|
Характеристика
|
Сплавление
|
Прессование
|
|
Гомогенность пробы
|
Высокая
|
Средняя
|
|
Матричные эффекты
|
Незначительные
|
Требуют коррекции
|
|
Возможность использования глобальной калибровки
|
Да
|
Нет
|
|
Стоимость в расчете на 1 пробу
|
> 50 р. + амортизация тиглей
|
< 10 р.
|
|
Время подготовки пробы
|
15-25 мин
|
7-10 мин
|
Авторами в работе
[19] предложен метод изготовления реперных образцов, заключающийся в
прессовании порошка вещества, используемого для изготовления образцов, со
связующим материалом и последующим нагреванием таблетки. В качестве связующего
материала используется полистирол эмульсионный ГОСТ 202822-74. Полистирол и
материал образца, взятые в пропорциях, определяемых экспериментальным или
расчетным путем, тщательно перемешиваются. Полученная смесь прессуется при
давлении ~ 800 кгс/м2. Изготовленные таблетки помещаются в
термостат, где выдерживаются в течение двух часов при температуре 150 0С.
Предложенный способ отличается
довольно высокой производительностью (один человек за семичасовой рабочий день
может изготовить до 40-50 образцов), простотой технологии, небольшим
количеством используемых технических средств, низкой стоимостью связующего
материала, очень высокой прочностью получаемых таблеток, возможностью
обеспечить однородность образца, устойчивостью к воздействию рентгеновского излучения
и экологической чистотой [4, 14-18].
Для сплавления обычно применяют
тетраборат лития или смесь тетрабората с метаборатом. Соотношение флюс / проба
варьируется от 5/1 до 12/1 в зависимости от типов флюса и пробы. Сплавление
ведут в Pt/Au тиглях в газовых (FluXana Vulcan) или электропечах (Katanax) при
температурах около 1100 oС. Стоит отметить, что при сплавлении
огнеупорных материалов износ тиглей значителен - переработка тиглей требуется
уже через несколько тысяч плавок. Фото плавильной электропечи для подготовки
образцов Katanax K1 Prime представлено на рис. 2.

Рис. 2. Плавильная
электропечь для подготовки образцов
Истирание
огнеупоров проводят в шаровых мельницах (Fritsch Pulverisette) или
виброистирателях (Herzog HSM), обычный материал гарнитуры - WC. Для прессования
можно использовать ручные, полуавтоматические или автоматические гидравлические
пресса (например, Herzog HTP40), достаточное усилие прессования для таблетки
диаметром 40 мм составляет 25 - 40 т, причем качество проб может быть повышено
путем введения связки (например, поливинилового спирта, крахмала, целлюлозы).
Одним из основных факторов выбора пресса является программируемая нагрузка и
выдержка при прессовании [13].
Для определения
оптимальной конфигурации рентгенофлуоресцентного спектрометра необходимо
сформулировать, в первую очередь, требования по производительности
аналитической системы. Современные спектрометры (рис. 3) обеспечивают
минимальный уровень инструментальной погрешности, что, зачастую, позволяет
использовать надежные и эргономичные инструменты с малой мощностью источника.

Рис. 3. Линейка
волновых рентгенофлуоресцентных спектрометров производства Thermo Fisher ARL
Products
Для построения калибровочных
кривых обычно используют стандартные образцы предприятия (СОП), аттестованные
по содержанию компонентов с помощью классических методов. Для построения
калибровочных кривых обычно требуется 5 - 10 СОП: число стандартных образцов
рассчитывают по формуле 2N+1, где N - число варьируемых параметров в регрессии.
В литературе [17]
доказано, что калибровочные кривые для плавленых проб имеют высокую степень
линейности в широком диапазоне концентраций, однако экспрессность анализа
уменьшается как за счет более сложной подготовки пробы, так и за счет более
низкой интенсивности характеристической линии для разбавленной флюсом пробы.
При этом прямая калибровка в широком диапазоне концентраций не обеспечивает
требуемой по ГОСТ 2642 точности в области малых содержаний компонентов.
Необходимая точность может быть достигнута использованием глобальной оксидной
калибровки ARL с фиксированными a - коэффициентами и типовыми стандартами.
Авторы [20]
показывают, что выбор корректного метода пробоподготовки и обработки спектральных
данных позволяет обеспечить необходимые метрологические характеристики
аналитического процесса как при анализе узкого сортамента огнеупорных
материалов, так и при исследовании большого числа типов различных огнеупоров.
Для экспрессной
оценки типа и состава огнеупорного материала возможно использование метода
фундаментальных параметров, реализованного в ПО UniQuant 5 (UQ5). Очевидным
достоинством метода является возможность проводить исследования даже при
отсутствии в лаборатории релевантных СО. В ряде случаев, при наличии
ограниченного количества СО, в параметры UQ5 могут быть внесены изменения для
повышения достоверности определений. Вместе с тем, использование UQ5 для
рутинного анализа не может быть рекомендовано к внедрению в лабораторную практику
из-за сложности обеспечения и контроля требуемых метрологических характеристик.
Использование
рентгенофлуоресцентной спектроскопии (РФС) позволяет решить задачу
экспресс-анализа химического состава огнеупорных материалов и обеспечить
метрологические характеристики аналитического процесса, определяемые
требованиями существующей нормативной документации (ГОСТ 2642). Выполнение
определений возможно как на приборах малой и средней мощности (ARL Optim’X),
так и на приборах большой мощности (ARL Perform’X, ARL 9900) - выбор типа
прибора диктуется, скорее, требованиями экспрессности, т.к. требуемая точность
обеспечивается всеми типами оборудования. Введение в действие перспективного
ГОСТ на рентгеноспектральный анализ огнеупорных материалов дополнительно упростит
внедрение метода в лаборатории предприятия и аккредитацию лаборатории [21].
Основным
преимуществом РФС является широкий динамический диапазон (10-4-102
мас.%). Можно одновременно и из одной пробы определять элементы, содержащиеся
на уровне процентов и на уровне миллионных долей [22]. Другим преимуществом в
сравнении с прочими аналитическими методами является анализ твердых проб без
растворения. Это позволяет осуществлять полный многоэлементный анализ за
несколько минут при использовании многоканального прибора РФСВД.
Сложная процедура
обработки результатов измерений и относительно высокая стоимость приборов - вот
основные недостатки РФС [23].
Атомно-эмиссионная
спектрометрия
Качество продукции, выпускаемой
металлургическими предприятиями, во многом определяется результатами
химического анализа как металлов и сплавов, так и вспомогательного сырья.
Современные методики анализа должны обеспечивать относительно быстрое и
экономичное определение нормируемых компонентов с высокой воспроизводимостью и
правильностью. До настоящего времени в лабораториях металлургических
предприятий для анализа сырья и материалов ГОСТ и ТУ рекомендованы методы
гравиметрии, титриметрии, спектрофотометрии, характеризующиеся невысокой
селективностью, значительной продолжительностью и трудоёмкостью, требующие
использования больших объёмов реагентов и индивидуальных приёмов
пробоподготовки при определении отдельных элементов. Во многих случаях
необходимо предварительное выделение определяемого элемента, отделение или
маскирование мешающих компонентов. Кроме того, установление состава ряда
труднорастворимых материалов, таких как огнеупоры, сопряжено с трудностью
переведения образцов в раствор [6, 24].
Метод АЭС ИСП положительно
зарекомендовал себя при анализе металлов и сплавов, поскольку имеет хорошие
метрологические характеристики, позволяет определять как макро-, так и
микроконцентрации компонентов. При спектральном анализе твёрдых проб, с целью
устранения помех связанных со структурой, для упрощения градуировки, как
правило, требуется переведение объектов анализа в раствор. В настоящее время,
растворение проб металлургических объектов осуществляется с применением
значительных объёмов кислот. Для деструкции труднорастворимых материалов
рекомендуется неоднократное и продолжительное упаривание, доплавление
нерастворённого остатка, и, в некоторых случаях, высокотемпературное
сплавление. При многоступенчатом разложении в открытых сосудах возрастает риск
потерь летучих компонентов. Но самое главное, пробоподготовка продолжает
оставаться самым узким местом анализа проб металлургического назначения [25].
В то же время, деструкция
труднорастворимых материалов в автоклавах в условиях МВ-нагрева положительно
зарекомендовала себя при разложении агломератов, объектов геологической
природы, металлов и сплавов, силикатов. Отмечено, что применение МВ-излучения
позволяет значительно повысить скорость переведения пробы в раствор, снизить
температуру разложения, а работа в герметичных системах исключает потери
летучих веществ [23, 26].
Однако систематические исследования
по автоклавному разложению в МВ-поле таких важнейших материалов, как огнеупоры,
в сочетании с АЭС ИСП ранее не проводились. Изучение таких подходов и
разработка методик анализа представляется актуальной с научной и, самое
главное, практической точки зрения, поскольку их внедрение в практику служб
аналитического контроля металлургических предприятий позволит унифицировать
процедуру прецизионного анализа и упростить его схему, повысить
воспроизводимость измерений, экономичность и производительность [27].
Метод АЭС-ИСП обеспечивает комплекс
уникальных метрологических характеристик: возможность одновременного
определения большого числа элементов в широком диапазоне концентраций - от
ультрамалых до 100%, хорошую воспроизводимость измерений (Бг не более 0,01),
незначительные матричные влияния, линейность градуировочных графиков в пределах
3-6 порядков величины. Пределы обнаружения большинства элементов в водных
растворах составляют 1 - 10 мкг/л. Метод не требует стандартных образцов -
соответствующие растворы сравнения могут быть смоделированы.
Индуктивно-связанная плазма (ИСП)
является одним из самых эффективных источников возбуждения в атомно-эмиссионной
спектрометрии (АЭС) [28].
Такие достоинства метода
атомно-эмиссионной спектрометрии с индуктивно связанной плазмой (ИСП)
обусловили стремительное внедрение данного метода анализа в практику работы
многих исследовательских и промышленных лабораторий во всем мире. Постоянно
продолжается увеличение его применения к анализу самых различных объектов, в том
числе исходного минерального сырья, продуктов и отходов черной металлургии.
Достаточно широкое применение этого метода на предприятиях черной металлургии
наблюдается и в нашей стране.
Разложение проб
Чаще всего при атомно-эмиссионном
спектральном анализе с индуктивно связанной плазмой (ИСП-АЭС. ICP-OES, 1CP-AES) пробы вводят в разряд
в виде аэрозолей растворов. При использовании техники гидридообразования
элементов в сочетании с ИСП-АЭС также необходимо предварительное получение
растворов проб. Состав растворов, вводимых в плазму разряда, должен адекватно
отражать состав исходных образцов испытуемых материалов по определяемым
элементам и не создавать помех для правильного измерения их аналитических
сигналов. Поэтому операции разложения твердых проб должны быть тщательно
продуманы и оптимизированы. При этом, в первую очередь, необходимо соблюдать
следующие основные правила разложения проб:
- достижение полного (100%) перевода определяемого элемента из
исходной пробы в анализируемый раствор:
- предотвращение загрязнения конечного раствора по определяемым
элементам;
- минимальные кислотность и засоление конечного раствора [29].
Неполное извлечение изучаемого
элемента в анализируемый раствор неизбежно ведет к занижению получаемых
результатов определения. Отмечается, что некоторые примесные элементы после
проведения операций вскрытия пробы могут присутствовать в растворе в виде
мелкодисперсных нерастворенных соединений, которые достаточно успешно
распыляются совместно с анализируемым раствором и попадают в плазму разряда.
Загрязнение раствора по определяемым
элементам вызывает, естественно, завышение результатов анализа. Для
предотвращения подобных погрешностей анализа необходимо использовать при
разложении проб чистые и обязательно проверенные по изучаемым элементам
реактивы, а также чистую посуду.
Высокая степень засоления раствора
может приводить как к завышению, так и занижению аналитических сигналов
определяемых элементов [30], ухудшению воспроизводимости результатов анализа.
Поэтому при подборе среды растворения необходимо придерживаться общепринятой в
аналитических работах очередности:
- использование разбавленных индивидуальных кислот;
- использование концентрированных индивидуальных кислот;
- использование смесей кислот;
- максимально возможное растворение пробы в кислотах и разложение
сплавлением нерастворимого остатка;
- разложение сплавлением всей пробы.
Минимальные расход реагентов и
уровень потерь определяемых элементов обеспечивает техника автоклавного
вскрытия с микроволновым или традиционным термическим нагревом.
Разлагаемая навеска пробы должна
обязательно обеспечивать ее представительность. При определении в сплавах
элементов, склонных к сегрегации (например, фосфор), рекомендуется большое
внимание уделять отбору средней пробы [19,31]. Для уменьшения расхождения
результатов между параллельными образцами авторы [28] советуют применять
аналитические навески не менее 1 г.
Степень разбавления конечного
раствора пробы должна быть оптимизирована для обеспечения необходимых пределов
обнаружения элементов. Возможности одновременного определения многих элементов
из одного раствора и минимизации матричных помех от основных компонентов пробы
и реактивов, используемых при разложении пробы (особенно в случае сплавления).
Обычно используемая концентрация анализируемого раствора по матричным
компонентам (без отделения основы) составляет СМа1 = 1-20 мг/мл
[32].
Предварительное
отделение и концентрирование элементов
Низкие содержания в материалах
черной металлургии некоторых примесных элементов не позволяют проводить их прямое
определение методом ИСП-АЭС. Кроме того, наличие в этих объектах анализа
высоких содержаний железа и других элементов, обладающих сложными атомными
спектрами, является причиной многочисленных спектральных наложений, что снижает
точность анализа, и, при использовании менее интенсивных аналитических линий,
не испытывающих спектральных помех, обуславливает увеличение пределов
обнаружения определяемых элементов. Поэтому часто оценивают возможность
применения и используют операции предварительного отделения и концентрирования
для повышения чувствительности определения следовых количеств элементов.
Применяемые для подобных целей методы и способы разделения и концентрирования
достаточно разнообразны.
Сочетание проточно-инжекционного
анализа (flow injection) с методом ИСП-АЭС обеспечивает высокую скорость проведения
анализа, отсутствие засорения капилляра распылителя при анализе
концентрированных растворов, возможность решения широкого круга аналитических
задач анализа материалов черной металлургии.
В обзоре [31] рассмотрены некоторые
проблемы и приемы концентрирования микроэлементов, в том числе определяемых в
материалах черной металлургии.
Ввод проб в плазму
разряда
Введение растворов
Наиболее часто при анализе
металлургических материалов методом ИСП-АЭС используют для ввода пробы в разряд
традиционное пневматическое распыление ее раствора до мелкодисперсного аэрозоля
с помощью потока аргона [33]. Для получения аэрозоля применяют концентрические
или поперечно-потоковые (уголковые) распылители, генерирующие полидисперсный
аэрозоль. В работе [24, 34] установлено, что для концентрических распылителей
Мейнхарда и распылителей уголкового типа получаются близкие аналитические
характеристики. Развиваемые в последние годы для метода ИСП-АЭС пневматические
распылители прямого ввода, малого расхода и распылители высокосолевых
растворов, а также ультразвуковые распылители крайне редко применяются для
анализа материалов и продуктов черной металлургии.
Для удаления из полидисперсного
аэрозоля, генерируемого пневматическими распылителями, крупных частиц, не
успевающих полностью испариться в плазме разряда, распылитель обычно состыкован
с распылительной камерой. Выходящий из распылителя поток аэрозоля в
распылительной камере резко уменьшает свою скорость крупные капли осаждаются за
счет сил гравитации на стенке камеры и уходят в слив. Поэтому эффективность
использования анализируемого раствора в методе ИСП-АЭС обычно составляет менее
1-2%. Время установления стабильного режима поступления аэрозоля в плазму
разряда обычно достаточно продолжительно - не менее 20 с. Для равномерной
генерации аэрозоля во времени и снижения влияния физико-химических свойств
раствора на характеристики распыления ввод пробы в распылитель осуществляют
принудительно с помощью перистальтического насоса.
Введение пробы в разряд в виде
растворов имеет следующие основные преимущества:
- возможность
устранения помех, связанных со структурой твердой пробы;
- хорошая
воспроизводимость результатов анализа;
- простота
градуирования с использованием модельных растворов;
- возможность
введения внутреннего стандарта.
К недостаткам этого способа введения
пробы нужно отнести:
- длительность и трудоемкость процедуры перевода и пробы в раствор;
- неизбежное разбавление пробы при растворении:
- возможность внесения загрязнений с используемыми для растворения
реактивами;
- возможность потерь определяемых элементов при пробоприготовлении.
Интересен прием введения растворов в
плазму разряда с помощью проточно-инжекционной системы, когда порции
анализируемой пробы и растворителя, разделенные пузырьком воздуха, двигаются к
распылителю по трубопроводу.
Возможен ввод растворов проб в
плазму разряда с использованием электротермического испарения [8, 27] в
графитовой печи, аналогичной применяемым в атомно-абсорбционной спектрометрии с
электротермической атомизацией. В этом случае пары пробы доставляются в горелку
небольшим потоком аргона. Это позволяет расходовать малые объемы проб,
устранять спектральные помехи матричных элементов за счет оптимизации
температурно-временной программы нагрева, разделяющей моменты испарения
определяемых и матричных элементов.
Введение суспензий
При изучении возможностей метода
ИСП-АЭС в приложении к материалам черной металлургии аналитики не обошли
вниманием и достаточно хорошо известный способ введения пробы в плазму разряда
в виде суспензий. Ввод проб в виде суспензий применим лишь для ограниченного
круга анализируемых объектов черной металлургии.
Прямой анализ твердых
проб
Прямой анализ твердых образцов имеет
ряд преимуществ по сравнению со способами ввода растворов: отсутствие
разбавления пробы, малый расход анализируемого материала, простота операций,
высокая скорость проведения анализа, отсутствие загрязнений от реактивов или
потерь определяемых элементов, простота и экономичность градуирования с использованием
минимального количества твердых стандартных образцов состава [12, 23]. Но
прямое термическое испарение давало неудовлетворительные результаты, что
свидетельствует о малой перспективности анализа.
Значительно большее распространение
получили способы прямого анализа металлических образцов в ИСП путем их
предварительной электрической или лазерной эрозии.
Искровая абляция
Воздействие каждого отдельного
импульса конденсированного искрового разряда обрабатывает на поверхности
металлического образца небольшой участок площадью в десятые доли квадратного
миллиметра. Глубина поражения поверхности образца составляет десятки
микрометров. Но последовательность сотен импульсов искрового разряда
статистически обрабатывает уже значительную зону на поверхности образца,
обеспечивая соответствующее число зон поражения на электродах общей площадью в
несколько квадратных миллиметров. Таким образом, несмотря на локальность
каждого отдельного импульса, общая площадь поражения за время экспозиции в
несколько секунд или десятков секунд является относительно большой. По этой
причине многоимпульсные искры обеспечивают необходимую представительность
результатов атомно-эмиссионного анализа [33].
Перевод под воздействием искрового
разряда части металлической пробы в состояние аэрозоля связан со сложными
физико-химическими процессами, обусловленными высокой температурой в зоне
взаимодействия искры с поверхностью электрода-пробы (в зоне поражения
электрода). В первую очередь это обусловлено быстрым локальным разогревом
участка образца и его взрывным испарением. Продукты испарения малым потоком
аргона направляют в горелку ИСП. где конденсированные частицы испаряются,
газообразные молекулы диссоциируют до атомов, атомы ионизируются и происходит
возбуждение спектров эмиссии. Так реализуется способ ИСП-АЭС с непосредственным
вводом металлической пробы с помощью искровой абляции [28].
Способ искровой абляции проб
металлов и сплавов резко увеличивает скорость проведения анализа, так как
устраняется обычная ля метода ИСП-АЭС процедура перевода конденсированной пробы
в раствор. При этом ликвидируются возможность загрязнения анализируемых
растворов примесями из реактивов и вероятность потери некоторых элементов при
пробоприготовлении.
При анализе металлов и сплавов
искровой разряд обычно возбуждают между вольфрамовым стержнем и анализируемой
пробой. Искровую абляцию можно применять для больших образцов с максимальным
диаметром более 15 мм. а также, используя специальные адаптеры, для тонких
металлических листов толщиной не менее 0,1 мм и проволоки диаметром не менее 1
мм [6, 27].
Оптимальные операционные параметры
(напряжение и ток разряда, частота пробоев за полупериод, скорость подачи газа
носителя, время предварительного обыскривания. время интегрирования сигнала)
для системы искровой абляции зависят от матричного состава анализируемого
образца и достаточно сильно влияют на пределы обнаружения, воспроизводимость
результатов, эффект памяти.
При способе искровой абляции в
сочетании с ИСП реализуется гораздо более широкий динамический диапазон градуировочных
графиков, чем при использовании атомно-эмиссионной спектрометрии с
высоковольтным искровым разрядом. Это упрощает процесс градуирования и
позволяет обходиться минимальным количеством стандартных образцов или образцов
сравнения для количественного анализа [35]. Данное качество является особенно
ценным в реальных условиях наличия только ограниченного количества образцов для
градуирования.
При анализе металлических образцов с
применением метода искровой абляции необходимо учитывать качество обработки
поверхности образца. Слишком гладкая (полированная) или грубая поверхность
образца не приводит к поступлению одинакового количества материала образца в
плазму разряда. После шлифования поверхность образцов необходимо промывать
соляной кислотой, водой и органическим растворителем, типа ацетон или спирт.
Погрешности количественного анализа
могут быть обусловлены «эффектом памяти», возникающим в результате осаждения
частиц аэрозоля на подставном вольфрамовом электроде и в трубопроводе во время
транспортировки пробы от ячейки искровой абляции к горелке ИСП. Такие
погрешности становятся особенно заметными, если проводится последовательное
определение элемента, содержащегося в пробе в следовых количествах, сразу же
после анализа образца, в котором тот же самый элемент содержался в высокой
концентрации. Для устранения этих погрешностей необходима смена подставного
электрода и увеличение времени предварительного обыскривания образца [19, 36].
Таким образом, необходимо отметить
суммарно следующие основные достоинства искровой абляции в методе ИСП-АЭС:
- возможность прямого анализа металлов и сплавов:
- низкий уровень первоначальных затрат на приобретение оборудования:
- простота эксплуатации приборов;
- относительно короткое время предварительного обыскривания (по сравнению
с лазерной абляцией);
- хорошая чувствительность определений:
- простота градуирования.
Основными недостатками способа
ИСП-АЭС
с искровой абляцией можно считать:
- необходимость использования для градуирования образцов сравнения,
идентичных с анализируемыми пробами:
- необходимость более качественной подготовки поверхности образца,
чем для способа лазерной абляции:
- применимость для анализа только общего (среднего) содержания
элементов в пробах.
Авторы [33] приходят к заключению,
что искровая абляция для металлических материалов черной металлургии является
хорошим компромиссом между первоначальными инвестициями и достигаемой
чувствительностью анализа.
Лазерная абляция
При воздействии на поверхность
твердой пробы сфокусированного излучения оптического квантового генератора
(ОКГ) происходит взрывообразное локальное разрушение материала пробы (лазерная
абляция) с образованием тонкодисперсного аэрозоля (твердые и жидкие частицы), а
также газообразных продуктов. Продукты разрушения потоком аргона направляют в
горелку ИСП, где конденсированные частицы испаряются, молекулы диссоциируют до
атомов, атомы ионизируются и происходит возбуждение спектров эмиссии. Так
реализуется способ ИСП-АЭС с непосредственным вводом пробы с помощью лазерной
абляции.
Скорость лазерной абляции твердых
металлических образцов V и размер частиц аэрозоля d зависят от рабочих параметров
лазера (мощность и частота импульсов, длина волны излучения). Значения этих
характеристик обычно составляют V = 1,0-16,7 мкг/си d>100 нм. По оценкам авторов [26]
эффективность поступления образца в плазму при лазерной абляции на 30-50%
меньше, чем при использовании пневматического распылителя (при условии
одинаковых плазменных горелок). Чугун и сталь являются материалами, наиболее
трудными для лазерной абляции [37].
Известно [8, 32], что скорости
лазерной абляции чистых металлов достаточно близки между собой. Но скорость
абляции сплавов не соответствует поведению составляющих чистых элементов и
зависит от типа сплава. Это вызывает серьезные трудности при градуировании,
когда определенный тип стандартных образцов состава желательно использовать для
количественного анализа других типов материалов. Использование внутреннего
стандарта не позволяет устранить это затруднение.
Применение лазеров, излучающих в ультрафиолетовой
(УФ) области спектра (эксимерные лазеры). минимизирует возможное различие в
скорости распыления материалов и повышает эффективность абляции по сравнению с
инфракрасными (ИК) лазерами (чаще всего твердотельные ОКГ Nd:YAG с основной длиной волны
X = 1064 нм). Кроме того, эксимерные лазеры обеспечивают лучшую
чувствительность определения, поскольку ИК лазеры имеют ограниченную мощность в
УФ диапазоне (используется третья или четвертая гармоника генерации)
Таким образом, можно отметить
суммарно следующие достоинства способа ИСП-АЭС с лазерной абляцией:
- возможность прямого анализа металлов и сплавов без химического
разложения проб, что снижает опасность загрязнения реактивами и/ или потери
определяемых элементов;
- предел обнаружения ниже, чем при искровой абляции (при высокой
скорости повторения лазерных импульсов):
- более низкие требования по качеству подготовки поверхности пробы,
чем при искровой абляции;
- возможность анализа образцов сложной формы, определения содержания
элементов в слоях и включениях:
- возможность проведения локального микроанализа.
К недостаткам данного способа ввода
пробы необходимо отнести:
- высокий уровень первоначальных затрат на приобретение
оборудования;
- необходимость применения для градуирования образцов сравнения,
идентичных с анализируемыми пробами;
- большая сложность эксплуатации приборов:
- длительное время предварительной лазерной абляции (до установления
стабильного сигнала), по сравнению со временем обыскривания при искровой
абляции [33].
Ввод газообразных проб
В принципе, наиболее удобным
способом является ввод газообразных проб в ИСП. Однако такой прием достаточно
сложно применять при анализе материалов черной металлургии. Но в некоторых
случаях подобный способ ввода пробы уже нашел свое применение.
Наиболее часто при анализе
металлургических объектов используют газообразный ввод проб в виде гидридов.
Спектральные помехи
Основные трудности при анализе
материалов черной металлургии методом атомно-эмиссионной спектрометрии с
индуктивно связанной плазмой связаны с наличием разнообразных спектральных
помех, присущих методу ИСП-АЭС [28]:
- изменение
интенсивности непрерывного фона;
- частичное или
полное перекрывание аналитической линии атомными или ионными линиями
сопутствующих элементов и аргона;
- наложение компонент
молекулярных полос:
- рассеянный свет в
спектрометре.
Градуирование
Метод ИСП-АЭС является относительным
(сравнительным), то есть требует предварительного точного градуирования
прибора: получение с помощью образцов сравнения или стандартных образцов
состава функциональных зависимостей аналитических сигналов (амплитуда
интенсивности аналитической спектральной линии в максимуме пика или площадь под
пиком, соотношение интенсивностей аналитической линии и линии сравнения,
суммарная интенсивность многих спектральных линий одного элемента) определяемых
элементов от их концентрации. Поэтому градуирование является очень важным
этапом аналитической процедуры, от которого в значительной степени зависит
правильность результатов анализа. Неизбежные долговременные дрейфы
измерительной системы прибора требуют периодического контроля градуирования и,
при необходимости, проведения своевременного переградуирования прибора.
Градуировочные графики в методе
ИСП-АЭС обычно линейны в интервале от 4 до 6 порядков концентрации
определяемого элемента (линейный динамический диапазон). Это проявляется и при
анализе объектов черной металлургии. Как правило, при аксиальном способе
наблюдения плазмы достигается меньший линейный диапазон градуировочной
зависимости. чем при радиальном способе наблюдения. В ИСП нелинейность
градуировочных графиков обычно наблюдается только при высоких концентрациях
определяемого элемента, что обусловлено самопоглощением спектральных линий. Это
обстоятельство серьезно упрощает операцию получения градуировочного графика.
так как. зачастую, можно использовать минимальное число точек градуирования.
Например. в случае анализа растворов проб можно измерять лишь раствор холостой
пробы и один-два стандартных раствора. Следовательно, сокращается время на проведение
градуирования и уменьшается расход образцов сравнения. Кроме того, большой
линейный динамический диапазон градуировочных графиков позволяет использовать
меньшую степень разбавления образца [34].
Ввод проб в ИСП в виде суспензий, с
помощью искровой, дуговой или лазерной абляции требует максимального подобия
анализируемых проб и градуировочных образцов по химическому и структурному
составу. В этом случае наиболее приемлемы для градуирования аттестованные
стандартные образцы состава изучаемых продуктов.
Сравнение с другими
аналитическими методами
В литературе неоднократно сравнивали
возможности метода ИСП-АЭС с другими аналитическими методами, в первую очередь
с атомно-абсорбционной спектрометрией (ААС), рентгенофлуоресцентной
спектрометрией (РФС), атомно-эмиссионной спектрометрией с другими источниками
возбуждения и пр. Проводились подобные сравнения и в отношении анализа
различных объектов черной металлургии [13, 31].
Кроме того, поскольку в ИСП-АЭС для
анализа используют растворы проб, то это позволяет использовать образцы, не
пригодные для прямого измерения на оптических (с искровым возбуждением спектра)
и рентгеновских квантометрах: стружка, образцы с трещинами и др. [12, 25].
Пределы обнаружения легирующих элементов в методе ИСП-АЭС. как правило, ниже,
чем для метода пламенной ААС.
Авторы [6, 13, 23] приходят к
заключению, что метод ИСП-АЭС по сравнению с пламенным атомно-абсорбционным
методом позволяет снизить предел обнаружения трудновозбудимых элементов.
Преимуществами являются также возможность одновременного определения многих
элементов и незначительный матричный эффект, больший динамический диапазон
градуировочных графиков. Современные приборы пламенной атомной абсорбции
обеспечивают меньшую погрешность воспроизводимости, чем спектрометры ИСП-АЭС, имеют
более низкую стоимость и дешевле в эксплуатации [38].
Обзор литературы показал
перспективность анализа огнеупоров методом АЭС ИСП с микроволновой
пробоподготовкой. Такой подход для анализа кремнеземистых материалов ранее не
изучался.
2. Экспериментальная
часть
.1 Химические реактивы,
основное и вспомогательное оборудование
Растворы готовили
на дистиллированной воде (ГОСТ 6709).
Для построения
калибровочного графика применяли следующие стандартные растворы:
стандартный раствор
с массовой концентрацией оксида алюминия 0,0001 г./см3 и 0,00001
г./см3.
стандартный раствор
с массовой концентрацией оксида кремния (IV) 0,001 г./см3.
стандартный раствор
с массовой концентрацией оксида магния 0,0001 г./см3.
стандартный раствор
с массовой концентрацией оксида железа (III) 0,0001 г./см3
и 0,00001 г./см3.
стандартный раствор
с массовой концентрацией оксида кальция 0,0001 г./см3 и 0,00001
г./см3.
стандартный раствор
с массовой концентрацией оксида натрия 0,0001 г./см3.
стандартный раствор
с массовой концентрацией оксида калия 0,0001 г./см3.
Государственные
стандартные образцы (ГСО): внутренний стандарт Y с концентрацией 0,001 г./см3;
внутренний стандарт Cd с концентрацией 0,001 г./см3; огнеупор динасовый - к1в
(табл. 5).
Таблица 5. Состав стандарта огнеупор
динасовый
|
Стандарт
|
Содержание массовых долей, %
|
|
К1в
|
Al2O3
|
SiO2
|
Fe2O3
|
CaO
|
|
0,55
|
96,1
|
1,36
|
1,35
|
Для приготовления стандартных
растворов и растворов внутренних стандартов использовались реактивы: кадмий
(ГОСТ 1467 или ГОСТ 22860); иттрий металлический высокой чистоты; натрий
хлористый (ГОСТ 4233); калий хлористый (ГОСТ 4234); оксид магния марки 11-2
(ГОСТ 4526), х. ч. или ос. ч.; железа оксид по нормативной документации, х. ч.,
или железо карбонильное (ГОСТ 13610); алюминий марки А995 (ГОСТ 11069),
стружка; смесь для сплавления: натрий углекислый, натрий тетраборнокислый
безводный и калий углекислый смешивают в соотношении 1: 1: 1; кислота
кремниевая водная (ГОСТ 4214); кальций углекислый (ГОСТ 4530), раствор молярной
концентрации 0,05 моль/дм3.
Для стандартных методов анализа
динасовых огнеупоров применялись следующие реактивы: аммоний виннокислый
средний по нормативной документации, раствор с массовой долей 25%; раствор
ацетатный буферный с рН 4,8-5,0; кислота сульфосалициловая 2-водная (ГОСТ
4478), раствор с массовой долей 30%; натрия гидроксид (ГОСТ 4328), х.ч.,
раствор с массовой долей 30%; аммиак водный (ГОСТ 3760), разбавленный 1:1; медь
(II) сернокислая 5-водная (сульфат меди) (ГОСТ 4165), раствор молярной
концентрации 0,025 моль/дм3 и 0,05 моль/дм3; индикатор 1,2
- (пиридил-азо) - 2-нафтол (ПАН), спиртовый раствор с массовой долей 0,2%; соль
динатриевая этилендиамин-N, N, N', N'-тетрауксусной кислоты 2-водная (трилон Б)
(ГОСТ 10652), раствор молярной концентрации 0,025 моль/дм3; кислота
щавелевая (ГОСТ 22180) и насыщенный раствор; калий пиросернокислый (ГОСТ 7172);
соль лантана, раствор с массовой концентрацией лантана 0,1 г/см3.
Для проведения экспериментальных
исследований применялось следующее оборудование:
· атомно-эмиссионный
спектрометр с индуктивно-связанной плазмой «iCAP 6500 Duo» фирмы «Thermo Scientific» (США). Характеристики
«iCaP
6500 Duo» - плазменный генератор с максимальной мощностью, подводимой к
плазме - 1.6 кВт; частота - 27.12 МГц; 4-канальный, 12-роликовый
перистальтический насос, управляемый компьютером; охлаждающий, вспомогательный
и распылительный (транспортирующий) потоки контролируются масс-контроллерами
(рис. 4).

Рис. 4. Атомно-эмиссионный
спектрометр c индуктивно связанной плазмой Thermo Scientific Corp. iCAP - 6500 (USA)
Данный прибор
характеризуется оптическим диапазоном 166 - 847 нм; двойным (аксиальным и
радиальным) наблюдением плазмы; оптическим разрешением (при λ = 200 нм) менее 0,007 нм,
которое дает возможность анализировать материалы с самыми сложными спектрами;
что касается чувствительности, то 10 элементов имеют пределы обнаружения
0,001-0,01 мкг/л, 17 элементов имеют пределы обнаружения 0,01 - 0,1 мкг/л и 39
элементов имеют пределы обнаружения 0,1 - 1 мкг/л. Спектрометр включает:
систему для работы с высоко засоленными растворами, плавиковой кислотой,
органическими растворителями и легколетучими органическими растворителями;
плазменный генератор с максимальной мощностью, подводимой к плазме - 1,6 кВт;
частота - 27,12 МГц; 4-канальный, 12-роликовый перистальтический насос,
управляемый компьютером; охлаждающий, вспомогательный и распылительный
(транспортирующий) потоки контролируются масс-контроллерами. Продувается
минимальным количеством аргона, термостатируется при температуре 38ºС с точностью 0,1ºС. Регистрация спектра -
на полупроводниковом детекторе нового поколения CID86. Вся система
управляется и контролируется программным обеспечением iTEVA - программным
обеспечением на русском языке для спектрометров с индуктивно-связанной плазмой,
совместимым с операционной системой WINDOWS XP Pro. Все параметры
плазменного источника оптимизируются автоматически. Возможно подключение
приставок твердого пробоотбора - искровой (SSEA) или лазерной.
· атомно-эмиссионный
спектрометр с индуктивно-связанной плазмой «OPTIMA 4300DV» фирмы «Perlin Elmer» (США): напряжение
220В; потребляемая мощность - 2,8 кВт; используемый газ - аргон; расход 25
л/мин; чистота газа 99,996%). Атомно-эмиссионный спектрометр (рис. 5)
представляет собой прибор, в котором реализован метод эмиссионного
спектрального анализа с возбуждением спектра пробы в аргоновой плазме,
возбуждаемой, в свою очередь, с помощью ВЧ разряда (рис. 5). Его спектральный
диапазон составляет от 165 до 782 нм, а спектральное разрешение - 0,004 нм.
Спектрометр состоит из источника возбуждения спектра, спектрального блока,
системы регистрации. Источник возбуждения спектра состоит из плазменной
горелки, распылителя, индуктора, перистальтического насоса и твердотельного (не
содержащего лампы) радиочастотного генератора с регулируемой мощностью от 750
до 1500 Вт с автоматической стабилизацией, работающего на частоте 40 МГц.

Рис. 5. Атомно-эмиссионный
спектрометр c индуктивно связанной плазмой Perkin Elmer Optima 4300 DV ICP-OES
Спектрометр позволяют осуществлять
два способа проектирования на входную щель спектрального блока факела плазмы -
радиальный и аксиальный. В аксиальной схеме на входную щель проектируется торец
факела, что позволяет увеличить интенсивность аналитического сигнала и в
определенных случаях уменьшить порог обнаружения до нескольких раз (для
образцов, в которых влиянием возрастания фонового излучения можно пренебречь).
Оптическая система имеет эшелле-полихроматор со скрещенной дисперсией.
Дисперсия в перпендикулярном направлении осуществляется с помощью двух
дифракционных решеток 374 штр/мм. Регистрация спектра осуществляется с помощью
двух линеек. Осуществляется одновременная регистрация в ультрафиолетовой и
видимой областях спектра. Управление процессом измерения и обработки выходной
информации осуществляется от IBM-совместимого компьютера с помощью специального
программного комплекса. Система охлаждения и компрессор позволяют поддерживать
оптимальную температуру и давление.
· микроволновая
система («SpeedWave four» фирмы «Berghoff» (Германия) с автоклавами DAK 100/4) (мощность
магнетрона 1450 Вт макс.; частота 2450 МГц; частота вращения ротора 6 об/мин;
температура до 300°С; давление до 150 атм.; напряжение до 230В; вес прибора 62
кг + контроллер 3 кг; размеры 640x570x440 мм + контроллер 220x240x150 мм).
Система SpeedWave four сконструирована для автоклавного разложения проб при температуре
до 230°C в длительных процессах (кратковременно при температуре до 300°C/572°F) и, в зависимости от применяемых
автоклавов, при давлении, достигающем 100 бар (1450 psi). Система представляет
собой микроволновую печь с вертикальной загрузкой, сделанную из нержавеющей
стали 1.4301 (SS 304), с шарнирной крышкой, электромеханическим замком и
цилиндрической камерой для равномерного распределения микроволнового излучения.
Камера имеет химически высоко стойкое покрытие из перфторалкокси-сополимера (PFA), защищающее ее от
коррозии. Необходимую безопасность эксплуатации обеспечивают три температурные
блокировки и три блокировки крышки. Камера системы постоянно продувается
специальным вентилятором. В микроволновую печь встроен инфракрасный термометр
(пирометр), который позволяет быстро определять и контролировать температуру
содержимого автоклавов. Заданные и фактические температуры для всех автоклавов
отображаются на экране системы в реальном времени и сохраняются в памяти
компьютера. Как показано на следующих рис. 6 и 7, система speedwave four имеет
различные элементы управления и коммуникации.

Рис. 6. Камеры системы

Рис. 7. Камеры системы

Рис. 8. Автоклавы DAK-100/4
Автоклавы DAK-100/4 (рис. 8)
представляют собой сосуды и крышки, сделанные из фторопласта TFM. Кроме того, автоклав
имеет защитную керамическую оправу и вкладыш из фторопласта TFM.
8 автоклавов из фторопласта TFM
Ротор
Система улавливания газов
· атомно-абсорбционный
спектрометр «SPECTR AA220» («Varian», Австралия): напряжение 220В; потребляемая мощность - 0,4 кВт; используемый
газ - ацетилен, сжатый воздух; расход ацетилена - от 0,1 до 10 л/мин; давление
- 0,76 атм.
· колориметр
фотоэлектрический концентрационный КФК-2-УХЛ4.2.
· весы лабораторные
электронные специального класса точности (BP221S фирмы «Сартогосм» (Россия,
Санкт-Петербург)); технические весы 3-го класса точности.
· печь муфельная с
терморегулятором, обеспечивающая нагрев до температуры 1000-1100°С.
· шкаф сушильный с
автоматическим регулированием температуры.
· колбы мерные
полиэтиленовые вместимостью 250 см3, 100 см3; стаканы
фторопластовый или полиэтиленовые вместимостью 50 см3, 100 см3,
200 см3; тигли платиновые №100-7 и 100-9 (ГОСТ 6563);
водяная баня; градуированная пипетка вместимостью 2 см3, 5 см3,
10 см3; пипетки Мора вместимостью 1,2,5,10 и 25 см3;
мерная колба вместимостью 100 см3, 200 см3, 250 см3,
500 см3 и 1000 см3; коническая колба вместимостью 300 и
500 см3; стаканы стеклянные лабораторные (ГОСТ 25336-82); бюретка
для титрования вместимостью 25 см3; штатив лабораторный; воронка
диаметром 5 см; стеклянный или деревянный шпатель; стеклянная палочка с
резиновым наконечником; индикаторная бумага Конго; фильтровальная бумага «синяя
лента» (d=90 мм).
2.2 Объекты анализа
В качестве объектов
анализа выбраны огнеупоры кремнеземистые с 80-% содержанием основного
компонента оксида кремния (IV) (табл. 6).
Таблица 6. Состав
исследуемых огнеупоров
|
Материал
|
Нормативный документ
|
Применение огнеупора
|
Содержание массовых долей, %
|
|
|
|
Al2O3
|
SiO2
|
Fe2O3
|
CaO
|
|
Siltis NS
|
ТУ 1523-001-97976618-2011
|
для теплоизоляции зеркала металла в чугуновозных
(чугунолитейных), сталеразливочных и промежуточных ковшах при непрерывной
разливке стали, а также для утепления головной части слитков и прибыльных
надставок
|
≤5
|
85,0-95,0
|
≤5
|
-
|
|
Инсулекс (Innsulekx)
|
ТУ 1522-001-99958856-2011
|
для изоляции зеркала жидкой стали в сталеразливочном ковше,
изложнице, промежуточном ковше МНЛЗ
|
0,1-3,5
|
80,0-95,0
|
0,2-2,2
|
0,1-1,7
|
|
Forward SS-G
|
ТУ 1523-019-77889395-2013
|
для покрытия металла в сталеразливочных и чугуновозных ковшах,
изложницах
|
≤3
|
85,0-95,0
|
≤3
|
-
|
Массовая доля
компонентов марки огнеупора Forward SS-G определялась в пересчете на сухое вещество (полученные данные
необходимо было делить на коэффициент k). Поэтому для данного огнеупора было
нужным рассчитать коэффициент k, связанный с потерей массы при прокаливании ( , рассчитываемый в соответствии с ГОСТ 2642.2-86 по формуле 1:
, рассчитываемый в соответствии с ГОСТ 2642.2-86 по формуле 1:
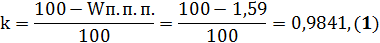
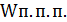 для Forward SS-G равна 1,59%.
для Forward SS-G равна 1,59%.
.3
Пробоподготовка
Отбор проб для
химического анализа производился в соответствии с нормативно-технической
документацией (таблица 6). Масса усредненной пробы для химического анализа
составляла 20г. Пробу измельчали до полного прохождения через сетку №0063 по
ГОСТ 6613-86 в карбидвольфрамовых ступках.
Навеску пробы
предварительно высушили и прокалили при температуре 950°С для удаления
органических примесей и свободного углерода в соответствии с ГОСТ 2642.0-86.
.4
Стандартные методы анализа динасовых огнеупоров
Ускоренный
метод определения оксида кремния (IV) в кремнеземистых огнеупорах (при массовой
доле свыше 90%)
Метод основан на
разложении навески пробы щавелевой и фтористоводородной кислотами и
гравиметрическом определении оксида кремния (IV) по разности масс навески пробы
и остатка после удаления кремния в виде SiF4.
Анализ проводился
следующим образом (ГОСТ 2642.3-97): в платиновый тигель, доведенный до
постоянной массы, помещали навеску массой 0,5 г, прибавляли 0,2 - 0,3 г
щавелевой кислоты и 10 см3 раствора фтористоводородной кислоты.
Тигель помещали на
электроплитку со слабым нагревом. Выпаривали при периодическом помешивании до
состояния влажных солей. В остывший тигель добавляли 5 см3
фтористоводородной кислоты и выпаривали досуха.
Сухой остаток
смачивали 1 см3 насыщенного раствора щавелевой кислоты и снова
выпаривали досуха до полного удаления налета щавелевой кислоты. Остаток
прокаливали в муфельной печи при температуре 1000 ± 50°С в течение 10 - 15 мин,
охлаждали в эксикаторе и взвешивали. Прокаливание и взвешивание повторяли до
достижения постоянной массы. Остаток в тигле сплавляли с пиросернокислым
калием, растворяли в горячей воде с добавлением 20 см3 соляной
кислоты (1: 3) и использовали для определения оксидов железа, алюминия,
кальция.
Комплексонометрический
метод определения оксида алюминия (при массовой доле оксида алюминия от 0,5 до
70%)
Сравнительное
определение оксида алюминия проводилось в соответствии с ГОСТ 2642.4-97.
Данный метод
основан на комплексонометрическом определении оксида алюминия после
предварительного отделения оксида кремния (IV) в кремнеземистых огнеупорных
материалах и изделиях.
Определение оксида
алюминия: навеску материала массой 0,5 г помещали в платиновый тигель,
смачивали несколькими каплями воды, прибавляли 0,5 см3 серной
кислоты, 10 см3 фтористоводородной кислоты и выпаривали досуха на
электроплитке со слабым нагревом при периодическом помешивании с помощью щипцов.
Сухой остаток, полученный после отделения оксида кремния (IV) нагревали
в муфельной печи при температуре 600 ± 20°С до полного удаления паров серной
кислоты. Осадок в тигле смешивали с 3 - 5 г пиросернокислого калия и сплавляли
в муфельной печи до получения прозрачного расплава. Сплав помещали в стакан,
растворяли в горячей воде с добавлением 20 см3 раствора соляной
кислоты (1: 3) (раствор 1).
Полученный
прозрачный раствор охлаждали до 40 - 50°С, нейтрализовали раствором гидроксида
натрия до красного цвета бумаги Конго и добавляли 15 - 20 см3 в
избыток. Раствор с осадком кипятили 3 - 5 мин, охлаждали, переводили в мерную
колбу вместимостью 250 см3, доводили водой до метки и перемешивали.
Раствор фильтровали через сухой складчатый фильтр, отбросив первые две порции
фильтрата, затем отбирали аликвотную часть раствора 100 см3,
помещали в коническую колбу вместимостью 300 см3, нейтрализовали
соляной кислотой до синего цвета бумаги Конго и добавили 3 - 5 см3 в
избыток.
В полученный кислый
раствор приливали 15 см3 0,025 моль/дм3 раствора трилона
Б и нагревали до кипения, после давали остыть до 70 - 80°С, нейтрализовали
раствором аммиака до переходного цвета бумаги Конго, приливали 15 - 20 см3
ацетатного буферного раствора, 5 - 7 капель раствора индикатора ПАН и титровали
избыточное количество трилона Б раствором сернокислой меди до перехода
желто-зеленой окраски в сине-фиолетовую.
Фотометрический
метод определения оксида железа (III) с сульфосалициловой кислотой (при
массовой доле от 0,05 до 6%)
Определение оксида
железа (III) проводили фотометрическим методом.
Этот метод основан
на измерении оптической плотности образующегося в аммиачной среде комплекса
трисульфосалицилата железа при использовании сульфосалициловой кислоты в
качестве комплексообразователя.
Для определения
оксида железа (III) использовали аликвотную часть раствора, полученного по ГОСТ
2642.3, разделы 4, 7, 9 после определения оксида кремния (IV) в соответствии с
п. II.4.1.
Объем аликвотной
части растворов 1 или 2, в зависимости от массовой доли оксида железа (III)
составил 10 см3.
Аликвотную часть
раствора помещали в мерную колбу вместимостью 100 см3, прибавляли 2
см3 виннокислого аммония, 15 см3 раствора
сульфосалициловой кислоты, раствор аммиака до появления устойчивой желтой
окраски и еще 3 см3 в избыток. Раствор охлаждали, доводили до метки
водой и перемешивали, в случае выпадения осадка или появления мути раствор
отфильтровывали.
Оптическую
плотность раствора измеряли на фотоколориметре с синим светофильтром (область
светопропускания 400 - 450 нм) в кювете толщиной слоя 30 мм. В качестве
раствора сравнения использовали раствор контрольного опыта, содержащий все
применяемые реактивы.
Массу оксида железа
(III) в граммах находили по градуировочному графику. Для его построения в
мерные колбы вместимостью 100 см3 помещали аликвотные части
градуировочного стандартного раствора Б: 1,0; 2,5; 5,0; 10,0; 15,0; 20,0; 25,0;
30,0 см3, что соответствовало 0,00002; 0,00005; 0,0001; 0,0002;
0,0003; 0,0004; 0,0005; 0,0006 г. оксида железа (III).
Атомно-абсорбционный
метод определения оксида кальция в кремнеземистых материалах (при массовой доле
от 0,02 до 15%)
Определение оксида
кальция проводили атомно-абсорбционным методом.
Сущность метод
заключается на разложении пробы смесью фтористоводородной и серной кислот и
измерении атомной абсорбции кальция в пламени воздух - ацетилен при длине волны
422,7 нм.
До начала анализа
готовили стандартный раствор оксида кальция массовой концентрации оксида
кальция 0,001 г./см3: 1,7852 г. углекислого кальция, предварительно
высушенного при температуре 105 ± 5°С в течение 1 ч, помещали в стакан
вместимостью 350 - 400 см3, приливали 100 см3 воды, затем
по каплям раствор соляной кислоты (1: 1), нагревали до полного растворения.
Раствор кипятили в течение 3 - 4 мин, охлаждали, переводили в мерную колбу
вместимостью 1 дм3, доливали водой до метки, перемешивали (раствор
А).
Затем стандартный
раствор оксида кальция массовой концентрации оксида кальция 0,00005 г./см3:
25 см3 стандартного раствора А помещали в мерную колбу вместимостью
500 см3, доливали до метки водой, перемешивали (раствор Б).
Серии
градуировочных стандартных растворов оксида кальция готовили следующим образом.
В мерные колбы вместимостью 100 см3 помещали 0,5; 1,0; 1,5; 2,0;
3,0; 4,0; 8,0; 12,0; 16,0; 20,0 см3 стандартного раствора Б оксида
кальция, что соответствовало 0,000025; 0,000050; 0,000075; 0,000100; 0,000150;
0,000200; 0,000400; 0,000600; 0,000800; 0,001000 г. оксида кальция. К растворам
добавляли 5 см3 раствора лантана, доливали до метки водой и
перемешивали.
Навеску пробы
массой 0,5 г помещали в чашку из стеклоуглерода, смачивали водой, приливали 15
см3 фтористоводородной кислоты, 2 - 3 см3 серной кислоты
и выпаривали на закрытой электроплитке при периодическом помешивании до
появления паров серного ангидрида. Чашку охлаждали, обмывали стенки водой и
выпаривали содержимое чашки досуха. Сухой остаток прокаливали в муфельной печи
при температуре 600 ± 50°С в течение 2 - 3 мин. Остаток при нагревании
растворяли в 20 - 30 см3 раствора соляной кислоты (1:1), переводили в
мерную колбу вместимостью 250 см3, тщательно очищая дно и стенки
чашки стеклянной палочкой с резиновым наконечником. Раствор кипятили 8 - 10
мин, охлаждали, доводили водой до метки и перемешивали. Затем фильтровали через
двойной сухой фильтр «синяя лента», отбрасывая первые две порции фильтрата.
В мерную колбу
вместимостью 100 см3 помещали 10 см3 аликвотной части
фильтрата, добавляли 5 см3 раствора лантана, доливали до метки водой
и перемешивали.
Измеряли абсорбцию
кальция в пламени ацетилен - воздух при длине волны 422,7 нм. Измерение
проводили методом ограничивающих растворов. После каждого измерения распыляли
воду и проверяли нуль прибора.
.5 Статистическая
обработка результатов
Промахи - 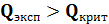
Среднее для выборки из n результатов
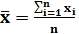 (2)
(2)
Дисперсия, характеризующая рассеяние
результатов относительно среднего V
V = (3)
(3)
Стандартное отклонение
S=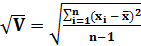 (4)
(4)
Относительно стандартное отклонение,
характеризующее воспроизводимость результатов химического анализа
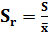 (5)
(5)
Доверительный интервал измеряемой
величины для заданной доверительной вероятности
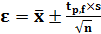 (6)
(6)
Для проверки правильности результата
использовали простой тест Стьюдента (7):
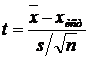 , (7)
, (7)
где t - коэффициент Стьюдента, 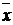 - среднее значение
выборочной совокупности,
- среднее значение
выборочной совокупности, 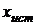 -
истинное значение определяемой величины, п - число измерений.
-
истинное значение определяемой величины, п - число измерений.
Далее экспериментальное
значение 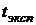 сравнивали
с табличным значением
сравнивали
с табличным значением  .
Если >,
то расхождение между и
значимо.
.
Если >,
то расхождение между и
значимо.
Результаты, полученные двумя
различными методами, сравнивали с применением распределения Фишера. Для этого
рассчитывали Fэксп, равное отношению
большей дисперсии к меньшей (8):
 (8)
(8)
Полученное
значение Fэксп сравниваем с табличными данными.
Если Fэксп ≤ Fкрит, то расхождение между
дисперсиями незначимо, то в этом случае оценивалась статистическая значимая
разница между результатами анализов с использованием модифицированного теста
Стьюдента. Для этого рассчитывали среднее взвешенное двух дисперсий
 , (9)
, (9)
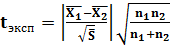 (10)
(10)
Критическим значением tкрит служит коэффициент Стьюдента t (P, f = f1 + f2). Если tэксп ≤ tкрит, то расхождение между
средними незначимо, т.е. выборки принадлежат одной и той же генеральной
совокупности и, следовательно, данные серий можно объединить и рассматривать
как одну выборочную совокупность из 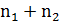 результатов.
результатов.
3. Результаты и
обсуждение
.1 Микроволновая пробоподготовка
Кислотное разложение матрицы образца
подчиняется множеству сложных зависимостей, которые были приняты во внимание.
Кислоты и их смеси выбирали так, чтобы они эффективно разлагали данную матрицу.
Обычно при разложении огнеупоров в открытых сосудах применяют азотную,
хлористоводородную, фосфорную и фтористоводородную кислоты. Информация о
матрице образца, составляющих ее макрокомпонентах и соединениях существенна для
выбора подходящей кислоты, обеспечивающей полное растворение пробы. Смеси
кислот выбирали в соответствии со способностью каждой кислоты эффективно
разлагать индивидуальные компоненты определенной матрицы. При этом учитывали,
что одна кислота эффективна как агент для разложения, а другая образует с
определенными элементами растворимые в воде комплексные соли.
Так как в основе пробы присутствуют
кремнийсодержащие компоненты, то для «высвобождения» следовых количеств
микроэлементов, которые могут удерживаться кремниевой кислотой, к азотной
кислоте добавляли фтористоводородную. Силикаты превращаются в SiF4, который, улетучиваясь, высвобождает другие определяемые
элементы. Небольшие количества фтористоводородной кислоты использовали в смесях
с другими кислотами для предотвращения связывания кремниевой кислотой
микроэлементов. Из-за низкой температуры кипения (106°С при концентрации 49
масс%) и высокого давления паров фтористоводородная кислота легко
улетучивается, а в закрытых контейнерах ее парциальное давление составляет
примерно 8 атм при 180°С. Часто образуются довольно устойчивые фторидные комплексы
металлов, которые при достаточно высокой концентрации фторид - ионов могут
удерживаться в растворе. Фтористоводородная кислота поглощает мощность
микроволнового излучения с частотой 2450 МГц примерно так же, как азотная
кислота.
Другим важным аспектом является
взаимодействие кислоты с материалом контейнера для разложения.
Фтористоводородную кислоту нельзя применять при работе со стеклом и кварцем.
При непосредственном взаимодействии микроволнового излучения с кислотой,
помещенной в закрытый сосуд из прозрачной для излучения пластмассы, учитывались
и дополнительные факторы. Хотя большинство минеральных кислот, традиционно
используемых для разложения, хорошо поглощают микроволновое излучение, все же
прежде перед тем, как провести разложение в закрытой системе, мы приняли во
внимание и другие свойства кислот: устойчивость в микроволновом поле, давление
паров и возможность взаимодействия с другими кислотами, присутствующими в
смеси.
Разложение с помощью азотной кислоты
относится к числу наиболее распространенных способов разложения. Азотная
кислота - одна из многих кислот, для которых достижима сверхвысокая степень
чистоты, что необходимо при определении малых содержаний элементов, она
идеально ведет себя под воздействием микроволнового излучения. В закрытом
контейнере при давлении около 5 атм азотная кислота может достичь температуры
176°С, что более чем на 50°С превышает температуру ее кипения. При повышении
температуры возрастает окислительный потенциал, и реакции протекают быстрее.
Методы микроволнового разложения в
закрытых системах получают дополнительные преимущества, когда кислоты
нагреваются до относительно высокой температуры в смесях друг с другом.
Азотная, хлористоводородная и фтористоводородная кислоты имеют относительно
низкие температуры кипения и большие парциальные давления паров. Фосфорная
кислота при сопоставимых температурах имеет низкое парциальное давление паров и
относительно высокие температуры кипения. Комбинируя по одной кислоте из каждой
группы, получили смеси с парциальным давлением, которое несколько ниже, чем
давление паров кислоты с более низкой температурой кипения. Результатом
смещения кислот является необходимое снижение давление паров над раствором.
Смешение фосфорной кислоты с азотной
кислотой приводит к значительному снижению парциального давления последней. При
температуре около 180°С смесь азотной и ортофосфорной кислот создает в сосуде
суммарное давление < 4 атм (4∙105 Па). Давление же паров
азотной кислоты во фторопластовом сосуде (неравновесные условия) при той же температуре
составляет несколько более 5 атм (5∙105 Па).
Азотная кислота в смеси с
фтористоводородной в соотношении 3:5 или 1:1 характеризуется кривыми
температуры и давления, которые весьма напоминают подобные кривые для
индивидуальных азотной или фтористоводородной кислот. Кривая зависимости
давления от температуры для этой кислотной смеси резко растет с ростом
температуры. Совпадение кривых нагрева и охлаждения свидетельствуют, что эти
кислоты не взаимодействуют с образованием продуктов разложения.
Царская водка, смесь азотной и
хлористоводородной кислот, является эффективным окислителем. Ее эффективность
обусловлена, в частности, образованием нитрозилхлорида (NOCl), одного из активных
компонентов этой смеси:
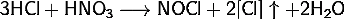 (1)
(1)
При нагревании нитрозилхлорид
диссоциирует с образованием токсичного газообразного хлора, обладающего
сильными корродирующими свойствами. Помимо хлорирующего эффекта, обусловленного
этими газами царская водка окисляет многие материалы более эффективно, чем хлористоводородная
или азотная кислота в отдельности. Находясь в контейнере, эти агрессивные газы
продолжают воздействовать на пробу и позволяют осуществить эффективное
разложение царской водкой в закрытой системе. Свежеприготовленная царская водка
(HNO3:НС1 = 1:3 по объему)
при температуре около 180°С обеспечивает внутри сосуда давление, превышающее 7
атм (7∙105 Па).
Анализ зависимостей температуры и
давления от времени для системы модифицированной царской водки (HNO3:НС1 = 3:5 по объему) при разложении пробы огнеупора и для той же
смеси кислот без пробы показывает, что в обоих случаях достигаются практически
одни и те же условия. Поглощение микроволновой энергии кислотой является
основным фактором, определяющим характер кривой нагрева, и не зависит
существенно от типа образца. Зависимость давления от температуры для обычной
царской водки демонстрирует возможность управлять кривой нагрева, несмотря на
образование газообразных веществ. Давление насыщенного пара внутри автоклава,
содержащего царскую водку выше, так как образуются NO и Cl2.
По окончании растворения образца
огнеупора необходимо добавлять борную кислоту с целью связывания избыточного
количества HF в комплексную борофтористоводородную кислоту HBF4. Присутствие в смеси соляной, азотной и плавиковой кислот
вследствие их летучести способствует увеличению давления внутри сосуда, поэтому
превышение их количеств может приводить к разгерметизации сосуда и потере
летучих фторидов кремния и бора. В то же время уменьшение содержания плавиковой
кислоты в смеси способствует образованию аморфного осадка кремниевой кислоты. HF является основным
компонентом, способствующим переведению Si в растворимые фторидные
комплексы. Превышение количеств низкокипящих кислот, создаёт избыточное
давление внутри сосудов с реакционной смесью, приводит к разгерметизации
автоклавов и, как следствие, потере компонентов проб. Для того чтобы снизить
давление внутри сосудов в смесь вводилась высокотемпературная фосфорная
кислота.
В основе метода лежит растворение в
кислотах проб огнеупоров в автоклаве под воздействием МВ-нагрева. Применение
МВ-излучения обеспечивает нагрев пробы «изнутри» по всему объему. Механизм
взаимодействия микроволнового излучения (МИ) с веществом заключается в
поглощении последним энергии электромагнитного излучения и рассеивании ее в
виде тепла.
Экспериментальным путем было
установлено, что для переведения в раствор навески проб огнеупора марки «Siltis NS», «Forward SS-G», «Innsulekx» и ГСО К1в массой
0,1000 г. растворяли в автоклаве под действием микроволнового излучения в 11 мл
смеси концентрированных кислот HF:HCl:HNO3:H3PO4 (6:3:1:1) с добавлением 2,1 г Н3ВО3.
Помимо состава
кислотной смеси на полноту деструкции огнеупоров влияют температурно-временные
параметры микроволнового воздействия (табл. 7, рис. 9), Во избежание вскрытия
автоклавов вследствие активного газообразования и потери кремния в виде SiF4 предложено осуществлять ступенчатый нагрев реакционной смеси с
невысокой скоростью подъема температуры и выдержкой при промежуточных
температурах в течение фиксированного времени. Поскольку некоторые компоненты
огнеупоров и продукты их взаимодействия с кислотами (например, MgO, СаО, Fe2О3 и Si(OH)4∙2Н2О) легко растворимы уже при 100
- 150°С на первом и втором этапе скоростью нагрева равнялась 12,5°С/мин.
Снижение скорости нагрева на третьем этапе до 10°С/мин, и на четвертом - до
8,5°С/мин было обусловлено возрастанием давления внутри автоклава из-за
повышенного газообразования. Параллельно на 3-м этапе наблюдается эффективное
взаимодействие медленноразлагаемого А12О3 Н3РО4,
присутствующей в кислотной смеси, а на 4-ом - кислотная деструкция Fe2O3. Полная деструкция проб происходит при пяти стадийном нагреве до
250°С. Оптимальные промежутки времени стабилизации температуры между отдельными
стадиями нагрева составляют: 3-15 мин (при нагреве свыше 150°С). Таким образом
продолжительность микроволновой деструкции огнеупоров составляет 88 мин.
Таблица 7. Условия деструкции
проб огнеупоров в микроволновом поле (мощность - 80%)
|
Шаг
|
Температура, °С
|
Продолжительность отдельных стадий, мин
|
|
|
нагрев
|
стабилизация
|
|
1 2 3 4 5
|
50 100 150 200 250
|
4 5 15 15 20
|
3 3 3 5 15
|

Рис. 9. Графическое
изображение микроволнового разложения
Таким образом, наилучшие результаты,
при использовании концентрированных кислот для навески образца массой 0,1 г,
показывает смесь состава 3,0 мл НСl, 1,0 мл HNO3, 6,0 мл HF, 1,0 мл Н3РО4
с добавлением 2,1г Н3В03.
Выбранные условия позволили
перевести в раствор все компоненты пробы. При разложении пробы огнеупоров
протекали следующие химические реакции, обеспечивающие полную деструкцию пробы:
) в автоклавах после прибавления
соляной кислоты
А12ОЗ + 6НС1(конц.
гор) = 2А1С13 + ЗН2О (2)
Fe2О3 + 6HCl = 2FeCl3 + 3H2О (3)
) после прибавления
плавиковой кислоты:
SiО2 + 6HF = H2[SiF6] + 2H2О (4)
H2SiF6 + А12О3
= 2AIF3 + SiО2 + Н2О
(5)
A1F3 + 3HF =
H3[A1F6] (6)
) после прибавления фосфорной
кислоты Fe3+ и А13+
образуют фосфатные комплексы:
Fe3+ + 2НРО42-
= [Fe(HPО4)2]
- (7)
Fe3+ + 4Н2РО4-
= [Fe(HPО4)4]
- (8)
А13++ Н2РО4-=
[А1 (Н2РО4)]2+ (9)
4) на последней стадии
пробоподготовки происходит связывание избытка плавиковой кислоты борной
кислотой:
3BО3 + 4HF = HBF4 + 3H2О (10)
.2 Атомно-эмиссионный
анализ с индуктивно связанной плазмой
Атомно-эмиссионный анализ позволяет
определять присутствие макрокомпонентов, таких как SiO2, Al2O3, Fe 2O3, CaO после
микроволнового разложения. Перед определением макрокомпонентов необходимо было
провести следующие дополнительные исследования:
) выбрать аналитические линии
для определения оксидов и провести градуировку прибора;
) правильно выбрать
внутренний стандарт, исключающий наложения спектральными линиями;
) изучить влияние разбавление
пробы на аналитический сигнал.
Замер прибора
происходил по абсолютной интенсивности элементов. Стандартные растворы и
добавки готовили из элементов в пересчёте на оксид (табл. 7). Коэффициент
пересчета рассчитывали по уравнению:
 (11)
(11)
где М -
молекулярная масса соответствующих веществ, г/моль.
Начальная
концентрация стандартных растворов 0,001 г./см3. Растворы для
внесения добавок готовили последовательно, разбавляя исходные до нужных
концентраций. Таким образом, так как график задавали в оксидной форме, то и
результаты были получены в аналогичной форме.
Построение
градуировочной зависимости осуществлялось с использованием следующих
стандартных растворов (табл. 8).
Таблица 8. Приготовление
стандартных растворов для анализа элементов (Начальная концентрация стандартных
растворов Cd, Y, SiO2 равна 0,001 г./см3, а Al2O3,
Fe2O3, CaO, MgO, Na2O, K2O - 0,0001
г./см3)
|
Стандартный раствор
|
Приготовление стандартного раствора
|
Коэффициент пересчета элемента на оксид
|
Нормативный документ
|
|
Cd
|
В стакан вместимостью 250 см3 помещали навеску кадмия
массой 0,1000 г., растворяли при нагревании в 15 - 20 см3 азотной
кислоты, разбавленной 1:1, кипятили раствор 3 - 5 мин, охлаждали, переводили
в мерную колбу вместимостью 1000 см3, приливали 50 см3
азотной кислоты, разбавленной 1:1, и доливали до метки водой
|
1,14
|
ГОСТ 13047.16-2002
|
|
Y
|
0,2 г металлического иттрия помещали в коническую колбу
вместимостью 250 см3, добавляли 25 см3 раствора соляной
кислоты (1:1) и растворяли при слабом нагревании. После растворения иттрия
раствор охлаждали до комнатной температуры, переносили в мерную колбу
вместимостью 200 см3, доливали водой до метки и перемешивали
|
0,7874
|
ГОСТ 11739.22-90
|
|
SiO2
|
0,5 г кремниевой кислоты, предварительно прокаленной в течение 1
ч при температуре (1000 ± 50)°С, сплавляли в платиновом тигле с 5 г смеси для
сплавления. Сплав растворяли в полиэтиленовом сосуде в 300 см3
воды с добавлением 20 г. гидрооксида натрия. После охлаждения раствор
переводили в мерную колбу вместимостью 500 см3, доводили водой до
метки и перемешивали
|
2,14
|
ГОСТ 2642.3-97
|
|
Al2O3
|
Точную навеску массой 0,529г металлического алюминия растворяли
в 150 см3 соляной кислоты (1:1), переводили в мерную колбу
вместимостью 1000 см3, доводили до метки водой и перемешивали
|
1,89
|
ГОСТ 2642.4-97
|
|
Fe2O3
|
0,112 г. высушенного при (110 ± 5)°С в течение 1 ч железо
карбонильное помещали в коническую колбу вместимостью 500 см3,
приливали 50 см3 соляной кислоты (1:1) и, накрыв колбу, нагревали
на водяной бане до полного растворения. Затем раствор охлаждали, переводили в
мерную колбу вместимостью 1000 см3, доводили водой до метки и
перемешивали
|
0,05
|
ГОСТ 2642.5-97
|
|
CaO
|
1,7847 г. углекислого кальция, высушенного при температуре (110
± 5)°С до постоянной массы, осторожно растворяли в стакане в 30 см3
соляной кислоты (1:1). Углекислый газ удаляли кипячением. Раствор охлаждали,
переводили в мерную колбу вместимостью 1000 см3, доводили до метки
водой, перемешивали
|
0,56
|
ГОСТ 2642.7-97
|
|
MgO
|
1 г оксида магния, прокаленного при температуре (950 ± 50)°С до
постоянной массы, растворяли в 40 см3 соляной кислоты (1:1) при
нагревании. Раствор охлаждали, переводили в мерную колбу вместимостью 1000 см3,
доливали водой до метки, перемешивали
|
0,33
|
ГОСТ 2642.8-97
|
|
Na2O
|
1,886 г. хлористого натрия, предварительно прокаленного при
температуре 500°С до постоянной массы, помещали в стакан вместимостью 400 см3
и растворяли в 200 см3 воды. Раствор переводили в мерную колбу
вместимостью 1000 см3, доводили водой до метки и перемешивали
|
0,53
|
ГОСТ 2642.11-97
|
|
K2O
|
1,583 г. хлористого калия, предварительно прокаленного при
температуре 500°С до постоянной массы, помещали в стакан вместимостью 400 см3
и растворяли в 200 см3 воды. Переводили раствор в мерную колбу
вместимостью 1000 см3, доводили водой до метки и перемешивали
|
0,63
|
ГОСТ 2642.11-97
|
Анализируемые стандартные растворы,
содержащие минимальную концентрацию определяемого элемента и максимальные
концентрации сопутствующих элементов и основы пробы, нормируемых ТУ для
построения градуировочного графика представлены в табл. 9.
Таблица 9. Процентное содержание
минимальной концентрации определяемого элемента и максимальной концентрации
сопутствующих элементов и основы пробы
|
Определяемый компонент
|
Объём аликвоты, мл
|
Процентное содержание, %
|
Концентрация, г/см3
|
|
Cd
|
1,0
|
0,1
|
0,001
|
|
Y
|
1,0
|
0,1
|
0,001
|
|
SiO2 max
|
9,5
|
95,0
|
0,001
|
|
SiO2 min
|
8,0
|
80,0
|
0,001
|
|
Al2O3max
|
5,0
|
5,0
|
0,0001
|
|
Al2O3min
|
1,0
|
0,1
|
0,00001
|
|
Fe2O3max
|
3,5
|
3,5
|
0,0001
|
|
Fe 2O3min
|
2,0
|
0,2
|
0,00001
|
|
CaO max
|
2,5
|
2,5
|
0,0001
|
|
CaO min
|
1,0
|
0,1
|
0,00001
|
|
MgO max
|
2,0
|
3,5
|
0,0001
|
|
Na2O max
|
3,5
|
3,5
|
0,0001
|
|
K2O max
|
3,5
|
3,5
|
0,0001
|
Выбор длин волн
При выборе длин волн (нм)
аналитических линий и линий внутреннего стандарта (нм) руководствовались
значениями интенсивности (табл. 10 - 13) и отсутствием значимых спектральных наложений,
которые выявляли путем анализа водных растворов, содержащих минимальную
концентрацию определяемого элемента и максимальные концентрации сопутствующих
элементов и основы пробы.
Таблица 10. Выбор аналитических
линий для оксида кремния (при минимальной концентрации SiO2 в
растворе - 80%)
|
SiO2 (212,4)
|
SiO2 (250,6)
|
SiO2 (251,6)
|
SiO2 (288,1)
|
|
холостая
|
855,3
|
32,78
|
7751
|
5269
|
|
SiO2(80,0%)
|
3775
|
119,5
|
33990
|
22220
|
|
Cd (1ml0,001)
|
901,1
|
35,17
|
7777
|
5767
|
|
Al2O3(5,0%)
|
892,4
|
33,71
|
7802
|
5686
|
|
Fe2O3(3,5%)
|
884,7
|
33,44
|
7879
|
5546
|
|
CaO (2,5%)
|
870,9
|
27,39
|
7847
|
5414
|
|
MgO (2,0%)
|
849,9
|
21,21
|
8122
|
5090
|
|
K2O (3,5%)
|
817,3
|
22,23
|
7572
|
4730
|
|
Na2O (3,5%)
|
807,5
|
22,41
|
7567
|
4754
|
|
Y (1ml0,001)
|
861,5
|
22,62
|
8086
|
Таблица 11. Выбор
аналитических линий для оксида алюминия (при минимальной концентрации Al2O3
в растворе - 0,1%)
|
Al2O3 (308,2)Al2O3
(309,2)Al2O3 (394,4)Al2O3
(396,1)
|
|
|
|
|
|
холостая
|
327,8
|
768,6
|
645,3
|
1543
|
|
Cd (1ml0,001)
|
330,9
|
771,0
|
679,8
|
1602
|
|
Al2O3(0,1%)
|
359,2
|
838,3
|
772,4
|
1831
|
|
Fe2O3(3,5%)
|
334,5
|
784,3
|
691,9
|
1690
|
|
CaO (2,5%)
|
330,5
|
777,9
|
683,5
|
1693
|
|
SiO2(95,0%)
|
241,0
|
631,8
|
415,8
|
1055
|
|
MgO (2,0%)
|
336,6
|
781,3
|
699,1
|
1634
|
|
K2O (3,5%)
|
318,9
|
744,8
|
658,4
|
1533
|
|
Na2O (3,5%)
|
319,1
|
745,0
|
660,2
|
1563
|
|
Y (1ml0,001)
|
317,0
|
752,0
|
692,3
|
1640
|
Таблица 12. Выбор аналитических
линий для оксида железа (при минимальной концентрации Fe2O3 в
растворе - 0,2%)
|
Fe2O3 (238,2)
|
Fe2O3 (239,5)
|
Fe2O3 (240,4)
|
Fe2O3 (259,9)
|
Fe2O3 (275,5)
|
|
холостая
|
53,13
|
46,61
|
36,84
|
91,2
|
24,40
|
|
Cd (1ml 0,001)
|
54,07
|
47,92
|
37,30
|
92,48
|
26,28
|
|
Al2O3(5,0%)
|
65,97
|
57,28
|
41,61
|
116,2
|
28,84
|
|
Fe2O3(0,2%)
|
324,3
|
294,2
|
224,1
|
568,5
|
147,7
|
|
CaO (2,5%)
|
59,56
|
55,99
|
37,69
|
100,8
|
30,14
|
|
SiO2(95,0%)
|
126,5
|
118,0
|
82,90
|
208,6
|
57,43
|
|
MgO (2,0%)
|
97,03
|
93,20
|
62,68
|
159,8
|
40,00
|
|
K2O (3,5%)
|
53,87
|
48,60
|
32,16
|
90,69
|
25,71
|
|
Na2O (3,5%)
|
52,65
|
49,17
|
31,39
|
85,30
|
25,74
|
|
Y (1ml 0,001)
|
48,40
|
48,27
|
35,91
|
81,27
|
23,48
|
Таблица 13. Выбор аналитических
линий для оксида кальция (при минимальной концентрации СаО в растворе - 0,1%)
|
СаО (315,8)
|
СаО (317,9)
|
СаО (318,1)
|
СаО (393,3)
|
|
холостая
|
2235
|
5007
|
501,1
|
602300
|
|
Cd (1ml 0,001)
|
2238
|
5079
|
500,7
|
603800
|
|
Al2O3(5,0%)
|
2248
|
5103
|
510,6
|
619400
|
|
Fe2O3(3,5%)
|
2316
|
5180
|
535,2
|
640600
|
|
CaO (0,1%)
|
4704
|
10280
|
1070
|
1264000
|
|
SiO2(95,0%)
|
1740
|
3950
|
409,9
|
465700
|
|
MgO (2,0%)
|
2308
|
4871
|
547,0
|
672100
|
|
K2O (3,5%)
|
2151
|
4547
|
514,2
|
629600
|
|
Na2O (3,5%)
|
2121
|
4521
|
500,5
|
621300
|
|
Y (1ml 0,001)
|
2365
|
6364
|
550,8
|
687800
|
В случае присутствия в образце
железа, аналитическая спектральная линия (λFe = 251,625 нм) не мешает
точному определению оксида кремния (IV) (λSi = 251,611 нм), так как она менее интенсивная, и
интерферент содержится в меньшей концентрации, чем анализируемый компонент.
По сравнению с однокомпонентными
растворами линия кадмия 251,622 нм находится приблизительно на том же уровне,
что и растворы, не содержащие кадмий. Кроме того, наблюдается отсутствие
мешающего влияния линий кадмия (λСd = 251,622 нм)
на выбранную аналитическую линию кремния (λSi = 251,611 нм). Поскольку интенсивность линии
интереферента значительно меньше интенсивности минимального содержания кремния
на выбранной длине волне.
Не смотря на то, что линия кальция (λСа = 393,633 нм) имеет большую интенсивность по сравнению
с линией (λСа = 317,933
нм), ее использование нецелесообразно, поскольку при высоких разбавлениях не
соблюдается линейность градуировочного графика (рис. 10). Вследствие этого,
определение кальция необходимо проводить при длине волны 317,933 нм.
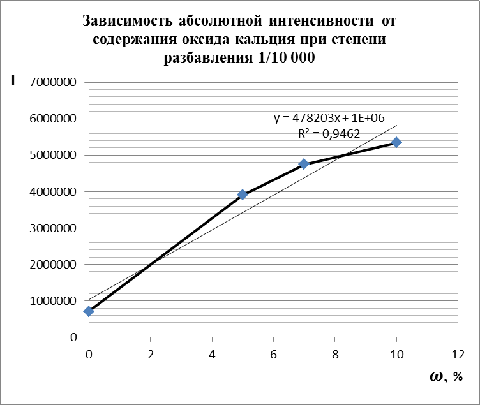
Рис. 10. Зависимость
абсолютной интенсивности при степени разбавления 1/10 000 от содержания оксид
кальция
Сопоставление результатов,
представленных в табл 9 - 12 показало отсутствие значимых спектральных
наложений аналитических линий компонентов огнеупоров, поэтому все выбранные
длины волн пригодны для определения оксидов (табл. 14).
В табл. 14 приведены уравнения
градуировочных функций для определения А12О3, SiО2, CaO, Fe2О3, Cd при оптимальных длинах волн, оценены диапазоны линейности
градуировочных функций, соответствующих содержанию оксидов в реальных образцах.
Значимость коэффициентов градуировочных функций проверена с использованием
критерия Стьюдента.
Таблица 14. Условия определения
нормируемых оксидов в огнеупорах методом АЭС ИСП
|
Аналит
|
Марка огнеупора
|
l, нм
|
Градуировочная функция y = а ∙ x + b
|
Диапазон градуировки, % мас.
|
|
SiО2
|
Siltis NS
|
251,611
|
y = 368,99x + 4480,3
|
0-100
|
|
Инсулекс (Innsulekx)
|
|
|
|
|
Forward SS-G
|
|
|
|
|
Al2O3
|
Siltis NS
|
396,152
|
y = 2016,3x + 1454,4
|
0-10
|
|
Инсулекс (Innsulekx)
|
|
|
|
|
Forward SS-G
|
|
|
|
|
Fe2O3
|
Siltis NS
|
259,940
|
y = 2252,8x + 172,93
|
0-10
|
|
Инсулекс (Innsulekx)
|
|
|
|
|
Forward SS-G
|
|
|
|
|
CaO
|
Инсулекс (Innsulekx)
|
317,933
|
y = 3672,7x + 4039,8
|
0-10
|
Выбор внутреннего
стандарта
Для улучшения воспроизводимости
результатов в АЭС широко применяют метод внутреннего стандарта. Внутренний
стандарт в АЭС представляет собой компонент, содержание которого во всех
образцах, применяемых для градуировки, а также в анализируемом образце,
одинаково. Чаще всего это компонент основы (содержание которого во всех
образцах можно приближенно считать равным 100%). При отсутствии подходящего
компонента внутренний стандарт во все образцы вводят специально. Сущность
метода внутреннего стандарта в том, что в качестве аналитического сигнала
вместо абсолютной интенсивности линии определяемого элемента используют
отношение I/I0 двух одновременно
измеряемых интенсивностей линий - определяемого элемента (I) и внутреннего стандарта (I0). Если колебания температуры (а также других условий анализа)
влияют на величины I и I0 в равной степени, то
при вычислении отношения I/I0, эти влияния взаимно
компенсируются и воспроизводимость результатов значительно улучшается.
Внутренний стандарт в методе ИСП-АЭС
предназначен также для снижения и устранения неспектральных помех. Поэтому
перед количественными измерениями на первой стадии подбирались аналитические
линии, свободные от спектральных помех, точки на спектре и способ учета фона.
Внутренний стандарт должен обладать малолинейчатым спектром, для того, чтобы
оказывать минимальное мешающее влияние на определяемый компонент. Таким
условиям удовлетворяет иттрий, кадмий и т.д.
Раствор иттрия и кадмия,
используемый в качестве внутреннего стандарта, в одинаковых количествах (1
мг/дм3) вводили до разбавления в «холостой» и градуировочные
растворы, а также в растворы проб. В качестве аналитического сигнала
используется отношение интенсивностей анализируемого элемента и внутреннего
стандарта:
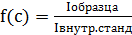 (11)
(11)
Для определения более подходящего
внутреннего стандарта из предложенных провели сравнительный анализ. Из
полученных ранее значений интенсивностей для каждого внутреннего стандарта
(таблицы 9 - 12) можно сделать заключение, несмотря на то, что иттрий по
сравнению с кадмием показывает большие значения интенсивности, возможно
наложение его спектральной линии (λY = 317,933) на
спектральную линию кальция (λСа = 317, 933 нм).
Аналитические линии кадмия не имеют наложения на аналитические линии ни одного
исследуемого элемента, поэтому в дальнейшем в качестве внутреннего стандарта
был использован кадмий.
Выбор степени
разбавления
При изучении условий определения
макрокомпонентов в огнеупорах большое значение имеет правильный выбор степени
разбавления пробы.
Перед определением нормируемых
компонентов в пробе, полученной путем микроволнового разложения, методом АЭС
ИСП необходимо обеспечить устойчивость растворов, провести разбавление до
концентраций, позволяющих наиболее надежно определять все аналиты в огнеупорах.
Были изучено влияние степени
разбавления пробы на интенсивность аналитических сигналов определяемых
элементов и их соответствие линейной области градуировочной функции. В табл. 15
представлены значения абсолютных интенсивностей определяемых компонентов в
диапазоне концентраций, отвечающих ТУ огнеупоров.
Для каждой степени разбавления были
построены градуировочные графики в координатах Iабс - концентрация определяемого элемента (Приложение 1-5).
Таблица 15. Абсолютные интенсивности
для различных степеней разбавления
|
Степень разбавления
|
Процесс разбавления
|
Абсолютная интенсивность определяемых элементов при их различном
массовом содержании
|
|
|
SiO2 I - 251,611
|
Al2O3 I - 396,152
|
Fe2O3 II - 259,940
|
CaO II - 317,933
|
|
1/1000
|
mн=0,1000 г. растворили
в Vк=100 мл
|
0%-78840 20%-85606,59 50%-214016,48 100%-428032,96
|
0%-17560 5%-109730 7%-155018 10%-219459
|
0%-2107 5%-113735 7%-159229 10%-227470
|
0%-49220 5%-209175 7%-292845 10%-418350
|
|
1/2000
|
mн=0,1000 г. растворили
в Vк=200 мл
|
0%-39420 20%-42803,295 50%-107008,24 100%-214016,48
|
0%-8780 5%-54865 7%-77509 10%-109729,5
|
0%-1053,5 5%-56867,5 7%-79614,5 10%-113735
|
0%-24610 5%-104587,5 7%-146422,5 10%-209175
|
|
1/10 000
|
mн=0,1000 г. растворили
в Vк=250 мл, затем взяли Va=25 мл и
разбавили до Vк=100 мл
|
0%-7884 20%-8560,659 50%-21401,648 100%-42803,296
|
0%-1756 5%-10973 7%-15501,8 10%-21945,9
|
0%-210,7 5%-11373,5 7%-15922,9 10%-22747
|
0%-4922 5%-20917,5 7%-29284,5 10%-41835
|
|
1/12 500
|
mн=0,1000 г. растворили
в Vк=250 мл, затем взяли Va=20 мл и разбавили
до Vк=100 мл
|
0%-6307,2 20%-6848,527 50%-17121,318 100%-34242,637
|
0%-1404,8 5%-8778,4 7%-12401,44 10%-17556,72
|
0%-168,56 5%-9098,8 7%-12738,3 10%-18197,6
|
0%-3937,6 5%-16734 7%-23427,6 10%-33468
|
|
1/25 000
|
mн=0,1000 г. растворили
в Vк=250 мл, затем взяли Va=10 мл и
разбавили до Vк=100 мл
|
0%-3153,6 20%-3424,264 50%-8560,659 100%-17121,318
|
0%-702,4 5%-4389,2 7%-6200,72 10%-8778,36
|
0%-84,28 5%-4549,4 7%-6369,16 10%-9098,8
|
0%-1968,8 5%-8367 7%-11713,8 10%-16734
|
Выбор степени разбавления пробы
осуществляли с учетом коэффициента корреляции градуировочных зависимостей всех
исследуемых огнеупоров. При степени разбавления 1/1000, 1/2000 соблюдается
линейность градуировочных функций (приложение 1 - 2) для примесных элементов и
не соблюдается для элемента основы, а при степени разбавления 1/12 500 и
1/25000 (приложение 4 - 5) соблюдается только для элемента основы.
В то же время, при разбавлении в 10
000 раз (приложение 3) линейность градуировочного графика выполняется как для
элемента основы, так и для примесных элементов.
На рис. 11 показано, что наиболее
высокий коэффициент корреляции градуировочной зависимости наблюдается при
разбавлении пробы в соотношении 1/10 000.
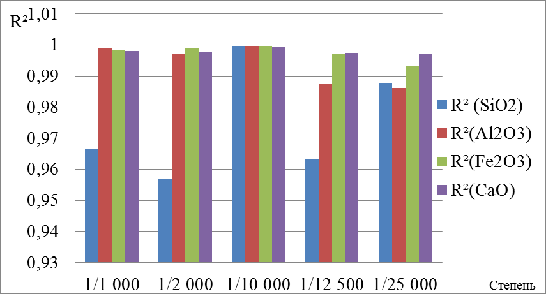
Рис. 11. Зависимость
коэффициента корреляции от степени разбавления для определяемых компонентов
Проверка правильности
определения оксидов А12О3, SiО2, CaO,
Fe2О3
в огнеупорах
Полноту микроволнового
разложения огнеупорных материалов и возможные потери определяемых элементов
устанавливали путем анализа ГСО (табл. 16). Статистический анализ с применением
простого теста Стъюдента не выявил систематической погрешности и подтвердил
правильность разработанной методики ( < ).
Воспроизводимость анализа (Sr)
не превышает величин, установленных стандартами для анализа огнеупоров.
Таблица 16. Результаты
анализа ГСО огнеупора динасового к1в (п=11; Р = 0,95)
|
Компонент
|
ГСО
|
Содержание, % масс.
|
|
tэксп
|
tтабл
|
|
|
аттестованное
|
найденное 
|
|
|
|
|
SiО2 Al2O3 Fe2O3 CaO
|
к1в
|
96,1 0,55 1,36 1,35
|
96,4 ± 0,6 0,54 ± 0,08 1,33 ± 0,08 1,35 ± 0,13
|
0,008 0,013 0,029 0,005
|
1,29 2,08 2,19 0,82
|
2,23
|
Разработанная методика анализа
огнеупорного сырья после микроволнового разложения в автоклаве методом АЭС ИСП
апробирована при определении А12О3, SiО2, CaO, Fe2О3 в производственных образцах огнеупоров. Полученные
результаты (табл. 17) сопоставлены с данными определения оксидов методами
гравиметрии, титриметрии, спектрофотометрии (ГОСТ 2642.0, ГОСТ 2642.3-97, ГОСТ
2642.7-97).
Критическое значение Fкрит равно коэффициенту Фишера для доверительной вероятности Р = 0,95 и
соответствующих степеней свободы 10 и 10 Fкрит = F (Р=0,95; f1 = 10; f2 = 10) = 2,90.
Критическое значение tкрит для этих серий равно коэффициенту Стьюдента для доверительной
вероятности Р = 0,95 и степени свободы 20 tкрит(Р=0,95; f = 20) = 2,09.
Таблица 17. Результаты
определения А12О3, SiО2, CaO, Fe2О3 в пробах огнеупорного сырья по стандартным и
разработанной методикам (n = 11; Р = 0,95) (Fкрит = 2,90; tкрит = 2,09)
|
Образец
|
Стандартный метод
|
АЭС ИСП
|
Fэксп
|
tэксп
|
|

|
|
|
|
|
|
|
SiO2
|
|
Siltis NS
|
90,9 ± 0,6
|
0,0007
|
90,7 ± 0,6
|
0,0008
|
1,07
|
0,66
|
|
Инсулекс (Innsulekx)
|
94,1 ± 0,6
|
0,0009
|
94,6 ± 0,6
|
0,0007
|
1,10
|
0,33
|
|
Forward SS-G
|
89,6 ± 0,6
|
0,0017
|
88,9 ± 0,6
|
0,0010
|
1,46
|
0,46
|
|
Al2O3
|
|
Siltis NS
|
5,5 ± 0,08
|
0,0307
|
5,6 ± 0,08
|
0,0126
|
1,78
|
0,07
|
|
Инсулекс (Innsulekx)
|
0,70 ± 0,08
|
0,0202
|
0,66 ± 0,08
|
0,0107
|
1,42
|
0,05
|
|
Forward SS-G
|
3,0 ± 0,14
|
0,0175
|
3,5 ± 0,15
|
0,0202
|
1,77
|
0,35
|
|
Fe2O3
|
|
Siltis NS
|
2,98 ± 0,07
|
0,0036
|
3,00 ± 0,08
|
0,0028
|
1,19
|
0,03
|
|
Инсулекс (Innsulekx)
|
0,0122
|
0,57 ± 0,08
|
0,0071
|
1,32
|
0,01
|
|
Forward SS-G
|
2,16 ± 0,07
|
0,0033
|
2,20 ± 0,08
|
0,0032
|
1,01
|
0,04
|
|
CaO
|
|
Инсулекс (Innsulekx)
|
1,10 ± 0,13
|
0,0068
|
1,04 ± 0,13
|
0,0064
|
1,00
|
0,18
|
С применением критерия Фишера было
установлено, что воспроизводимость результатов, полученных по разработанной
методике и с помощью альтернативных методов одинакова (Fтабл > Fэксп). Следовательно для
оценки правильности методики можно использовать модифицированный тест
Стьюдента. Рассчитанные значения tэкс < t табл. показывают, что
систематическая ошибка в результатах новой методики отсутствует.
Таким образом, разработанная
методика определения оксидов А12О3, SiО2, CaO, Fe2О3 в кремнеземистых огнеупорах методом АЭС ИСП с
микроволновой пробоподготовкой может быть рекомендована для анализа и других
огнеупоров этого класса. Кроме того, применение разработанной методики
позволяет существенно сократить время пробоподготовки (табл. 18), повысить
экономичность анализа за счет уменьшения количества концентрированных кислот и
улучшить экологических климат лабораторий.
Таблица 18. Сравнительная
характеристика разработанных ГОСТированных методик
|
Марка огнеупора
|
Определяемый компонент
|
Сокращение
|
|
|
продолжительности деструкции
|
общего времени анализа
|
расхода концентрированных кислот
|
|
Siltis NS
|
Si4+
|
в 8 раз
|
в 4 раза
|
в 4 раза
|
|
Al3+
|
в 10 раз
|
в 3 раза
|
в 12 раз
|
|
Fe3+
|
в 8 раз
|
в 2,5 раза
|
в 4 раза
|
|
Инсулекс (Innsulekx)
|
Si4+
|
в 8 раз
|
в 4 раза
|
в 4 раза
|
|
Al3+
|
в 10 раз
|
в 3 раза
|
в 12 раз
|
|
Fe3+
|
в 8 раз
|
в 2,5 раза
|
в 4 раза
|
|
Ca2+
|
в 14 раз
|
в 4 раза
|
в 4 раза
|
|
Forward SS-G
|
Si4+
|
в 8 раз
|
в 4 раза
|
в 4 раза
|
|
Al3+
|
в 10 раз
|
в 3 раза
|
в 12 раз
|
|
Fe3+
|
в 8 раз
|
в 2,5 раза
|
в 4 раза
|
Выводы
1. Исследованы условия
автоклавного вскрытия проб огнеупорных материалов металлургического назначения
в условиях микроволнового нагрева. Оптимизированы температурно-временные
параметры МВ-вскрытия проб огнеупоров. Экспериментально установлены составы и
объёмы кислотных смесей для эффективной деструкции огнеупоров.
2. Оптимизированы условия
определения методом АЭС ИСП SiO2, СаО, Al2О3 и Fe2О3 в
кремнеземистых огнеупорах после их МВ-деструкции. Изучено влияние степени
разбавления пробы при определении макрокомпонентов в огнеупорах, выбраны
внутренние стандарты, исключающие спектральные помехи.
. Разработана методика
анализа кремнеземистых огнеупоров, включающая МВ-пробоподготовку в автоклаве и
определение SiO2, СаО, Al2О3 и Fe2О3 методом
АЭС ИСП. Правильность разработанной методики подтверждена путём анализа ГСО,
сопоставлением результатов анализа с данными, полученными альтернативными
методами. Разработанная методика АЭС ИСП анализа кремнезёмистых огнеупорных
материалов с микроволновой пробоподготовкой характеризуется высокой
прецизионностью, экономичностью, безопасностью и может быть рекомендована для
лабораторий металлургических предприятий.
. Разработанная методика
позволяет существенно сократить продолжительность анализа (с 8 ч до 1,5 ч),
улучшить экономические и экологические показатели (сокращается объем
используемых кислот, разложение проводится в закрытом сосуде).
Библиографический список
1. Воронов Г.В., Старцев В.А. Огнеупорные материалы и изделия
в промышленных печах и объектах вспомогательного назначения // учебное пособие.
Екатеринбург: УГТУ-УПИ, 2006. 303 с.
. Кубракова И.В. Микроволновое излучение в аналитической
химии: возможности и перспективы использования / И.В. Кубракова // Успехи
химии. Т.71, №4. 2002. С. 1-14.
. ГОСТ 24717-2004. Огнеупоры и огнеупорное сырье.
Маркировка, упаковка, транспортировка и хранение.
. Современные огнеупоры: ресурсосбережение и применение в
металлургических технологиях: Сб. научн. тр. / под ред. проф., д.т.н. А.Н.
Смирнова. - Донецк: ДонНТУ. 2013. 200 с.
. Кащеев И.Д. Свойства и применение огнеупоров: Справочное
издание. - М.: Теплотехник. 2004. 352 с.
. Пупышев A.A., Луцак А.К. Современное состояние методов
атомного спектрального анализа // Аналитика и контроль. 2000. Т. 4, №2. С. 141
- 146.
. Беккер Ю. Спектроскопия / пер. с нем. J1.H. Казанцева;
под ред. A.A. Пупышева, М.В. Поляковой. - М.: Техносфера, 2009. 528 с.
. Некоторые аспекты подготовки проб к атомно-эмиссионному
спектральному и масс-спектрометрическому определению микроэлементов / А.И.
Сапрыкин, И.Р. Шелпакова, Т.А. Чанышева и др. // Журнал аналитической химии.
2003. Т.58, №3. С. 273-280.
. Сычь Л.Г., Сенина Е.А., Галыгина О.С. Разложение
материалов металлургического производства в микроволновой системе MARS-5 //
Заводская лаборатория. Диагностика материалов. 2007. Т.73, №02. С. 5-6
. Корда, М.Г. Демидова и др. // Журнал аналитической химии.
2009. Т.64, №6. С. 611-619.
11. J.M. Chalmers, ed., Spectroscopy in Process Analysis. Boca
Raton, FL: CRC Press. 2000.
. Пупышев А.А. Аналитика и контроль. // А.А. Пупышев / Журнал
аналитической химии. Т. 12, №1-2. 2008. 1-63 с.
. Аналитическая химия. Проблемы и подходы: В 2 т: Пер. с
англ./ Под ред. Р. Кельнера, Ж.-М. Мерме, М. Отто, М. Видмера. - М.: «Мир»: ООО
«Издательство АСТ». (Лучший Заребежный учебник). 2004. Т.2. 728 с.
. Смагунова А.Н. Контроль корректирующих коэффициентов при
рентгеноспектральном анализе способом калибровки / А.Н. Смагунова, С.Д.
Паньков, Н.Ф. Лосев, Р.И. Плотников // Журнал аналитической химии. 1977. Т. 32,
№1. С. 15-20.
. Попова В.И. Оценка возможности использования различных
устройств при подготовке порошковых проб к рентгеноспектральному анализу / В.И.
Попова, С.Ф. Розова, А.Н. Смагунова // Аппаратура и методы рентгеновского
анализа. Вып. 20, Л.: «Машиностроение».1981. С. 17-22.
. Мосичев В.И. Изготовление градуировочных образцов состава
сплавов методом порошковой металлургии / В.И. Мосичев, Н.В. Першин, А.А.
Хомутников, С.А. Мишина, В.М. Шушканова // Аппаратура и методы рентгеновского
анализа. Вып. 25. Л.: «Машиностроение», 1981. С. 21-25.
. Симаков В.А. Сплавление порошковых проб с получением
совершенных излучателей для рентгенофлуоресцентного анализа / В.А. Симаков,
Н.С. Вахонин, В.Е. Исаев // Заводская лаборатория. 1996. Т. 62, №8. С. 19-21.
. ОСТ 48-165-80. Метод рентгеноспектрального анализа руд и
продуктов обогащения на обогатительных фабриках Минцветмета СССР. Общие
требования к разработке методик и оценке их метрологических характеристик. М:
Минцветмет СССР, 1980. 28 с.
. Карпов Ю.А., Савостин А.П. Методы пробоотбора и
пробоподготовки. - М.: БИНОМ. Лаборатория знаний. 2003. 243 с.
. Лосев Н.Ф. Количественный рентгеноспектральный
флуоресцентный анализ. М.: «Наука». 1969. 336 с.
21. D.F. Swinehart, «The Beer-Lambert Law», J. Chem. Ed., 39. 1962. 333p.
. Смагунова А.Н. Примеры применения математической теории
эксперимента в рентгенофлуоресцентном анализе / А.Н. Смагунова, В.А. Козлов
Иркутск: изд. ИГУ. 1990. 230 с.
23. Thompson М., Walsh N. Handbook of inductively coupled plasma spectrometry /
N.Y.:Blackie. 1989. 316p.
24. Бок P. Методы разложения в аналитической химии / М.: Химия.
432 с.
25. Брицке М.Э., Сукач Ю.С., Филимонов Л.Н. Индукционный ВЧ
разряд и его применение в эмиссионном спектральном анализе // Журнал прикладной
спектроскопии. 1976. Т.25, №1. С. 5-11.
. Высокочастотный индуктивно-связанный плазменный разряд в
эмиссионном спектральном анализе / Под ред. Зильберштейна Х.И.П.: Наука. 1987.
230 с.
. Чудинов Э.Г. Атомно-эмиссионный анализ с индукционной
плазмой / Итоги науки и техники ВИНИТИ. 1990. Т.2. С. 3-251.
. Пупышев А.А., Данилова Д.А. Использование
атомно-эмиссионной спектрометрии с индуктивно связанной плазмой для анализа
материалов и продуктов черной металлургии // Аналитика и контроль. Т.11, №2-3.
2007. 131-181c.
. Томпсон М., Уолш Дж. Руководство по спектрометрическому
анализу с индуктивно-связанной плазмой / М.: Недра. 1988.
. Атомно-спектроскопические методы определения следов
элементов / Под ред. Вайнфорднера Дж.М.: Мир. 1979.
. Харламов И.П. Еремина Г.В. Атомно-абсорбционный анализ в
черной металлургии.-М. Металлургия, 1982. 168 с.
. Томпсон M., Уолш Д.Н. Руководство по спектрометрическому
анализу с индуктивно-связанной плазмой / Пер. с англ. Н.И. Гулько; под ред.
В.Б. Белянина.-М.: Недра. 1988. 288 с.
. Пупышев А.А., Данилова Д.А. Атомно-эмиссионный
спектральный анализ с индуктивно связанной плазмой и тлеющим разрядом по
Гримму. - Екатеринбург: ГОУ ВПО УГТУ-УПИ. 2002. 202 с.
. Карпов Ю.А., Орлова В.А. Современные методы автоклавной
пробоподготовки в химическом анализе веществ и материалов // Заводская
лаборатория. Диагностика материалов. 2007. Т. 73, №1. С. 4-11.
. Основы аналитической химии. В 2 т. Т.2: учеб. для студ.
Учреждений высш. проф. образования/[Н.В. Алов и др.]; под ред. Ю.А. Золотова. -
4-е изд., перераб. и доп. - М.: Издательский центр «Академия». 2010. 416 с.
. Кузьмин Н.М., Кубракова И.В. Микроволновая
пробоподготовка // Журнал аналитической химии. 1996. Т.51, №1. С. 44-48.
. Орлова В.А., Шерстнякова С.А., Карпов Ю.А. Современные
возможности автоклавной химической подготовки аналитических проб // Заводская
лаборатория. Диагностика материалов. 1993. Т.59, №9. С. 1-7.
38. Васильев В.П. Аналитическая химия. В 2 частях. Часть 2. -
М.: Высш. шк. 1989. 320 с.