анализируют, как стандартные
растворы.
Полученные полярограммы обрабатывают графически и
путем построения полулогарифмического графика рассчитывают E½ и число электронов, участвующих в
электродном процессе. Значения E½ для полярограммы неизвестного раствора сравнивают с
найденными значениями E½ для стандартных растворов и табличными данными. Делают
вывод о качественном составе анализируемого раствора.
.3 Определение ионов Cd2+ и Zn2+
Для титрования используют реакции осаждения:
5Cd2+ + 4K4[Fe(CN)6]
→ K6Cd5[Fe(CN)6]4↓
+ 10K+,
3Zn2+ + 2K4[Fe(CN)6]
→ K2Zn3[Fe(CN)6]4↓
+ 6K+
с регистрацией тока электрохимического окисления
титранта на вращающемся платиновом аноде:
[Fe(CN) 6]4- →
[Fe(CN)6]3- + e-.
В качестве катода выбирают насыщенный каломельный
электрод.
Приборы и реактивы:
Амперометрическая установка любого типа
Платиновый вращающийся микроэлектрод
Насыщенный каломельный электрод
Мерная колба вместимостью 50 мл
Градуированная пипетка вместимостью 5 мл
Мерный цилиндр вместимостью 50 мл
Микробюретка вместимостью 5 мл
Стаканы вместимостью 50 мл
Раствор сульфата калия, 0,5 М
Раствор гексацианоферрата II калия, 0,03 М
Анализируемый раствор сульфата цинка (кадмия), ≈10-3
М
Выполнение работы
Перед титрованием выбирают потенциал электрохимического
окисления K4[Fe(CN)6], для чего необходимо
зарегистрировать вольтамперную кривую. Очищают поверхность платинового
электрода погружением его в раствор HNO3
(1:1), затем несколько раз рабочую поверхность электрода обмывают
дистиллированной водой. Вводят в электролизер 1мл раствора титранта, добавляют
30 мл фонового электролита, замыкают цепь, включают электромотор, вращающий
рабочий микроэлектрод, и постепенно изменяя внешнюю э.д.с. в интервале 0−2,0В
через каждые 0,2В, регистрируют показания микроамперметра. Строят
функциональную зависимость I-Е и
выбирают значение потенциала, соответствующее предельному току окисления K4[Fe(CN)6].
Анализируемый раствор ZnSO4 (CdSO4) помещают в мерную колбу вместимостью 50 мл, раствор
разбавляют дистиллированной водой до метки, перемешивают, микробюретку
заполняют раствором K4[Fe(CN)6].
В электролизер пипеткой вносят 5 мл этого раствора, 30 мл раствора
фонового электролита, погружают электроды, включают электромотор, устанавливают
потенциал, соответствующий предельному току окисления титранта, и титруют
раствором K4[(Fe(CN)6], прибавляя его
порциями по 0,1мл и регистрируя показания микроамперметра. Титрование повторяют
несколько раз. По результатам титрования строят кривую в координатах I-VK4[Fe(CN)6], по излому на этой кривой
определяют объем раствора K4[(Fe(CN)6],
соответствующий точке эквивалентности и рассчитывают содержание цинка (кадмия)
в анализируемом раствором.
1.4 Определение ионов Zn2+
Определение основано на реакции осаждения ионов цинка
гексацианоферрат (II)-ионами,
генерируемыми из гексацианоферрат (III)-ионов на платиновом катоде в кислых растворах. При этом протекают
следующие реакции:
Fe(CN)63-
+ e- → Fe(CN)64- на
электроде,
Zn2++2Fe(CN)64- + 2K+→ K2Zn3[Fe(CN)6]2↓ в
растворе.
Конечную точку титрования определяют
потенциометрическим или амперометрическим методом с двумя поляризованными
платиновыми электродами.
Приборы и реактивы:
Кулонометрическая установка с потенциометрической
индикацией конечной точки титрования
Генераторный и индикаторный серебряные электроды
Вспомогательный электрод - стальной стержень
Хлорсеребряный электрод сравнения
Мерная колба вместимостью 100 мл
Пипетка вместимостью 10 мл
Раствор K3Fe(CN)6, 0,2 М в буферной смеси рН = 1 - 3
(вспомогательный реагент)
Анализируемый раствор соли цинка, 10-2 М
Выполнение работы
Исследуемый раствор разбавляют дистиллированной водой
в мерной колбе до метки и перемешивают. Переносят пипеткой 10 мл в
кулонометрическую ячейку, добавляют 15 мл раствора вспомогательного реагента,
опускают генераторный и индикаторный электроды. Титрование ведут при величине
тока 5мА.
По данным титрования строят кривую зависимости
потенциала от времени при потенциометрической индикации или тока от времени при
амперометрической индикации и находят конечную точку титрования. Содержание
цинка (в мг) рассчитывают по формуле:
G = 0,01036ItMfVk/Vп,
где I- ток,- время генерации, с;
M - молекулярная масса цинка;
fэкв - фактор эквивалентности;
Vk - вместимость мерной колбы;
Vп - вместимость пипетки.
1.5 Разделение и определение меди и цинка
Определение основано на электроосаждении меди на катоде из азотнокислого
раствор и взвешивании полученного осадка:
Cu2+ + 2e- → Cu
После отделения меди ионы цинка определяют
потенциометрическим титрованием исследуемого раствора стандартным раствором NaOH в водно-ацетоновой среде (1:2). При
этом наблюдаются два скачка потенциалов, первый из которых соответствует
оттитровыванию сильной кислоты, присутствующей в растворе, а второй -
оттитровыванию цинка:
HNO3 + NaOH →NaNO3
+ Н2О(NO3)2 + 2NaOH →
Zn(OH)2 + 2NaNO3
Приборы и реактивы:
Установка для электроосаждения металлов
Потенциометр: рН-метр - милливольтметр рН = 12
Магнитная мешалка
Аналитические весы
Электроды для электролиза: катод и анод - платиновые
цилиндрические сетки
Стеклянный индикаторный электрод
Хлорсеребряный электрод сравнения
Мерная колба вместимостью 250 мл
Микробюретка вместимостью 2 мл
Пипетки вместимостью 10 и 50 мл
Стаканы вместимостью 100 мл
Раствор HNO3> (1:1)
и 2 M
Раствор NaOH,
0,1 М и 25%-й
Этиловый спирт
Ацетон
Анализируемый раствор смеси солей меди и цинка, ≈0,1М
Выполнение работы
Определение меди. Поверхности катода и анода
предварительно очищают, погружая в раствор HNO3 (1:1) (под тягой), после чего их тщательно промывают
дистиллированной водой. Катод ополаскивают этиловым спиртом, высушивают в
сушильном шкафу при 110°С, охлаждают в эксикаторе и взвешивают на аналитических
весах.
Катод и анод закрепляют в электродержателях,
присоединяя к источнику тока соответственно полюсам. В электролизер помещают
анализируемый раствор, добавляют 1 мл 2 М раствора НNOз, погружают в него электроды и разбавляют исследуемый
раствор таким количеством дистиллированной воды, чтобы часть катода (5-7 мм)
выступала над поверхностью раствора. Это необходимо для проверки в дальнейшем
полноты осаждения меди. Электроды не должны касаться друг друга, а также дна и
стенок стакана. Включают магнитную мешалку и регулируют перемешивание раствора.
Включают ток и проводят электролиз 35-40 мин, контролируя напряжение (2-2,5 В)
или ток (1- 0,5 А) по прибору. По мере осаждения меди катод окрашивается в
красный цвет, а раствор постепенно обесцвечивается. Затем для проверки полноты
осаждения меди приливают в электролизер 10-15 мл дистиллированной воды. Если
через 10 мин на вновь погруженной поверхности катода не наблюдается дальнейшего
выделения меди, электролиз заканчивают. В противном случае электролиз
продолжают еще 10-15 мин.
По окончании электролиза, не выключая ток, извлекают
электроды из раствора, поднимая осторожно электродержатель, и промывают
электроды струей дистиллированной воды над стаканом с исследуемым раствором.
Промывание электродов под током необходимо во избежание химического растворения
выделенной меди. Выключают ток, ополаскивают катод этиловым спиртом, высушивают
в сушильном шкафу при 110°С охлаждают в эксикаторе и взвешивают на
аналитических весах. Содержание меди в исследуемом растворе определяют по
разности между массой катода с осадком и массой чистого катода.
Определение цинка методом потенциометрического
титрования. После электроотделения Сu2+ раствор
из электролизера количественно переносят в мерную колбу вместимостью 250 мл,
доводят объем до метки дистиллированной водой и тщательно перемешивают.
Отбирают пипеткой аликвотную часть полученного раствора в стакан для
титрования, прибавляют 20 мл ацетона, опускают в раствор индикаторный электрод
и электрод сравнения, включают магнитную мешалку и потенциометр. Титруют 0,1 М
раствором NaOH порциями по 0,1 мл, записывая
показания потенциометра по шкале потенциалов (мВ). Титрование повторяют до получения
трех сходящихся результатов.
Строят кривые потенциометрического титрования
(интегральную и дифференциальную), по которым находят конечные точки титрования
азотной кислоты (V1) и суммы HNO3+Zn2+(V2). Разность объемов титранта (V2-V1) соответствует содержанию ионов
цинка в исследуемом растворе. Содержание Zn2+ рассчитывают по формуле:
gZn2+ = CNaOH(V2-V1) AZn2+fэквZn2+Vk/1000Vп
Определение цинка на платиновом катоде, предварительно
покрытом медью. После отделения меди электролизом, как описано выше, раствор
переносят в мерную колбу вместимостью 250 мл и доливают до метки
дистиллированную воду. Отбирают 50 мл раствора в электролизер, добавляют 25%-й
раствор NaOH в количестве, необходимом для
растворения гидроксида цинка, и еще 5 мл избытка щелочи. Осаждение проводят на
взвешенном омедненном платиновом катоде, проверяют полноту осаждения и
промывают электроды, как описано выше. Катод ополаскивают этиловым спиртом,
высушивают при 110°С в сушильном шкафу, охлаждают в эксикаторе и взвешивают на
аналитических весах. Содержание цинка вычисляют по формуле:
gZn =(m1- m2)VK/Vп.
2. Методы определения Fe3+-ионов
.1 Роданильный метод. Люминисцентный
анализ
Ионы железа (III) в кислой среде образуют красные родонильные комплексы.
Это один из наиболее часто применяемых методов, хотя
он имеет много недостатков. Действительно, роданоферратные комплексы
малоустойчивы, полученные окраски зависят от концентрации этих комплексов,
ионной силой связывающих железо (III) в
комплексы, например хлорид- и сульфат- ионов. Зависят окраски и от величины рН
раствора.
Интенсивность получаемой окраски можно повысить,
уменьшая диэлектрическую постоянную среды: добавляя ацетон или диоксан.
Ионы Fe3+ постепенно восстанавливаются
роданид-ионами, быстрее на свету и особенно, когда колориметрическую кювету
освещают сильными пучками света. Этого можно избежать, прибавляя окислитель
(персульфат, перекись водорода).
Можно стабилизировать комплекс в отношении света,
добавлением смеси метилэтилкетона и ацетона.
Роданид железа Fe(CNS)3
можно экстрагировать эфирами и спиртами; он также имеет красный цвет.
Точность и чувствительность:
Молярный коэффициент светопоглощения ε = 7000 при λ = 480 нм и ε = 15000 в присутствии ацетона.
Результаты воспроизводимы только при работе в точно одинаковых условиях.
Большое значение имеет соблюдение одинаковой температуры.
Мешающие ионы:
Некоторые ионы мешают определению. Следующие вещества,
присутствуя в концентрации, в 250 раза превышающие концентрацию ионов железа,
могут привести к ошибке, которая не будет больше 2%: ацетат-, арсенат-,
бромид-, цитрат-, хлорид-, нитрат-, сульфат-, и тартрат-ионы, синильная,
муравьиная, фосфорная и кремневая кислоты, ионы алюминия.
Медь (II),
кобальт, висмут, титан, рутений, осмий и молибден (VI) образуют с роданит-ионам окрашенные комплексы.
Вольфрам (VI) осаждается в виде вольфрамовой кислоты, медь (I) и серебро осаждаются в виде родонитов, ртуть (II) связывает роданид-ионы в комплекс.
Кислотность раствора должна быть постоянной. Роданид обычно вводят в
концентрации, равной 0,3М. Если надо провести определение с ошибкой, не
превышающей 1%, то указанная концентрация роданида должна соблюдаться с
отклонениями в пределах +1%.
Реактивы:
Роданид калия, 15% раствор. Когда определяют очень малые количества
железа, надо проверить реактив на чистоту. Для этого к небольшому количеству
(объёму) роданида калия прибавляют равный объём ацетона.
Если появиться розовое окрашивание, прибавляют немного соли алюминия и
осаждают аммиаком алюминий, который захватывает с собой и примесь железа (III), которая была в реактиве, осадок
отфильтровывают и отбрасывают; раствор нейтрализуют.
Соляная или азотная кислота. Проверяют реактивы на
чистоту. Если для анализа взяли азотную кислоту, пропускают через неё некоторое
время воздух, чтобы удалить окислы азота.
Ход определения:
К 20 мл анализируемого раствора, содержащего 50-300 мкг железа (III), прибавляют 5 мл концентрированной
соляной кислоты и 12 мл раствора роданида калия. Разбавляют водой до 50 мл и
определяют оптическую плотность раствора при λ = 480 нм.
2.2 Определение с сульфосалициловой кислотой. Фотометрический
метод
Молярный коэффициент светопоглащения ε = 6000 при λ = 430 нм. Сульфосалициловая
(5-моносульфосалициловая) кислота образует в аммиачном растворе жёлтое
соединение как с ионами железа (III),
так и с ионами железа (II).
Это одно из значительных преимуществ данного метода: при определении общего
содержания железа не требуется предварительного окисления, в то же время
иметься возможность раздельного определения обеих форм железа.
Интенсивность получаемой окраски практически не
зависит от количества прибавленного аммиака. В кислых растворах тот же реактив
образует красное соединение только с железом (III). Интенсивность окраски зависит от концентрации
кислоты, необходимо добавление буферного раствора. При различном значении рН
сдвигается, и максимум светопоглащения (при рН 1,5 максимум находиться у λ=500
нм, при рН 5,0 - у λ = 460 нм.)
Фосфор не мешает, даже если его содержание превышает
содержание железа более чем в 100 раз. Не мешает и умеренный избыток солей
аммония. Мешает влияние кальция и магния (устраняется добавлением ортофосфата).
Сильные окислители должны отсутствовать. Медь, кобальт и никель мешают цветом
своих ионов, но ионы меди можно связать в бесцветный комплекс добавлением небольшого
количества цианида.
Чаще всего определяют общее содержание железа в
щелочной среде. В этих условиях мешающее влияние марганца может быть преодолено
добавлением солянокислого гидроксиламина. При большом содержании марганца
отделяют от него железо окисью цинка. Большие количества алюминия и магния
образуют комплексы с реактивом. В этих случаях рекомендуется добавление солей
аммония и увеличение количества реактива; кроме того, в стандарт вводят
соответствующее количество этих мешающих солей.
Реактивы:
Сульфосалициловая кислота 10%-й раствор, или
сульфосалицилат натрия, насыщенный раствор. Аммиак, разбавленный (2:3) раствор.
Соляная кислота, разбавленный (3:2) раствор.
Ход определения общего содержания
железа:
Анализируемый раствор должен содержать в 10 мл от 1 до
10 мкг железа. Более концентрированные растворы предварительно разбавляют в
мерной колбе так, чтобы отобранная аликвотная часть в 10 мл содержала железо в
указанных пределах. Раствор должен быть нейтральным или слабокислым. Прибавляют
5 мл раствора сульфосалициловой кислоты или сульфосалицилата натрия, 5 мл
раствора аммиака, перемешивают и через 10 мин измеряют оптическую плотность при
λ
= 420 - 430 нм.
Определение железа (III):
Анализируемый раствор, содержащий в 10 мл от 1 до 10 мкг железа (III) сначала нейтрализуют по конго
красному (необходимое количество кислоты или щёлочи находят титрованием по
этому индикатору другой порции пробы), приливают 0,1 мг разбавленной соляной
кислоты, 5 мл раствора реактива и через 10 мин измеряют оптическую плотность
при λ = 500-520 нм. Результаты определения
находят по калибровочным кривым, которые строят в тех же условиях, пользуясь
стандартным раствором соли железа (III) и применяя те же светофильтры. По разности между общим содержанием
железа и содержанием железа (III)
находят содержание железа (II).
2.3 Определение с 1,10-фенантролином
(о-фенантролином). Фотометрический метод
Железо (III)
надо предварительно восстановить до железа (II). Для этого применяют солянокислый гидроксиламин или
гидрохинон.
Ион Fe2+ образуют с 1,10-фенантолином красные
комплексные ионы, в которых на один ион Fe2+ приходиться три молекулы 1,10-фенантролина. Получаемая
окраска устойчива более шести месяцев. Интенсивность её не зависит от рН в
границах от 2 до 9. Комплекс очень устойчив. Однако, надо учитывать влияние
некоторых факторов, которые могут оказаться причиной небольших ошибок, это:
порядок прибавление реактивов, время, температура, рН раствора, мешающие ионы.
Чувствительность метода:
Молярный коэффициент светопоглощения ε = 10600 при λ = 490 нм и ε = 10900 при λ = 505 нм.
Мешающие ионы:
Мешают многие ионы, образуя или осадки, или окрашенные
соединения. В осадок могут выпасть гидроксиды и фосфаты многих элементов, но
часто этого можно избежать, добавляя цитрат. Висмут мешает определению, но в
его присутствии определение возможно при добавлении ЭДТА. Следующие элементы и
ионы не мешают, по крайней мере, в концентрациях ниже 500мг/л: ацетат-,
хлорид-, хлорат-, бромид-, иодид-, нитрат-, сульфат-, сульфит-, роданид- и
цитрат ионы, мышьяк (V),
мышьяк (III), алюминий, свинец, марганец,
щелочноземельные и щелочные металлы.
Реактивы:
Гидроксиламин солянокислый, 10% раствор. Ацетат
натрия, 25% -ный (2М) раствор. 1,10-фенантролин солянокислый, 0,5%-й раствор.
Ход определения:
Стандартный раствор железа помещают в мерную колбу
ёмкостью 50 мл. Готовят 5-6 растворов с концентрацией железа 25-125 мг/мл путём
разбавления точно отмеренного объёма железа в мерной колбе на 50 мл. Пользуясь
другой аликвотной порцией пробы, определяют сколько надо прибавить раствора
ацетата, чтобы привести рН к 3,5 (по бромфеноловому синему) и требуемое
количество ацетата прибавляют в мерную колбу. Приливают 1 мл раствора
солянокислого гидроксиламина и 1 мл 1,10-фенонтролина. Дают постоять 1 час,
разбавляют водой до метки и определяют оптическую плотность раствора при
490-520 нм.
.4 Определение высокочастотным титрованием
Ионы Fe3+ образуют устойчивые комплексные соединения с ЭДТА
(Na2H2Y) при рН=2-3 (lg βFeY=14,33) и могут быть определены
методом высокочастотного титрования. При этом происходят следующие реакции:
3+ + H3Y-↔FeY- + 3H+,
H++H3Y-↔H4Y(pКа1 = l,0×10-2)
На кривой титрования наблюдается два излома - первый соответствует
количественному связыванию ионов Fe3+ комплексоном, второй -
указывает на завершение кислотно-основного взаимодействия.
Приборы и реактивы:
Высокочастотный титратор; магнитная мешалка; мерная колба вместимостью 50
мл; пипетка вместимостью 5 мл; бюретка вместимостью 25 мл; стакан для
титрования вместимостью 100 мл; раствор ЭДТА, 0,1 М; раствор H2S04,
1 М.
Ход определения:
К анализируемому раствору в мерной колбе прибавляют 20 мл 1 М раствора H2S04,
до метки доливают дистиллированную воду и тщательно перемешивают. В стакан для
титрования отбирают пипеткой 5 мл приготовленного раствора, доливают
дистиллированную воду на 3-5 мм выше верхнего электрода и при перемешивании
титруют раствором ЭДТА, приливая его порциями по 0,1 мл и регистрируя показания
прибора. Строят кривую титрования. По первому излому на кривой определяют объем
ЭДТА, затраченный на титрование ионов Fe3+. Пользуясь формулами
титриметрического анализа, рассчитывают содержание железа.
2.5 Определение железа в растворе. Потенциометрическое
титрование
) Титрование смеси 2-х восстановителей (Sn2+ и Fe2+)
раствором К2Сr2О7
Определение основано на предварительном восстановлении Fe3+ до
Fe2+ хлоридом олова SnCl2 и последующем титровании смеси
двух восстановителей (Sn2+ и Fe2+) раствором К2Сr2О7.
При этом сначала титруется ион Sn2+, а затем Fe2+:
SnCl42- + Cr2O72- + 14Н+ + 6С1-→3SnCl62- + 2Cr3+ + 7H2О
6Fe2+ + Cr2O72- +
14H+→6Fe3+ + 2Cr3+ + 7H2О
Кривая титрования характеризуется двумя скачками: первый соответствует
окислению Sn2+, а второй окислению Fe2+. Объем (V2-V1)
соответствует объему дихромата калия, израсходованному на титрование железа.
Реактивы:
Дихромат калия К2Сr2О7,
0,1М (1/6 К2Сr2О7) титрованный раствор.
Хлорид олова SnCl2, раствор с массовой долей 5%. Хлороводородная
кислота НС1, разбавленный раствор (1:1).
Посуда:
Колба мерная (100 мл), пипетка (10 мл), цилиндр мерный (50 мл), бюретка
(25 мл).
Аппаратура:
Установка для потенциометрического титрования, индикаторный электрод -
платиновый, электрод сравнения - хлорсеребряный.
Выполнение работы:
. Приготовление раствора титранта. Рассчитывают массу навески К2Сг2О7,
необходимую для приготовления 100 мл 0,1М (1/6 К2Сг2О7)
раствора дихромата калия. Навеску взвешивают на аналитических весах,
количественно переносят в мерную колбу вместимостью 100 мл, растворяют в
небольшом объеме воды и доводят до метки водой.
2. Анализ исследуемого раствора. В мерную колбу вместимостью 100 мл
отбирают анализируемый раствор, доводят до метки водой, тщательно перемешивают.
Аликвоту 10 мл этого раствора переносят в электрохимическую ячейку, добавляют
мерным цилиндром 50 мл разбавленного раствора (1:1) НСl и нагревают раствор почти до кипения. К горячему раствору
прибавляют по каплям из бюретки раствор SnCl2 до полного
обесцвечивания, а затем еще 1-2 капли раствора SnCl2.
Подготавливают к работе установку для потенциометрического титрования. В
горячий раствор помещают платиновый электрод и замыкают цепь с помощью солевого
мостика, заполненного насыщенным раствором КС1.
Бюретку заполняют раствором К2Сг2О7,
включают магнитную мешалку и титруют анализируемую смесь, приливая титрант
медленно по каплям. После каждого приливания записывают объем титранта и
показания прибора. Когда олово (П) будет оттитровано (первый скачок
титрования), раствор К2Сг2О7 приливают более
крупными порциями (по 0,2 мл), а вблизи второго скачка титрования добавляют
титрант снова по каплям. Строят интегральную и дифференциальную кривые
титрования и находят объемы титранта, соответствующие первой (V1) и
второй (V2) точкам эквивалентности. Титрование повторяют два-три
раза. По полученным данным рассчитывают массу ионов Fe3+ в растворе.
2) Комплексонометрическое титрование раствором ЭДТА
Содержание железа (III) можно определить также комплексометрическим
титрованием раствором ЭДТА (двунатриевая соль этилендиаминтетрауксусной кислоты
- Na2H2Y). Образующийся при титровании комплексонат
железа характеризуется константой устойчивости βFeY- = 1025,1.
Приборы и реактивы:
Установка для потенциометрического титрования; платиновый индикаторный
электрод; хлорсеребряный электрод сравнения; пипетка вместимостью 1 мл; раствор
СНзСООН, 50%-й; стандартный раствор ЭДТА, 0,05 М.; анализируемый раствор FeCl3,
≈ 0,03 М.
Выполнение работы:
Раствор соли железа (III) в ячейке для титрования разбавляют 100 мл
дистиллированной воды, добавляют 1 мл 50%-го раствора СН3СООН,
погружают в раствор электроды и измеряют ЭДС цепи. Прибавляют ЭДТА из бюретки
порциями по 1 мл, измеряя ЭДС цепи. По кривой Е - V определяют точку
эквивалентности. Содержание железа рассчитывают по формулам титриметрического
анализа.
химический роданильный фотометрический нефелометрический
3. Методы определения хлора
.1 Нефелометрическое определение хлорид-ионов в электролите
никелирования
Электролит никелирования имеет вид NiSO4*7H2O-200г/л, NaCl-20г/л,H3BO3-15-20г/л. Определение хлорид-ионов основано на измерении
кажущейся оптической плотности взвеси хлорида серебра, образующегося по
реакции:
Cl-+Ag+=AgCl↓.
Стабилизатором суспензии служит раствор желатина.
Посуда и реактивы:
- NaCl,стандартный раствор с содержанием Cl- 0,5 мг/мл
HNO3-0,1М
раствор
AgNO3,0,005М
раствор
желатин 0,5%-й раствор.
- мерные
колбы-50 и 100 мл
пипетка-2 мл
бюретка -25мл.
Аппаратура: абсорбциометр-нефелометр ЛМФ-69 или фотометр ЛМФ-72.М.1
Выполнение работы:
1) построение градуировочного графика. Для этого приготовляют серию
разбавленных стандартных растворов NaCl в воде.
Для этого в мерную колбу на 100 мл пипеткой помещают 20 мл стандартного
раствора NaCl доводят до метки водой и тщательно
перемешивают (раствор 1). Приготовленный раствор наливают в бюретку. В мерные
колбы на 50 мл вносят по 10 мл HNO3, добавляют
по 2 мл раствора желатина и разбавляют примерно до 25 мл водой. Затем добавляют
по 10 мл AgNO3, вводят в каждую колбу последовательно при
перемешивании точно отмеренные объемы раствора 1 (из бюретки) = 10,8,6,4,2 мл.
Каждый из стандартных растворов рекомендуется готовить не раньше, чем за 5
минут до начала измерений. Содержимое колб доводят до метки водой, тщательно
перемешивают, оставляют стоять в темноте (AgCl на свету разлагается), затем переносят в кювету
прибора (l = 5 см) и ровно через 5 минут после
приготовления измеряют кажущуюся оптическую плотность раствора при зеленом
светофильтре.
Измерения начинают с раствора, имеющего наиболее высокую концентрацию
хлорид-ионов (10 мл раствора 1). Помещают растворов кювету прибора и измеряют
кажущуюся оптическую плотность раствора. Таким же образом измеряют АКАЖ
остальных приготовленных растворов. Строят гралуировачный график в координатах
АКАЖ-lg c(Cl-).
) анализ исследуемого раствора. В мерную колбу вместимостью 100 мл
пипеткой помещают 2 мл анализируемого раствора, доводят до метки
дистиллированной водой. В мерную колбу на 50 мл вносят 10 мл HNO3,добавляют по 2 мл раствора желатина и разбавляют
примерно до 25 мл водой. Затем добавляют по 10 мл AgNO3 и 5 мл разбавленного анализируемого раствора, доводят
до метки водой и перемешивают. Ровно через 5 минут измеряют кажущуюся
оптическую плотность раствора.
По графику находят концентрацию хлорид-ионов в анализируемом растворе.
Вычисляют массовую концентрацию хлорида натрия в исходном растворе электролита.
3.2 Кондуктометрическое титрование хлороводородной и уксусной
кислот
Определение основано на последовательном взаимодействии с раствором
сильного основания NaOH кислот, отличающихся друг от друга степенью ионизации.
В первую очередь взаимодействует сильная кислота, что вызывает понижение
электрической проводимости раствора вследствие связывания высокоподвижных
водородных ионов. При титровании слабой кислоты, проводимость обычно
возрастает, так как вместо слабого электролита образуется хорошо диссоциирующая
соль. И наконец, после точки эквивалентности проводимость резко возрастает
благодаря появлению в растворе гидроксильных ионов, обладающих высокой
подвижностью. Объем щелочи V1 соответствует оттитровыванию HCl, а
объем V2-оттитровыванию суммы HCl и СН3COOH.
Посуда и реактивы:- NaOH-0,1М раствор
HCl-0,5М титрованный раствор
HNO3-разбавленный
раствор 1:1
- колбы
мерные -50 мл и пипетки-10 мл
Аппаратура: установка для кондуктометрического титрования в комплексе с
кондуктометром КЭЛ-1М.
Выполнение работы:
1) подготовка прибора к работе. Собирают установку для
кондуктометрического титрования. Получают у лаборанта электролитическую ячейку
и промывают платиновые электроды. Для этого наливают в ячейку азотную кислоту
(1:1) до полного погружения электродов и выдерживают их в растворе 2-3 минуты.
Затем кислоту сливают в склянку, в которой она хранится, а электроды и ячейку
промывают под струей водопроводной воды, после чего дважды ополаскивают
дистиллированной водой. Прибор включают в сеть и подготавливают его к работе.
Бюретку моют и заполняют раствором NaOH.
) стандартизация раствора NaOH по HCl. В мерную колбу вместимостью 50 мл
помещают 10 мл титрованного раствора HCl, доводят до метки водой и тщательно
перемешивают. Отбирают 10 мл полученного раствора в электролитическую ячейку,
добавляют дистиллированную воду до полного погружения электродов, включают
магнитную мешалку и начинают титрование, приливая по 0,5 мл раствора NaOH.
После приливания каждой порции титранта измеряют электрическую проводимость в
растворе, титрование продолжают до тех пор, пока не обнаружат излом на кривой
титрования, после чего снимают показания еще в 4-5 точках. По полученным данным
строят кривую титрования в координатах показания прибора - объем титранта.
Находят объем титранта в точке эквивалентности и рассчитывают концентрацию
раствора NaOH.
) анализ исследуемого раствора: Исследуемый раствор, содержащий смесь HCl
и СН3COOH,помещают в
мерную колбу вместимостью 50 мл и доводят до метки водой. Пипеткой отбирают 10
мл раствора в электролитическую ячейку, добавляют дистиллированную воду до
полного погружения электродов и включают магнитную мешалку. Титруют раствором
NaOH, приливая его порциями по 0,5 мл и в каждой точке записывая значения
электрической проводимости раствора. Титрование прекращают после того, как будут
обнаружены 2 излома на кривой титрования-то резкого падения показаний к
плавному, а затем резкому росту значений. Строят кривую титрования, по которой
находят V1 и V2-объемы титранта в первой и второй точках
эквивалентности: V1-нейтрализация HCl, а V2- V1-
нейтрализация СН3COOH.рассчитывают
массу HCl и СН3COOH
во взятом на анализ образце.
.3 Потенциометрическое определение иодид- и хлорид-ионов в их
смеси
Определение основано на последовательном осаждении I- и Cl- нитратом серебра AgNO3. Вследствие различия в растворимости
ПР(AgCl)=1,78*10-10, ПР(Agl)=8,3*10-17, в первую
очередь оттитровываются иодид-ионы, а затем хлорид-ионы: в моменты завершения
первой и второй реакций наблюдается резкое изменение потенциала серебряного
электрода. Электродом сравнения служит насыщенный каломельный электрод. Так как
ЕН.К.Э>ЕAgI/I-,Ag, при титровании иодид-ионов
индикаторный электрод подключают к клемме со знаком (-),а н.к.э. - клемме со
знаком (+). Когда иодид-ионы оттитрованы и начинается реакция между ионами
серебра и хлорида, потенциал индикаторного электрода становится больше
потенциала н.к.э., поэтому необходимо переключить электроды к соответствующим
клеммам: индикаторный электрод подключают к (+), а н.к.э - к (-), что следует
учитывать при записи результатов.
Необходимо помнить, что равновесный потенциал индикаторного электрода
устанавливается во времени. Поэтому показания потенциометра, особенно вблизи
точки эквивалентности, следует фиксировать только после того, как ЭДС примет
постоянное значение в пределах 0,1 мВ, после этого можно добавлять следующую
порцию титранта.
Для уменьшения адсорбции иодид-ионов осадком Agl титрование ведут в присутствии постороннего
электролита (обычно нитрата или ацетата бария). Соединительный мостик заполняют
раствором сульфата калия, но не хлорида, во избежание диффундирования Cl- из соединительного мостика в титруемый раствор.
Посуда и реактивы:- AgNO3, 0,02М титрованный раствор.
Ва(NO3)2, 10%-й раствор
Na2S2O3*5H2O, насыщенный раствор
- мерные
колбы-50 мл, пипетка-10 мл, мерный цилиндр-25 мл
Аппаратура: установка для потенциометрического титрования в комплексе с
рН-метром в режиме милливольтметра.
Индикаторный электрод - серебряный,
электрод сравнения - н.к.э.
Выполнение работы: приготавливают к работе серебряный электрод. Очищают его от
пленки галогенидов серебра, для чего опускают электрод в раствор Na2S2O3*5H2O на 10-15 минут, а затем тщательно
промывают дистиллированной водой, высушивают поверхность электрода
фильтровальной бумагой.
После этого собирают установку для титрования. Настраивают потенциометр в
соответствии с инструкцией к прибору данного типа.
Анализируемый раствор смеси иодид- и хлорид-ионов доводят до метки водой
в мерной колбе на 50 мл и тщательно перемешивают. Берут пипеткой 10 мл в стакан
для титрования и прибавляют цилиндром 10 мл раствора Ва(NO3)2. Бюретку заполняют растворм AgNO3. Включают магнитную мешалку и начинают
ориентировачное титрование галогенидов, приливая из бюретки в стакан по 0,2 мл
раствора AgNO3.После каждой порции записывают объем прилитого
раствора титранта и соответствующее ему значение ЭДС цепи (Е,мВ). При этом не
следует забывать о переключении электродов в тио момент, когда ЭДС начнет
изменяться в противоположном направлении.
Обнаружив два скачка потенциала, выполняют с новой порцией анализируемого
раствора точное титрование, прибавляя раствор AgNO3 по 0,1 мл, а около точки эквивалентности - по 2
капли. По данным, полученным в результате второго титрования, строят две
кривые: в координатах Е-V(AgNO3) и
дифференциальную в координатах ΔЕ/ΔV - V(AgNO3). Находят объемы в точках эквивалентности, V1 соответствует
объему AgNO3,затраченному на титрование I-, а V2-
объему AgNO3,затраченному на титрование суммы I- и Cl-. Разность V2- V1 дает объем
раствора AgNO3,затраченному на титрование Cl-. Рассчитывают массу иодид- и хлорид-ионов в исходной смеси.
3.4 Фотометрическое определение по окраске роданоферрата (III)
Используется реакция обмена между роданидом ртути (II) и хлорид ионами, приводящая к
образованию комплексного хлорида ртути (II) и вытеснению роданид ионов. Последние определяют
колориметрически в виде роданоферрата. Так можно определить до 0,05мг/л хлорид
ионов. Число мешающих ионов очень велико: мешают все ионы, образующие комплексы
с ртутью (II); их можно определить этим же
способом.
Реактивы:
Hg(SCN)2
,насыщенный 0,07%-й раствор
- Fe(ClO4)3, 6%-й раствор в 4н хлорной кислоте. Этот
раствор может быть приготовлен следующим образом: растворяют 14,0 г Fe в разбавленной HNO3, прибавляют 120 мл HClO4 и выпаривают до появления белых паров
последней. Продолжают нагревать, пока раствор не станет пурпурный, и тогда
разбавляют водой до 1000 мл
Мерные колбы на 50 мл
Мерная колба на 1000 мл
Пипетки градуированные на 10,5 и 2 мл
Аппаратура: фотоэлекроколориметр
Выполнение работы: В мерную колбу на 50 мл помещают 10
мл анализируемого раствора, содержащего 5 мг/л Cl-, прибавляют 5 мл 60%-й HClO4, 1 мл раствора Hg(SCN)2 и
2 мл раствора Fe(ClO4)3. Разбавляют водой до 50 мл,
перемешивают, дают постоять 10 минут и определяют оптическую плотность при λ=480нм.
3.5 Фотометрическое определение по цвету хлорферрата (III)
Бесцветные ионы Fe3+ образуют с хлорид ионами жёлтые комплексные соединения FeCl2+, поглощающие ультрафиолетовое
излучение. Оптимальная концентрация - 5 мг/л хлорид ионов.
Мешают все ионы, образующие комплексы с хлорид ионами,
также многочисленные анионы, дающие окрашенные комплексы с ионами Fe3+: сульфат-ионы, роданид-ионы и т.п.
Не мешают те анионы, которые дают с Fe3+, бесцветные комплексы: фторид-ионы.
Приборы и реактивы:
Fe(ClO4)3. Твёрдую соль очищают
взбалтыванием с концентрированной хлорной кислотой, отфильтровывают перхлорат
железа (III) и растворяют, обрабатывая 120 г его
смесью 540 мл 60% - хлорной кислоты и 460 мл воды
Мерные колбы на 50 мл и 100 мл
Пипетки градуированные - 10 мл
Аппаратура: фотоэлекроколориметр
Выполнение работы: К 4,9 мл реактива приливают 5мл анализируемого раствора,
содержащего не более 100 мг/л хлорид ионов, и разбавляют до 10 мл. Определяют
оптическую плотность полученного раствора при λ=348 нм. Проводят «холостой» опыт. Учитывают
влияние температуры.
4. Методы определения натрия
.1 пламенно-фотометрический метод определения окисей натрия и
калия
Сущность метода: Метод основан на введении раствора сернокислых солей натрия и
калия в пламя горелки в виде аэрозоля и измерении интенсивности излучения
методом спектрофотометрии пламени: натрия - при длине волны (589 ± 5) нм, калия
- при (768 ± 5) нм.
Аппаратура, реактивы и растворы:
Фотометр пламенный типа ПФ-102 или другие, обеспечивающие требуемую
точность измерения;
Кислота серная;
Кислота фтористоводородная, раствор с массовой долей 40%;
Аммоний щавелевокислый, насыщенный раствор;
Натрий сернокислый безводный;
Калий сернокислый;
Аммиак водный, раствор с массовой долей 25 %;
Чашка платиновая;
Выполнение работы: Навеску материала массой 0,5 г помещают в платиновую чашку,
смачивают водой, прибавляют 1 см3 серной кислоты, 10-15 см3
фтористоводородной кислоты (если в пробе содержатся органические вещества или
свободный углерод, то навеску пробы предварительно прокаливают при (700±20)°С в
окислительной среде), выпаривают на закрытой электроплитке при периодическом
помешивании до получения влажного остатка, затем приливают 10 см3
фтористоводородной кислоты и выпаривают досуха.
Сухой остаток прокаливают в муфельной печи при 700-800°С в течение 5-10
мин. Остаток в чашке обрабатывают горячей водой, приливают раствор аммиака до
появления легкого запаха. Содержимое в платиновой чашке нагревают до кипения,
отфильтровывают, собирая фильтрат в стакан вместимостью 100-150 см3.
Остаток на фильтре промывают 5-6 раз горячей водой, фильтрат нагревают до
кипения, приливают 15-20 см3 насыщенного раствора щавелевокислого
аммония и оставляют на 1,5 ч в теплом месте.
Раствор вместе с осадком после охлаждения переводят в мерную колбу
вместимостью 100 см3, доводят до метки водой, перемешивают и
фильтруют через сухой фильтр. Первые порции фильтрата отбрасывают. 25-30 см3
раствора используют для определения массы окислов натрия и калия на пламенном
фотометре. Анализируемый раствор подносят к всасывающему капилляру пламенного
фотометра, измеряют интенсивность излучения при соответствующем определяемому
элементу светофильтре: натрия - при длине волны (589±5) нм, калия - при (768±5)
нм.
Массу окислов натрия и калия в граммах определяют по градуировочному графику.
Построение градуировочного графика
Стандартный раствор сернокислых солей натрия и калия: 0,5730 г
сернокислого натрия и 0,4625 г сернокислого калия, предварительно высушенных в
течение 2 ч при 100°С, растворяют в стакане вместимостью 150-200 см3
и переносят в мерную колбу вместимостью 1000 см3, доводят до метки
водой и перемешивают.
Стандартный раствор в пересчете на окислы с массовой концентрацией окиси
натрия 0,00025 г/см3 и массовой концентрацией окиси калия 0,00025
г/см3.
В мерные колбы вместимостью 100 см3 отбирают пипеткой
аликвотные части стандартного раствора: 1,0; 3,0; 5,0; 7,0; 9,0; 11,0; 13,0;
15,0; 17,0; 20,0 см3, доводят до метки водой и перемешивают.
Приготовленные растворы переносят в стаканчики непосредственно перед их
измерением, затем растворы последовательно распыляются и снимаются показания
гальванометра по всей серии растворов при установке ручки диска светофильтров в
положение измеряемого элемента (натрия или калия).
Для каждого раствора производят три измерения. По средним значениям этих
измерений строят градуировочный график в координатах: масса окиси натрия или
калия в граммах в 100 см3 раствора - показания шкалы гальванометра
(число делений).
Перед началом работы проверяют градуировочный график по одному или двум
стандартным растворам.
Обработка результатов: Массовую долю окиси натрия или окиси калия (X) в процентах вычисляют по формуле
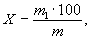
где m1 - масса окиси натрия или окиси калия, найденная по
градуировочному графику, г;
m -
масса навески пробы, г.
4.2 Пламенно-фотометрический метод определения окисей натрия
и калия (в огнеупорных материалах и изделиях с массовой долей двуокиси циркония
до 65%, кроме бадделеитовых)
Сущность метода: Измеряют на пламенном фотометре излучения, соответствующие
массовым долям щелочных металлов, возникающие при распылении анализируемого
раствора в ацетилено-воздушном пламени (длина волны для натрия - 589 нм, для
калия - 768 нм).
Междуэлементное влияние и взаимное спектральное влияние щелочных металлов
на результаты измерений подавляют с помощью добавляемых растворов, содержащих в
избытке конкурирующий катион и комплексообразующий реагент.
Аппаратура, реактивы и растворы:
Чашка платиновая;
Кислота серная, разбавленная 1:1;
Кислота фтористоводородная, раствор с массовой долей 40 %;
Кислота соляная;
Калий хлористый;
Натрий хлористый;
Аммоний фосфорнокислый двузамещенный;
Дополнительный раствор для определения окиси натрия: 100 г двузамещенного
фосфорнокислого аммония и 9,4 г хлористого калия растворяют приблизительно в
500 см3 воды, прибавляют 250 см3 соляной кислоты,
переводят в мерную колбу вместимостью 1000 см3, доводят до метки
водой и перемешивают.
Дополнительный раствор для определения окиси калия: 100 г двузамещенного
фосфорнокислого аммония и 9,4 г хлористого натрия растворяют приблизительно в
500 см3 воды, прибавляют 250 см3 соляной кислоты, раствор
переводят в мерную колбу вместимостью 1000 см3, доводят до метки
водой и перемешивают.
Стандартный раствор хлористого натрия: взвешивают после предварительного
высушивания в течение 24 ч при температуре (110±2)°С 0,3772 г хлористого натрия
и растворяют в воде в мерной колбе вместимостью 1000 см3, доводят
водой до метки и перемешивают.
Стандартный раствор хлористого натрия с массовой концентрацией окиси
натрия 0,0002 г/см3.
Стандартный раствор хлористого калия: после предварительного высушивания
в течение 24 ч при (110±2)°С взвешивают 0,3166 г хлористого калия, растворяют в
воде в мерной колбе вместимостью 1000 см3, доводят водой до метки и
перемешивают.
Стандартный раствор хлористого калия с массовой концентрацией окиси калия
0,0002 г/см3.
Дополнительные и стандартные растворы следует хранить в полиэтиленовых
сосудах.
Выполнение работы: Для приготовления исходного раствора навеску материала массой
1,0 г помещают в платиновую чашку вместимостью 50 см3, осторожно
приливают 1 см3 раствора серной кислоты (1:1) и 10 см3
раствора фтористоводородной кислоты. Нагревают на песчаной бане до прекращения
выделения паров серной кислоты. Чашку с сухим остатком прокаливают в течение
10-15 мин при (700±10)°С.
Остаток после охлаждения обрабатывают 5 см3 соляной кислоты и
20 см3 воды, накрывают чашку часовым стеклом, растворяют остаток при
умеренном нагреве на песчаной бане примерно 15-20 мин.
Если раствор остался мутным, его фильтруют через фильтр «синяя лента».
Фильтрат собирают в мерную колбу вместимостью 100 см3, доливают
водой до метки, перемешивают и переливают в сухой полиэтиленовый сосуд.
Для определения массовой доли окиси натрия отбирают пипеткой 25 см3
исходного раствора в мерную колбу вместимостью 50 см3, прибавляют 10
см3 дополнительного раствора для определения окиси натрия, доводят
водой до метки и перемешивают. Измеряют интенсивность излучения на пламенном
фотометре при длине волны 589 нм и регистрируют соответствующие показания
гальванометра. Проводят три измерения и вычисляют среднее значение.
Массу окиси натрия в граммах определяют по градуировочному графику,
построенному в тех же условиях.
Для определения массовой доли окиси калия отбирают пипеткой 25 см3
исходного раствора в мерную колбу вместимостью 50 см3, прибавляют 10
см3 дополнительного раствора для определения окиси калия, доводят
водой до метки и перемешивают. Измеряют интенсивность излучения на пламенном
фотометре при длине волны 768 нм и регистрируют соответствующие показания
гальванометра. Производят три измерения и вычисляют среднее значение.
Массу окиси калия в граммах определяют по градуировочному графику,
построенному в тех же условиях.
Допускается проводить определение щелочных металлов по методу
ограничивающих растворов.
Построение градуировочного графика
Для построения градуировочного графика, по которому определяют окись
натрия, в шесть из семи мерных колб вместимостью 50 см3 отбирают из
бюретки 0,5; 1,0; 5,0; 10,0; 20,0 и 35,0 см3 стандартного раствора
окиси натрия. Прибавляют в каждую колбу 10 см3 дополнительного
раствора для определения окиси натрия, доводят водой до метки и перемешивают.
Для построения градуировочного графика, по которому определяют окись
калия, в шесть из семи мерных колб вместимостью 50 см3 отбирают из
бюретки 0,5; 1,0; 5,0; 10,0; 20,0 и 35,0 см3 стандартного раствора
окиси калия. Прибавляют в каждую колбу 10 см3 дополнительного
раствора для определения окиси калия, доводят водой до метки и перемешивают.
После настройки прибора распыляют контрольный раствор средней
концентрации определенного щелочного элемента и устанавливают диафрагму так,
чтобы показания гальванометра соответствовали всей шкале или ее определенной
части.
Затем устанавливают при распылении раствора контрольного опыта нулевое
значение измерительного прибора (гальванометра). После этого последовательно
распыляют градуировочные растворы, начиная с наименьшей концентрации. Каждый
раствор измеряют по три раза. По средним значениям показания гальванометра и
соответствующим им массовым концентрациям окиси натрия (окиси калия) в граммах
в 50 см3 строят градуировочные графики для каждого соответственно.
Перед анализом градуировочный график подлежит проверке не менее чем по
одному контрольному раствору.
Для определения окисей натрия и калия при массовой доле менее 0,1%
рекомендуется проводить определение из всей навески пробы массой в 1,0 г.
Обработка результатов: Массовую долю окиси натрия или окиси калия (X1) в процентах вычисляют по формуле
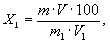
где m - масса окиси натрия (калия),
найденная по градуировочному графику, г;
V -
объем исходного раствора, см3;
V1 - объем аликвотной части раствора,
см3;
m1 - масса навески, г.
.3 Метод определения натрия и калия в
водной вытяжке
Настоящий стандарт устанавливает метод определения натрия и калия в
водной вытяжке из засоленных почв при проведении почвенного, агрохимического,
мелиоративного обследования угодий, контроля за состоянием солевого режима
почв, а также при других изыскательских и исследовательских работах.
Суммарная относительная погрешность метода составляет:
,5% - при определении натрия;
% - при определении калия.
Сущность метода заключается в определении
интенсивности излучения атомов определяемых элементов с помощью пламенного
фотометра. Натрий определяют по аналитическим линиям 589,0 и 589,9 нм, калий -
по аналитическим линиям 766,5 и 769,9 нм.
Аппаратура,
материалы и реактивы
Для
проведения анализа применяют:
Пламенный фотометр с монохроматором или
интерференционными светофильтрами с максимумом пропускания в области 588-590 нм
для определения натрия и 766-770 нм для определения калия (допускается
использование газовой смеси состава пропан - бутан - воздух и сетевой газ -
воздух);
Весы лабораторные 2-го класса точности с наибольшим
пределом взвешивания 200 г и 4-го класса точности с наибольшим пределом
взвешивания 500 г по гост 24104-80;
Натрий хлористый по гост 4233-77, х. Ч.;
Калий хлористый по гост 4234-77, х. Ч.;
Посуду мерную лабораторную 2-го класса точности по
гост 1770-74;
Пипетки и бюретки 2-го класса точности по гост
20292-74;
Воду дистиллированную по гост 6709-72.
подготовка
к анализу
Приготовление
раствора концентрации натрия с (Na+) = 0,1 моль/дм3 и калия с
(К+) = 0,01 моль/дм3
5,845 г хлористого натрия и 0,746 г хлористого калия,
прокаленных до постоянной массы при температуре 500°С, взвешивают с
погрешностью не более 0,001 г, помещают в мерную колбу вместимостью 1000 см3
и растворяют в дистиллированной воде, доводя объем раствора до метки.
Приготовленный раствор тщательно перемешивают.
Приготовление растворов сравнения
В мерные колбы вместимостью 250 см3
помещают указанные в таблице объемы раствора, приготовленного по п. 3.1, и
доводят объемы до меток дистиллированной водой. Приготовленные растворы
тщательно перемешивают. Растворы хранят в склянках с притертыми пробками не
более 1 мес.
Растворы сравнения используют для градуировки пламенного фотометра в день
проведения анализа.
|
Характеристика раствора
|
Номер раствора сравнения
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
|
Объем раствора,
приготовленного по п. 3.1, см3
|
0
|
5,0
|
10
|
20
|
30
|
40
|
50
|
|
Концентрация натрия:
|
|
|
|
|
|
|
|
|
в растворе сравнения,
моль/дм3
|
0
|
0,002
|
0,004
|
0,008
|
0,012
|
0,016
|
0,02
|
|
в пересчете на 100 г почвы,
ммоль
|
0
|
1,0
|
2,0
|
4,0
|
6,0
|
8,0
|
10
|
|
Концентрация калия:
|
|
|
|
|
|
|
|
|
в растворе сравнения,
моль/дм3
|
0
|
0,0002
|
0,0004
|
0,0008
|
0,0012
|
0,0016
|
0,002
|
|
в пересчете на 100 г почвы,
ммоль
|
0
|
0,1
|
0,2
|
0,4
|
0,6
|
0,8
|
1,0
|
Выполнение
работы: Приготовление
вытяжки из почвы
Для анализа используют фильтраты вытяжек,
приготовленных по ГОСТ 26423-85.
Определение
натрия и калия. Пламенный фотометр настраивают на измерение концентрации натрия
или калия в соответствии с инструкцией по его эксплуатации. Растворы сравнения
и анализируемые вытяжки вводят в пламя и регистрируют показания прибора.
Обработка
результатов
По результатам фотометрирования растворов сравнения
строят градуировочный график. По оси абсцисс откладывают концентрации натрия
или калия в растворах сравнения в пересчете в миллимоли в 100 г почвы, а по оси
ординат - соответствующие им показания прибора.
Количество эквивалентов натрия или калия в анализируемых
почвах определяют непосредственно по градуировочному графику.
За результат анализа принимают значение единичного
определения натрия и калия.
Если результат определения выходит за пределы
градуировочного графика, определение повторяют, предварительно разбавив
фильтрат дистиллированной водой. Результат, найденный по графику, увеличивают
во столько раз, во сколько был разбавлен фильтрат.
Массовую долю натрия в анализируемой почве (X) в процентах вычисляют по формуле:
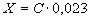 ,
,
где С - количество эквивалентов натрия в почве, ммоль
в 100 г;
,023 - коэффициент пересчета в проценты.
Массовую долю калия в анализируемой почве (X1) в процентах вычисляют по формуле:
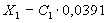 ,
,
где С - количество эквивалентов калия в почве, ммоль в
100 г;
,0391 - коэффициент пересчета в проценты. Результаты
анализа выражают в мг·экв на 100 г почвы и в процентах с округлением до трех
значащих цифр.
При проведении массовых анализов вместо построения
градуировочного графика допускается градуирование шкалы прибора по растворам
сравнения в день проведения анализа. Допускаемые относительные отклонения при
доверительной вероятности Р = 0,95 от среднего арифметического результатов
повторных анализов при выборочном статистическом контроле составляют:
% - для определения натрия;
% - для определения калия.
4.4 Определение содержания натрия и калия, растворимых в
разбавленной соляной кислоте, методом экстракции
Сущность метода: Пробу бурого угля или лигнита подвергают экстрагированию в
кипящей соляной кислоте 0,005 н. и полученный экстракт центрифугируют.
Содержание натрия и калия в растворе определяют любым точным
аналитическим методом (например, гравиметрическим или пламенно-фотометрическим
методом).
Реактивы: При проведении анализа используют только реактивы
квалификации не ниже ч.д.а. и дистиллированную воду или воду эквивалентной
чистоты.
Соляная кислота, 0,005 н. раствор.
Этиловый спирт, 95 %-й по объему.
Оборудование: Все калиброванные приборы должны быть максимально точными.
Мерная колба с одной отметкой объема вместимостью 250 см3.
Лабораторная центрифуга со скоростью вращения 2000 об/мин.
Полиэтиленовая бутыль минимальной вместимостью 250 см3.
Весы с точностью взвешивания до 0,1 мг.
Проба: Лабораторную пробу помещают на противень и доводят до воздушно-сухого
состояния. Измельчают испытуемую пробу до размера частиц, проходящих через сито
с размером квадратных ячеек 212 мкм. Приготовленную таким образом пробу для
анализа хранят в закрытом пробкой сосуде, наполнив его более чем на 80% его
объема.
Выполнение работы: Точно отвешивают (1,5±0,05) г пробы и переносят в химический
стакан вместимостью 250 см3. Смачивают пробу 3 см3 этилового спирта, добавляют
100 см3 соляной кислоты и медленно кипятят в течение 15 мин. Стакан должен быть
открытым, чтобы этиловый спирт мог свободно испаряться. Суспензию охлаждают и
переносят количественно в сосуды центрифуги и центрифугируют в течение 5 мин со
скоростью вращения 2000 об/мин. Сливают раствор в мерную колбу. Остаток смывают
в тот же химический стакан, используя 100 см3 соляной кислоты, вновь кипятят в
течение 15 мин и снова центрифугируют.
Переливают раствор в ту же мерную колбу, остаток в стакане промывают
теплой соляной кислотой, вновь центрифугируют и сливают промывную воду в ту же
мерную колбу. Охлаждают содержимое колбы до комнатной температуры и доводят
объем до метки соляной кислотой. Хранят в полиэтиленовой бутыли.
Определяют содержание натрия и калия в растворе любым точным методом
(например, гравиметрическим методом или методом пламенной фотометрии).
Определяют содержание влаги в отдельной порции пробы методом, указанным в
ГОСТ 27314.
Рассчитывают содержание натрия и калия на сухую массу по формулам,
предназначенным для методов, принятых для данных испытаний.
Примечание. Раствор может быть светло-желтого цвета вследствие
присутствия в нем органических кислот, но на результаты определений это не
влияет.
.5 Метод определения концентрации обменных катионов натрия и
калия
Аппаратура
и реактивы:
Спектрофотометр или пламенный фотометр
Аммоний хлористый по ГОСТ 3773, раствор 1 моль/дм3
Стандартный раствор калия
,98 г хлористого калия растворяют в воде в мерной колбе вместимостью 1 дм3,
доливают водой до метки и перемешивают.
Стандартный раствор натрия
,542 г хлористого натрия растворяют в воде в мерной колбе вместимостью 1
дм3, доливают водой до метки и перемешивают.
Стаканы химические вместимостью 250-300 см3 по ГОСТ 23932
Колба мерная вместимостью 500 см3 по ГОСТ 1770
Выполнение работы:
Навеску глины массой 5 г помещают в стакан вместимостью 250-300 см3,
приливают 150 см3 раствора хлористого аммония, перемешивают в
течение 5-10 мин, дают отстояться и отфильтровывают через фильтр «синяя лента»
диаметром 12-14 см в мерную колбу вместимостью 500 см3. Обработку
хлористым аммонием заканчивают после получения 500 см3 фильтрата.
Раствор в мерной колбе перемешивают и на спектрофотометре измеряют
интенсивность излучения натрия и калия. Натрий определяют по интенсивным
резонансным линиям 589,0-589,6 нм, калий - по линиям 766,5-769,9 нм. По
интенсивности излучения натрия или калия в растворе исследуемой пробы
(измеренной в делениях шкалы прибора) находят их содержание по градуировочному
графику.
Для построения градуировочного графика в мерные колбы вместимостью 1 дм3
отмеривают 5, 10, 25, 50, 75 и 100 дм3 стандартного раствора,
содержащего равные количества натрия и калия, доводят водой до метки,
перемешивают и измеряют интенсивность излучения.
Обработка
результатов:
Массовую долю катионов натрия и калия Х в процентах вычисляют по формуле
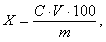 (1)
(1)
где С - концентрация катионов натрия или калия по градуировочному
графику, мг/дм3;- объем раствора анализируемой пробы, см3;-
масса навески глины, г.
Концентрацию обменных катионов натрия или калия X1, мг/экв на
100 г глины, вычисляют по формуле
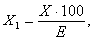 (2)
(2)
где Е - эквивалентная масса катионов натрия или калия.
Результат анализа рассчитывают до третьего и округляют до второго
десятичного знака.
Точность определения: Расхождение результатов двух параллельных определений не
должно превышать 0,05% при массовой доле оксида натрия (или калия) до 1% и
0,10% - при массовой доле оксида натрия (или калия) свыше 1%. Если расхождение
превышает указанные значения, то испытание повторяют.
За результат испытания принимают среднее арифметическое результатов трех
испытаний.
5. Экспериментальная часть
. Определение ионов Zn2+
Определение основано на реакции осаждения ионов цинка
гексацианоферрат (II)-ионами,
генерируемыми из гексацианоферрат (III)-ионов на платиновом катоде в кислых растворах. При этом протекают
следующие реакции:
Fe(CN)63-
+ e- → Fe(CN)64- на
электроде,
Zn2++2Fe(CN)64- + 2K+→ K2Zn3[Fe(CN)6]2↓ в
растворе.
Конечную точку титрования определяют
потенциометрическим или амперометрическим методом с двумя поляризованными
платиновыми электродами.
Приборы и реактивы:
Кулонометрическая установка с потенциометрической
индикацией конечной точки титрования
Генераторный и индикаторный серебряные электроды
Вспомогательный электрод - стальной стержень
Хлорсеребряный электрод сравнения
Мерная колба вместимостью 100 мл
Пипетка вместимостью 10 мл
Раствор K3Fe(CN)6, 0,2М в буферной смеси рН = 1 - 3
(вспомогательный реагент)
Анализируемый раствор соли цинка, 10-2 М
Выполнение работы
Исследуемый раствор разбавляют дистиллированной водой
в мерной колбе до метки и перемешивают. Переносят пипеткой 10 мл в
кулонометрическую ячейку, добавляют 15 мл раствора вспомогательного реагента,
опускают генераторный и индикаторный электроды. Титрование ведут при величине
тока 5мА.
По данным титрования строят кривую зависимости
потенциала от времени при потенциометрической индикации или тока от времени при
амперометрической индикации и находят конечную точку титрования. Содержание
цинка (в мг) рассчитывают по формуле:
g = 0,01036ItMfVk/Vп,
где I-
ток, t- время генерации, с;
M - молекулярная масса цинка;
fэкв - фактор эквивалентности;
Vk - вместимость мерной колбы;
Vп - вместимость пипетки.
Заключение
Главной целью данной курсовой работы являлся самостоятельный поиск
методик по определению ионов, как электрохимическими, так и оптическими
методами.
При написании курсовой работы, мною были изучены
методики определения ионов: ZnCl2, FeCl3, NaCl, которые изложены в теоретической
части курсовой работы.
Для определения любого иона используют различные методы. Каждый из
методов в чем-то уникален и имеет свою специфику - у одних это возможность
количественного определения геометрических параметров молекулы, у других -
определение электрических свойств, у третьих - энергетических состояний или спектральных
характеристик.
Однако при определении, какого либо иона предпочтение отдают тому или
другому методу. Выбор метода зависит от целей и задач, поставленных в основе
анализа, а также от теоретической базы данного метода.
Выбор метода может определяться наличием используемых приборов и
реактивов, а также задачами, поставленными в ходе анализа.
Использование одного метода возможно для определения большого количества
ионов, но данные полученные одним методом являются оценочными или косвенными.
Совокупность этих данных не дает полного представления обо всех необходимых
характеристиках иона. Лишь комплексное изучение ионов несколькими
физико-химическими методами сделает возможным повышение надежности результатов
анализа.
Совершенствование экспериментальной техники исследований, повышение
качества и новизна характера получаемых данных расширяют круг тех задач,
которые могут ставиться и решаться при использовании того или другого метода,
что, в свою очередь, стимулирует развитие теории физико-химических методов
исследования.
Список литературы
1. Золотов
Ю.А. Основы аналитической химии: учеб. для хим. фак. ун-тов/ Ю.А. Золотов. -
2-е изд., перераб. и доп. - М.: Высш. шк., 1999. - 493 с.
2. Дорохова
Е.Н. Аналитическая химия: учебник для вузов / Е.Н. Дорохова, Г.В. Прохорова;
под ред. С.С. Трапезникова. - М.: Высш. шк.,1991. - 255 с.
. Васильев
В.П. Аналитическая химия: учеб. для хим. фак. ун-тов / В.П. Васильев. - 2-е
изд. - М.: Высш. шк., 1989. - 383 с.
. Марченко
З. Фотометрическое определение элементов: учебник для вузов / З. Марченко. -
М.: Мир, 1971. - 230 с.
. Пешкова
В.М. Практическое руководство по физико-химическим методам анализа./ В.М
Пешкова, М.И. Громова - М.: Высш. шк.,1965. - 230 с.
. Барковский
В.Ф. Практикум по физико-химическим методам анализа./В.Ф. Барковский, С.М.
Горелик - М.: Высш. шк., 1963. - 350 с.
. Крешков
А.П. Основы аналитической химии: учеб. для хим. фак. ун.-тов / А.П. Крешков . -
4 е изд., перераб. - М.: Химия, 1976. - 480 с.
. Коренман
И.Н. Аналитическая химия калия: учеб. для хим. фак. Ун-тов / И.П. Коренман. -
М.: Наука, 1964. - 235 с.
9. Гурецкий
И.Я., Кузнецов В.В., Кузнецова Л.Б. и др. Практикум по физико-химическим
методам анализа/ Под ред. О.М. Петрухина. М.: Химия, 1987. 248с.
10. В.П.
Живописцев, Е.А. Селезнева. Аналитическая химия цинка. Издательство «Наука»,
М., 1975.