Міністерство освіти і науки,
молоді та спорту України
Сумський
державний педагогічний університет ім. А.С. Макаренка
Кафедра
хімії та методики навчання хімії
ІНДЗ
з дисципліни
Хімія
координаційних сполук
на
тему «Реакції комплексоутворення»
Підготувала:
Сагайдак А.В.
Суми
- 2011
План
Вступ
1. Зовнішні ознаки реакцій
комплексоутворення та найпростіші методи виявлення координаційної сфери
2. Методи синтезу
координаційних сполук
3. Взаємний вплив
координованих групп
Висновки
Список використаної літератури
Вступ
Не становить таємниці той факт, що
комплексні сполуки та реакції комплексоутворення мають широке застосування в
аналітичній хімії, в методах якісного і кількісного аналізу, перш за все
завдяки властивостями комплексних сполук: стійкість, інтенсивність забарвлення,
мала розчинність.
Як свідчить назва, комплексна
сполука (комплекс) - це складна частинка, що містить декілька складових частин
(іонів, молекул), здатних до самостійного існування. Комплексні сполуки
виділені в окремий клас речовин за певними ознаками: багатокомпонентність
складу; здатність складових частин до самостійного існування; здатність
дисоціювати на складові частини за гетеролітичним механізмом; наявність стійкої
просторової структури, утвореної за донорно-акцепторним механізмом.
Центральний атом
(комплексоутворювач) координує частинки тобто безпосередньо зв’язує і утворює
внутрішню сферу комплексу. Йони, безпосередньо не зв’язані з центральним
атомом, утворюють зовнішню сферу.
Ліганди є донорними частинками, центральний
атом - акцептором. Центральний атом характеризується координаційним числом - це
число зв’язків, утворених з центральним атомом у комплексній сполуці. Якщо
ліганд утворює зв’язок тільки з одним донорним атомом, координаційне число
дорівнює числу приєднаних лігандів.
Ліганди характеризуються
дентатністю. Дентатність - це число донорних атомів ліганду, які утворюють
координаційні зв’язки з центральним атомом. Ліганди, що утворюють тільки один
зв’язок ( Сl, I, СN, NH3, ОН, Н2O та інші), називаються
монодентатними, а ті, що утворюють декілька зв’язків (як правило молекули
органічних сполук), -полідентатними.
Будь-який ліганд повинен мати хоча б
один донорний атом. Тому ліганди - це аніони або полярні молекули.
Найважливішими неорганічними лігандами є NH3, ОН, Н2O,галогенід-іони,
СN, SCN, NO3, NO2, SO4, СО3.
В органічних лігандах донорні атоми
входять до складу функціональних груп. Як правило, це функціональні групи, що
містять атом О, N, S.
У сучасних умовах актуальними
постають питання утворення комплексних сполук, розробки технології урегулювання
та методів утворення комплексу.
.
Зовнішні
ознаки реакцій комплексоутворення та найпростіші методи виявлення
координаційної сфери
Проходження реакції
комплексоутворення в розчині можна на якісному рівні зафіксувати за появою в
розчині властивостей, притаманних утвореному комплексу. Так, утворення
комплексів може призводити до зміни кольору розчину, наприклад, додавання
роданіду до слабко-жовтого розчину солей F3+ приводить до появи
яскраво-червоного забарвлення:
Fe3+
+ NCS- = [Fe(NCS)]2+
а додавання амоніаку до
світло-блакитного розчину солі Cu2+ - до утворення темно-синього
комплексу:
Cu2+ + 4NH3 =
[Cu(NH3)4]2+
Нерідко результатом
комплексоутворення є розчинення або утворення осодів, наприклад хлорид срібла
розчиняється при додаванні NH3 , а солі Cu2+ при
додаванні ацетил ацетону утворюють осад нерозчинної КС.
AgCl↓ + 2 NH3 =
[Ag(NH3)2]Cl(CH3CO2)2 +
2Hacac = Cu(acac)2 ↓ + 2 CH3CO2H
Комплексоутворення може призводити
до зміни кислотності розчину. Наприклад, при додаванні етиленгліколю або
гліцерину до розчину тетраборату натрію (рН = 9,18) середовище стає близьким до
нейтрального, оскільки комплексна кислота, що утворюється, є сильнішою
порівняно з боратною кислотою.
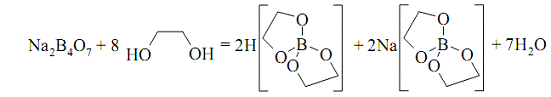
Рис. 1
У цілому зовнішні ознаки, за якими
можна говорити про проходження реакцій комплексоутворення, такі ж самі, як і
для будь-яких хімічних реакцій. Однак, слід відзначити, що теплові ефекти
реакцій комплексоутворення невеликі, тому звичайно за зміною температури
реакційної суміші неможливо встановити, чи відбувається утворення комплексів.
Також ознакою комплексоутворення є
зникнення чи послаблення властивостей, зумовлених наявністю незв'язаних іонів
металу або ліганду. Якщо комплекс досить стійкий, центральний атом і ліганди не
вступають у якісні хімічні реакції, що використовуються при визначенні іонів,
які входять до складу комплексу. Наприклад, комплекс [FeF]3- не
забарвлює розчин роданіду, а [TiF]2- не утворює оранжевого комплексу
при обробці пероксидом водню. Тому якісні реакції можна застосувати і для
встановлення, знаходиться ліганд у внутрішній чи в зовнішній сфері комплексу.
Так, сполука [CoCl(NH3)5 ](SO4 ) миттєво
утворює осад при додаванні Ba(NO3)2, а при додаванні AgNO3
осад утворюється досить повільно. Обернену картину маємо для сполуки [Co(SO4)(NH3)5]Cl.
Для встановлення молярної маси КС
можуть бути застосовані методи кріо- та ебуліоскопії. Вимірювання
електропровідності розчину КС дає змогу встановити, на скільки іонів дисоціює
досліджувана сполука. Так, значення електропровідності 10-3 M
розчинів сполук, що дисоціюють на певну кількість іонів, наведено в табл. 1.
Таблиця 1

2. Методи синтезу координаційних
сполук
Перш за все, реакції, за якими
одержують координаційні сполуки, можна розділити з погляду кінетики на
термодинамічно-контрольовані (або рівноважні) й кінетично-контрольовані (або
нерівноважні). Відмінність між цими реакціями можна проілюструвати рис. 1.

Рис.1. Енергетичні профілі
термодинамічно (1) й кінетично (2) контрольованих реакцій комплексоутворення
Якщо реакція є рівноважною, то
утворюється найстійкіший продукт реакції з можливих, його будова визначається
лише складом реакційної суміші і ніяк не пов'язана з будовою вихідних сполук.
Так, при додаванні надлишку флуорид-іонів до солей алюмінію ( Al(NO3)3
9H2O , AlCl3 6H2O ) і більшості його
комплексів (наприклад, K3 [Al(C2O4)3],
[Al3 (-μ-CH3CO2)6
-μ3
-O]Cl, комплекс з алізарином) утворюються аніони AlF63-,
оскільки флуоридний комплекс алюмінію є стійкішим за всі вищенаведені сполуки.
Якщо ж реакція нерівноважна,
утворюється такий продукт, швидкість утворення якого максимальна. Будова цього
продукту пов'язана зі структурою вихідних реагентів, тому іноді методи синтезу,
що базуються на кінетично-контрольованих реакціях, називають
"генеалогічними". Наприклад, при обробці сполуки K2[PtCl4]
амоніаком утворюється цис-ізомер, а при дії хлороводню на [Pt(NH3 )4
]Cl2- транс-ізомер сполуки [PtCl2 (NH3 )2
].
Слід відзначити, що проходження
кінетично-контрольованих реакцій можливе лише при утворенні інертних
комплексів, оскільки для лабільних комплексів значення енергії активації Еа
несуттєво мале, отже утворюється продукт, що є термодинамічно найбільш
вигідним. Тому більшість методів синтезу КС базуються на рівноважних реакціях.
Однак, слід відзначити, що рівноважні реакції часто є такими лише до певної межі.
Наприклад, досліджуючи систему Fe3- − C2O42-
, слід пам'ятати, що найстійкішими з термодинамічного погляду продуктами є СО
та CO2 , оскільки щавлева кислота - метастабільна сполука й має
самочинно розкладатися за реакцією:
H2C2O4
→ H2O + CO + CO2 , ∆G2980 =
-76,6 кДж∙моль-1.
Але за нормальних умов процес Fe3++
3C2 O4 ↔ [Fe(C2O4) ]3-
є рівноважним.
Найпростішими та найбільш
дослідженими є реакції одержання комплексних сполук, що полягають у заміщенні
лігандом молекули розчинника або іншого ліганду у внутрішній координаційній
сфері. Загалом, без урахування зарядів, таку реакцію можна записати як:
[M(Solv)n ] + mL = [MLm]
+ nSolv ;
Найчастіше реакції заміщення
проводять у водних розчинах шляхом додавання ліганду до відповідної солі,
наприклад:
CuSO4 + 4NH3 =
[Cu(NH3)4 ]SO4 ;
[Cr(H2 O)6]Cl3
+ 6KNCS = K3[Cr(NCS)6] + 3KCl + 6H2O;
Для завершення синтезу роданідного
комплексу хрому реакційну суміш кип'ятять зі зворотним холодильником, оскільки
аквакомплекси хрому(ІІІ) досить інертні.
Нерідко процеси заміщення лігандів
супроводжуються зміною ступеня окиснення центрального атома:
4CoCl2 + 20NH3
+ 4NH4 Cl + O2 = 4[Co(NH3)6 ]Cl3
+ 2H2O ;
або хімічними перетвореннями,
пов'язаними з самими лігандами:
K2 PtCl4 + C2H5OH
= K[PtCl3(C2H4)] + KCl + H2O ;4Fe(CN)6
+ 4HNO3 = K2 [Fe(CN)5 (NO)] + NH4NO3
+ 2KNO3 + CO2;
Якщо сполука має досить велике
значення константи утворення, складнішу проблему становить не її синтез, а
виділення у чистому вигляді. У наведених вище прикладах [Cu(NH3)4]SO4
"висолюють" із водного розчину етиловим спиртом, комплекс K3[Cr(NCS)6]
екстрагують тим самим розчинником із реакційної суміші після випарювання води,
а комплекс феруму кристалізують у вигляді Na2[Fe(CN)5(NO)],
додаючи до реакційної суміші Na2CO3 і спирт.
Умовою кристалізації сполуки є
близькість розмірів катіона й аніона. Оскільки комплексні іони мають досить
великі розміри, як протиіони нерідко використовують великі за розміром катіони
( NH3+, Cs+, Tl+, N(CH3)4+,
PPh4+ ) та аніони ( ClO4- , BF4-,
BPh4- , PF6- , [Cr(NCS)4
(NH3)2 ]- ).
Для синтезу сполук, що є
недостатньостійкими, застосовують підхід, який отримав назву "метод
примусового зсуву рівноваги".
Так, хлорид-іон координується до Pt+2
краще, ніж вода. Однак, додавання водного розчину нітрату срібла до cis-[PtCl2(NH3)2]
дає змогу отримати [PtCl(NH3)2 (H2O)](NO3)
, оскільки хлорид вилучається з реакції у вигляді AgCl. Ацетонітрил
координується до купруму(ІІ) слабкіше за воду. Однак, кип'ятіння [Cu(H2O)](BF4)2
у суміші ацетонітрилу з бензолом призводить до відгонки потрійного азеотропну
CH3CN:C6H6:H2O, який при збиранні
його у водовідділювач (рис. 2), розшаровується на воду та суміш ацетонітрилу з
бензолом. Тривале кип'ятіння з водовідділювачем призводить до вилучення всієї
води із системи й утворення цільової сполуки [Cu(CH3CN)6](BF4)2
.
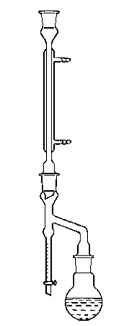
Рис. 2. Пристрій для кип'ятіння з
водовідділювачем.
Близьким до методу примусового зсуву
рівноваги є екстракційний синтез КС. Так, у водному розчині кобальт(ІІ)
взаємодіє з роданідіоном за схемою:
[Co(H2O)6]2++
NCS-= [Co(NCS)(H2O)5]+ + H2O
Однак, якщо до системи додати
ізоаміловий спирт, в органічній фазі утворюється сполука [Co(NCS) ]2-
синього кольору. Причиною цього є погана розчинність води в ізоаміловому спирті
й екстракція KNCS або NH4NCS в органічну фазу. Обидва ці чинники
зсувають рівновагу реакції в бік утворення цільового продукту. Аналогічно було
отримано ряд роданідних комплексів Fe3+ , включаючи [Fe(NCS) ]3-
, хоча у водних розчинах стійким є лише [Fe(NCS)(H2O)5]2+
.
Недоліком синтезу КС із водних
розчинів є досить сильна координація води до іонів металів і можливість
проходження реакції гідролізу. Для того, щоб позбутися дії цих чинників,
синтези КС проводять у неводних розчинниках. Оскільки більшість КС - полярні
сполуки, для їхнього синтезу використовують полярні розчинники: спирти й
різноманітні апротонні розчинники (ацетон, диметилформамід (ДМФА),
диметилсульфоксид (ДМСО), ацетонітрил, нітрометан, ефір, 1,4-діоксан,
тетрагідрофуран (ТГФ) тощо). Апротонними ці розчинники називаються через неможливість
їхньої дисоціації з утворенням протону, а отже і неможливість сольволізу іонів
металів. Усі вищенаведені розчинники координуються до більшості іонів металів
слабше, ніж вода, тому їхнє використання дає змогу одержати різноманітні КС, що
є нестійкими у водних розчинах. Так, у ацетонітрильному розчині легко одержати
комплекс алюмінію K3[Al(NCS)6] , у ефірному - [Cr(en)3]Cl3
, спроба ж одержати відповідні комплекси у воді призводить до утворення
гідрооксидів металів.
Сполуки металів у аномально-низьких
ступенях окиснення та метал-органічні сполуки звичайно синтезують у неводних
розчинниках за відсутності кисню повітря, оскільки більшість подібних сполук
або проміжних речовин для їхнього одержання легко розкладаються водою й
окиснюються.
2C5H6 + 2Na =
C5H5Na + H2 ↑ ;
C5H5Na + MnCl2
= Mn(C5H5)2 + 2NaCl ;
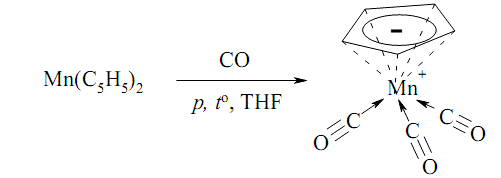
Усі реакції проводять у безводному
ТГФ у атмосфері сухого азоту або аргону.
Синтез "комплексу
Уілкінсона" RhCl(PPh3)3, що є каталізатором
гомогенного гідрування, проводять в етиловому спирті, а синтез реактиву
Гриньяра C2 H5 MgCl , що застосовується в органічному
синтезі - в ефірі або в ТГФ.
RhCl3 + 4PPh3
+ H2O = RhCl(PPh3)3 + 2HCl + O=PPh3;2H5Cl
+ Mg = C2H5 MgCl .
Крім включення у внутрішню
координаційну сферу молекул води або інших розчинників, нерідко має місце координація
протиіонів, таких як галогеніди, сульфат тощо. Позбутися цього можна,
синтезуючи комплексні сполуки з солей металів з аніонами, що слабко
координуються. Для більшості металів це нітрати та солі з аніонами великого
розміру ( ClO- , BF- , BPh - , PF6-
, [Cr(NCS)4 (NH3)2 ]), що вже згадувалися
вище.
Ще один спосіб виключення
координації аніонів - застосування як вихідних речовин для синтезу
координаційних сполук не солей, а самих металів або їхніх оксидів. Цей підхід
отримав назву прямий синтез, його можна проводити в розчині, у газовій і навіть
твердій фазі. Наприклад, карбоніли металів одержують шляхом взаємодії порошків
металів із газоподібним СО при нагріванні під тиском:
Fe + 5CO = Fe(CO)5
Взаємодія металу або оксиду з
розчином ліганду відбувається на поверхні частинок за рахунок їхнього
поступового розчинення. Тому методом прямого синтезу в рідкій фазі було
одержано ряд сполук, які неможливо або дуже важко отримати при безпосередній
взаємодії солей металів із лігандами. Це, зокрема, сполуки металів у низьких
ступенях окиснення,

а також багатоядерні й різнометальні
комплекси, синтез яких активно досліджується на кафедрі неорганічної хімії під
керівництвом проф. В.М. Кокозея. Наприклад, при взаємодії металічної міді,
оксиду цинку, хлориду або броміду амонію та N,N-диметилетаноламіну ( HMe2
Ea ) у метанольному розчині з виходом близько 70 % утворюються комплекси [Cu2
ZnX3(NH3)(Me2 Ea)3 ] .
2Cu + ZnO + 3NH4X + 3HMe2Ea
+ O2 =
= [Cu2 ZnX3
(NH3 )(Me2Ea)3 ] + 2NH3 + 3H2O
До недоліків прямого синтезу слід
віднести неконтрольованість процесів окиснення металу за рахунок взаємодії з
лігандом або киснем повітря та, часто, непередбачуваність утворення тих чи
інших сполук.
Широкого використання для одержання
комплексних сполук знаходять процеси електросинтезу. Анодне розчинення металу в
присутності ліганду є близьким до розглянутого вище прямого синтезу, однак при
цьому процес окиснення контролюється значеннями прикладеного потенціалу
окиснення та густини струму, тобто є більш керованим. Якщо розчинення
відбувається в неводному середовищі (звичайно використовують CH3CN
), до розчинника додають (CH3)4N+ ClO4-
або інші солі тетраалкіламонію для збільшення його електропровідності.
За допомогою електросинтезу звичайно
одержують амінні комплекси, сольватокомплекси, алкоксиди, ацетилацетонати,
безводні галогеніди, комплекси з аніонами слабких кислот і металоорганічні
сполуки:
Cu + 2NH3 - e-
= [Cu(NH3 )2]+
Ta + 5C2H5OH −
5e- = Ta(OC2 H5 )5 + 5H+
Cu + 2Hacac − 2e- =
Cu(acac)2 + 2H+
Pb + 4C2H5MgCl
− 4e-= Pb(C2 H5)4 + 4Mg++
4Cl+
Реакції електрохімічного відновлення
відбуваються на катоді. Наприклад, катодне відновлення RhCl(PPh3)3
у ацетонітрильнотолуольному розчині у присутності надлишку PPh3 призводить
до утворення Rh(PPh3)4 . При відновленні на металічному
катоді ліганда-окисника можуть проходити реакції розчинення катоду, наприклад:
Hg + 2(CH3)2C=O
+ 6H++ 6e-= Hg(CH(CH3)2)2
+ 2H2 O+ 4H2C=CH-CN + 4H++ 4e-=
Sn(CH2CH2CN)4
Типовим для координаційної хімії є
темплатний синтез - процес, що включає в себе перетворення лігандів, зумовлені
їх певною, визначеною іоном металу геометричною орієнтацією. Шляхом темплатного
синтезу одержано різноманітні макроциклічні сполуки, імовірність одержання яких
за відсутності іонів металу дуже мала:
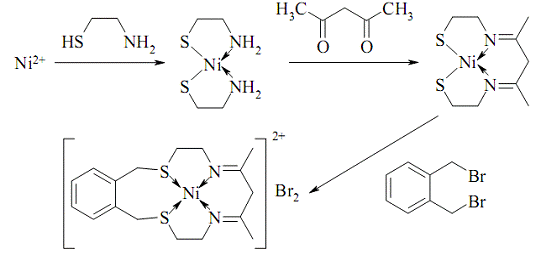
Рис. 3
Підхід темплатного синтезу
використовують і для одержання супрамолекулярних і клатрохелатних сполук,
наприклад, саркофагінату кобальту:
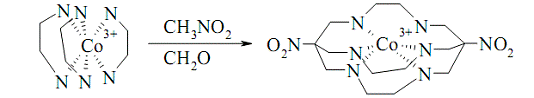
Рис. 4
Одержання фталоціаніну купруму
ілюструє комбінацію підходів прямого та темплатного синтезу:
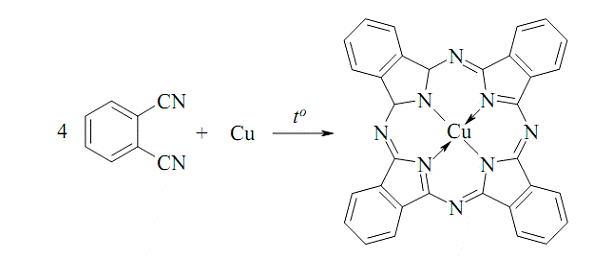
Рис. 5
Ця сполука має яскраво-синій колір і
використовується як барвник. Вона витримує нагрівання до 500 ºC
на повітрі, не розчинна у воді й більшості органічних розчинників, стійка до
дії лугів, концентрованих кислот (крім нітратної) та сонячного світла.
3. Взаємний вплив координованих груп
Найбільш вивченою формою взаємного
впливу лігандів є ефект транс-впливу. Він полягає в тому, що легкість заміщення
ліганду у внутрішній координаційній сфері інертних комплексів (плоско-квадратні
комплекси Ni2+, Pd2+,Pt 2+, октаедричні
комплекси Co3+, Rh3+, Ir3+, Pt4+)
пов'язана із природою ліганду у транс-положенні до ліганду, що заміщується. За
силою транс-впливу (тобто лабілізуючої дії на ліганд у транс-положенні) ліганди
можуть бути розташовані в такий ряд:

Розглянемо явище транс-впливу на
прикладі взаємодії K2 PtCl4 з амоніаком. На першій стадії
реакції амоніак заміщує один хлоридний ліганд, і при цьому утворюється сполука
K[PtCl3(NH3)]. Транс-вплив хлориду значно більший
порівняно з амоніаком, тому на другій стадії відбувається заміщення хлоридного
ліганду, що знаходиться у транс-положенні до іншого хлориду.

Рис. 6
Утворена сполука cis-[PtCl2(NH3)2]
взаємодіє з амоніаком лише в достатньо жорстких умовах (висока концентрація
амоніаку, нагрівання), тому легко може бути виділена з реакційної суміші.
Транс-ізомер [PtCl2 (NH3)2 ] синтезують за
реакцією [Pt(NH3)4]Cl2 із хлороводнем. На
першій стадії утворюється сполука [PtCl(NH3)3]Cl .
Оскільки транс-вплив хлориду сильніший за транс-вплив амоніаку, другий
хлоридний ліганд координується в транс-положення до першого.
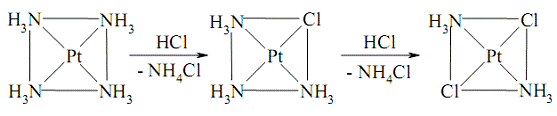
Рис. 7
Явище транс-впливу можна пояснити
двома основними чинниками. По-перше, це зміщення електронної густини в ланцюжку
L1 → M → L2 , аналогічне до відомого в
органічній хімії індуктивного ефекту. Тобто, чим сильніше координується до
металу ліганд L1 , тим більше послаблюється зв'язок між металом і
лігандом L2. За І.І. Черняєвим, що у 1926 р. сформулював правила
транс-впливу, це твердження звучить так: "найбільший транс-вплив має
ліганд, що утворює з центральним атомом найміцніший зв'язок". Еквівалентне
пояснення транс-впливу із залученням поляризаційних уявлень було запропоновано
Б.В. Некрасовим. Послаблення зв'язку між металом і лігандом L2
призводить до симбатного збільшення швидкості заміщення цього ліганду.
Друга причина транс-впливу,
досліджена Дж. Чаттом і Л. Оргелом, має суто кінетичну природу. Якщо ліганд L1
є π-акцептором , він
відтягує на свою π-орбіталь
електронну густину з орбіталі dyz центрального атома. Це полегшує взаємодію
між зв'язком M-L2 і лігандом L, що заміщує L2 (рис.3).

Рис.8. Пояснення явища транс-впливу
за Дж. Чаттом і Л.Оргелом
Безумовно, на швидкість реакцій
заміщення впливають не тільки транс, але і цис-розташовані ліганди. Так,
швидкість заміщення хлорид-іона, що знаходиться в цис-положенні до амоніаку у
сполуці [PtCl(NH3)3]Cl приблизно втричі вища, ніж у K2
PtCl4 , де він знаходиться в цис-положенні до хлориду. Отже, NH3 ,
що перебуває у цис-положенні до Cl-, збільшує рухливість останнього.
.А. Грінберг і Ю.М. Кукушкін назвали це явище цис-ефектом і встановили, що ряд
цис-впливу в загальних рисах є оберненим до ряду транс-впливу.
Висновки
Знання про сутність, природу
виникнення комплексних сполук важливе в практичній роботі педагога, хіміка.
Для з'ясування суті комплексних
сполук важливо виділити його основні ознаки, сформулювати необхідні й достатні
умови його виникнення. Доцільно будь-яку комплексну сполуку розглядати в
статиці ( як систему взаємозалежних структурних елементів) і в динаміці ( як
процес).
Комплексні, або координаційні
сполуки - це складні сполуки, що утворюються внаслідок взаємодії між собою
більш простих неорганічних речовин: солей, кислот, основ.
Для опису хімічного зв'язку в
комплексних сполуках використовують З підходи: Теорія валентного зв'язку (
ТВЗ). Метод молекулярних орбіталей (ММО). Теорія кристалічного поля ( ТКП). Всі
ці підходи доповнюють один одного і використовуються в залежності від того, яка
комплексна сполука розглядається.
В 30 роках XX століття підходи до
опису хімічного зв'язку в комплексних сполуках розроблені Полінгом, згідно чого
хімічний зв'язок в комплексних сполуках з позиції теорії валентного зв'язку
включає такі положення:
Центральний йон зв'язаний з
лігандами донорно-акцепторним ковалентним зв'язком, де функцію донора виконують
ліганди.
Перед утворенням хімічного зв'язку
атомна орбіталь центрального йона гібридизується. Тип гібридизації залежить від
числа і природи, а також електронної структури лігандів. Тип гібридизації
визначає геометрію утворених структур.
В молекулі комплексної сполуки може
бути і додаткове зв'язування центрального йона за рахунок перекривання вільних
орбіталей ліганда і центрального йона, де виникають пі-звязки поряд з уже
сформованими сігма-звязками.
Якщо у комплексній сполуці є
неспарений електрон, то вона буде парамагнітною. Йони зовнішньої сфери зв'язані
з внутрішньою сферою зв'язком, який близький до йонного. Отже, з точки зору
ТВЗ: у внутрішній сфері зв'язок ковалентний полярний, а у зовнішній сфері -
близький до йонного.
Недоліки ТВЗ в комплексних сполуках:
Він не розглядає багато центральних
сполук, тому описує їх межі.
Не враховує збудженого стану атома,
тому не пояснює і не передбачає оптичних властивостей комплексних сполук.
Не описує і не оцінює енергії
зв'язку, тому не може пояснити чому комплекси плоскотрикутникової та плоско
квадратної конфігурації що є менш стабільними, енергетично існують і досить
стійкі і не переходять в більш стійкі (тетраедричні).
У водних розчинах комплексні сполуки
( крім тих, що складаються з однієї внутрішньої сфери) дисоціюють практично
повністю, тобто є сильними електролітами.
Внаслідок дисоціації у розчині
з'являються комплексні йони ( катіони або аніони). Значно меншою мірою
розпадаються комплексні йони.
Як рівноважний процес розпад
комплексного йона можна характеризувати константою рівноваги, що називається
константою нестійкості комплексу.
Комплексні сполуки відіграють
важливу роль в житті живих організмів.
комплексоутворення
ліганд координаційний
Список використаної літератури
1. Григорович О.В. Хімія,
повний курс. - X.: Видавництво «Ранок», 2010 р. - 400с.
. Глинка Н.Л. Общая химия. -
Санкт-Петербург: Узд. «Химия», 1967 г.
. Середа А.С. Аналітична хімія.
Якісний аналіз. - ЦУЛ, 2002 р.
5. Некрасов В.В. Основы общей химии
т.1. - Москва: Изд. «Химия», 1973 г.
. Некрасов В.В. Основи общей химии
т.2. - Москва: Изд. «Химия», 1973 г.
. Энциклопедия для детей т. Химия. -
Москва: Изд. «Аванта плюс», 2000 г.
. Гликина Ф.Б. и Ключников Н.Г.
Химия комплексних соединений. Учеб. Пособие для пед. Ин-тов м., «Просвещение»
1967. 166с.
. Данильченко В.Є., Фрадіна Н.В.
Хімія. Навч. Посібник.- X.: Країна мрій, 2003.-220с.
. Григорович О.В. Хімія: Довідник/О.В.
Григорович, Б.А. Мареха, О.Ю. Мацаков. - X.: Факт, 2008. - 160с.
. Москаленко Л.М., Чамата Н.М.
Хімія. X.: Країна мрій, 2004. - 60с.
. Уткіна О.А. Хімія комплексних
сполук. Харків: Країна мрій, 2006. - 64с.