Основные химические свойства полимеров и реакции в полимерных цепях
Контрольная работа
ОСНОВНЫЕ ХИМИЧЕСКИЕ СВОЙСТВА
ПОЛИМЕРОВ И РЕАКЦИИ В ПОЛИМЕРНЫХ ЦЕПЯХ
Содержание
1. Особенности химических реакций в полимерах
2. Деструкция полимеров под действием тепла и химических сред
3. Химические реакции, протекающие при действии света и
ионизирующих излучений
4. Механохимические реакции в полимерах
5. Реакции полимеров с кислородом и озоном
6. Формирование сетчатых структур в полимерах
Библиографический список
1.
Особенности химических реакций в полимерах
Химические реакции полимеров изучают с целью решения важных
задач модификации и стабилизации их свойств,
которые могут ухудшаться при воздействии теплоты, света, воздуха и других
агрессивных воздействий, защита от которых удлиняет сроки эксплуатации изделий.
Задачи модификации и стабилизации полимеров могут тесно переплетаться, так как
в результате модификации могут быть получены более стабильные полимеры. Принцип
Флори, по которому реакционная способность функциональной группы не зависит от
длины молекулы, качественно соблюдается и в полимерах. Количественные же оценки
выявляют большие различия в активности низко - и высокомолекулярных соединений
одной химической природы. Для полимера характерно неполное превращение
функциональных групп из-за малой подвижности сегментов или трудности доступа
реагента к ним, из-за свернутости в клубок макромолекулы или какой-либо
надмолекулярной организации структуры. Скорость химической реакции может
ограничиваться также диффузией или скоростью растворения реагирующих веществ.
Макромолекулярная природа полимеров существенно изменяет и
характер реакций. При взаимодействии низкомолекулярных олефинов с серой или
кислородом образуются низкомолекулярные сульфиды, альдегиды, кетоны и другие
соединения, а у высокомолекулярных аналогов (полидиенов) - сетчатые структуры.
При хорошей растворимости низкомолекулярного реагента в полимере химическая
реакция является гомогенной. Если же скорость химической реакции
значительно превышает скорость растворения или диффузии низкомолекулярного
реагента, или он находится в избытке по сравнению с концентрацией насыщения и
образует в полимере отдельную фазу, то реакция становится гетерогенной.
Часто химические реакции в эластомерах приобретают признаки топохимических,
локализуясь в микрообъемах системы, например у поверхности твердых частиц
оксидов или солей металлов.
В продукте реакции присутствуют измененные и неизмененные
звенья, придающие макромолекулам и полимеру композиционную неоднородность
по составу в целом и распределению прореагировавших групп по длине цепей.
Неоднородность может иметь разный характер сочетания измененных и неизмененных
звеньев: статистический по длине цепи или наличие блоков. ММР также влияет на
характер размещения композиционно неоднородных звеньев. Наряду с более
прореагировавшими цепями присутствуют менее прореагировавшие цепи.
Композиционно неоднородный продукт можно получить и при синтезе сополимеров -
при равном содержании звеньев бутадиена и стирола каучук СКС-30АРК резко
отличается по свойствам от термоэластопласта ДСТ-30 (блок-сополимер). Топохимические
реакции, в отличие от гомогенных реакций, локализуются в определенных
микрообъемах вещества. Это относится и к твердофазным реакциям,
протекающим не только в стеклообразных или кристаллических полимерах, но и в
системах, содержащих твердые оксиды или соли металлов. Из-за многостадийности
им присущи сложные зависимости скорости и степени превращения от времени.
Разделение их на кинетическую и диффузионную зоны проявляется, например, в
экстремальной зависимости скорости от степени превращения реагентов.
Основные отличия реакций веществ в полимерном состоянии
от реакций их низкомолекулярных аналогов:
. реакции, присущие только полимерному состоянию -
распад макромолекул на более мелкие образования или до исходных молекул
мономеров и межмакромолекулярные реакции ("эффект цепи");
2. конфигурационные эффекты, связанные с изменением
механизма или скорости химической реакции вследствие присутствия в
макромолекулах звеньев иной пространственной конфигурации ("эффект
соседа");
полимер полимерная цепь реакция
3. конформационные эффекты, связанные с изменением
конформации макромолекулы после того, как прошла химическая реакция;
4. концентрационные эффекты, влияющие на скорость
реакции вследствие изменения концентрации реагирующих групп около макромолекулы;
. надмолекулярные эффекты, связанные с распадом или
формированием новых надмолекулярных структур в массе либо в растворе полимера,
способные изменить скорость реакции и структуру конечных продуктов.
Предложенные классификации химических реакций в
полимерах:
1. Классификация по аналогии с известной
для низкомолекулярных углеводородов и их производных - реакции замещения,
присоединения и т.д.
2. Классификация по видам воздействия на полимеры
(молекулярная природа реагентов при различной их химической природе) - полимер
и низкомолекулярное вещество, функциональные группы в одной макромолекуле или
разных макромолекул, деструкция макромолекул. В основе лежит исходная природа
реагентов: высокомолекулярная или низкомолекулярная.
. Классификация по видам химических превращений
макромолекул (по изменениям их химической структуры): реакции
полимераналогичные, внутримолекулярные и межмакромолекулярные. Полимераналогичные
реакции изменяют только химический состав и природу функциональных групп в
полимере без изменения исходной длины макромолекул. Если изменяется длина
исходной макромолекулярной цепи (чаще в сторону уменьшения) или в ней
появляются циклические структуры, то такие реакции называются внутримолекулярными.
Если макромолекулы соединяются друг с другом химическими связями, то реакции
называются межмакромолекулярными. Они приводят к образованию
полимеров сетчатой структуры, физико-механические свойства которых существенно
отличаются от свойств аналогичных по природе полимеров, состоящих из
изолированных макромолекул.
. Классификация смешанная - по видам
превращений макромолекул и видам воздействия на них, т.к. один вид воздействия
в зависимости от химической природы полимера приводит к разным изменениям
структуры макромолекул. Высокие температуры вызывают деструкцию полистирола,
полипропилена и полиизопрена, циклизацию полиакрилонитрила и образование
сетчатых структур в 1,2-полибутадиене и БСК. При облучении полиэтилена
одновременно идут реакции сшивания и распада макромолекул.
Примеры полимераналогичных превращений. Поливиниловый спирт (ПВС)
получают гидролизом или метанолизом поливинилацетата:

Длина макромолекулы не меняется, при этом реагируют не все
сложноэфирные группы, поэтому конечный продукт композиционно неоднороден.
Поливинилацетат и ПВС могут участвовать в различных полимераналогичных
превращениях с органическими кислотами с образованием новых сложных эфиров,
которые используются в качестве лаков различного назначения. Перерабатываемость
полиамидов улучшают реакцией с формальдегидом:

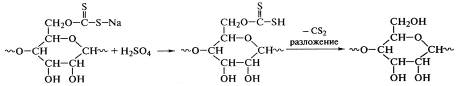
Перевод природной целлюлозы в вязкотекучее состояние путем
нагревания невозможен из-за опасности химического разложения, поэтому
перерабатывают вискозным методом (ряд полимераналогичных
реакций):

Эти реакции не охватывают все звенья макромолекул целлюлозы -
лишь одна из шести гидроксильных групп образует ксантогенат натрия, но и такая
степень превращения достаточна для нарушения регулярности ее строения,
уменьшения плотности упаковки макромолекул и перевода их в раствор. Последующий
гидролиз ксантогенатов серной кислотой приводит к разложению ксантогеновой
кислоты и регенерации целлюлозы, которая нерастворима в воде и поэтому легко
формуется в пленку или волокно:
 .
.
Применяют и другие полимераналогичные превращения целлюлозы. Ацетилирование
целлюлозы проводят взаимодействием ее гидроксильных групп с уксусной
кислотой в присутствии катализаторов (H2SO4, HClO4):
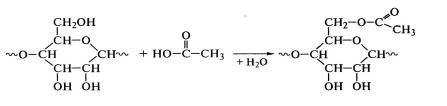
Реакцией гидрирования водородом в присутствии
металлического никеля, кобальта и других металлов в качестве катализаторов при
270оС и давлении 3 МПа из полиизопрена получают чередующийся
сополимер этилена и пропилена, а из полибутадиена - полиэтилен:
 .
.
Примеры внутримолекулярных реакций. Реакцией ПВС с
альдегидами получают поливинилбутираль - пленку для многослойных стекол
"триплекс":

Реакцией ПВС с кетонами получают пленочный материал
поливинилкеталь:

Термообработкой ПВС получают термостойкие волокна
"винол" в результате внутримолекулярных реакций дегидратации с
образованием системы сопряженных двойных связей и циклических эфиров:

Из полиакрилонитрила при 200оС получают теплостойкий
полимер "черный орлон" с полупроводниковыми свойствами, линейные
макромолекулы которого образуют циклы и систему полисопряженных двойных связей:

Он применяется для производства термостойких (до 800оС)
волокон и пленок.
Эпоксидированием полидиенов надкислотами
получают химически активные вещества, используемые для проведения дальнейшей
модификации:
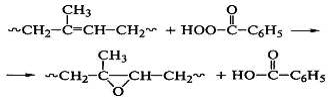
Эпоксидированные полимеры обладают повышенной адгезией к
металлам и другим полярным поверхностям и применяются в качестве покрытий и
клеев.
Галогенирование полимеров - наиболее широко
применяющийся на практике способ их модификации. Полиэтилен реагирует с хлором
в темноте при 150оС в отсутствие инициатора по радикальному механизму:
Cl2 → 2Cl*; ~CH2-CH2~ + Cl* → ~C*H-CH2~ + HCl; ~C*H-CH2~ +Cl2 → ~CHCl-CH2~ + Cl*.
Чаще хлорируют в растворе или водной суспензии полиэтилена в
присутствии инициаторов радикальных реакций, под действием УФ - или γ-излучения. Свойства продукта реакции изменяются от
каучукоподобного до жесткого в зависимости от степени хлорирования. Для
полиэтилена распространена также реакция сульфохлорирования путем
обработки его раствора газообразной смесью хлора и диоксида серы до 27%
связанного хлора и 1,5% серы:

Присоединение хлора и хлорсульфогрупп нарушает регулярность и
исключает кристаллизацию макромолекул, что придает хлорсульфополиэтилену
свойства эластомера. Хлорирование ПВХ проводят до содержания хлора 66% теми же
способами, что и для полиэтилена, с целью получения покрытий, стойких к
агрессивным средам, и повышения адгезии к полярным субстратам.
Реакция замещения водорода у α-метиленовых групп с образованием хлороводорода наиболее ярко
выражена при хлорировании полиизопрена:
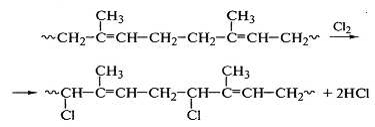
Вследствие гибкости макромолекул полиизопрена после
связывания 35% хлора развивается реакция образования циклических структур,
затем идут реакции хлора с образовавшимися циклами - замещение водорода в
оставшейся метиленовой группе и присоединение к двойной связи:
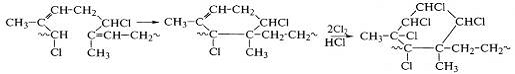
При хлорировании полибутадиена циклизация практически
отсутствует, но происходит транс-присоединение хлора по двойным связям и
сшивание макромолекул с образованием сетчатой структуры. Хлорированный
полиизопрен сохраняет линейную циклизованную структуру макромолекул, остается
растворимым во многих растворителях, является твердым продуктом до 70оС,
кристаллизуется при растяжении, обладает хорошей адгезией к полярным поверхностям,
высокой коррозионной стойкостью и клеящей способностью. Хлорирование
полихлоропрена до 68% связанного хлора позволяет получать
высококачественные клеи для крепления резин к металлам. Хлорирование
применяется также для модификации малоненасыщенных сополимеров (бутилкаучук и
тройной этиленпропиленовый сополимер), что позволяет усовершенствовать приемы
их вулканизации, повысить химическую стойкость и улучшить другие свойства резин
на их основе.
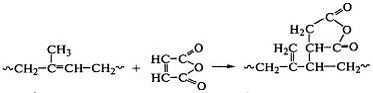
Реакции с ненасыщенными низкомолекулярными
соединениями - распространенный способ модификации полидиенов. Реакция
с малеиновым ангидридом протекает при температуре выше 180оС
и инициируется теплом:
В присутствии радикальных инициаторов реакция протекает при
100оС и ниже. Радикал инициатора отрывает водород от
метиленовых групп полидиена:
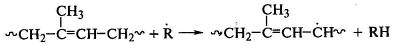 ,
,
и полимерный радикал реагирует с молекулой малеинового
ангидрида:
 .
.
Продукт присоединения, являясь свободным радикалом, отрывает
водород от макромолекулы полидиена или рекомбинирует с полимерным радикалом. В
последнем случае возникает химическая связь между двумя макромолекулами.
Реакция сшивания более вероятна при использовании низкомолекулярных соединений
с двумя малеиновыми фрагментами в молекуле, например бисмалеимидами.
Такие соединения применяются для вулканизации каучуков.
Реакции двухатомных фенолов с аминосоединениями занимают особое место
при модификации эластомеров. При прогреве смеси резорцина с
гексаметилентетрамином (молификатор РУ-1) протекают реакции конденсации:
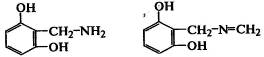 .
.
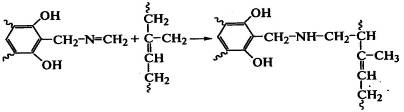 .
.
Такие олигомерные фрагменты на макромолекулах могут содержать
на концах активные аминогруппы, способные к реакциям друг с другом или с
макромолекулами полидиенов. Частично протекают реакции сшивания, что вместе с
образованием продуктов конденсации в полимерной матрице ведет к росту прочности
резиновых смесей и вулканизатов и прочности связи модифицированного неполярного
полимера с полярными волокнами. Модификаторы РУ-1, АРУ, алрафор широко
используются в композициях на основе СК для повышения длительности эксплуатации
шин, рукавов и др.
Некоторые химические реакции сопровождаются изменением
конфигурации макромолекул, т.е. их изомеризацией. В ненасыщенных полимерах
(1,4-полидиенах) возможны переходы цис-структуры в транс-структуру,
перемещение двойных связей вдоль цепи макромолекулы или в боковые группы, что
также влияет на свойства конечных продуктов. Обратимость реакций приводит к
существованию равновесия между цис - и транс-формами.
Например, нагревание полиизопрена с диоксидом серы при
температуре 140оС приводит к получению продукта с равновесным
соотношением цис - и транс-форм. В основном же происходит
изомеризация энергетически менее выгодной цис-формы в устойчивую транс-форму.
Интенсивная циклизация полидиенов происходит при нагревании их в присутствии
протонных и апротонных кислот, т.е. при развитии реакций катионного типа. Циклокаучук
представляет собой твердое вещество, переходящее в пластическое состояние при
100оС. Протекающая одновременно деструкция снижает его молекулярную
массу до 3000-4000 и делает пригодным для химически стойких покрытий и типографских
быстросохнущих красок.
Таким образом, при всём сходстве с реакциями
низкомолекулярных аналогов основным правилом макромолекулярных реакций,
связанным со спецификой полимерного состояния вещества, является неполное
превращение реагирующих групп. Это приводит к получению конечных продуктов,
неоднородных по молекулярному составу.
Примеры некоторых реакций модификации полимеров показывают
широкие возможности изменения их химической природы и создания на их основе
материалов с новыми свойствами. На примерах галогенирования насыщенных и
ненасыщенных углеводородных полимеров показано, как существенно меняются
структура их макромолекул и основные физические и механические свойства.
Полимеры с двойными связями в главной цепи макромолекул способны к реакциям присоединения,
что широко используют при глорировании, гидрохлорировании, гидрировании,
эпоксидировании и в реакциях химической модификации низкомолекулярными
полярными соединениями.
2. Деструкция
полимеров под действием тепла и химических сред
Среди физических факторов, способных инициировать химические
реакции в полимерах, тепловое воздействие занимает важное место и определяет их
термостабильность - верхнюю температурную границу пределов
эксплуатации.
Термический распад макромолекулярной структуры является одной
из причин старения полимеров - ухудшения их механических свойств
до опасного для дальнейшей эксплуатации уровня и выделения токсичных
низкомолекулярных продуктов. Часто снижает сроки службы изделий из полимеров
совместное воздействие теплоты и агрессивных сред.
При действии высоких температур большинство полимеров
претерпевает деструкцию, т.е. распад макромолекул на более мелкие
молекулы. В зависимости от химического строения макромолекул одни полимеры деполимеризуются
полностью, т.е. разлагаются до мономеров, в других происходит случайный разрыв
связей и образование устойчивых макромолекул пониженной молекулярной массы.
Иногда отщепляются низкомолекулярные продукты за счет реакций боковых групп без
существенного изменения исходной молекулярной массы, или происходит
беспорядочное сшивание макромолекул с образованием разветвленных и сетчатых
структур. Скорости реакций радикальной полимеризации и деполимеризации
возрастают с температурой и при некоторой температуре могут стать равными. Это
можно установить по равенству вязкости растворов продуктов полимеризации и
деструкции, что свидетельствует об одинаковой их молекулярной массе.
Установлена важная зависимость характера продуктов деструкции от теплоты
полимеризации. Деполимеризуются в основном до мономера полимеры с четвертичными
атомами углерода в цепи и низкой теплотой полимеризации (поли-α-метилстирол; 39,7 кДж/моль). Если полимер содержит в
цепях вторичные и третичные атомы углерода и имеет высокое значение теплоты
полимеризации (выше 60 кДж/моль), то при термической деструкции мономер
практически не образуется, а процесс заканчивается образованием устойчивых
макромолекул пониженной молекулярной массы. Цепной радикальный процесс
термической деструкции включает в себя стадии инициирования, роста, передачи
и обрыва реакционной цепи. Радикальные реакции передачи цепи идут
за счет отрыва водорода от макромолекулы полимеров, содержащих атом водорода у
третичного атома углерода или α-метиленовые группы у
двойных связей. Такие полимеры, например полиметилакрилат без
двойных связей на концах макромолекул, почти не дают мономеров при термическом
распаде:
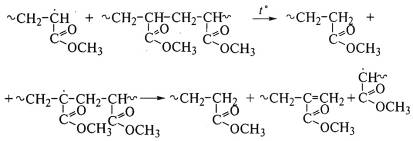
Частный случай цепной деструкции - цепная
деполимеризация путем последовательного отщепления мономерных звеньев
от конца цепей вплоть до полного перехода полимера в мономер. При этом
молекулярная масса полимера последовательно уменьшается. Так идет термическая
деструкция полиметилметакрилата с двойными связями на концах
макромолекул - продукта полимеризации при обрыве цепи диспропорционированием:
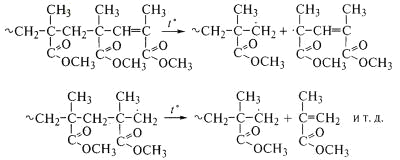
Таким образом, реакцонная способность радикала, образующегося
при разрыве макромолекулы, и легкость отрыва атома водорода определяют
дальнейшее направление деструкции. Из-за невозможности передачи цепи
термодеструкция поли-α-метилстирола и
политетрафторэтилена проходит с высоким выходом их мономеров. Чтобы это
исключить, применяют метод сополимеризации подобных мономеров с мономером,
склонным к реакции передачи цепи, например метилметакрилата с 2% акрилонитрила.
При термическом воздействии на полиэтилен резко уменьшается его
молекулярная масса из-за распада цепей в местах разветвлений. Далее с
уменьшением ММ стабильность осколков макромолекул возрастает, и скорость
распада снижается. В линейном полиэтилене возможен разрыв цепи за счет
внутримолекулярной реакции передачи цепи:
~СН2-СН2-СН2-СН2-СН2-СН2-СН2-С*Н2~
→
→ ~СН2-С*Н2
+ СН2=СН2-СН2-СН2-СН2-СН2~.
При термическом воздействии на полиизопрен также имеет место
передача цепи, а распад идет по ослабленной диаллильной связи:

Высокая гибкость цепи позволяет прореагировать концевому
свободному радикалу с соседней двойной связью и оторвать димер изопрена -
циклический дипентен, являющийся одним из основных продуктов распада
полиизопрена:
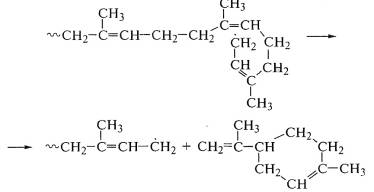
В атмосфере инертного газа полиизопрен начинает разлагаться
при 200оС. Превращение макромолекулы в макрорадикал может
привести к разрыву соседней С-С-связи с выделением молекулы изопрена и образованием
нового радикала, который на конце остатка цепи вызовет новый акт распада с
выделением мономера вплоть до полной деполимеризации полимера. Проявлением
"эффекта клетки" - затруднения выхода продуктов распада из зоны
реакции и рекомбинации их в исходные продукты объясняют более высокую
температуру начала термического распада закристаллизованного полимера по
сравнению с аморфным.
При термической деструкции полиамидов разрывается N-C связь и развиваются
последующие реакции рекомбинации макрорадикалов:
~CO-NH-CH2- (CH2)
4-CO-NH-CH2- (CH2) 4-CO-NH~→
→~CO-NH-CH2- (CH2)
4-CO-N*H + C*H2-
(CH2) 4-CO-NH~→
→ ~CO-NH-CH2- (CH2)
4-CO-NH2 + CH2=CH- (CH2)
3-CO-NH~→
→ ~CO-NH-CH2- (CH2)
4-CN + H2O + CH2=CH- (CH2)
3-CO-NH~
Термическое воздействие на поливинилхлорид и некоторые другие
полимеры с функциональными подвесками приводит к выделению низкомоле-кулярных
веществ и образованию двойных связей без распада цепей:
~CH2-CHCl-CH2-CHCl~ → ~CH2-CHCl-CH=CH~ + HCl → ~CH=CH-CH=CH~ + HCl.
Выделяющийся HCl катализирует дальнейшее дегидрохлорирование
("эффект соседа") ускоряя выделение HCl по соседству с возникшей
двойной связью. При удалении HCl реакция распада замедляется, а дополнительном
его введении - ускоряется. В результате возникает система сопряженных двойных
связей, придающая макромолекулам свойства полупроводников. При близком контакте
происходит сшивание двух макромолекул. Возможна внутримолекулярная циклизация
звеньев с тремя сопряженными двойными связями с образованием циклодиенильных
комплексов и выделением молекул бензола. Поскольку скорость термодеструкции ПВХ
обратно пропорциональна его ММ, вероятнее всего инициирование идет у концов
цепей. Стабилизация ПВХ бариевыми и кадмиевыми солями карбоновых кислот
связана, возможно, с обменом его подвижных атомов хлора на карбоксильные группы
стабилизаторов. Поливинилацетат при нагревании окрашивается и выделяет уксусную
кислоту, а образующаяся двойная связь также облегчает ее отщепление от
соседнего звена с развитием деструкции как автокаталитической цепной реакции.
Таким образом, даже при одном термическом воздействии
наблюдается большое разнообразие реакций и получающихся продуктов в зависимости
от химического строения полимеров. Термическое воздействие может приводить к
получению ценных и теплостойких полимеров (ПВХ) и образованию систем
сопряженных двойных связей в линейных макромолекулах (ПВХ, поливинилацетат),
т.е. не к ухудшению, а к улучшению их качества.
Стойкость полимеров к термическому старению (термостабильность)
оценивают по потере массы при нагревании и по составу летучих продуктов.
Скорость термодеструкции характеризуется величиной массы полимера,
разлагающегося за 1 мин при 350оС, или температурой
полураспада, при которой происходит потеря 50% исходной массы за 40 мин.
Термостабильность повышается при наличии шестичленных или конденсированных
циклов или системы сопряженных кратных связей в основной цепи полимеров. Важная
характеристика термической стойкости полимеров - температура начала
термораспада. Сопоставление этих температур, полученных в инертной среде
или вакууме, с теплотами полимеризации указывает на их корреляцию: чем меньше
теплота полимеризации, тем ниже температура начала термораспада (табл.1).
Кислород резко активизирует термодеструкцию полимеров.
Таблица 1.
Теплоты полимеризации и температуры начала термораспада для
различных полимеров
|
Полимер
|
Теплота
полимеризации, кДж/моль
|
Температура
начала термораспада, оС
|
|
Полиэтилен
|
91,9
|
320
|
|
Полистирол
|
71,0
|
310
|
|
Полипропилен
|
71,3
|
300
|
|
Полиакрилонитрил
|
71,1
|
298
|
|
Полиметилакрилат
|
83,6
|
292
|
|
Поли-α-метилстирол
|
39,7
|
260
|
Под старением полимеров понимается
комплекс химических и физических изменений, ухудшающих механические свойства и
снижающих работоспособность изделий. Это может быть изменение молекулярной,
надмолекулярной или фазовой структуры полимеров в процессе хранения или
эксплуатации. В реальных условиях на изделия часто действуют различные факторы
комбинированно, а процессы деструкции классифицируют и по видам
вызывающих их энергетических воздействий, и по характеру
протекания химических реакций, подобно классификации процессов их
синтеза.
К первой группе относят реакции распада макромолекул,
приводящие к единичным актам разрыва в результате концентрации энергии
разрушающего воздействия на какой-либо одной связи, которая рвется по
случайному закону независимо от другой. Образующиеся осколки макромолекулы
существуют как устойчивые молекулы, т.е. деструкция идет ступенчато и может
дойти до образования мономеров. При действии химических агентов на гетероцепные
полимеры с функциональными группами, широко используемые в производстве волокон
и пленок и способные подвергаться гидролизу, ацидолизу, аминолизу и другим
химическим превращениям, наблюдается беспорядочная деструкция.
Глубина деструкции зависит от количества и продолжительности действия
низкомолекулярного реагента, и она может быть остановлена на любой стадии путем
снижения температуры, удаления реагента или доведена до образования устойчивых
молекул мономеров. Целлюлоза распадается под каталитическим действием кислот по
случайному закону:
 .
.
Реакция может пройти до образования мономера (глюкоза) и
далее:
 .
.
Аналогично распадаются белки и полиамиды под действием кислот
и щелочей:
 .
.
Статистическое рассмотрение беспорядочного гидролитического
расщепления макромолекул приводит к следующим количественным зависимостям. Если
все связи, способные к расщеплению, одинаково реакционноспособны, то степень
деструкции α выражается зависимостью:
α=Р/ (No-1), где Р-число
разорвавшихся при гидролизе связей за время t; Nо-число звеньев в исходной
макромолекуле полимера; (No-1) - число способных к
расщеплению связей в исходной макромолекуле. В зависимости от времени реакции
степень деструкции выражается как α=1-е-kt, где k - константа скорости
мономолекулярной реакции расщепления. Тогда P= (No-1) (1-e-kt). Длина полимерной цепи в
момент времени реакции t: Nt=No/ (P+1). Если степень деструкции
невелика, т.е. значение Nt достаточно велико по
сравнению с No, то можно получить довольно простую
зависимость для скорости гидролитической деструкции: 1/Nt-1/No=kt. Величины 1/Nо и 1/Nt пропорциональны
соответственно начальной концентрации концевых групп (no) и мгновенной их концентрации
к моменту времени t (nt). Тогда nt-nо=k*t, т.е. скорость
деструкции представляет собой разность мгновенной и начальной концентраций
концевых групп в полимере. При линейном строении макромолекул длина
(молекулярная масса) цепи и концентрация концевых групп однозначно связаны с
характеристической вязкостью растворов, по изменению которой определяется
степень деструкции.
Ко второй группе реакций деструкции относятся цепные
реакции, при которых на один акт разрыва макромолекулы под действием
какого-либо фактора приходится несколько актов распада в других местах цепи.
Как и цепная полимеризация, цепная деструкция может протекать по радикальному
или ионному механизму. Инициирование цепной деструкции происходит под влиянием
факторов, вызывающих образование радикалов или ионов в цепях полимера -
теплоты, света, излучений, кислорода и веществ, распадающихся на свободные
радикалы (пероксиды) или ионы. Цепная деполимеризация как частный случай цепной
деструкции рассмотрена выше на примере полиметилметакрилата, содержащего
двойные связи на концах макромолекул.
3. Химические
реакции, протекающие при действии света и ионизирующих излучений
При воздействии света с длинами волн 230-410 нм и
проникающих излучений изменяются химическое строение и физико-механические
свойства полимеров. Направление химических превращений - разрыв С-С-связей в
главных цепях полимеров с образованием свободных радикалов и отрыв водорода от
углеродных атомов. Дальше развиваются химические реакции, приводящие к
деструкции, сшиванию, отщеплению боковых групп и другим химическим изменениям
макромолекул. При повышении температуры ускоряется процесс деструкции,
называемый в этом случае фотолизом.
Полиизопрен при действии солнечного света
размягчается и становится липким. При облучении его кварцевой лампой в вакууме
при комнатной температуре выделяются летучие продукты распада, состоящие на 80%
из молекулярного водорода. При облучении ультрафиолетовым светом снижается
вязкость и ММ толуольных растворов полиизопрена. В концентрированных растворах
после этого отмечен рост ММ, что вызвано формированием нерастворимой фракции
(геля) с сетчатой структурой макромолекул. Из этого следует, что главный
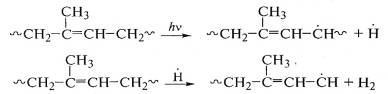
химический процесс при действии ультрафиолетового света на полиизопрен - отрыв
водорода с образованием свободных радикалов:
 .
.
Отщепление водорода происходит от α-метиленовых групп полиизопреновой цепи, где за счет
сопряжения с двойной связью энергия связи С-Н уменьшена на 42 кДж/моль.
Образующийся свободный радикал аллильного типа может изомеризоваться, вызывая
деструкцию макромолекулы полиизопрена:
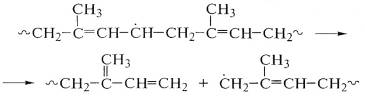 .
.
Он может взаимодействовать также с другим макрорадикалом, что
приведет к сшиванию макромолекул с образованием вначале разветвленных, а затем
и сетчатых структур, т.е. с ростом ММ и образованием геля:
 .
.
Особенно чувствительны к действию светового излучения тонкие
пленки из полиэтилена, полиметилметакрилата, целлюлозы. С ростом интенсивности
светового потока ускоряется реакция фотодеструкции, что наблюдается при
эксплуатации полимеров в южных районах, в горах, где имеет место повышенная
интенсивность световых потоков и ультрафиолетовых лучей. Под действием
ультрафиолетового облучения меняется окраска целлюлозы и ее эфиров, уменьшается
их прочность и вязкость растворов, выделяются летучие продукты (СО и СО2).
В присутствии атмосферного кислорода развиваются окислительные реакции. При
облучении целлюлозы в инертной среде вязкость растворов либо остается
неизменной, либо снижается незначительно, тогда как на воздухе и в кислороде
она падает резко.
Глубокие химические изменения происходят в полимерах при
действии радиационных излучений - рентгеновских и γ-лучей, быстрых и медленных нейтронов, быстрых электронов, α-частиц, протонов и других продуктов ядерных реакций. Энергия этих
излучений значительно больше энергии химических связей в полимерах, поэтому они
способны вызвать разрыв связей в цепи. Под действием излучений высоких энергий
происходят деструкция и сшивание цепей, образование двойных связей, циклизация,
отщепление боковых функциональных групп и разрушение кристаллических структур.
Макромолекулы полимера ионизируются и возбуждаются, распадаясь на два свободных
радикала, что и будет актом деструкции: Р → Р+ + е-;
Р+ + е - → Р*; Р*→
R1+R2, где Р-полимерная
макромолекула, Р+-ионизованная макромолекула, Р*-возбужденная
макромолекула, е - электрон. Выделяющийся при
радиолизе вторичный электрон с относительно низкой скоростью может не только
рекомбинировать с образовавшимся ионом полимера (реакция в "клетке"),
но и реагировать с другими молекулами (выход из "клетки"), образуя
новые ионы. Указанные изменения происходят очень быстро (10-12с).
Время жизни полимерных ионов или радикалов зависит от подвижности макромолекул
и при низких температурах составляет недели и месяцы.
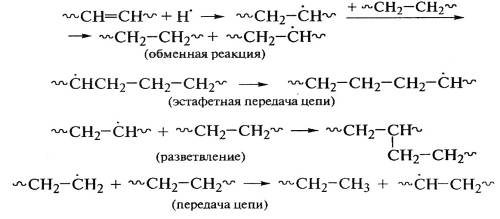
При облучении линейного полиэтилена выделяется
до 99% водорода, а возникновение разветвленности сопровождается образованием
бутана:

 .
.
Реакции деструкции и сшивания протекают одновременно с
преобладанием одной из них в зависимости от химического строения полимера. В полипропилене
могут протекать реакции деструкции:
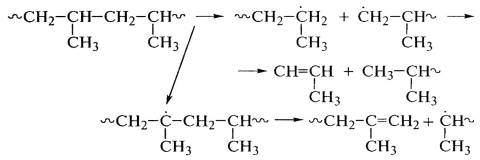 .
.
Деструкции подвергаются α-замещенные этиленовых
углеводородов и другие полимеры с низкими значениями теплоты полимеризации
(полиметил-метакрилат, полиизобутилен, поли-α-метилстирол,
полиэтилентерефталат, бутилкаучук, целлюлоза и др.), а при их пиролизе
образуется большое количество мономера.
Радиолиз полиизобутилена, например, идет по
схеме:
 .
.
Быстрая рекомбинация в "клетке" легко
образовавшихся радикалов часто затруднена из-за стерического фактора. Поэтому
они успевают выйти из "клетки", закрепляя акт деструкции
макромолекулы полиметилметакрилата:
 .
.
Полимеры с высокой теплотой полимеризации, малым выходом
мономера при пиролизе, без четвертичных атомов углерода в цепи, при действии
излучений в основном сшиваются (полиэтилен, полистирол, поливинилацетат,
полиизопрен, полибутадиен, полиметилакрилат). Разрывы цепей при облучении
происходят по случайному закону, а число разрывов или сшивок пропорционально
дозе облучения и не зависит от его интенсивности. В присутствии кислорода при
облучении часто развивается процесс окисления полимера. Стойкость к облучению
увеличивается при наличии в структуре ароматических колец, что связано с
значительным рассеянием энергии и называется "эффектом губки".
Это явление используют для защиты полимеров от нежелательного действия
излучений при радиационном старении, а вещества, препятствующие развитию
деструктивных процессов, называются антирадами. Они содержат в
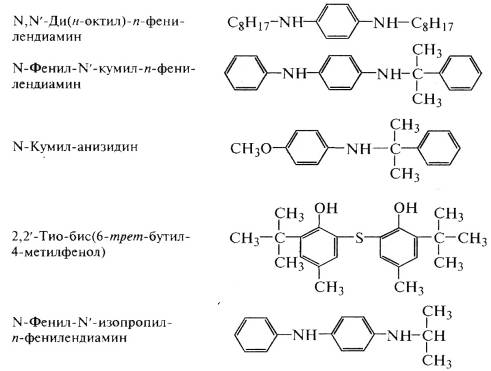
структуре ароматические кольца (например, N. N! - дифенил-n-фенилендиамин,
1,4-нафтохинон, 2-нафтиламин и др.).
Сшивание макромолекул при облучении используют для
радиационной вулканизации полиизопрена, полибутадиена, бутадиен-стирольного и
бутадиен-нитрильного сополимеров, полиэтилена, полиамидов и полиакрилонитрила.
Химические связи интенсивно образуются после перехода из стеклообразного
состояния, в котором полимер подвергался облучению, в высокоэластическое,
облегчающее подход сегментов друг к другу на расстояния, равные длине
химической связи между атомами углерода соседних макромолекул. Сшивание при
облучении облегчается и тем, что возникший при отрыве водорода свободный
радикал передает неспаренный электрон вдоль цепи, отчего увеличивается
вероятность его нахождения по соседству с таким же свободным радикалом другой
макромолекулы. Минимальная доза облучения, при которой образуется единая
сетчатая структура полимера, называется дозой гелеобразования Dг и выражается формулой: Dг = 1/αМ (2-β/2), где α-константа скорости сшивания; М-ММ исходного полимера;
β-константа деструкции.
Таким образом, основная часть низкомолекулярных соединений
(до 90%), выделяющихся при радиолизе углеводородных полимеров, составляет
водород. При радиолизе политетрафторэтилена выделяется и CF4, а полиакрилонитрила - HCN, что следует учитывать
при эксплуатации изделий из этих полимеров. Анализ превращений полимеров при
термическом и радиационном воздействиях позволяет выявить в них существенные
различия. При термическом воздействии у полиэтилена и полиамидов преобладает
деструкция, у полиизопрена - деполимеризация, у полиакрилонитрила - циклизация,
а при радиационном воздействии все они структурируются. Ряд полимеров
эксплуатируется в атомной промышленности и космосе, где действуют интенсивные
потоки различных радиоактивных излучений, и от поведения их в этих условиях
зависят сроки эксплуатации изделий.
4.
Механохимические реакции в полимерах
При перемешивании воды или бензола в какой-либо емкости
никакие химические реакции не идут. Протекание химических реакций при воздействии
механических напряжений характерно только для макромолекул и связано с
превышением суммарной энергии слабых физических взаимодействий между их
звеньями над энергией химической связи в главной цепи. Величины энергии и длины
различных типов связей (табл.2) позволяют сделать вывод, что суммарная энергия
даже слабых ван-дер-ваальсовых физических связей на протяжении 100-150 звеньев
макромолекулы малополярных полимеров превысит энергию ковалентной С-С-связи в
основной цепи. Очевидно, при сближении отдельных участков макромолекулы в поле
механических напряжений возрастает суммарная энергия межмолекулярного
взаимодействия их друг с другом (рис.1). Когда суммарная энергия связей
сблизившихся участков цепи превысит энергию ковалентной С-С-связи, последняя
разорвется.

Рис.1. Конформация макромолекулярного клубка исходного
аморфного полимера (а) и при разрушении флуктуационной сетки (б)
в поле сдвиговых напряжений (растяжение по оси О-О` и сжатие по оси Р-Р`).
Механодеструкция приводит не только к снижению ММ полимера до
некоторой величины, которая определяется соотношением суммарной энергии
физических межмолекулярных взаимодействий и энергии химической связи в основной
цепи. Выравниваются размеры макромолекул до этой величины, т.е. изменяется вид
кривой молекулярно-массового распределения (рис.2). Молекулы малых размеров
участвуют лишь в механическом перемешивании.

Рис.2. Снижение молекулярной массы (а) и изменение ММР
(б) полимера в результате деструкции от времени механической обработки τ: 1-исходное ММР; 2-4-ММР после пластикации на вальцах при
различной продолжительности τ (τ2> τ3> τ4).
Процессы механодеструкции протекают при переработке полимеров
на вальцах, в экструдере или резиносмесителе, где в поле сдвиговых напряжений
интенсивно снижаются ММ и вязкость, что облегчает их последующую переработку.
Эти процессы ускоряются реакциями макрорадикалов с низкомолекулярными
акцепторами свободных радикалов, специально вводимых в полимерную матрицу или
присутствующих в ней в виде примесей. Предельно низкое значение ММ (М∞),
достигаемое при механической обработке, называется пределом
механодеструкции. Полимеры сильно различаются по эффективности
механодеструкции, которая тесно связана с плотностью энергии когезии (табл3).
Таблица 3.
Энергия когезии участка цепи длиной 0,5 нм и число звеньев на
отрезке цепи длиной 5 нм для ряда промышленных полимеров
|
Полимер
|
Энергия
когезии, кДж/моль
|
Число звеньев
|
|
Полиэтилен
|
4, 19
|
3
|
|
Полиизобутилен
|
4,51
|
1
|
|
Полиизопрен
|
5,44
|
1
|
|
Поливинилхлорид
|
10,87
|
2-3
|
|
Поливиниловый
спирт
|
17,56
|
2-3
|
|
Полиамид
|
24,25
|
1
|
|
Белок (шелк)
|
41,00
|
1
|
На разрушение физических связей влияет температура, и от нее
сильно зависит эффект механодеструкции. При низких температурах механическим
силам труднее преодолевать силы взаимодействия между макромолекулами полимера
из-за отсутствия проскальзывания между ними, а с повышением температуры эффект
скольжения макромолекул возрастает. Следовательно, механодеструкция имеет
отрицательный температурный коэффициент, т.е. число актов разрывов химических
связей в главных цепях растет с понижением температуры. Наиболее существенные
факторы, влияющие на эффективность механодеструкции полимеров, суммированы в
табл.4.
Таблица 4.
Влияние различных факторов на эффективность механодеструкции.
|
Факторы,
влияющие на механодеструкцию
|
Показатели
эффективности механодеструкции
|
|
|
Скорость
механодеструкции
|
Предел
механодеструкции
|
|
Рост
температуры
|
Снижается
|
Увеличивается
|
|
Рост ММ
(вязкости)
|
Растет
|
Не влияет
|
|
Интенсивность сдвига
|
Растет
|
Уменьшается
|
Наибольшая скорость механодеструкции достигается в
стеклообразном состоянии полимеров, средняя - в высокоэластическом состоянии и
наименьшая - в вязкотекучем. В таком же порядке уменьшается величина
механических напряжений, которые надо приложить к полимеру, чтобы вызвать
разрыв или проскальзывание его макромолекул. Следовательно, в полимерах можно
осуществить прямое превращение механической энергии в химическую энергию, так
как образующиеся активные осколки макромолекул (радикалы) инициируют реакции
полимеризации мономеров с активными участками других макромолекул, с кислородом
или низкомолекулярными добавками. В ряде случаев могут образовываться
разветвленные и сшитые структуры. Путем механической обработки смесей полимеров
или полимеров с жидкими мономерами получают блок - и привитые сополимеры. Таким
образом, этот сравнительно недорогой и доступный и при этом весьма эффективный
прием обработки позволяет проводить химическую модификацию полимеров.
Механические напряжения могут активировать химические реакции
в полимерах, когда они и не разрывают макромолекулы. Например, образцы
эластомеров и их вулканизатов быстро разрушаются в присутствии небольших
количеств озона, если находятся в растянутом состоянии. При приложении многократных
деформирующих напряжений быстрее протекает взаимодействие полимеров с
кислородом, приводящее к разрыву макромолекул. Механическая активация
химических реакций в полимерах объясняется изменением их направления (например,
распада озонидов) и ускорением роста трещин. Помимо разрыва основной цепи, при
механическом воздействии могут разрываться химические поперечные связи в
сетчатых полимерных структурах с образованием осколков, которые уже могут
растворяться. На этом принципе основан один из методов регенерации резин в
целях получения пластичного формуемого материала, который может
перерабатываться в изделия наравне с исходными полимерами. Принцип
механического измельчения с механодеструкцией широко используют для переработки
полимерных отходов с целью придания им второй жизни в новых полимерных
изделиях.
В полибутадиене развитие
механохимических реакций начинается с образования первичных радикалов, которые
участвуют в химических реакциях друг с другом и рекомбинируют с образованием
поперечных связей между макромолекулами. Идут также реакции присоединения
радикалов по двойным связям соседних макромолекул с образованием ответвлений
или реакции отрыва водорода от макромолекул с образованием новых радикалов:
~СН2-СН=СН-С*Н2
+ ~СН2-СН=СН-СН2~ → ~СН2-СН=СН-СН3
+ ~С*Н-СН=СН-СН2~
В обоих случаях образуются полимерные свободные радикалы,
которые могут участвовать в дальнейших реакциях образования разветвленных
структур:
или рекомбинации с образованием поперечной связи между макромолекулами:
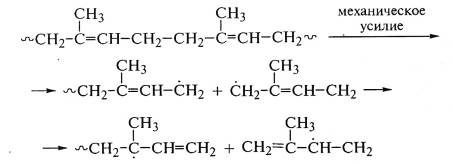
В полиэтилене механокрекинг приводит к
химическим реакциям:
~СН2-СН2-СН2-СН2~→~СН2-С*Н2
+ С*Н2-СН2~→~СН=СН2
+ СН3-СН2~
или ~СН2-С*Н2
+ ~СН2-СН2~→~СН2-СН3
+ ~СН2-С*Н~.
При механическом смешении полимеров с наполнителями свободные
радикалы образуют химические связи полимер-наполнитель. Так, химические связи
эластомера с техуглеродом приводят к образованию гелеобразных структур.
Первичные радикалы могут стабилизироваться ограничением подвижности
стеклообразным состоянием, сохранив способность к последующим реакциям:
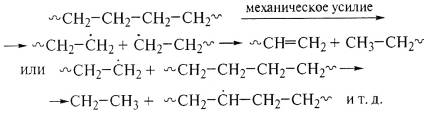
Барамбойм предложил классификацию механохимических процессов
по направленности превращений полимеров и результатам этих превращений:
. механодеструкция - разрыв линейных макромолекул со
снижением молекулярной массы и полидисперсности и развитие реакций
разветвления;
2. механосшивание - соединение вторичных макрорадикалов
и формирование сетчатой структуры полимера;
. механосинтез - присоединение к первичным и вторичным
радикалам полимеризующихся молекул мономера, свободных радикалов другого
полимера, химическое присоединение полимера к частицам наполнителя;
. механоактивация химических реакций в полимерах
(разложение, замещение, присоединение и др.), когда механические напряжения
лишь снижают энергию активации таких реакций;
. механохимическое течение - разрушение сетчатых
полимеров в поле механических сил, приводящее к образованию новых структур в
таких полимерах и позволяющее вовлекать их в процессы переработки наряду с
линейными полимерами.
Помимо рассмотренных наиболее простых механических
воздействий на полимеры, приводящих к развитию в них химических реакций,
существует и ряд других видов воздействий, вызывающих механическое разрушение
химических связей. Сюда относятся процессы дробления и измельчения, действие
ультразвука, высоких давлений и др. В зависимости от вида механического
воздействия наблюдаются различия в явлениях, которые они вызывают. При
одноосной деформации преобладают сдвиговые и растягивающие усилия, приводящие в
итоге к разрыву макромолекул. При смешении, вальцевании и экструзии действуют
различные скорости сдвига. При высокоскоростных воздействиях преобладают
ударные эффекты, например при измельчении полимеров. При действии ультразвука
развиваются высокочастотные колебания. Все эти воздействия приводят к
деструкции макромолекул. Таким образом, механохимические процессы и их конечные
результаты зависят от природы источников механических усилий.
5. Реакции
полимеров с кислородом и озоном
Химическое взаимодействие полимеров с кислородом лежит в
основе реакций окисления, которые могут ускоряться под действием
теплоты (термоокислительное старение), света, излучений, солей металлов
переменной валентности и механических воздействий. В реальных условиях изделия
из полимеров испытывают совместное действие многих из перечисленных и ряда
других факторов. Скорость термоокислительной деструкции углеводородных
полимеров выше скорости их термического распада, что снижает предельные
температуры их эксплуатации до 120-150оС.
Термоокислительная деструкция полимеров сложна по химической
природе и протекает по механизму цепных реакций с вырожденными разветвлениями в
соответствии с теорией цепных химических реакций академика Н.Н.
Семенова. Основную роль в реакциях окисления играют пероксидные и
гидропероксидные соединения, образующиеся при взаимодействии полимеров с
кислородом и легко распадающиеся на свободные радикалы. Реакции идут с
выделением летучих продуктов, а состав и строение конечных продуктов зависят от
условий ее проведения и природы полимера. Механизм реакций окисления можно
представить следующей схемой:
) зарождение цепи (образование полимерных радикалов R*)
RH + O2 → R* +
HOO*; 2RH + O2 → 2R* + H2O
) развитие цепи (взаимодействие пероксидного радикала с
молекулой)
R*+ O2 → ROO*;
ROO* + RH → ROOН + R*;
) вырожденное разветвление цепи
ROOН → RO* + НО*; ROOН + RH → RO* + R* + Н2O;
ROOН → RОO* + RO* + Н2O;
) обрыв цепи: 2R* → R-R; 2RОO*→ ROOR + O2; ROО* + R*→ ROOR.
Анализ кинетики окисления полимеров также
указывает на признаки наличия цепных радикальных реакций. На кривой химического
связывания кислорода полимером имеется участок I быстрого начального
присоединения кислорода, участок II индукционного периода ингибированного окисления,
удлиняющийся при введении ингибиторов; участок III свободнорадикальной
реакции по цепному механизму (автокатализ) и участок IV снижения скорости
окисления вследствие исчерпания активных центров (рис.3). Окисление ускоряется
при освещении, а после удаления источника света проявляется
"постэффект" действия света. При повышении температуры индукционный
период сокращается, а скорость окисления возрастает. Зарождение цепи окисления
состоит в образовании полимерных радикалов R* при распаде макромолекул
в результате различных энергетических воздействий. На стадии зарождения цепи
дополнительные свободные радикалы, легко реагирующие с кислородом, могут
образовываться также при действии света, излучений высоких энергий,
механических напряжений и распада полимерных пероксидов и гидропероксидов (RO* и ROO*). Развитие реакционной
цепи происходит в результате взаимодействия пероксидного радикала ROO* с молекулой полимера.
Пероксидный радикал RОO* стабилизируется, отрывая подвижный атом водорода от макромолекулы
полимера, но при этом вновь образуется полимерный радикал, взаимодействующий с
молекулярным кислородом.
а б в
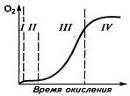
Рис. 3. Кинетическая кривая окисления полимеров (а) и
влияние на нее солнечного света при низких температурах (б) и
температуры (в): 1-без освещения; 2 - при освещении; 3-удаление света в
точке А; t1>t2>t3
Вырожденноразветвленный характер окисления состоит в том, что
образующийся гидропероксид ROOН нестабилен и распадается с образованием новых
свободных радикалов RO*, НО*, RОO*, которые также отрывают атомы водорода от молекул полимера,
возбуждая вторичные окислительные цепи:
RO* + RH → ROН + R*; НO* + RH → R* + Н2O.
В результате скорость присоединения кислорода и окисления
полимера резко возрастает, и это явление называется автокатализом.
Длина кинетической цепи реакции окисления представляет собой число звеньев
макромолекулы, приходящееся на каждый свободный радикал в реакциях зарождения и
вырожденного разветвления цепей. Скорость распада гидропероксидов зависит от
структуры полимеров и условий окисления. Обрыв цепи происходит при
взаимодействии радикалов друг с другом по реакциям их рекомбинации или
диспропорционирования с образованием веществ, которые не распадаются на
свободные радикалы. Водород отрывается от макромолекулярных цепей в тех
участках их структуры, которые содержат ослабленные Н-С-связи, например СН2-группа
в α-положении к двойной связи диенов или водород у третичного атома
углерода в полистироле или полипропилене. Образование функциональных
групп при окислении полимеров подтверждается исследованиями ИК-спектров
(рис.4). Кинетическая кривая накопления гидропероксидов имеет максимум. Между
количеством химически связанного кислорода и степенью деструкции полимера
наблюдается линейная зависимость. Наиболее активными катализаторами окисления
являются соли металлов переменной валентности (меди, железа, кобальта, марганца
и др.), разлагающие пероксиды на свободные радикалы:
ROOH + Fe2+ → RO* + Fe3+ + OH͞; ROOH + Fe3+ → ROО* + Fe2+ + ОН+.
Эффективность их действия растет с увеличением растворимости
или коллоидного диспергирования в полимерах, а нежелательное действие можно
подавить связыванием ионов металлов в виде недиссоциирующих или нерастворимых
комплексных соединений.
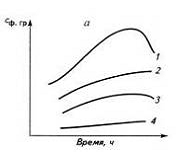
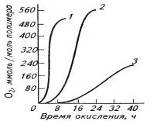
Рис. 4. Кинетические кривые образования в НК гидропероксидных
(1), гидроксильных (2), кетонных (3) и пероксидных (4) функциональных групп (а)
и окисления НК (б) при 120оС с 1,5 (1) и
0,5% (2) стеарата железа и без него (3).
Константы скоростей химических реакций связаны с
сегментальной подвижностью полимерной матрицы, поэтому различают диффузионный
режим реакции, когда она определяется скоростью диффузии реагентов, и кинетический,
который лимитируется скоростью взаимодействия реагентов. Медленный физический
процесс диффузии затрудняет рекомбинацию макро-радикалов, поэтому реализуется
"эстафетный" механизм передачи цепи вдоль макромолекулы, что
происходит при температурах ниже температуры стеклования полимера. Диффузионный
режим химической реакции легче реализуется в высокоэластическом и вязкотекучем
состояниях полимеров.
В большинстве случаев термоокислительное воздействие приводит
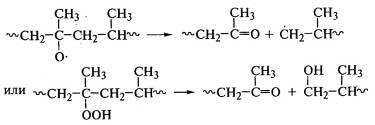
к деструкции макромолекул с резким снижением прочностных свойств полимеров.
Деструкция макромолекул протекает при окислении полиэтилена:
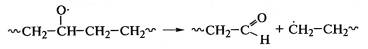 , полипропилена:
, полипропилена:
 , полистирола:
, полистирола:
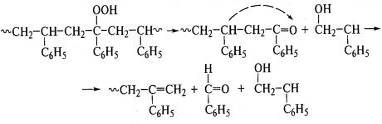 ,полиизопрена:
,полиизопрена:
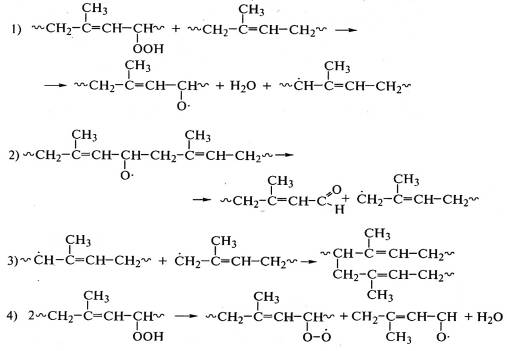 .
.
В присутствии кислорода увеличивается скорость
дегидрохлорирования ПВХ, так как свободные радикалы облегчают реакцию отрыва
атома хлора:
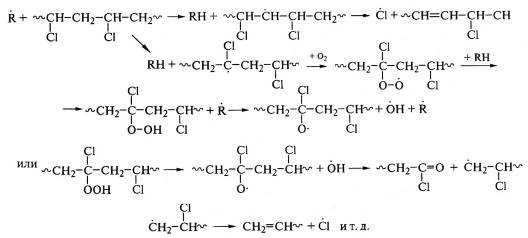
Окисление полиамидов также является цепной реакцией и
начинается у водорода в α-положении к NH-группе:
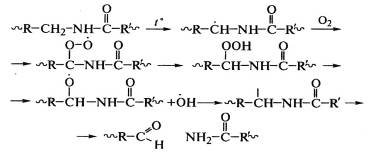
При рекомбинации макрорадикалов могут образоваться поперечные
химические связи между макромолекулами:

Аналогично протекает термоокислительная деструкция
полиэфиров. Процесс термоокислительной деструкции полиэтилентерефталата
(лавсана) при 170-220оС идет через образование оксидов и пероксидов
у метиленовых групп:

После распада гидропероксида молекула лавсана
деструктируется. Могут образоваться также неустойчивые циклические оксиды и
пероксиды, которые при разрыве образуют карбонильные и карбоксильные группы,
ослабляющие эфирную связь и облегчающие ее разрыв. Окисление полимеров в твердой
фазе осложняется трудностями диффузии кислорода и надмолекулярными эффектами.
Наиболее устойчивы к термоокислительной деструкции фторорганические и
кремнийорганические полимеры (табл.5).
Таблица 5.
Стойкость полимеров к термоокислительной деструкции.
|
Полимер
|
Потери массы за
34 ч при 350оС на воздухе, %
|
|
Политрифторхлорэтилен
|
98,9
|
|
Полиамид
(капрон)
|
94,3
|
|
Полиэтилентерефталат
(лавсан)
|
91,2
|
|
Полиакрилонитрил
|
72,0
|
|
Фенолоформальдегидная
смола
|
68,0
|
|
Полидиметилфенилсилоксан
|
22,8
|
|
Полидиметилфенилалюмосилоксан
|
|
Политетрафторэтилен
(тефлон)
|
2,1
|
Окислительный и термический виды деструкции, усиливающиеся
при одновременном действии света и протекающие по механизму цепных радикальных
реакций, являются одной из основных причин старения полимеров, поэтому проблема
их защиты является комплексной. Меры защиты полимеров в первую очередь
направлены на подавление этих реакций. Высокомолекулярная природа полимеров
позволяет использовать для их защиты малые добавки низкомолекулярных
ингибиторов, выполняющих функцию стабилизаторов. Они часто
называются еще противостарителями, так как служат в качестве антиоксидантов,
т.е. веществ, препятствующих развитию в полимерах окислительных реакций.
Окисление можно ингибировать двумя путями: обрывать цепной процесс путем
взаимодействия полимерных радикалов с молекулой или радикалами ингибитора и
исключать развитие реакций, приводящих к образованию таких радикалов. В
соответствии с этим по механизму действия все антиоксиданты делятся на две
группы:
· вещества, обрывающие окислительную цепь
реакций, т.е. ингибиторы, реагирующие со свободными радикалами на стадии их
образования;
· вещества, предотвращающие разложение
пероксидов по радикальному механизму, т.е. разрушающие гидропероксиды до
неактивных продуктов.
Антиоксиданты первой группы характеризуются наличием
в их молекуле подвижного атома, который отрывается и участвует в радикальных
реакциях легче, чем активные атомы водорода молекул полимера:

Образующиеся при этом свободные радикалы ингибитора
малоактивны и не могут вызвать продолжение цепи радикальных реакций.
Эффективность их действия зависит не только от их химического строения, но и от
типа полимера и температуры. В реакции ингибированного окислении ингибитор
расходуется:
ROO* + InH → ROOH + In*; R* + InH → RH + In*.
Концентрация пероксидных радикалов при этом резко снижается,
и цепной процесс окисления не развивается. Для того, чтобы эти реакции успешно
конкурировали с реакцией развития цепи, энергия связи In-H в ингибиторе должна быть
меньше энергии связи R-H в полимере. Радикал ингибитора In* малоактивен и не может
оторвать водород от полимера, поэтому дезактивирует полимерные радикалы или
дезактивируется сам:
ROO* + In*→ROO-In; R*
+ In*→R-In; In* + In* → In-In.
Защитное действие ингибитора характеризуется длиной
индукционного периода (ИП) на кинетической кривой окисления при данной
температуре. Существует его критическая концентрация (скр)
в полимере, ниже которой защитное действие не проявляется, и оптимальная
концентрация (сопт), при которой индукционный
период имеет наибольшую длину (рис.5). Ниже и выше оптимальной концентрации
величина индукционного периода снижается.


Рис. 5. Влияние концентрации ингибитора на ИП окисления
полимера (а) и кинетические кривые окисления полипропилена при 140оС
(б): 1-без ингибитора; 2-бензидин; 3-дифениламин; 4-фенил-β-нафтиламин.
Антиоксиданты второй группы (сульфиды,
дитиокарбаматы, тиофосфаты) отличаются от аминов и фенолов превентивным
действием, разлагают пероксиды и гидропероксиды с образованием стабильных
продуктов:
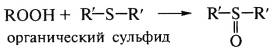 ,
,

Важным для практики является эффект взаимоусиления действия смеси
двух антиоксидантов первой и второй групп, называемый синергизмом
(рис.6). Если применить два слабых антиоксиданта из этих групп раздельно, то
величина индукционного периода будет невелика. Смесь же этих антиоксидантов при
постоянной общей концентрации увеличивает по сравнению с аддитивной величиной
индукционный период до максимума, достигаемого при их эквимолярном соотношении.
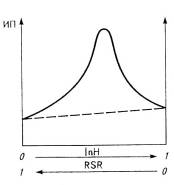

Рис.6. Проявления синергизма защитного действия аминного (InH) и сульфидного (RSR) антиоксидантов (а) и пассивации металлов переменной
валентности диэтилдитиокарбаматами (б): 1 - кадмия; 2 - никеля; 3-цинка;
4-аминный антиоксидант; 5-без добавки.
Особенно важно, что превентивные антиоксиданты образуют с солями
металлов переменной валентности комплексы, не влияющие на развитие
окислительной цепи. Амины и фенолы более эффективны при низких температурах
окисления, а превентивные антиоксиданты - при высоких.
Синергизм действия смеси амина и сульфида объясняется тем, что
первый компонент при обрыве цепи окисления образует гидропероксидную группу, а
второй разрушает ее, увеличивая время жизни амина, поскольку уменьшается число
окислительных цепей. Таким действием обладает также смесь ионола и
дилаурилтиодипропионата для стабилизации полипропилена или смесь фенола и
фосфата для стабилизации ненасыщенных полимеров. Превентивный антиоксидант
диэтилдитиокарбамат цинка образует комплекс, например, с ионом меди, который не
влияет на развитие окислительной цепи:
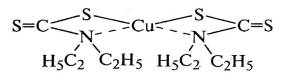
Кроме этих классов антиоксидантов, эффективно защищают от
окисления соединения с системами сопряжения, где электроны
делокализованы и свободно перемещаются по всей молекуле, и стабильные
радикалы, например азот-оксидные радикалы алифатических и ароматических
соединений:
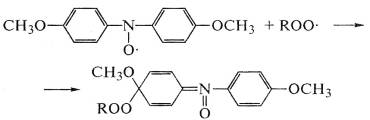 .
.
Последние обрывают окислительную цепь реакций, дезактивируя
радикалы ROO* и R*. Их применение рационально для полимеров,
которые не содержат подвижных атомов водорода в макромолекулах (полиформальдегид,
полиамид), так как азот-оксидные радикалы могут отрывать подвижный атом
водорода полимера и инициировать цепной процесс окисления. Современные методы
исследования дали возможность изучить механизмы старения и стабилизации
полимеров и предложить для многих из них меры комплексной защиты от теплового,
термоокислительного, светоозонного и радиационного старения. При этом
эффективность оценивается не только по активности противостарителей в
химических реакциях, но и по растворимости их в полимере, летучести,
термостабильности и другим факторам.
Полиэтилен устойчив к старению при эксплуатации, но для защиты его
от термодеструкции при переработке применяют небольшие количества (0,01%)
фенольных или аминных антиоксидантов, а для защиты его тонких пленок от
ультрафиолетового света - бисфенолы. Полипропилен защищают от старения при
эксплуатации фенилендиаминами. При стабилизации ненасыщенных полимеров
(каучуков) достаточно эффективны аминные противостарители или их сочетание с
превентивными антиоксидантами. Для стабилизации ПВХ применяют 5-6 компонентов -
стеараты свинца или кадмия (для поглощения HCl), бензофенолы (защита от ультрафиолетовых лучей), фосфиты
(разложение пероксидов) и вещества, связывающие продукты реакции этих
стабилизаторов.
Волокнообразующие полимеры (полиамиды и особенно полиэфиры)
достаточно термостойки, но от светового и ультрафиолетового излучения они
нуждаются в защите смесью диариламинов с аминокетонами. При освещении
полиамидных пленок образуются свободные радикалы и гидропероксиды:

Защита полиамидов от старения под действием солнечного света -
сложнейшая проблема, так как затем идут процессы деструкции макромолекул с
выделением оксида углерода, обнаруженного в продуктах их деструкции:
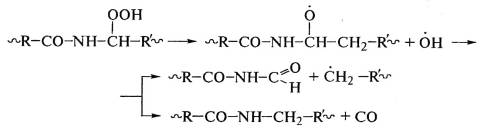 .
.
Полиэтилентерефталат устойчив к солнечному свету, но при
переработке в волокна протекает его термоокислительная деструкция с выделением
диоксида углерода, воды, формальдегида и уксусного альдегида:
 .
.
Поэтому его стабилизируют бисфенолами и ароматическими аминами,
которые обрывают окислительные цепи.
Из приведенных примеров видно, что проблема стабилизации полимеров
- весьма сложная и многоплановая.
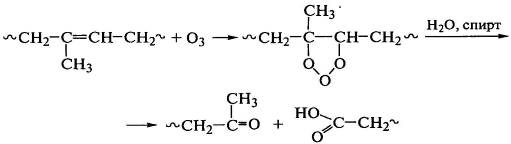
Озон является значительно более сильным окислителем диеновых полимеров
по сравнению с кислородом, присоединяясь к двойным связям основной цепи с
образованием циклических структур, которые перестраиваются в эпоксидные группы
с выделением молекулярного кислорода или распадаются по месту бывшей двойной
связи:
 .
.
Деструкция полидиена протекает по случайному закону как результат
атаки озоном двойной связи и последующего распада озонидов:
 .
.
По продуктам разложения озонидов можно судить о строении
ненасыщенного полимера. Скорость реакции озона с простой С-С-связью значительно
меньше, чем с двойной связью. Поэтому насыщенные полимеры на несколько порядков
более устойчивы к озону и используются при создании озоностойких полимерных
материалов. При этом разные полимеры реагируют по-разному. Полиизобутилен более
устойчив к озону, чем полиэтилен, полипропилен или полистирол.
Свободно-радикальная реакция начинается по схеме:
RH + O3 → R* + О2 + HO* или RO* + HO2* или RO2* + HO*.
Радикалы реагируют с насыщенными полимерами с образованием
алкильных радикалов R*, которые реагируют с кислородом по рассмотренным выше
схемам. С увеличением концентрации озона растет скорость этих реакций. В
полиэтилене, например, идут следующие реакции:

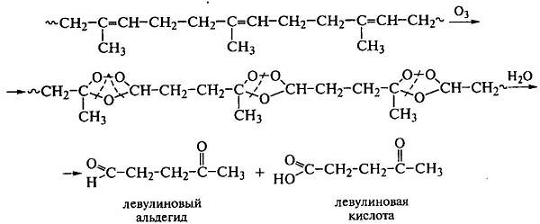
Наиболее уязвимыми для атаки озоном из углеводородных полимеров
являются полидиены и их сополимеры с виниловыми мономерами из-за большого
количества двойных связей. Реакции этих полимеров с озоном сопровождаются
деструкцией с образованием поверхностных и внутренних трещин и разрушением
материалов. Схема реакции полиизопрена с озоном:
 .
.
Из озонидов полиизопрена с большим количеством 1,4-звеньев
образуется много левулиновых производных. Обнаружение в продуктах реакции
диоксида углерода, малых количеств уксусной кислоты и альдегида, янтарной и
муравьиной кислот свидетельствует еще и о линейном строении макромолекул.
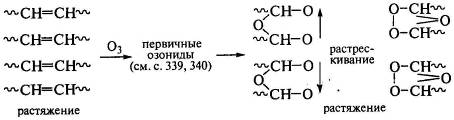
Особенно активно идут реакции в напряженных полимерах, в частности в пленках,
которые быстро растрескиваются, так как растет вероятность расхождения концов
распавшихся под действием озона макромолекул. Озониды перестраиваются под
действием напряжений, закрепляя структуру из двух соседних макромолекул с
трещиной между ними, что и приводит последующему быстрому разрушению этого
участка:
 .
.
В толстых изделиях сначала озонируется поверхностный слой,
растрескивание которого затем распространяется на внутренние слои полимера. Для
протекания этих процессов достаточно долей процента озона. Защита полимеров от
разрушающего действия озона осуществляется несколькими способами. Физические
способы защиты от проникновения озона включают нанесение покрытий из воска или
устойчивых к озону пленок. Химические способы состоят во введении в полимеры антиозонантов
(аналогично антиоксидантам) - соединений класса вторичных аминов и фенолов:
 .
.
6.
Формирование сетчатых структур в полимерах
Межмакромолекулярные реакции в полимерах занимают
особое место, так как создают новую сетчатую систему химически связанных друг с
другом макромолекул, не способных к растворению, проскальзываниям и
пластическим деформациям, т.е. к необратимым перемещениям при действии теплоты,
механических напряжений и растворителей. Образование сетчатых структур за счет
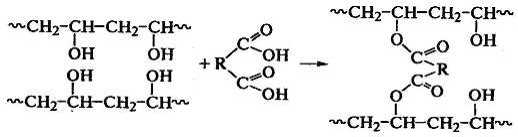
соединения макромолекул возможно по двум направлениям: реакции функциональных
групп разных макромолекул и свободнорадикальные реакции низкомолекулярных
веществ с макромолекулами полимера.
По функциональным группам сшивают
поливиниловый спирт двухосновными кислотами: или альдегидами:
 ,
,
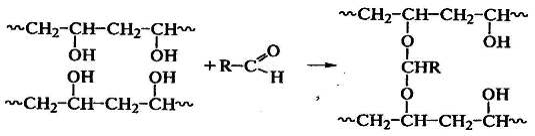
или проводят отверждение эпоксидных смол
диаминами:
 .
.
Реакции формальдегида с амидными группами белков или
полиамидов приводят к образованию ковалентных поперечных химических связей в
этих полимерах и используются в процессах дубления белков и кож:
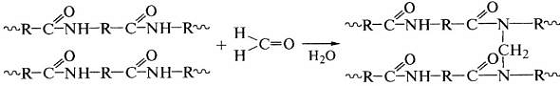 .
.
К формированию сетки приводит также сложная реакция
аллильного хлора в 1-2-звеньях полихлоропрена с оксидом или хлоридом цинка:
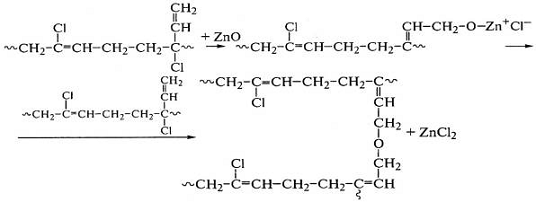
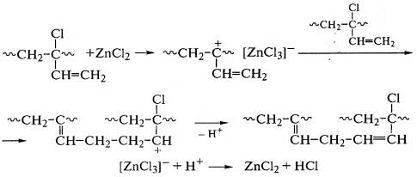
Свободнорадикальные реакции сшивания цис-1,4-полиизопрена
с дву-трет-бутилом используют в производстве теплостойких резин:
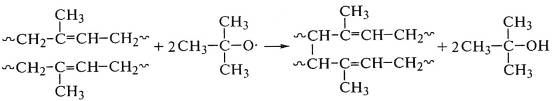
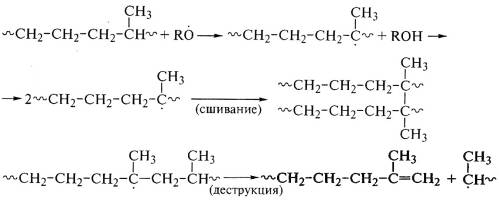
При взаимодействии этилен-пропиленового каучука с пероксидами
идут одновременно и сшивание, и деструкция макромолекулярных цепей:
Радикальные реакции пероксидных функциональных групп
позволяют при температуре 120-150оС формировать сетчатые структуры в
тройных сополимерах бутадиена и стирола с 2-10% диметилвинилэтинил-трет-бутилпероксида
(каучуки СКС-30П-2, СКС-25П-5 и СКС-20П-10) или их комбинациях с другими
диеновыми каучуками. Эластомерные пероксиды при температурах переработки до 100оС
по пласто-эластическим и технологическим свойствам идентичны
бутадиен-стирольному каучуку СКС-30АРК.
Дисульфиды способны сшивать полидиены в присутствии
серы и без нее:
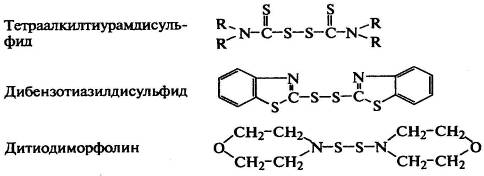 .
.
R-S-S-R ↔ 2RS*; КаН + RS* →Ка* + RSH; Ка* + Ка*
→ Ка-Ка (сшивка);
RS*+RSSR→RSR+RSS*; Ка*+RS*→КаSR; Ка*+RSS*→КаSSR (модификация цепи);
КаSSR→КаS*+RS*+КаН→КаS*+Ка*+RSH→КаSКа (сульфидная сшивка).
Продукты превращения дисульфидных ускорителей (меркаптаны RSH) могут присоединяться к
двойным связям эластомеров, т.е. модифицировать цепи.
Молекулярная сера является главным
вулканизующим агентом в составе большинства серно-ускорительных групп, а
реакции сшивания ненасыщенных каучуков серой наиболее интересны. Кроме
указанных выше дисульфидов, в качестве ускорителей применяют и другие вещества.
В отсутствие серы они не способны сшивать эластомеры, но образуют с серой промежуточные
активные полисульфиды, которые легко распадаются и сшивают каучук:
RSSR+S8→RSS8SR+КаН→КаSxSR+RS8-xH→ КаSx*+RS*+RS8-xH;
КаSx*+RS*+КаН→КаSx*+Ка*+RSH→КаSxКа (полисульфидная
сшивка);
RSH+S8→ RSxH→RS*x-y+S*yH;
КаН+Ка*+S*yH→
КаS*y-1+Ка*+H2S→
КаSy-1Ка (полисульфидная сшивка).
Промежуточные гидрополисульфиды могут также присоединяться к
двойным связям полидиенов, химически модифицируя макромолекулы. Из таких
ускорителей наиболее распространены
меркаптобензотиазол
и получаемые на его основе сульфенамидные производные:
циклогексил-бензотиазил-сульфенамид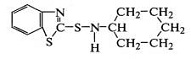 ,
,
трет-бутил-бензотиазил-сульфенамид  ,
,
морфолил-бензотиазил-сульфенамид  .
.
В присутствии ускорителей диэтилдитиокарбамата цинка и
дифенилгуанидина:
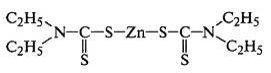
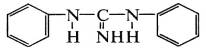
протекают ионные реакции с гетеролитическим распадом кольца
серы по схеме S8 → - S-S6-S+. В присутствии оксидов или жирнокислотных солей металлов
реакции распада ускорителей и получения активных промежуточных продуктов
протекают на их поверхности и имеют топохимический характер, что сказывается на
сетчатой структуре вулканизата:

При обобщенном подходе можно выделить три стадии сшивания
серой:
· взаимодействие серы, ускорителей, оксида
или соли цинка (или другого металла) между собой с образованием активных
промежуточных соединений полисульфидной природы (мономерные полисульфиды);
· взаимодействие промежуточных соединений с
макромолекулами эластомера по его реакционным центрам с образованием
реакционноспособных участков (подвесков) на макромолекулах (полимерные
полисульфиды);
· реакции активных центров между собой или с
активными центрами других макромолекул с образованием поперечных связей - тетрафункциональных
узлов сетки различной сульфидности. Общая схема этих реакций,
протекающих по свободнорадикальному механизму, приведена выше.
В общем виде кинетика вулканизации описывается кривой
изменения модуля сдвига, а изменение основных механических свойств -
экстремальными кривыми, по которым определяют оптимальное время вулканизации
(рис.7). При дальнейшем прогреве или эксплуатации изделий серные поперечные
связи могут распадаться, образовывать дополнительные узлы (поствулканизация)
или превращаться в циклические структуры в пределах одной макромолекулы и таким
образом уменьшать плотность химической сетки (реверсия).
 (а)
(а)  (б)
(б)
Рис.7. Периоды кинетики вулканизации (а):
1-индукционный; 2-главный; 3-реверсия. Изменения основных свойств эластомера
при вулканизации (б): 1-разрывная прочность; 2-напряжение при заданной
деформации; 3-относительное удлинение.
Наилучшим вариантом является сохранение сформированной сетки
при длительных тепловых и механических воздействиях (плато вулканизации).
Наиболее четко выраженное разделение индукционного периода образования
полисульфидов и главного периода сшивания поперечными связями на кинетической
кривой процесса вулканизации обеспечивают сульфенамидные ускорители с
серой (рис.8). Они наиболее эффективны в применении, т.к. удлиняют
индукционный период и повышают скорость структурирования, а достигнутая
плотность сетки сохраняется при дальнейшем прогреве системы.
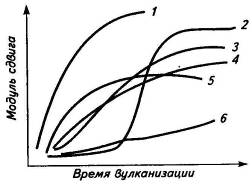
Рис.8. Кинетика серной вулканизации эластомеров в присутствии
ускорителей: 1-диалкилдитиокарбамат цинка; 2-сульфенамид;
3-дибензотиазилдисульфид; 4-дифенилгуанидин; 5-меркаптобензотиазол; 6-сера без
ускорителя.
Активаторы образуют в эластомерной матрице
нерастворимые или диспергированные мицеллярные структуры, в которых
адсорбируются и хемосорбируются реагенты и участки макромолекул по их активным
центрам. В результате этого реакции сшивания протекают в микрообъемах
образовавшихся реакторов, что усложняет их, придает им топохимический характер
и приводит к образованию, наряду с тетра - и гексафункциональными узлами сетки,
полифункциональных узлов. Таким образом, формирование сетчатых
структур в эластомерах серно-ускорительными системами представляет собой
комплекс сложных многостадийных химических реакций, в которых участвуют
практически все компоненты резиновой смеси.
Реакции формирования сетчатых структур из мономеров и
олигомеров с концевыми функциональными группами приводят к образованию более
совершенных сетчатых структур по сравнению с реакциями вулканизации, так как
фрагменты между узлами сетки задаются структурой мономера или олигомера и поэтому
являются более однородными.
При формировании таких сеток можно соотношением реагирующих
компонентов регулировать количество свободных концов вплоть до полного их
исключения. В реакциях олигомеров сетчатая структура формируется по схеме:
 .
.
Так реагируют конденсационноспособные олигодиендиолы и
триизоцианаты:
 .
.
В этом случае участки между узлами сетки образуют эластичные
блоки олигодиенов. Полимеризационноспособные олигомеры (олигоэфиракрилаты)
сшиваются за счет концевых двойных связей, которые раскрываются при нагревании
с пероксидом и формируют сетчатую структуру:
 .
.
Такие системы применяются при изготовлении изделий путем
пропитывания армирующего материала жидким олигомером, который после отверждения
образует прочную конструкцию практически любой сложной конфигурации. Они имеют
большое значение в технологии реактопластов, где создание
материала и изделия из него совмещено в одном процессе - переход из жидкого в
твердое состояние и формирование изделия. Можно объединить оба пути
формирования сеток и создать взаимопроникающие сетки на основе
двух материалов, каждый из которых образует сетку по своей реакции.
Кристаллические структуры из регулярно построенных
макромолекул могут придавать полимерам те же свойства, увеличивая их стойкость
к действию температур, напряжений и растворителей. Однако при длительном
действии этих факторов они разрушаются, и полимер становится способным к
необратимым деформациям. В сетчатых же структурах такая способность появляется
лишь при химическом распаде макромолекулярных цепей или поперечных связей, что
повышает предел эксплуатационной устойчивости полимера до температур его
химического разложения. В этом состоит принципиальное отличие сетки химических
связей в структуре полимера от флуктуационной сетки физических связей между его
макромолекулами.
В структуре сетчатого полимера участок соединения
макромолекул поперечными химическими связями называется узлом сетки,
или поперечной связью. Каждый узел оканчивается двумя сшитыми
звеньями двух разных макромолекул полимера. Если размер поперечной связи
совпадает с размером элементарного звена макромолекулы полимера, т.е. проявляет
себя как жесткое структурное образование, то понятия узла сетки и поперечной
связи совпадают. Если же поперечная связь по размеру существенно больше
элементарного звена и сегмента, то узлами сетки называются сшитые звенья. Число
узлов вдвое больше числа поперечных связей.
Определяющей характеристикой сетчатой структуры полимера
является размер (молекулярная масса) участка цепи между
двумя сшитыми звеньями. От него зависит проявление свойств
индивидуальных макромолекул в сетчатой структуре полимера. Если эти участки
значительно больше размеров сегмента макромолекулы, то сетчатый полимер
сохранит свойства исходного полимера (например, реакционноспособность и
высокоэластичность). Такой сетчатый полимер будет ограниченно набухать в тех же
растворителях. Если же размер участка цепи между сшитыми звеньями близок к
размеру сегмента или меньше его, то свойства исходного полимера существенно
изменяются: уменьшается гибкость цепи и связанная с ней высокоэластичность,
снижается или теряется способность к набуханию. Существенную роль в проявлении
механических свойств сетчатого полимера играют не вошедшие в сетку концы
исходных макромолекул, так называемые свободные концы сетки. При
деформации сетки они не несут нагрузки и являются разбавителями в сетчатой
структуре, повышая ее дефектность и снижая уровень механических свойств.
Индекс сшивания показывает, сколько
сшитых звеньев приходится на одну среднестатистическую макромолекулу сшиваемого
полимера, и представляет собой величину γ=Мо/Мс,
где Мо - исходная средняя молекулярная масса полимера
в момент его сшивания; Мс - средняя молекулярная масса
отрезка между сшитыми звеньями (узлами сетки). Обозначим b число исходных
макромолекул, образовавших заданный фрагмент сетчатой структуры из 8
макромолекул, т.е. b=8 (рис.9). Число узлов сетки (v), или число поперечных
связей между двумя разными макромолекулами, равно 10, т.е. v=10. Два параметра сетки
заданы произвольно, а остальные уже зависят от них. Число свободных концов у 8
макромолекул равно 16, так как по принятым условиям формирования сетки сшивание
прошло по внутренним составным звеньям макромолекул. Тогда число отрезков цепей
между узлами сетки (nc), равное удвоенному
числу узлов за вычетом числа свободных концов, можно подсчитать по формуле: nc=2v-2b=20-16=4. Величина 2v в формуле соответствует
числу сшитых звеньев макромолекул. Поскольку каждый узел сетки (поперечная
связь) образован двумя сшитыми звеньями двух разных макромолекул, число сшитых
звеньев равно удвоенному числу узлов.
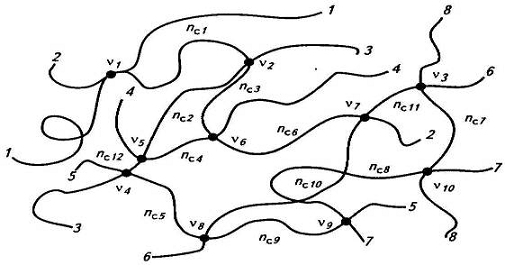
Рис. 9. Схема статистической полимерной сетки на примере
эластомера, подвергнутого пероксидному или радиационному сшиванию.
Свободные концы сетки относятся к дефектам ее структуры, так
как сопротивление внешним воздействиям оказывает лишь "активная"
часть сетки, расположенная между ее узлами. Вторым видом дефектов сетки
являются зацепления, которые при деформации играют роль
дополнительных узлов сетки, а в недеформированном состоянии участвуют в
тепловом движении наряду с другими участками цепей. Ограничение
подвижности участков цепей сетки у химических узлов математически
описано еще П. Флори. Зацепления физической природы (флуктуационная сетка)
вносят существенный вклад в свойства исходных полимеров и их сетчатых структур.
Среднее значение молекулярной массы между зацеплениями в расплаве линейного
полиэтилена составляет 4000, цис-1,4-полибутадиена - 7000, цис-1,4-полиизопрена
- 14000, полиизобутилена - 17000 и полистирола - 35000. Эти показатели
определяют механические свойства несшитых полимеров.
Важный показатель сетки сшитых полимеров - функциональность
узлов, которая определяется количеством эластически активных цепей,
исходящих из одного узла. В соответствии с этим узлы сетки делятся на три-,
тетра-, гекса - и в целом полифункциональные узлы (рис.10). Функциональ-ность
узлов как показатель входит в уравнения различных моделей сетчатых структур и
существенно влияет на зависимости свойств (модули, разрывная прочность,
разрывное удлинение и др.) от числа узлов сетки. Более всего такие зависимости
относятся к тетрафункциональным узлам, которые в простейшем случае представляют
собой С-С-связи между двумя составными звеньями сшитых макромолекул эластомеров
(реже - пластиков). Они образуются при радиационном и пероксидном сшивании
углеводородных полимеров.
Рис.10. Функциональность узлов в сетчатых полимерах: а-трехфункциональный
узел; б-тетрафункциональный узел; в-гексафункциональный узел; г-полифункциональный
узел.
Для достаточно плотно сшитых сеток, когда nc намного больше b, влиянием свободных
концов на структуру сетки можно пренебречь. Тогда для плотных сеток nc=2v, т.е. число отрезков
цепей между узлами сетчатой структуры равно удвоенному числу узлов сетки, и все
основные свойства сетчатой структуры определяются этим параметром. Так, модуль
сдвига при растяжении такой сетки прямо пропорционален nc или v. Эти положения
справедливы для сетчатых структур, в которых межмолекулярное взаимодействие
между узлами сетки мало и не влияет на свойства эластомеров. Если же
межмолекулярное взаимодействие велико (пластики, волокна), то оно и определяет
модуль сетки, на величину которого число химических узлов уже не влияет. С
повышением температуры тепловое движение сегментов макромолекул преодолевает
межмолекулярное взаимодействие, и механические свойства сетки определяются
числом ее узлов.
Любую систему макромолекул полимеров можно характеризовать
молекулярным, надмолекулярным и топологическим уровнем ее организации.
Молекулярный уровень организации полимеров -
это элементный состав повторяющихся звеньев макромолекул, их стереохимические
характеристики по расположению заместителей у основной цепи полимера или
расположению структурных элементов цепи относительно двойных связей, порядок
чередования химически и стереохимически различающихся звеньев в макромолекуле,
вид их присоединения, характер концевых групп макромолекул и их распределение.
Эти параметры определяются условиями получения полимера и механизмом синтеза
макромолекул.
Надмолекулярный уровень организации учитывает
межмолекулярное взаимодействие макромолекул друг с другом, степень
упорядоченности их взаимного расположения, что определяется уже не только
способом синтеза полимера, но и способом его переработки.
Топологический уровень организации полимера
характеризуется функцией молекулярно-массового распределения, а разветвленный и
сетчатый полимер - еще и функцией участков цепей между узлами разветвления или
сетки. Он является показателем связности элементов структуры полимера,
отвлекается от конкретного химического строения макромолекул и расположения их
элементов и поэтому может быть количественно описан.
Существуют два заметно различающиеся подхода к рассмотрению
теории эластичности: а) с позиций статистической механики, когда учитывается
только топология макромолекулярных цепей без их химического строения; б) с
учетом химического строения и конфигураций цепей. Первый подход используется в
двух классических моделях сетчатых эластомеров, которые сформулированы в 40-х
годах прошлого века, - аффинной и фантомной моделях сетки.
Аффинная модель (Кун) предполагает, что
узлы сетки перемещаются аффинно, т.е. так же, как при макроскопической
деформации всего образца эластомера. Согласно этой модели, которая недостаточно
подтверждена экспериментом, приращение свободной энергии эластической
деформации ΔАэл. эфф=1/2nckT (λx2+λy2+λz2-3), где nc плотность узлов сетки; k константа Больцмана; T абсолютная температура; λx, λy, λz - деформации в трех
измерениях.
Поэтому была сформулирована фантомная модель
(Джеймс, Гут), которая предполагает асимметричные флуктуации узлов сетки при ее
растяжении: ΔАэл. фант=1/2ξkT (λx2+λy2+λz2-3). Цепи сетки по
этой модели испытывают меньшие деформации, поэтому и величина модуля в ней
меньше, чем по аффинной модели. Для рассматриваемого случая сетки с
тетрафункциональными узлами, где каждый узел связывает 4 эластически активные
отрезка цепей, величина ξ=nc/2 названа фронт-фактором
фантомной модели сетки. Таким образом, аффинная и фантомная модели различаются
только множителем в формуле модуля (nckT или ξkT).
Большинство молекулярных теорий сетчатых эластомеров основано
на гауссовом распределении расстояний между узлами сетки. Однако
молекулярно-массовые распределения растянутых или коротких отрезков цепей
заметно отклоняются от функции Гаусса. Поэтому существуют еще и более сложные негауссовы
модели сеток. В 60-70-х годах в развитие теории фантомных сеток в
работах Флори предложено учитывать в выражении для свободной энергии
высокоэластической деформации вклад зацеплений в ограничения подвижности узлов
сетки. В диффузионно-ограничительной теории предлагается
распределение ограничений по контуру цепей. Развитие представлений о
негауссовой статистике рассмотрено Марком на сетчатых структурах из модельных
силоксановых олигомеров заданной структуры, состоящих из коротких и длинных
участков цепей между узлами.
Эпохальным событием в технологии переработки эластомеров было
открытие явления усиления эластомеров техуглеродом (1892г), превратившего
резину в уникальный конструкционный материал для техники 20-го века. С
применением техуглерода значительно повысились износостойкость, твердость и
другие важные свойства изделий из эластомеров и более чем на порядок -
прочность резины на основе некристаллизующихся каучуков. Причины этих эффектов
долгое время связывались с взаимодействиями на межфазной границе. К настоящему
времени установлена причастность явления усиления эластомеров к химическим
реакциям формирования сетчатых структур.
Наименьшей дисперсной единицей техуглерода при смешении с
эластомерами являются агрегаты химических сросшихся сферических
частиц диаметром 9-320 нм, входящие в состав более крупных физических
образований вторичной структурности - агломератов.
С увеличением степени разветвленности (открытости) агрегатов и количества
частиц в них растет первичная структурность техуглерода, напрямую
связанная с объемом межагрегатных пустот в агломератах, которые
при смешении способны окклюдировать эластомер (заполняться его
сегментами). На поверхности агрегатов содержатся ароматические соединения с
системой сопряженных двойных связей, включая графитоподобные плоскости, а также
могут находиться фрагменты неполностью разложившегося углеводородного сырья,
кислородсодержащие функциональные группы и различные по размеру поры.
При диспергировании техуглерод физически
связывает часть эластомера, уменьшая воздействием своей поверхности его
сегментальную подвижность до состояния, близкого к застеклованному (связанный
эластомер), и окклюдируя в межагрегатных пустотах (окклюдированный
эластомер). При воздействии сдвиговых напряжений часть макромолекул
связываемого эластомера разрывается, а образующиеся из них активные осколки
(макрорадикалы) инициируют реакции сшивания и прививки сшитых структур к
техуглероду с образованием нерастворимого углерод-каучукового геля.
В результате физических и механохимических взаимодействий с участием техуглерода
в эластомерной матрице формируется дисперсная фаза коллоидных углерод-каучуковых
частиц, окруженных переходными слоями пространственно сшитого
эластомера. Связанный эластомер на межфазной границе и
окклюдированный эластомер в межагрегатных пустотах образуют внутри частиц межфазную
область, существенно отличающуюся меньшей подвижностью и физическими
свойствами от эластомерной матрицы.
Углерод-каучуковые частицы способны физически
взаимодействовать с эластомером матрицы, набухая в его более подвижных сегментах,
или между собой с образованием цепочечных структур, когда высокая степень
механохимического сшивания препятствует их набуханию в матрице. Взаимодействия
частиц по сегментальному (с матрицей) или цепочечному
(между собой) механизмам с образованием более слабых сеток,
диссипируют (выравнивают) перенапряже-ния, которые возникают в макромолекулах
при механических воздействиях. Таким образом, техуглерод формирует в эластомере
своеобразную и сложную надмолекулярную структуру в виде микрогетерогенной
сетки проходных макромолекул, в которой углерод-каучуковые частицы
являются ее узлами, способными к слабым физическим взаимодействиям в
эластомерной матрице.
Электронообменными взаимодействиями полисопряженных структур
с азотсодержащими ускорителями вулканизации углеродная поверхность локализует
в межфазной области реакции сшивания, превращающие углерод-каучуковые частицы в
полифункциональные химические узлы сетки, которые сохраняют способность к
указанным выше физическим взаимодействиям в эластомерной матрице. Локализация в
частицах реакций сшивания приводит:
· к значительному увеличению
функциональности и прочности узлов сетки;
· к переходу единичных химических поперечных
связей (цепей) из категории прочных тетрафункциональных узлов в слабые элементы
полифункциональных узлов, которые своим разрушением при деформации резины
выравнивают напряжения в оставшихся цепях и их пучках;
· к многоуровневому характеру структуры
узлов сетки, придающему им способность деформироваться при
деформации резины и диссипировать перенапряжения с межфазной границы. Последнее
дает возможность направленного улучшения свойств резины путем изменения
конструкции узлов.
Деформируемость узлов повышается с ростом
структурности техуглерода, что увеличивает их каучукоемкость, а также с
применением гидрированных бутадиен-нитрильных каучуков, позволяющих уменьшить
долю химического и повышать долю полярного взаимодействия на межфазной границе.
Площадь межфазной границы в каучукоемком узле растет при комбинировании
высокоструктурного техуглерода с низкоструктурным, агрегаты которого способны
внедряться вместе со связанным каучуком в межагрегатные пустоты другого.
Указанные изменения структуры узлов позволяют увеличить статическую
прочность резин до 45 МПа, т.е. в полтора раза превзойти уровень
серийных резин. В межагрегатных пустотах техуглерода могут окклюдироваться
высокомолекулярные полярные модификаторы, которые уменьшают деформируемость
узлов, повышая тем самым динамическую выносливость резин на
основе СКИ-3 в особо жестких условиях эксплуатации.
В последние годы техуглерод на 75% заменяют в протекторе
экологически чистых "зеленых" шин коллоидной кремнекислотой. При этом
техуглерод, являясь сильным донором электронов кремнекислоты, облегчает ее
диспергирование в эластомере. Вхождение кремнекислоты в сетчатую структуру
агрегатов техуглерода в полифункциональных узлах сетки снижает деформируемость
этих узлов и уровень гистерезисных потерь, расходуемых на преодоление трения
между частицами наполнителей при эксплуатации шин. Новые возможности направленного
изменения структуры резины открывают нанонаполнители (соевая мука, слоистые
каолины, нанокарбонат кальция, углеродные нанотрубки, фуллерены и др.), которые
вводят в каучук при его синтезе на стадии водной дисперсии или раствора
мономеров.
Таким образом, изменение структуры эластомерных сеток с
помощью наполнителей и нанонаполнителей становится важным направлением развития
химии и технологии переработки полимеров, позволяющим получать конструкционные
материалы с принципиально новыми свойствами. Более подробно эти важные для
химии и технологии полимеры и материалы на их основе рассмотрены в
рекомендуемых литературных источниках.
Библиографический
список
1. Гаришин
О.К., Свистков А.Л., Герасин В.А., Гусева М.А. Моделирование
упругопластического поведения полиолефиновых нанокомпозитов с различной
структурой слоистого наполнителя. // Высокомол. соед., А. 2009. Т.51. №4. -
С.610-619.
2. Никитин
Ю.Н., Ходакова С.Я., Гиренко М.М., Корнев А.Е. Современные подходы к решению
проблемы усиления эластомеров. // Каучук и резина. 2008. №1. - С.33-39.
. Свистков
А.Л., Lauke B. Структурно-феноменологическое моделирование механического
поведения резин. // Высокомол. соед., А. 2008. Т.50. №5. - С.892.
. Никитин
Ю.Н., Ходакова С.Я. О взаимосвязи усиления с вулканизацией эластомеров. //
Каучук и резина. 2008. №2. - С.14-15.
. Межуев
С.В. Разработка технологии и организация производства полимерных композиционных
материалов на основе нанокомпозитов с повышенным в 1,5-2 раза сроком
эксплуатации. // Российские нанотехнологии. 2007. Т.2. №1-2. С.41.
. Хайретдинов
М.Г. Динамические термоэластопласты "кварта". // Химия и бизнес.
2006. №7-8. С.26.
. Морозов
И.А., Свистков А. Л.,Heinrich G., Lauke B. Структура каркаса из агрегатов частиц технического углерода
в наполненных эластомерных материалах. // Высокомол. соед., А. 2007. Т.49. №3.
- С.456; Kautsch.
Gummi. Kunststoffe. 2006. Bd.59. - S.642.
9. Kлюппель M., Хайнрих Г. Физика и технология усиленных эластомеров: от
молекулярных механизмов к применению в промышленности. // Мир шин. 2005. №12. -
С.22; Kautsch. Gummi, Kunststoffe. 2005. Bd.58. №5. - S.217-224.
. Гамлицкий
Ю.А., Швачич М.В. Методы описания напряженно-деформированного состояния и
прогнозирования поведения в эксплуатации шин. // Проблемы шин и резинокордных
композитов. 2005. №4. - С.51.
. Гамлицкий
Ю.А., Швачич М.В. Прочность резины. Модель и расчет. // Высокомол. соед., Серия
А. 2005. Т.47. №4. - С.660.
12. Гаришин О.К., Мошев
В.В. Структурная перестройка дисперсно-наполненных эластомерных композитов и ее
влияние на их механические свойства. // Высокомол. соед., А.
<http://elibrary.ru/issues.asp?id=7756> 2005. Т.47. № 4. - С.669.
. Любартович С.А.,
Шуманов Л.А., Веселов И.В. Полиуретановые шины. // Проблемы шин и резинокордных
композитов. 2005. №3. - С.5.
. Любартович С.А.,
Шуманов Л.А., Басс Ю.П. Резиновые шины с резиновым протектором и полиуретановым
каркасом. // Проблемы шин и резинокордных композитов. 2005. №2. - С.31.
. Гамлицкий Ю.А., Басс
Ю.П. К описанию явления усиления наполненных эластомеров. // ИФЖ. 2003. Т.76.
№3. - С.1.