Качественный и количественный анализ образца сплава
Министерство
по образованию РФ
Государственное
Образовательное Учреждение Высшего Профессионального Образования
Липецкий
Государственный Технический Университет
Кафедра
химии.
“Качественный
и количественный анализ образца сплава”
Выполнил: Паршин Ю.В.
Проверила: д.х.н., проф.
Ермолаева Т. Н.
Липецк
2010
Содержание
Введение
. Качественный
анализ
. Литературный
обзор
.1 Краткая
справка
.2 Химические
методы количественного определения кобальта
.2.1 Гравиметрические
методы
.2.2 Титриметрические
методы
.3 Химические
методы количественного определения вольфрама
.3.1 Титриметрические
методы
.3.2 Гравиметрические
методы
. Экспериментальная
часть
.1
Гравиметрическое определение кобальта
.2 Титриметрическое
определение вольфрама
Выводы
Литература
Введение
Цель работы.
Целью данной работы является выполнение
качественного и количественного анализа предложенного образца, а именно,
определение его состава и нахождение процентного содержания основных
составляющих элементов исследуемого образца по выбранным методикам.
1. Качественный анализ
Стружка магнитится, следовательно, в сплаве
находится железо.
В HCl
при нагревании сплав растворяется не полностью, остаются частички сплава.
При добавлении HNO3
сплав растворяется, но на дне колбы остается мелкий черный кристаллический
осадок. При добавлении Н2SO4
осадок растворяется полностью. Раствор приобретает зеленую окраску. Это
свидетельствует о присутствии хрома.
В ходе анализа было определено, что данный сплав
является сплавом железа. Металлы, которые могут содержаться в этом сплаве: Ni,
Cr, Mn,
Co, Cu,
Al, Ti,
V, Mo,
W.
1. WO3œ+HNO3"H2WO4œ+NO2 Выпадает
черный осадок.
Выпадает
черный осадок.
2. Cr3++NaOH"Cr(OH)3œ
(желто-зеленый осадок).
3. 2VO43-+3H+"V2O6(OH)3-+H2O
Раствор становится красно-коричневым, что
свидетельствует о присутствии ванадия.
4. MoO42-+SnCl42-+12H++2Cl-+10NCS-"2MoO(SCN)52-+6H2O
Образуется комплекс интенсивно-красного цвета,
что свидетельствует о присутствии молибдена
5. Co(CH3COO)2+7KNO2+2CH3COOH=K2Co(NO2)6œ+NO+H2O+4CH3COOK
Выпадает желтый осадок, что свидетельствует о
наличии кобальта.
2Mn(VO3)2+5PbO2+6HNO3=2HMnO4+5Pb(NO3)2+2H2O
Раствор становится красно-фиолетовым, что
свидетельствует о наличии марганца.
6. Fe(OH)3+HNO3+CH3COONa
кипение
Fe(OH)2CH3COOœ(желтый)
. Ni2++2C4H8N2"NiC8H14N4+2H+
Образуется комплексное соединение ало-красного
цвета, что свидетельствует о наличии никеля.
8. Cu2++4NH3"[Cu(NH3)4]2+
Раствор приобретает синюю окраску.
Вывод: сплав содержит Fe,
Ni, Cr,
Mn, Co,
Cu, V,
Mo, W.
Схема качественного определения состава данного
объекта.
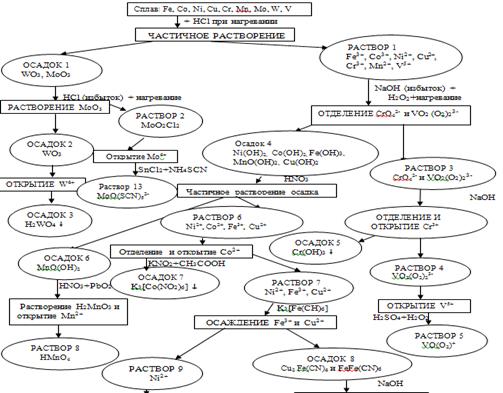
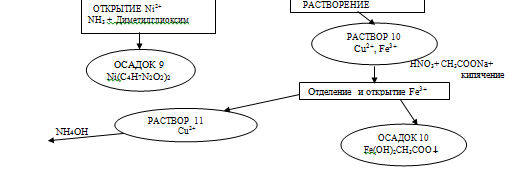
2. Литературный обзор
.1 Краткая справка
Возможны два пути приготовления пробы к анализу:
) Растворить точно взвешенное количество
сплава, довести до метки в мерной колбе и далее отбирать аликвоты на
титрометрический и гравиметрический анализ;
) Титрометрическое и гравиметрическое
определение проводить из отдельных частей образца.
В данной работе был выбран второй вариант.
Следующим этапом является растворение пробы. Обе части образца растворились в
смеси соляной, серной и азотной кислот.
.2 Химические методы количественного определения
кобальта
.2.1 Гравиметрические методы
Наиболее часто стали и сплавы кобальта
растворяют в соляной или серной кислоте и смеси этих кислот с азотной кислотой.
Рекомендуется применять хлорную кислоту в смеси с азотной, в частности, в тех
случаях, когда сталь содержит хром или ванадий, и если предполагают кобальт
титровать раствором феррицианида калия. Смесь азотной и хлорной кислот окисляет
хром и ванадий до высших степеней окисления, чем устраняется их мешающее
влияние при титровании кобальта феррицианидом. Высоколегированные стали с
высоким содержанием хрома растворяют в разбавленной серной кислоте или в смеси последней
с фосфорной кислотой, после чего прибавляют для окисления двухвалентного железа
азотную кислоту.
Для отделения мешающих определению элементов от
кобальта или для устранения их влияния применяют следующие методы:
) Осаждение железа и хрома (и др. элементов)
окисью цинка, карбонатом бария или пиридином в присутствии хлоргидрата
пиридина;
) Связывание железа в пирофосфатный,
тартратный или оксалатный (вместе с медью) комплексы;
) Окисление хрома и ванадия хлорной
кислотой до ионов хромата и ванадата;
) Осаждение марганца в виде двуокиси
смесью азотной и хлорноватой кислот;
) Осаждение кобальта в виде K2Na[Co(NO2)];
) Осаждение кобальта
фенилтиогидантонновой кислотой или тиогликолевой кислотой.
). Отделение железа от кобальта. К раствору,
содержащему ионы трехвалентного железа и двухвалентного кобальта, прибавляют
раствор нитрата кальция, раствор нитрата аммония до концентрации 5 моль/л и
концентрированный раствор гидроокиси аммония до рН 9 и осаждают ионы кальция
раствором двузамещенного фосфата аммония. Осадок промывают раствором нитрата
аммония с рН 9. Железо полностью соосаждается с фосфатом кальция, в то время
как кобальт остается в растворе.
). Анализируемый материал должен содержать от
0,5 до 4 мг кобальта. Если отношение железа к кобальту больше, чем 10:1, то
железо предварительно экстрагируют диэтиловым эфиром из солянокислого раствора.
Раствор делают слабокислым и разбавляют до 200 мл. Далее прибавляют 10 мл
10%-ного раствора щавелевой кислоты, затем нейтрализуют раствором гидроокиси
аммонии и прибавляют некоторый избыток последней. Кипятят и осадок, содержащий
железо, отфильтровывают. Кобальт остается в растворе.
9).Отделение кобальта от железа
фторидом натрия основано на образовании плотного кристаллического осадка 5NaF
• 2FeF3
при прибавлении раствора фторида натрия к не содержащему свободных минеральных
кислот раствору соли железа. Осадок занимает небольшой объем. Кобальт полностью
остается в растворе. Метод применяют при анализе железных руд на кобальт.
).Отделение меди, ванадия,
вольфрама и молибдена от кобальта. К раствору солей, содержащему приблизительно
0,5 г. окислов металлов III
группы, прибавляют 20-25 г. хлорида аммония и нейтрализуют раствор аммиаком.
Объем раствора доводят до 150 мл, прибавляют бумажную массу и нагревают раствор
до 60° С. Далее прибавляют по каплям 6 мл солянокислого пиридина и начинают
пропускать умеренный ток сероводорода. Спустя 10-15 мин., прибавляют из
капельной воронки по каплям при взбалтывании 7 г уротропина в виде раствора (35
мл 20%-ного раствора), колбу переносят на электрическую плитку и продолжают
пропускать сероводород еще 45- 60 мин., не доводя раствор до кипения. Далее
снимают колбу с плитки и пропускают сероводород до полного охлаждения раствора
до комнатной температуры. Осадок сульфида отфильтровывают и промывают 3%-ным
раствором нитрата аммония, к которому прибавлено 0,5 мл свежеприготовленного
раствора сульфида аммония на каждые 100 мл. Сульфиды выделяются в плотной
кристаллической форме, хорошо отфильтровываются и практически не окисляются на
воздухе.
CoSО4+Na2WО4=CoWO4
+ Na2SO4.
Состав осадка соответствует
формуле. Метод применим в случае растворов, не содержащих других ионов,
образующих малорастворимые вольфраматы. Оптимальные условия осаждения: рН около
8, небольшой избыток раствора вольфрамата натрия, раствор должен содержать
около 50% метилового спирта. Осадок промывают после отстаивания в течение часа
50%-ным раствором метанола и сушат 3 часа при 1250 С. При
определении от 0,006 до 0,45 мг кобальта ошибка лежит в пределах от -2,38 до
0,84%.
Осаждение щавелевой кислотой.
Щавелевая кислота образует малорастворимые оксалаты с катионами многих
металлов. Оксалат аммония при рН -8 полностью осаждает ионы кальция, стронция,
скандия, иттрия, лантана, редкоземельных элементов, актиния, железа, золота,
висмута, индия, олова, ниобия, тантала; частично осаждает ионы лития, бериллия,
магния, бария, радия, титана, циркония, гафния, тория, марганца, кобальта, никеля,
ртути, таллия и свинца. При некоторых условиях осаждаются также ванадий и
вольфрам. При рН 3-4 полностью осаждаются ионы кальция, стронция, скандия,
иттрия, лантана, редкоземельных элементов, актиния, тория и золота; не
полностью осаждаются ионы бария, тантала, марганца, кобальта, никеля, меди,
серебра, цинка, кадмия, олова, свинца и висмута.
Определение кобальта в форме
оксалата неселективно. Весовая форма - СоС2О4'2Н2О
(после высушивания осадка при температуре 100-105°С). Кобальт осаждают из
уксуснокислых растворов прибавлением избытка раствора оксалата аммония. Осадок
промывают 50%-ным этанолом.
Определение в форме CoSO4.
Осадок нитрозонафтола кобальта прокаливают до Со3О4,
остаток в тигле смачивают 1-2 мл концентрированной азотной или соляной кислоты,
осторожно нагревают до растворения Со3О4 и удаления
избытка кислоты и охлаждают. Затем прибавляют 0,3-1 мл раствора серной кислоты,
осторожно нагревают на воздушной бане до удаления всей серной кислоты и
прокаливают несколько минут при температуре около 5000 С. В
охлажденный тигель приливают 1-2 капли воды, снова осторожно выпаривают и
прокаливают для удаления последних следов серной кислоты.
При отделении кобальта
фенилтиогидантоиновой кислотой полученный осадок переводят в сульфат
нагреванием с серной кислотой и перекисью водорода.
Если кобальт отделяют от других
металлов осаждением раствором нитрита калия, то промытый осадок
гексанитрокобальтната калия переносят вместе с фильтром в стакан, прибавляют
смесь серной и азотной кислот и выпаривают до появления паров серной кислоты;
снова прибавляют раствор азотной кислоты и выпаривают. Далее поступают, как в
предыдущем случае.
сплав
проба кобальт железо
2.2.2 Титриметрические методы
). Определение кобальта после
осаждения в виде гидроокиси трехвалентного кобальта. Метод основан на следующих
реакциях:
2CoS04+4NaOH
-H202=2Co(OH)3 + 2Na2S04;
2Co(OH)3+2FeS04+3H2S04=2CoS04+Fe2(S04)3+6H2О
К анализируемому раствору,
содержащему 80-100 мг кобальта, прибавляют 30 мл 0,2 N
раствора едкого натра и 80 мл 3% -ного раствора перекиси водорода, кипятят 25
мин., вводят 30 мл 0.1 N
раствора соли Мора в 6 N серной кислоте и 15 мл 5' N раствора серной кислоты,
перемешивают, охлаждают и оттитровывают избыток ионов двухвалентного железа 0,1
N раствором перманганата. Метод применим для определения кобальта в присутствии
небольших (до 10% от содержания кобальта) количеств железа и никеля.
). К нейтральному
приблизительно 0,05 М раствору соли кобальта приливают 2 мл ледяной уксусной
кислоты, 5 мл 20%-ного раствора ацетата аммония, 10 мл 1 М раствора оксалата
калия и вводят I г двуокиси
Свинца, не содержащей марганца. Через 5-10 мин. фильтруют, промывают остаток
двуокиси свинца. Прибавляют к фильтрату вместе с промывными водами титрованный
раствор FeS04
в небольшом избытке, что видно по переходу окраски из зеленой в желтую. Через 5
мин. прибавляют 1 мл раствора, содержащего 10 мл фосфорной кислоты и 25 мл
серной кислоты в 100 мл, и разбавляют водой приблизительно до 100 мл.
Прибавляют 3 капли 1%-ного раствора дифениламина и титруют 0,05 N
раствором бихромата калия. На титрование не должно пойти более 1 мл. Отмечают
показания обеих бюреток (с растворами FeSO4
и К2СГ2О7), приливают к оттитрованному
раствору столько раствора FeSC4,
сколько это приблизительно отвечает израсходованному на обратное титрование
объему раствора бихромата, и вторично титруют бихроматом.
3). Броматометрическое
определение кобальта после осаждения в виде 8-оксихинолината.
Осадок 8-оксихинолината
кобальта малопригоден для гравиметрического определения. Лучшие результаты дает
броматометрическое титрование 8-оксихинолина, связанного с кобальтом в осадке.
К нейтральному или очень
слабому уксуснокислому раствору соли кобальта. содержащему ацетат натрия (рН
5,1-5,2), прибавляют при 60° С небольшой избыток уксуснокислого раствора
8-оксихинолина. Раствор со светло-коричневым осадком 8-оксихинолината кобальта
кипятят до тех пор, пока осадок примет мясо-красный цвет и кристаллическую
форму. Затем осадок отфильтровывают, промывают теплой водой, растворяют в
15%-ном растворе соляной кислоты и титруют 0,1 N растворам бромид-брамата.
Определению кобальта этим
методом мешает большое число других ионов, осаждающихся 8-оксихинолином при
данных условиях.
2.3 Химические методы количественного
определения вольфрама
.3.1 Титриметрические методы
Титриметрические методы определения вольфрама
можно разделить на четыре группы: 1) методы кислотно-основного титрования; 2)
методы, использующие реакции осаждения, многие из которых применимы при
гравиметрическом определении вольфрама; 3) методы, основанные на реакциях
окисления- восстановления; 4) методы комплексообразования. Предложены косвенные
методы, основанные на различных принципах.
Конечную точку титрования
устанавливают визуально, потенциометрически и амперометрически. Последние два
способа, позволяющие определять вольфрам в непрозрачных и окрашенных растворах,
более объективны. Амперометрическое титрование очень перспективно, поскольку,
сочетая высокую чувствительность, объективность к экспрессивность, кроме того,
позволяет повысить селективность определения подбором фона и потенциала для
титрования.
Методы основанные на
кислотно-основных реакциях.
).Навеску вольфрамовой кислоты
0,2 г растворяют в избытке щелочи при нагревании в мерной колбе емкостью 50 мл.
К горячему раствору добавляют каплю раствора фенолфталеина, 3,5 М HCl
до обесцвечивания и снова NаОН до бледно-розовой окраски. Раствор охлаждают и
разбавляют водой до объема 50 мл. К 25 мл раствора прибавляют 0,2 М НС1 до рН
7.8 и 0,5г. маннита и титруют 0,2 М HC1
до рН 4,4 (величину рН контролируют по рН-метру).
).Навеску вольфрамата натрия
растворяют в 10-15 мл воды в колбе для титровании, добавляют по 0,15 г маннита
на каждые 100 мг вольфрамата, 1-2 капли раствора метилового красного и титруют
0,05 М НС1 до изменения окраски из красной в желтую.
Практически аналогичен метод,
позволяющий определять >2,5 10-3 г-ион/л W(VI)
с ошибкой <0,1% в вольфраматах щелочных металлов и вольфрамовом ангидриде.
).Для определения вольфрама в
вольфраматах щелочных металлов навеску 5.000г образца растворяют в конической
колбе емкостью 250 мл в 100 мг воды, вводят одну каплю раствора фенолфталеина и
подщелачивают раствором NaОH.
Титруют 1,0 Н H2SO4
до исчезновения розовой окраски, замечают объем H2SO4
в бюретке, вводят 20 г маннита, 10 капель раствора метилового красного и
титруют раствором 1,0 Н Н2О, до появления розовой окраски.
Параллельно проводят холостой опыт.
).Навеску стали 0,1-0.2 г
растворяют при нагревании в 5 мл НС1 (1 : 1), По окончании растворения вводят 4
мл HNO3
(пл. 1,4), помещают на песчаную баню и кипятят до изменения цвета осадка из
белого в желто-оранжевый. Осадок оставляют в темном месте на 1 час, разбавляют
горячей водой примерно до 15 мл, прибавляют 3 мл 0.1% раствора желатина и
оставляют на некоторое время. Смесь переносят в центрифужные пробирки,
центрифугируют, промывают осадок несколько раз 1% раствором NaNO3
и растворяют в 10 мл 0.06 М NаOH.
Раствор переносят в колбу, вводят фенолфталеин и титруют 0,06 М HCI
избыток щелочи.
Методы, основанные на реакциях
осаждения.
Используют образование
малорастворимых солей серебра, бария, ртути и свинца определенного состава.
Навеску стали 0,2 г растворяют
в 5 мл НСl (1 : 1) при
нагревании на песчаной бане, вводят 4 мл HNO3
(пл. 1,4) и нагревают до образования осадка желто-оранжевого цвета. Оставляют
осадок в теплом месте на 20-30 мин., разбавляют горячей водой до ~ 15 мл и
вводят 3 мл 0,1%-ного раствора желатина. Через некоторое время осадок отделяют
центрифугированием, промывают HNО3
(1 : 10) 2-3 раза. К осадку прибавляют 2 мл 0,5 М NaOH;
частицы осадка на стенках колбы растворяют в 1,5 мл 0,5 М NaOH,
оба раствора объединяют, переносят в мерную колбу емкостью 25 мл и разбавляют
водой до метки. Аликвотную часть раствора 5 мл переносят в кювету, вводят 1
каплю раствора фенолфталеина и нейтрализуют 0,2 М HNО3
до обесцвечивания. В кювету вводят ~ 0,01 М раствор Рb(СН3СОО)2,
измеряя оптическую плотность после прибавления каждой новой порции титранта.
Титруют до достижения постоянной оптической плотности. Точку эквивалентности
находят графически.
Методы, основанные на реакциях
окисления - восстановления.
).К раствору Na2WO4,
содержащему 25-150мг вольфрама, прибавляют 10- 30 мл 0,1H
Н2O2,
10-15 мл 3 М H2SO4,
5-20 мл 0,1 М раствора винной кислоты, разбавляют водой до объема 50-60 мл,
устанавливают на потенциометре потенциал 0,51 в и титруют свободную Н2О2
0,1 Н раствором NaSO3
до перемены направления тока. Устанавливают потенциал второй точки
эквивалентности (0,39 в) и продолжают титрование перекисного комплекса
вольфрама. В конце титрования каждую новую порцию титранта прибавляют через 1-2
мин. 2). К раствору, содержащему 15-100 мг W,
прибавляют 10-30 мл 0,1 N Н2О2,
-15 мл 4 М H2S04,
5-20 мл 0,1 М раствора винной кислоты и разбавляют
водой до 50-60 мл. Далее устанавливают потенциал на потенциометре раствором Ce(S04)2
до перемены направления тока, после этого устанавливают потенциал 0,85 в и
титруют до скачка, прибавляя вблизи скачка каждую порцию титранта с интервалом
1 мин.
Методы, основанные на реакциях
комплексообразования.
).К 0,5-5 мл раствора
вольфрамата, содержащего 1-18,4 мкг/мл W
в 0,1 М ацетатно-аммонийном буферном растворе с рН 3 в 30 объемн, % этанола,
прибавляют 2-10-5 М раствор реагента в 30%-ном этаноле до устойчивой
синей окраски.
Вольфрам(VI)
и комплексон III
взаимодействуют в молярном отношении 2 : 1, образуя соединение с устойчивостью,
достаточной для титриметрического определения вольфрама.
К раствору W(VI)
прибавляют в избытке стандартный раствор комплексна III,
1-4 мл концентрированной НCl;
образующийся осадок растворяют прибавлением раствора ацетата аммония до рН 5.
Вводят ксиленоловый оранжевый и оттитровывают избыток комплексона III
0,01 М раствором Т1С13 до появления красной окраски раствора.
). Навеску сплава 0,5 г
растворяют в 40 мл H2SO4
(1 : 3) при нагревании. По окончании растворения добавляют несколько капель
концентрированной HNO3
и упаривают до паров H2SO4.
Остаток
растворяют в 50 мл воды при нагревании, переносят в мерную колбу емкостью 500
мл, содержащую 30 мл 20%-ного раствора NaOH,
и разбавляют водой до метки. Раствор фильтруют в сухой стакан, аликвотную часть
10-15 мл переносят в ячейку для титрования, вводят Н3РО4
(1 : 10) до рН 3,5, 2 г KI
разбавляют водой до объема 35-40 мл и титруют на холоду 0,5%-ным раствором β-нафтохинолина
при потенциале -0,86 в.
.3.2 Гравиметрические методы
). Навеску 0,2-1 г руды или
минерала сплавляют в кварцевом тигле с 5- 6-кратным количеством K2S207;
сплав выщелачивают 40 мл 0,5 М винной кислоты, раствор нагревают, осадок
отфильтровывают и промывают 6 раз горячей водой. Фильтрат упаривают до объема
около 25 мл, вводят 40 мл концентрированной HN03
(в два приема), кипятят для разрушения тартрата, упаривают до объема 10-20 мл,
разбавляют водой до объема 50 мл и дают отстояться при нагревании на водяной
бане. Осадок отфильтровывают, промывают 6-8 раз горячей водой, прокаливают в
платиновом тигле при 750° С, выпаривают с HF
и несколькими каплями H2S04,
прокаливают и взвешивают. Осадок не требует переосаждения.
). Навеску образца около 0,1 г
обрабатывают 30 мл смеси НС1 -f-
HN03 (3 :
1), раствор упаривают до объема 5-6 мл, охлаждают, прибавляют 5 мл
концентрированной H2SO4
и дважды упаривают до паров H2S04.
При этом второй раз после предварительного прибавления 5 мл воды раствор
охлаждают, осторожно разбавляют водой, нейтрализуют 20%-пым раствором NaOH
по бумаге конго и прибавляют избыток NaOH
до полного растворения выпавшей вольфрамовой кислоты. Раствор с осадком
гидроокисей кипятят, фильтруют и промывают осадок на фильтре. Фильтрат и
промывные воды нейтрализуют НС1 по конго до появления фиолетового окрашивания
вводят, 0,5 мл 10%-ного раствора SnCl2
или 0,5 -ил 30%-ной Н202. После этого прибавляют
концентрированной НС1 по 7 мл на каждые 100 мл раствора, 5 мл 2%-ного раствора fi-нафтохинолина,
нагревают до кипения (при использовании перекиси водорода нагревают до 75-80°
С) и оставляют на 1,5-2 часа. Осадок отфильтровывают и прокаливают до WOs
3). Анализируемый раствор,
содержащий 15-35 мг W{VI),
разбавляют в стакане водой до объема 150-200 мл, вводят 2 мл концентрированной CH3COOH
и 3-5 г CH3COONH4.
Раствор нагревают до кипения и вводят 8-10 мл
4%-ного раствора ацетата свинца, нагретого до кипения. Раствор с осадком
кипятят 15-20 мин., оставляют на 1 час и отфильтровывают па фильтр «синяя
лента», промывая осадок на фильтре горячим 1%-ным раствором CH3CООNH4
до отрицательной реакции на свинец. Осадок прокаливают до постоянной массы.
). К нейтральному или щелочному
анализируемому раствору W(VI)
прибавляют в избытке 4%-ный этанольный раствор 8-оксихинолина, нагревают до
кипения и прибавляют разбавленную СН3СООН до слабокислой среды (если
исходный раствор содержал минеральные кислоты, их нейтрализуют 1 М раствором CH3COONH4).
Нагревают, фильтруют осадок в тигель с пористым дном, промывают горячей водой
до получения бесцветных промывных вод и высушивают при 120° С до постоянного
веса.
3. Экспериментальная часть
.1 Гравиметрическое определение кобальта
Осаждение кобальта
1-нитрозо-2-нафтолом (щелочная среда)
Для получения осадка
определенного состава сначала надо окислить кобальт до трехвалентного
состояния. Состав осадка очень близок к выражаемому формулой:
Co(C10H6ONO)3·2Н2О.
F
= М(Со) / М(осадка) = 0,096.
Результаты получаются с
точностью ± 2 %. При желании получить более точный результат осадок надо
превратить в окись кобальта или сульфат кобальта.
Осадок легкий, кристаллический,
поэтому оптимальная масса осажденной формы 0.1 г.
Приблизительное содержание
кобальта в сплаве 0.5%, тогда масса навески для анализа составит:
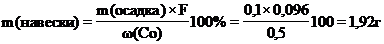
Мешающие ионы. В кислой среде вместе
с кобальтом осаждаются: железо (III), медь, уран (IV), хром (III), цирконий,
серебро, висмут, титан, ванадий (V), олово (IV), вольфрам,
молибден, палладий (II). Остаются в растворе: никель,
цинк, алюминий, марганец, фосфор (V), аммоний,
бериллий, щелочноземельные элементы. Мешают определению нитрат-ионы.
Если анализируемая проба содержит
много никеля (в количестве, более чем в два раза превышающем содержание
кобальта), осадок надо прокалить, растворить полученную золу в кислоте и
повторить осаждение.
Отделение железа от кобальта. К
раствору, содержащему ионы трехвалентного железа и двухвалентного кобальта,
прибавляется раствор нитрата кальция, раствор нитрата аммония до концентрации 5
моль/л и концентрированный раствор гидроокиси аммония до рН 9 и осаждаются ионы
кальция раствором двузамещенного фосфата аммония. Осадок надо промыть раствором
нитрата аммония с рН 9. Железо полностью соосаждается с фосфатом кальция, в то
время как кобальт остается в растворе.
Отделение меди, ванадия, вольфрама и
молибдена от кобальта. К раствору солей, содержащему приблизительно 0,5 г
окислов металлов III группы, надо прибавить 20-25 г
хлорида аммония и нейтрализовать раствор аммиаком. Объем раствора доводится до
150 мл, прибавляется бумажная масса, и раствор нагревается до 60° С. Далее
прибавляется по каплям 6 мл солянокислого пиридина и пропускается умеренный ток
сероводорода. Спустя 10-15 мин., прибавляется из капельной воронки по каплям
при взбалтывании 7 г уротропина в виде раствора (35 мл 20%-ного раствора),
колбу надо перенести на электрическую плитку и продолжить пропускать
сероводород еще 45- 60 мин., не доводя раствор до кипения. Далее снять колбу с
плитки и пропустить сероводород до полного охлаждения раствора до комнатной
температуры. Осадок сульфида отфильтровывают и промывают 3%-ным раствором
нитрата аммония, к которому прибавлено 0,5 мл свежеприготовленного раствора
сульфида аммония на каждые 100 мл. Сульфиды выделяются в плотной
кристаллической форме, хорошо отфильтровываются и практически не окисляются на
воздухе.
Реагенты. 1-нитрозо-2-нафтол.
Приготовление раствора: растворяют на холоде 8 г реактива в 300 мл чистой
уксусной кислоты и разбавляют водой до 500 мл (свежеприготовленный раствор).
Уксусная кислота, СН3СООН, концентрированная. Перекись водорода, Н202,
свежеприготовленная, 30%-ная. Едкий натр, NaOH, 2 Н
раствор. Азотная кислота, HN03,
концентрированная, ρ=
1,4 г/см.
Серная кислота, H2S04,
концентрированная, ρ=
1,84 г/см3.
Соляная кислота, НС1, концентрированная, ρ = 1,17 г/см3.
Посуда и оборудование. Стакан
300-400 мл; стеклянная палочка с резиновым наконечником, L = 30 см;
стеклянный фильтрующий тигель № 4; мерный цилиндр 25 мл; мерная колба 500 мл,
часовое стекло. Плитка электрическая. Сушильный шкаф. Промывалка.
Выполнение определения.
К 10-20 мл анализируемого слабокислого
раствора, содержащего 1 -30 мг сульфата, хлорида или нитрата кобальта, надо
прибавить 15-20 капель 30%-ной перекиси водорода и 2 Н раствор едкого натра до
начала осаждения черной гидроокиси кобальта (III). Затем
прилить 10-20 мл уксусной кислоты и, если осадок не растворился, нагреть до
растворения. Разбавить до 200 мл горячей водой, прибавить 10-20 мл осаждающего
реактива, нагреть, сильно перемешать, довести до кипения и отфильтровать через
стеклянный фильтрующий тигель. Промыть осадок разбавленной (1 ;2) горячей
уксусной кислотой, потом водой, высушить при 130 °С и взвесить.
|
M(фильтра),г
|
M(фильтра с осадком),г
|
M(осадка), г
|
|
0,8675
|
1.0632
|
0,1957
|
|
0,8675
|
1.0243
|
|
0,8675
|
0,9704
|
0,1029
|
M(опр.)=0,1029·0,096=0,00988г.
На основе этого массовая доля
кобальта составляет:

3.2 Титриметрическое определение вольфрама
Определение вольфрама объемным
щелочным методом
Необходимые реактивы
1.
Глицерин
нейтральный, ч. д. а.
2.
Кислота
азотная плотностью 1,4 г/см3, ч. д. а.
3.
Кислота
соляная, плотностью 1,19 г/см3, ч. д. а.
4.
Кислота
серная, титрованный раствор. 17 мл Н2S04
плотностью 1,84 г/см3, ч. д. а., приливается тонкой струей к 500 мл
воды, разбавляется водой до 5 л и хорошо перемешивается. Кислоту надо хранить в
бутыли, соединенной с бюреткой (см. рис. 15).
Установление соотношения К
между эквивалентными объемами титрованных растворов NаОН (см. п. 7) и Н2SО4.
В коническую колбу емкостью 500
мл надо налить из бюретки 25 мл раствора NаОН (см. п. 7), прибавить 2-3 капли
раствора фенолфталеина, разбавить свежепрокипяченной и охлажденной водой до
250-300 мл и титровать раствором Н2S04
до исчезновения фиолетово-красного цвета раствора. Величина К находится по
формуле
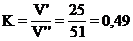
где V - число мл
раствора NаОН (25 мл);
V" -
число мл раствора Н2S04, пошедшего на
титрование 25 мл раствора NаОН. .
Это соотношение следует установить
три раза и принять среднее значение при условии хорошо совпавших результатов.
Для упрощения (ускорения) вычислений результатов определения содержания
вольфрама желательно, чтобы К = 1. Соотношение следует проверять через каждые
5-8 дней (см. стр. 100).
5.Маннит, ч.
д. а.
6.Натрий
азотнокислый (45%-ный раствор). Его надо готовить на свежепрокипяченной воде
для удаления из нее СO2.
Раствор готовится в количестве не свыше 2-3-дневной потребности и он должен
быть нейтральным. Для проверки нейтральности надо взять 5 мл этого раствора в
пробирку, прибавить 2 капли раствора фенолфталеина и 1 каплю титрованного
раствора NаОН (см. п. 7). Раствор в пробирке должен окраситься в яркий
фиолетово-красный цвет.
Натр едкий, титрованный
раствор. 27 г NаОН, ч. д. а., надо растворить в 1 л свежепрокипяченной
охлажденной воды и затем разбавить такой же водой до 5 л.
Выполнение определения
Навеска средней пробы стали в
виде мелкой стружки в количестве 0,5 г (2 г при содержании W
от 1 до 4 %, 0,5-1 г при содержании W
от 4 до 10 %, 0,5-0,3 г при содержании W
от 10 до 25 %) помещается в коническую колбу емкостью 500-300 мл и растворяется
соответственно взятой навеске в 30 мл разбавленной (1 :1) НС1.
Растворение необходимо
проводить при умеренном нагревании на песчаной бане до прекращения или
значительного замедления реакции; на дне колбы остается мелкий темный осадок
металлического вольфрама, его карбидов и карбидов других элементов. После этого
колбу надо снять с бани и прилить к горячему раствору осторожно, небольшими
количествами (по 0,3-0,5 мл) HNO3
плотностью 1,4 г\смъ для окисления вольфрама (металлического и его
карбидов) в вольфрамовую кислоту H2WO4,
двухвалентного железа в трехвалентное и для разложения карбидов других
элементов. Азотная кислота приливается до прекращения вспенивания раствора
сплава, сопровождающегося выделением бурых окислов азота (процесс окисления
проводится в вытяжном шкафу). Обычно в зависимости от навески сплава на
окисление требуется 3-6 мл HNО3.
После этого колбу надо вновь поставить на песчаную баню, накрыть ее часовым
стеклом и умеренно кипятить до тех пор, пока выпавший осадок вольфрамовой
кислоты из серо-зеленого (вследствие наличия в нем низших окислов W)
не перейдет в желтый с оранжевым оттенком (цвет осадка следует наблюдать со дна
колбы). Если переход окраски осадка совершается медленно, то следует прилить
еще 1-2 мл HNО3
плотностью 1,4 г/см3 и продолжать нагревание раствора.
После этого раствор
разбавляется 60-75 мл горячей воды, хорошо размешивается стеклянной палочкой,
стоит на песчаной бане еще 35-45 мин (лучше несколько часов), не надо доводить
его до кипения. Затем его надо профильтровать через плотный фильтр, содержащий
небольшое количество бумажной массы, или через цилиндрическую воронку со слоем
бумажной массы (см. рис. 12).
Осадок H2W04
смывается на фильтр горячей разбавленной (5:95) соляной кислотой и затем
промываются колба и фильтр с осадком вначале 5-6 раз такой же кислотой, а под
конец - 5-6 раз горячим 4%-ным нейтральным раствором NaNО3
для удаления со стенок колбы, из осадка и фильтра следов соляной кислоты. Для
проверки полноты удаления кислоты надо набрать в пробирку или на часовое стекло
0,5-1 мл стекающей с фильтра промывной жидкости, добавить 1-2 капли раствора
фенолфталеина и 1 каплю титрованного раствора NaOH;
раствор в пробирке должен окраситься в фиолетово-красный цвет.
Осадок вольфрамовой кислоты
обладает свойством плотно приставать к стенкам стеклянного сосуда, особенно
если его внутренняя поверхность несколько разъедена (шероховата).
В данном случае эту часть
осадка нет необходимости переносить на фильтр; важно лишь тщательно отмыть его
от маточного раствора (от кислоты, солей железа, хрома и др.).
Промытый осадок H2W04
вместе с фильтром надо поместить в ту же колбу, в которой проводилось
растворение навески сплава, прилить 15-20 мл свежепрокипяченной холодной воды
(лишенной углекислоты), закрыть колбу пробкой. Затем приливается из бюретки
35-45 мл титрованного раствора NaOH,
добавляется 2-3 капли раствора фенолфталеина. Закрыв горлышко колбы резиновой
пробкой, надо осторожно встряхнуть содержимое ее для разрыхления фильтра и
растворения осадка H2WO4.
Если фиолетово-красная окраска в колбе после встряхивания в течение некоторого
времени обесцветится, то прилить из бюретки еще 5-10 мл раствора NaOH
до появления указанной окраски и взбалтывать 2-3 мин до полного растворения
желтого осадка (раствор должен остаться окрашенным в фиолетово-красный цвет).
H2WО4
+ 2NaOH = Na2WО4
+ 2H2О.
Удалив пробку, надо ополоснуть
ее и стенки колбы свежепрокипяченной холодной водой, одновременно разбавляя
содержимое колбы до 150-250 мл.
Примечание. С целью повышения
кислотных свойств шестивалентного вольфрама перед тем, как приливать
титрованный раствор едкой щелочи, к содержимому колбы можно прилить 10-15 мл
нейтрального глицерина или добавить 0,5-1 г маннита и хорошо размешать.
Титрование избытка щелочи надо
производить раствором серной кислоты до наступления момента полного исчезновения
розовой окраски.
Примечание. Для проверки
правильности установления конца титрования к обесцвеченному раствору после
окончания титрования надо прилить из бюретки еще 2-3 мл раствора NaOH,
перемешать и вновь титровать раствором H2S04
до исчезновения розовой окраски. Добавленное количество мл раствора NaOH
и пошедшее на повторное (проверочное) титрование количество мл раствора H2SО4
суммируется соответственно с израсходованным при первом титровании числом мл NaOH
и H2SО4.
Вычисление содержания вольфрама
может быть проведено раздельно по данным первого титрования и по сумме мл
раствора NaOH и раствора H2SO4,
пошедших при первом и втором (проверочном) титровании.
Содержание вольфрама в
анализируемом образце вычисляют по формуле
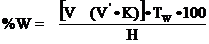 ,
,
где V - число мл
титрованного раствора NaOH (включая избыток), прилитого
для растворения осадка вольфрамовой кислоты;
V' - число мл
раствора H2SO4, пошедшего
на титрование избытка раствора NaOH;
К - коэффициент cоотношения
между эквивалентными объемами растворов NaOH и H2SО4,
расходуемых при их взаимном (непосредственном) титровании
Тw - титр
раствора NaOH на
вольфрам, г;
Н - навеска образца, г.
Результаты:
|
V(NaOH), мл
|
V(H2SO4), мл
|
|
40.0
|
20.8
|
|
40.0
|
20.9
|
|
40.0
|
21.0
|
|
<V>=40.0
|
<V>=20.9
|
Исходя их этих данных массовая доля вольфрама
составляет:

Вывод
В результате проведенного анализа
было установлено, что анализируемый объект представляет собой образец стали.
В качестве добавок присутствуют
кобальт, никель, хром, марганец, медь, вольфрам, молибден, ванадий.
Содержание кобальта определялось
гравиметрически. По результатам анализа была получена величина 0,52%.
Определение вольфрама проводилось
титриметрически. Была получена величина 5,95%.
Литература
1. Золотов
Ю.А. Основы аналитической химии М.; Высшая школа, 2001.
. Дымов
А. М. Технический анализ М.; Металлургия, 1964.
. Посыпайко
В.И. Васина Н. А. Аналитическая химия и технический анализ М.; Высшая школа,
1979
. Коростелёв
П. П. Химический анализ в металлургии М.; Металлургия, 1988.
. Пятницкий
И.В. Аналитическая химия кобальта М.; Наука, 1965.
. Бусеев
А.И., Иванов В. И., Соколова Т. А. Аналитическая химия вольфрама М.; Наука,
1976.
. Алексеев
В. Н. Качественный анализ М.; Химия,1972.