Фотометрический способ определения окислов азота
Введение
Борьба с загрязнением воздуха - одна
из основных проблем охраны окружающей среды, техники безопасности в
промышленности, обеспечения обитаемости людей в замкнутых помещениях.
Физические, химические и биологические факторы могут в той или иной степени
вызвать нежелательные функциональные сдвиги в организме человека, снижать
эффективность его работы, оказывать отрицательное влияние на здоровье.
В различных помещениях химические
факторы определяются газовым составом воздуха. Газовый состав воздуха в экологически
замкнутых помещениях претерпевает значительные изменения в результате
жизнедеятельности людей, работы машин и механизмов. При работе различных
механизмов, а также в результате обслуживания оружия - баллистических ракет
Р-29; Р-29Р; Р-29РМ; Р-39; Р-29РМУ могут выделяться компоненты ракетных топлив,
в том числе оксиды азота NxOy. В аварийных же случаях оксиды азота могут появляться в
концентрациях, превышающих допустимые нормы в тысячи и более раз и приводить к
серьезным поражениям личного состава и далее к гибели корабля. В настоящее
время нет достаточно эффективных средств определения компонентов ракетных
топлив. Среди определяемых соединений основными являются: НДМГ,
тетраметилтетразен, нитрозодиметиламин. Главным продуктом превращения НДМГ является
диметилгидразон формальдегида, средства, определения которого разработаны
недостаточно.
Для контроля за концентрациями
оксидов азота существует на ПЛА метод определения - с помощью индикаторных
трубок при использовании переносных средств анализа воздуха ПГА ВП и КГП ВП.
Недостатками данного метода
являются:
- значительная погрешность
измерения, составляющая почти 50 - 60%, поэтому индикаторные трубки на оксиды
азота фактически могут определять приблизительное количественное присутствие
окислов азота;
- малая надежность из-за
коротких сроков их годности; неудобство пользования при пожарах;
большое время,
затрачиваемое на измерения, составляющее от 12 до 13 минут;
малый диапазон измеряемых
концентраций (0,5-5) мг/м3.
Чтобы устранить эти недостатки, предлагается
исследование существующих методов линейно-колористического определения с целью
расширения диапазона измерения КРТ. Для этого предлагается среди известных
методов количественного анализа методы определения КРТ. Этому исследованию и
будет посвящена настоящая работа.
1. Аналитический обзор
.1 Общие сведения об
окислах азота
окисел азот бензидин
Оксиды азота - неорганические
бинарные соединения азота с кислородом. Это группа ядовитых газов, образующихся
при высоких температурах (более 1300°С) и высокой компрессии. Имеются много
видов окислов азота (NO, NO2, NO3 и т.д.). Окись азота представляет собой атом
азота и один или несколько атомов кислорода в соответствии с рисунком 1.
 Оксид
азота, N2O.
Оксид
азота, N2O.
 Оксид
азота, NO.
Оксид
азота, NO.
 Оксид
азота, N2O3.
Оксид
азота, N2O3.
 Оксид
азота, NO2.
Оксид
азота, NO2.
 Оксид
азота, (NO2)2 или N2O4.
Оксид
азота, (NO2)2 или N2O4.
 Азотный
ангидрид, N2O5.
Азотный
ангидрид, N2O5.
Рисунок 1 - Атомы различных окислов
азота
Содержание азота в воздухе не
меняется до тех пор, пока температура не достигнет 1370°С при высоком давлении.
При этих условиях азот начинает реагировать с кислородом и образуется оксид
азота (NO). В двигателе автомобиля эти условия наступают при полном открытии
дроссельной заслонки, ускорении и движении с максимальной скоростью. Под воздействием
солнечного света оксид азота разлагается на двуокись азота (NO2), озон (О3) и
взаимодействуя с парами воды - нитрат ион (NO3 -). NO2
представляет собой газ светло-коричневого цвета, обычно называемый «смогом».
Характеристика
физических и химических свойств оксидов азота
Окись азота (NO).
Для азота характерны устойчивые
оксиды с четным числом электронов NO и NO2. Оба оксида -
эндотермические соединения со стандартными теплотами образования DHo298 соответственно 90,4 и 33,9 кДж/моль. В обычных условиях оксид
азота (II) NO - бесцветный газ (tпл = минус 163,7о C, tкип = минус 151,7о C) с легким приятным запахом и
сладким вкусом. В лаборатории его получают действием разбавленной HNO3 на медь:
HNO3 + 3Cu = 2NO + 4H2O + 3Cu(NO3)2.
В промышленности - окислением NH3 на платиновом катализаторе, в технике - продуванием воздуха через
пламя электрической дуги. В отличие от всех остальных оксидов азота NO образуются также прямым
взаимодействием простых веществ на поверхности электрического разряда:
N2 + O2 → 2NO.
Вследствие повышенной кратности
связи молекула NO достаточно устойчива, и ее распад становится заметным лишь при t = 500о C. Оксид азота (II) - химически активное
соединение легко восстанавливается (при действии SО2, Cr2+) в растворах до NH2OH и H3N, с водородом образуют гремучую смесь. Легко окисляется
кислородом, галогенами, Cr2O3, HMnO7 и др. При окислении галогенами NO образует оксогалид
азота (III) NOHal.
При взаимодействии с наиболее
активными металлами в жидком аммиаке NO сам выступает как окислитель.
Диоксид азота (NO2).
Диоксид азота (IV) - красно-бурый газ,
имеющий характерный запах, токсичен и состоящий из собственно двуокиси азота и
ее бесцветного полимера - четырех окиси азота (N2O4) - азотноватого ангидрида. Двуокись азота при понижении температуры
легко сгущается в красно-бурую жидкость, кипящую при tкип. = 21,4о C и затвердевающую в бесцветные кристаллы при tпл = минус 11,2о C. Растворяется в воде с образованием
азотистой и азотной кислот.
Молекулы NO2 даже в парах частично димеризованы. Энергия ионизации у NO2 равна 9,78 эВ (может терять электроны) и сродство к электрону
1,62 эВ (может приобретать электроны).
Диоксид азота химически активен. В
его атмосфере горят уголь, сера, фосфор. В лаборатории NO2 получают взаимодействием меди с концентрированной азотной
кислотой, воздействием серной кислоты на нитриты калия или натрия.
Диоксид азота применяют как
нитрующий агент, в частности, для получения безводных нитратов.
Оксид азота III (N2O3).
Темно-синяя жидкость, затвердевающая
при t = минус 103оC в голубые кристаллы. Его получают охлаждением смеси NO2 и NO. Согласно данным, полученным с помощью инфракрасной спектрометрии
полагают, что кристаллы N2O3 состоят из стабильной
модификации ONNO2 и нестабильной ONONO.
Оксид азота (III) - ангидрид азотной кислоты,
легко поглощается щелочами, образуя нитриты.
При взаимодействии оксида азота (III) с концентрированными
кислотами можно получить солеподобные вещества tпл = 200о C [NO] Hal4, [NO]2SeO4.
Закись азота (N2O).
Закись азота, он же «веселящий газ»,
средство для ингаляционного наркоза (Nitrogenium oxydulatum) - бесцветный газ, тяжелее воздуха (удельный вес 1,527) с легким
характерным запахом и сладковатый на вкус, растворим в воде (1:2), tкип = минус 890 С, tзамерз= минус 1020 С. При t = 0 и давлении 30 атмосфер
сгущается в бесцветную жидкость. Не воспламеняется, но при высоких температурах
поддерживает горение. Смеси закиси азота с диэтиловым эфиром, пропаном, и
хлорэтилом, также как и смеси с кислородом, в определенных концентрациях
взрывоопасны.
Пятиокись азота (N2O5).
Пятиокись азота (азотный ангидрид) -
белое кристаллическое вещество (температура возгорания 32о C), образованное ионами NO2- и NO3-.
Это белое вещество (кристаллы) имеет
плотность 1,63, а при t = 30о C плавится в желтую, слегка разлагающуюся жидкость, разложение
усиливается при нагревании и при действии света, tкип = 50о C. С водой образует сильную, довольно устойчивую кислоту (азотную),
со щелочами - соли этой кислоты, нитраты.
Азотный ангидрид получают осторожным
обезвоживанием HNO3 (например, с помощью P2O5) или окислением NO2 озоном. В обычных
условиях N2O5 постепенно разлагается
на NO2 и O2, при нагревании взрывается.
При нагревании азосоединения
непосредственно соединяются со многими металлами, образуя нитриды металлов,
например: Li3N, Mg3N2, AlN и др. Многие из них
разлагаются водой с образованием аммиака.
Тетраоксид азота.
Тетраоксид азота в паре с
алкилгидразинами образует самовоспламеняющуюся ракетную топливную пару с
периодом задержки около 0,003 с. При обычных условиях тетраоксид азота
находится в равновесии с диоксидом азота:
N2O4↔2NO2+∆H.
Состав смеси зависит от температуры
и давления. С увеличением температуры равновесие смещается в сторону диоксида
азота - окислитель окрашивается в бурый цвет. При увеличении давления при постоянной
температуре степень диссоциации N2O4 уменьшается. N2O4 практически полностью диссоциирован при 1400 С. Далее
в, соответствии с таблицей 1, показаны физико-химические свойства отетраксида
азота.
Таблица 1 - Физико-химические
свойства тетраоксида азота
|
Физик- химические величины
|
Значения
|
|
Плотность жидкости при 293 К, кг/м3
|
1450
|
|
Температура, К:
|
|
|
кипения
|
294,3
|
|
кристаллизации
|
262,0
|
|
Давление насыщенных паров при 293 К, кПа
|
96,5
|
|
Физико - химические величины
|
Значения
|
|
Вязкость при 298 К, Мпа ▪ с
|
0,39
|
|
Параметры критического состояния:
|
|
|
температура, К
|
431,4
|
|
давление, МПа
|
10,1
|
|
Теплота, кДж/моль:
|
|
|
испарения при 294,3 К
|
38,1
|
Тетраоксид азота и вода имеют
ограниченную взаимную растворимость. Так, при 00С смесь
расслаивается. Растворяясь в воде, тетраоксид азота вступает с ней во
взаимодействие, образуя азотную и азотистую кислоты:
N2O4 + H2O → HNO3 + HNO2.
Образовавшаяся азотистая кислота
разлагается по реакции:
HNO2 ↔ HNO3 + 2NO + H2O.
В свою очередь, монооксид азота
также нестоек и легко окисляется до диоксида азота, что способствует
образованию новых количеств коррозионно-активной азотной кислоты. Это свойство
окислителя учитывается при его транспортировке и хранении, всячески исключается
контакт паров компонента с влагой воздуха. Добавление в тетраоксид азота оксида
азота смещает равновесие реакции влево и снижает коррозионную активность
окислителя.
Оксид азота взаимодействует с
тетраоксидом азота с образованием триоксида азота:
NO + N2O4 ↔ 2N2O3.
Триоксид азота придает окислителю
голубовато-зеленую окраску и снижает температуру кипения и кристаллизации.
Тетраоксид азота имеет малую вязкость (0,468…. 0,599 МПа▪с), примерно в
два раза ниже, чем окислители на основе азотной кислоты. Поэтому этот
окислитель легко перекачивается насосами или передавливается.
Тетраоксид азота - плохой проводник
электричества, он не подвергается ионизации, а растворимость в нем
неорганических соединений, в частности солей металлов, очень мала.
Многие органические соединения
хорошо растворяются в тетраоксиде азота - алифатические и ароматические
углеводороды, галогеноводороды, нитрофенолы, эфиры, кетоны и другие. Смеси
тетраоксида азота с органическими соединениями взрывоопасны. Тетраоксид азота
очень сильный окислитель, с гидразином и НДМГ образует самовоспламеняющиеся ракетные
топлива.
Большинство металлов при контакте с
тетраоксидом азота пассивируются вследствие образования на их поверхности
оксидной пленки и плохо растворимых солей металлов. Однако с увеличением
содержания воды в N2O4 всегда повышается
равновесное количество азотной кислоты и возрастает коррозионная активность
окислителя. В окислителе, содержащем не более 0,1% H2O, стойкими являются легированная и
углеродистая стали, никель, алюминий и его сплавы; нестойкими при обычных
температурах в условиях хранения и транспортирования - серебро, медь, цинк,
кадмий и свинец, титан и его сплавы значительно корродируют в этих условиях.
Добавление к окислителю до 1,0% NO снижает скорость коррозии указанных металлов и позволяет
использовать их для изготовления топливных баков космических аппаратов.
Тетраоксид азота является токсичным
и агрессивным веществом. При попадании на кожу человека он подобно
концентрированной азотной кислоте вызывает сильные ожоги. Особенно опасны пары
тетраоксида азота. Они сильно раздражают дыхательные пути и легкие, разрушают
роговицы глаз, зубы, а также оказывают общее отравляющее действие на организм.
В тяжелых случаях отравления происходит отек легких.
Для защиты баков ракет с
окислителем, которые изготавливаются из алюминия, используют йод, фтористый
водород, фосфорную кислоту. При их добавлении в баки происходит пассивация
поверхностей баков, которая надолго защищает алюминий от воздействия АТ. Йод в
добавках 1% лучше пассивирует сталь, хуже алюминий (рисунок 2). Фтористый
водород лучше защищает алюминий, образуя пленку иона F- В практике используют (0,5-0,75)% раствор плавиковой кислоты. С
увеличением воды в АТ защита становится неэффективной. Поэтому требования к
качеству АТ очень высоки.
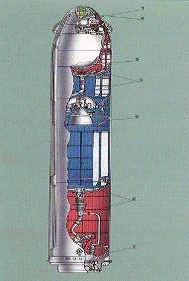
Рисунок 2 - Общий вид баков
окислителя и топлива баллистических ракет (баки с АТ - синего цвета).
При добавлении в баки раствора
фосфорной кислоты происходит образование хлопьев фосфорнокислого алюминия,
которые защищают поверхность баков.
Токсикологические
свойства оксидов азота
Оксиды азота образуются при
производстве и применении азотной кислоты (в процессе взаимодействия ее с
различными металлами или органическими веществами), в процессе термического
окисления азота воздуха при электро- и газосварке, работе дизельных и
карбюраторных двигателей. Предельно допустимая концентрация (ПДК) оксидов азота
для 2000-3000 часов работы в присутствии этих окислов установлена [4] 0,5 мг/м3.
Максимально допустимая концентрация (МДК) зависит от времени воздействия
оксидов азота на организм человека и приведена в таблице 2.
Таблица 2 - Максимальные допустимые
концентрации оксидов азота
|
МДК, мг/м3
|
Время воздейст., мин.
|
|
60
|
5
|
|
45
|
10
|
|
35
|
15
|
|
30
|
20
|
|
25
|
30
|
|
20
|
40
|
|
15
|
60
|
|
10
|
480
|
Общий характер действий нитрогазов
на организм зависит от содержания в газовой смеси различных окислов азота. В
основном раздражение протекает по нитритному типу действия. При контакте
окислов азота с влажной поверхностью легких образуются азотная или азотистая
кислоты, которые поражают легочную ткань, вызывая отек легких. Одновременно в
крови образуются нитраты или нитриты, непосредственно действующие на
кровеносную систему, ее сосуды, расширяя их и вызывая снижение кровяного
давления. Нитриты, взаимодействуя с оксигемоглобином, превращают его в
метимоглобин, вызывая метимоглобинемию. Общим следствием действия окислов азота
является кислородная недостаточность.
Рассмотрим следующие возможные
случаи и воздействия отдельных оксидов азота на человеческий организм.
Окись азота (NO).
Действует на центральную нервную систему,
угнетая гемоглобин (переводит оксигемоглобин в метимоглобин). При легком
отравлении окисью азота наблюдается общая слабость, сонливость, головокружения
(симптомы обратимы). При более тяжелом отравлении начальные симптомы
усиливаются, к ним присоединяется тошнота, иногда рвота, наступает
полуобморочное состояние, при отравлении средней тяжести резкая слабость и
головокружения продолжаются много часов, нередко наблюдаются сонливость,
синюшность слизистых оболочек и кожи, учащение пульса. При тяжелых отравлениях
начальные явления нередко стихают, но после трех дневной ремиссии повторяются
слабость и головокружение, наблюдается снижение кровяного давления, серо-синяя
окраска слизистых оболочек кожи, увеличение и болезненность печени; границы
сердца расширены, тоны глухие. Возникают полиневриты, полиневралгии. Кровь
становится шоколадно-бурого цвета, повышенной водности. Последствия тяжелого
отравления могут длиться более года: нарушения ассоциативных способностей,
ослабление памяти и мышечной силы, общая слабость, головная боль,
головокружение, быстрая утомляемость сопровождают самочувствие человека.
Диоксид азота (NO2).
Острое отравление начинается с
легкого кашля, в более тяжелых случаях - с сильного кашля, чувства стеснения в
груди, головной боли, иногда рвоты, саливации. Период относительно
удовлетворительного состояния длится от 2 до 18 часов. Затем появляются
признаки нарастающего отека легких: сильная слабость, увеличивающийся кашель,
боли в груди, цианоз, в легких много влажных хрипов, учащенное сердцебиение,
иногда озноб, повышение температуры. Нередки случаи значительных расстройств со
стороны желудочно-кишечного тракта; тошнота; рвота, сильные боли в верхней
части живота. Отек легких характеризуется тяжелым состоянием (резкий цианоз,
сильная одышка, учащенный пульс, кашель с пенистой мокротой, иногда с кровью).
Кровяное давление нестабильно, в крови уменьшение количества эритроцитов и
гемоглобина, лейкоцитоз, замедленная РОЭ. Рентгенологически - пониженная
прозрачность легочных полей. В обоих легких большое количество хлопьевидных
затемнений различной величины. Токсический отек легких сопровождается «синим»
типом гипоксимии, при осложнении компасом, наблюдается «серый» тип. Нередки
осложнения пневмонией. Возможен смертельный исход. На секции отек легких,
кровоизлияние в них, темная жидкая кровь в сердце и сосудах. Состояние
отравленных ухудшается, если пострадавшие до отравления страдали заболеваниями
сердца или легких.
При хронических отравлениях -
хронически воспалительное заболевание верхних дыхательных путей, хронические
бронхиты, эфизема, понижение кровяного давления, зеленоватый налет на губах,
разрушение коронок резцов. Кожа после контакта с двуокисью азота становится
выражено желтой - результат химического ожога.
Закись азота (N2O).
Большие ее концентрации вызывают шум
в ушах, асфиксию, потерю сознания. Смерть настает от паралича дыхательного
центра.
Азотный ангидрид, пятиокись азота (N2O5).
Действует на организм аналогично
окиси азота и другим его аналогам.
.2 Применение окислов
азота
Окись азота (NO).
Получение NO является одной из
стадий получения азотной кислоты. Других ссылок на применение не обнаружено.
Закись азота (N2O).
Используется в основном как средство
для ингаляционного наркоза, в основном в сочетании с другими препаратами (из-за
недостаточно сильного обезболивающего действия). В то же время это соединение
можно назвать самым безопасным средством для наркоза, так как после его
применения почти не бывает осложнений. Также иногда используется для улучшения
технических характеристик двигателей внутреннего сгорания
Малые концентрации закиси азота
вызывают чувство опьянения (отсюда название - «веселящий газ») и лёгкую
сонливость. При вдыхании чистого газа быстро развиваются состояние
наркотического опьянения, а затем асфиксия. В смеси с кислородом при правильном
дозировании вызывает наркоз без предварительного возбуждения и побочных
явлений. Закись азота обладает слабой наркотической активностью, в связи с чем
её необходимо применять в больших концентрациях. В большинстве случаев применяют
комбинированный наркоз, при котором закись азота сочетают с другими, более
мощными, средствами для наркоза, а также с миорелаксантами.
Применять закись азота, как и любое
средство для наркоза, необходимо с осторожностью, особенно при выраженных
явлениях гипоксии и нарушении диффузии газов в лёгких. Закись азота иногда
используется для улучшения технических характеристик двигателей внутреннего
сгорания. В случае автомобильных применений вещество, содержащее закись азота,
и горючее впрыскиваются во впускной (всасывающий) коллектор двигателя, что
приводит к следующим результатам:
снижает температуру
всасываемого в двигатель воздуха, обеспечивая плотный поступающий заряд смеси;
увеличивает содержание
кислорода в поступающем заряде (воздух содержит лишь ~21 масс.% кислорода);
повышает скорость
(интенсивность) сгорания в цилиндрах двигателя.
В пищевой промышленности соединение
зарегистрировано в качестве пищевой добавки E942, как пропеллент и упаковочный
газ.
Диоксид азота (NO2).
Применяется в производстве серной и
азотной кислот, в качестве окислителя в жидком ракетном топливе и
многочисленных смесевых взрывчатых веществ.
.3 Выводы по
аналитическому обзору
Из проведенного аналитического
обзора можно сделать следующие выводы:
1) из анализа химических свойств окислов
азота выявлено, что они являются очень реакционноспособными и по своей природе
являются долгоживущими радикалами NO• (трехвалентным); NO2• (пятивалентным);
2) методики приготовления
парогазовых смесей окислов азота в воздухе, инертных газах отработаны
недостаточно. Во всей рассмотренной литературе способов приготовления
статических постоянных концентраций не обнаружено;
) в промышленности методом
ЛКМ контролируются только сумма двуокиси азота по реакции с выделением
свободного йода и последующего взаимодействия с крахмалом в интервале
концентраций (0,5 - 5) мг/м3 (1-10 ПДК). Большие значения
концентраций не контролируются;
) новых направлений в
аналитическом контроле двуокиси азота пригодных для реализации пока не имеется;
) для рассмотрения возможных
методов контроля в настоящей дипломной работе был выбран способ
колориметрического;
) определение окислов азота
по реакции с бензидином и b-нафтолом;
7) токсикология оксидов азота
требует постоянного мониторинга за состоянием среды обитания, так как средства
непрерывного контроля на производствах отсутствуют;
) согласно установленных
санитарно-химических норм мониторинг оксидов азота осуществляется 1 раз в сутки
с помощью индикаторных трубок типа ИТМ-1Б, что явно недостаточно по причинам,
которые будут изложены в следующем разделе;
9) необходимо разработка
автоматизированных систем контроля по этим химически вредным веществам.
2. Цель и задачи работы
Целью работы является исследования
предложенного метода определения окислов азота с последующим установлением
диапазона их определения и возможности разработки спектрофотометрического
метода определения для повышения диапазона измерения и достоверности
определения.
В соответствии с целью данной работы
были определены следующие основные задачи исследования:
воспроизвести методику
колористического определения окислов азота по первоисточнику, определить
диапазон ее измерения;
исследовать способ
определения двуокиси азота колористическим методом, варьируя значения
концентраций, реагирующих компонентов и заменой растворителя;
разработать способ
приготовления стандартной паровоздушной концентрации двуокиси азота в воздухе;
исследовать возможность
определения высоких концентраций двуокиси азота спектрофотометрическим методом
на основе данного способа;
снять спектр образующегося
окрашенного соединения и построить градуировочный график на длине волны, на
которой наблюдается максимум поглощения;
определить линейный
диапазон определения.
3. Экспериментальная
часть
.1 Обоснование необходимости разработки темы
Одним из факторов, определившим
стремительное развитие и современное состояние химии ракетной техники -
компонентов ракетных топлив (КРТ), оказались уникальные значения
термодинамических и кинетических параметров реакций простейших гидразинов с
окислами азота. Благодаря этим особенностям, гидразин и его простейшие
производные стали одними из важнейших горючих, применяемых в современной
ракетной технике. При сжигании диметилгидразина в кислороде может быть получена
наибольшая для жидких реактивных топлив такого типа удельная тяга. Весьма
эффективными являются самовоспламеняющиеся топливные пары из гидразинов и
азотной кислоты или четырехокиси азота. Горючее на основе гидразинов
применяются и в мощных двигателях всех ступеней гигантских ракет-носителей, и в
двигателях систем маневрирования космических кораблей. Именно с помощью
двигателей, работавших на паре - диметилгидразин-АТ, были произведены посадка и
взлет лунной кабины системы «Апполон» при исторической высадке человека на
Луну.
Азотный тетраоксид - «амил»
исключительно широко применяется в качестве ракетного окислителя. Вместе с тем
эксплуатация ракет с двухкомпонентным топливом является чрезвычайно опасной. В
практике описаны тяжелые аварии с ракетным техникой и оружием, которые имели
место в космической и оборонной отраслях.
Следует отметить, что многие вопросы
обеспечения безопасности ракетного комплекса не решены должным образом:
· предаварийный
контроль за протечками АТ на уровне ПДК недостаточно эффективен;
· контроль окислов
азота имеет малый диапазон концентраций;
· контроль в
аварийных ситуациях осуществляется только с помощью газосигнализатора
АПГА-ВП-ВМ, который лишь сигнализирует о факте протечки.
По этой причине тема настоящей
дипломной работы является очень актуальной. Настоящая работа посвящена
химическим процессам определения АТ путем его взаимодействия с солянокислым
бензидином и в нафтолом, которая позволит проводить определение АТ в области
высоких концентраций.
.2 Анализ существующих
способов определения окислов азота
При анализе воздуха используется
комплекс газоаналитических средств, имеющий самые различные возможности. Обычно
средства имеющие высокую точность, селективность являются громоздкими и
дорогими устройствами. Для анализа воздушной среды на них необходимо доставлять
к ним пробы воздуха. Длительность процессов отбора и анализа исключает
возможность своевременной сигнализации об опасных концентрациях токсических
веществ. Поэтому в практике используются достаточно простые экспрессные методы,
которые позволяют быстро получить результат непосредственно на месте отбора
пробы на уровне 1-10 ПДК.
Данные методы получили исключительно
широкое распространение в практике, в первую очередь благодаря их
селективности, достаточной чувствительности, быстроты определения, простоты в
эксплуатации, доступности. Методы можно условно разделить на следующие группы:
·
колориметрия растворов по стандартным шкалам;
·
колориметрия осадков в растворах по стандартным шкалам;
·
колориметрия с применением индикаторной бумаги;
·
линейно-колористический метод, основанный на применении
индикаторных трубок (ИТ), которые представлены на рисунке 3.
Среди перечисленных методов в
практике химического контроля линейно-колористический метод (ЛКМ) получил
широкое распространение.
Метод основан на получении
окрашенного слоя индикаторного порошка.
Длина окрашенного слоя индикаторного
порошка пропорциональна концентрации исследуемого вещества в воздухе,
просасываемого через индикаторную трубку. В индикаторной трубке происходит
специфическая реакция между исследуемым веществом и реагентом, нанесенным на
инертный носитель - силикагель. Особенность ЛКМ заключается в том, что реакция
между определяемым веществом и реагентом протекает в динамических условиях.
Поэтому в основе ЛКМ должна лежать высокоспецифичная реакция, способная резко
изменять цвет наполнителя, содержащего эти реактивы. Существенными факторами
для получения правильных результатов в ЛКМ являются:
·
строгое соблюдение постоянства длины и диаметра индикаторной
трубки;
·
определенная процедура приготовления инертного носителя и
реактивного раствора;
·
порядок нанесения реактивного раствора на силикагель;
·
определенная насыпная плотность индикаторного порошка в трубке;
·
объем анализируемой смеси;
·
меры по защите реактивного вещества от окисления (разложения).
Приготовленные индикаторные трубки
способны измерять определенный диапазон концентраций вредного вещества и
обладают определенным сроком годности. Калибровка индикаторных трубок
осуществляется с помощью искусственных парогазовых смесей исследуемого вещества
с воздухом или азотом. С течением времени реактивное вещество в индикаторной
трубке разлагается (при этом концентрация реактивного вещества на единицу длины
падает) и индикаторная трубка начинает завышать показания. По мере уменьшения
концентрации реактивного вещества наблюдается увеличение показаний и уменьшение
интенсивности окрашивания. Затем индикаторная трубка теряет способность
реагировать на анализируемое вещество. Для продления срока годности
индикаторных трубок в состав реактивного вещества обычно вводят (если это
позволяет метод определения):
·
гидразин гидрат, связывающий кислород воздуха в трубке;
·
буферный кислотный раствор, повышающий устойчивость реагентов;
·
инертный газ, замещающий воздух в индикаторной трубке.
В качестве инертного носителя используется
силикагель - ангидрид кремниевой кислоты. Различают силикагели: крупнопористые
(диаметр пор 50А), мелкопористые (диаметр пор 15А). Силикагели выпускают
следующих марок:
·
КСМ - крупный, мелкопористый;
·
ШСМ - шихтовый мелкопористый;
·
МСМ - мелкий мелкопористый;
·
АСМ - активированный мелкопористый;
·
КСК - крупный крупнопористый;
·
ШСК - шихтовый крупнопористый;
·
МСК - мелкий крупнопористый;
·
АСК - активированный крупнопористый.
Диаметр зерен от 0,2 до 7 мм.
Реактивные растворы готовят в соответствии
с методикой определения. Порядок нанесения реактивного раствора является очень
ответственной процедурой, так как именно она определяет допустимую линейность и
интенсивность окраски. По этой причине все операции по нанесению реактивного
раствора на силикагель должны быть стандартизированы.
В работающей индикаторной трубке
различают отработанные слои и работающий слой (Рисунок 4). В отработанном слое
интенсивность окрашивания равномерна. В работающем слое окраска менее
интенсивна, а внешняя граница расплывчата. По мере своей отработки работающий
слой передвигается вдоль трубки. В некоторых трубках работающий слой окрашен, а
отработавшие слои бесцветны. На таких трубках оператор видит только пробегающий
в виде кольца рабочий слой.
Длина окрашенного слоя и размытость
работающего слоя зависят от зернения инертного носителя - силикагеля. Обычно
величина зернения в основном применяемая в ИТ лежит в диапазоне (0,1-0,4) мм. С
уменьшением зернения уменьшается длина окрашенного слоя на единицу концентрации
анализируемого вещества. С увеличением зернения длина окрашенного слоя
увеличивается. Размытость работающего слоя зависит от свойств реагента,
нанесенного на силикагель, зернения силикагеля (или соотношения определенных по
зернению фракций), скорости и функции изменения расхода анализируемой газовой
смеси. Чаще всего оптимальное соотношение диапазона измеряемой концентрации,
размера зернения силикагеля, концентрации реактивного вещества на нем для
определения конкретным типом аспиратора подбирают экспериментально.
Применение индикаторных трубок
ограничивают следующие факторы:
·
мешающее влияние вредных веществ, совместно присутствующих в
воздушной среде с определяемыми веществами (недостаточная селективность
метода);
·
низкие концентрации вредных веществ, подлежащих определению;
·
резкое изменение газового состава при пожарах, проливах различных
топлив, подаче фреона - огнегасителя в помещения;
·
сроки годности индикаторных трубок.
В настоящее время в промышленности
применяются около 70 наименований индикаторных трубок.
Описание приборов
газового анализа
ЛКМ широко применяется в
промышленности для быстрого определения газов и паров в воздухе
производственных помещений. Для этих целей были разработаны портативные
газоанализаторы марок УГ-1, 2 (универсальный газоанализатор), который
представлен на рисунке 5, АМ-5, АМ-5М, НП-3М и др. На базе этих
газоанализаторов был разработан также газоанализатор ПГА-ВПМ, которые
представляют собой устройство, прокачивающее воздушную среду через индикаторную
трубку. Каждый из этих приборов обладает своими достоинствами и недостатками.
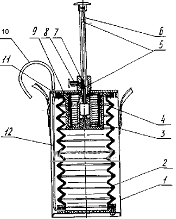
-корпус; 2-сильфон; 3-пружина;
4-кольцо распорное; 5-канавка с двумя углублениями; 6-шток; 7-втулка;
8-фиксатор; 9-плата; 10-трубка резинова; 11-штуцер; 12-трубка резиновая.
Рисунок 5 - Воздухозаборное
устройство УГ-2.
УГ представляет собой аспиратор
сильфонного типа. При сжатии сильфона к нему подсоединяется ИТ через резиновый
шланг. Шток ГА имеет выемки фиксирующие прокачиваемый объем. В состав ГА входят
штоки на различные объемы воздуха, которые применяются с определнным типом ИТ.
Газоанализатор АМ-5
представляет ручной сильфон работающий по принципу ГА фирмы «Дрегер». В верхней
части ГА расположено отверстие, куда вставляется ИТ (Рисунок 6).


Рисунок 6 - Газоанализатор АМ -
слева; газоанализатор АМ-5 с ИТ - справа
Требуемый объем воздуха
прокачивается определенным числом качаний, которое оговаривается в инструкции к
ИТ.
К АМ-5 выпускают по ТУ
12.43.01.166-86 различные типы ИТ, представленные в таблице 4.
Таблица 4 - Индикаторные трубки для
аспиратора АМ-5
|
Определяемый газ Срок годности, лет Кол-во в упаковке, шт.
Диапазон измерений (% об. доли) СО - 0,25 оксид углерода 3 24 0,0005-0,025
или 0,005-0,25 СО-5 оксид углерода 3 24 0,25-5,0 СО2-2 диоксид
углерода 2 24 0,25-2,0 СО2-15 диоксид углерода 2 24 1,0-15,0 СО2-50
диоксид углерода 2 12 5,0-50,0 SO2-0,007 диоксид серы 1 24
0,0002-0,007 Н2S - 0,0066 сероводород 3 24 0,00033-0,0066 СН2О
- 0,004 формальдегид 2 18 0,00002-0,0002 или 0,00004-0,004 NO+NO2-0,005
оксид азота 1 24 0,0001-0,005 О2-21 кислород 2 10 1,0-21,0 ТП
трубка защитная 2 24 улавливание углеводородов из газовой пробы
|
Прибор газового анализа вредных
примесей ПГА-ВПМ
ПГА-ВПМ предназначен для
определения концентрации вредных примесей в воздушной среде с помощью
индикаторных трубок и представлен на рисунке 7.
Время определения концентрации
вредных примесей приведено в таблице 5.
Таблица 5 - Время определения
концентрации вредных примесей
|
Наименование вредных примесей
|
Время
|
Наименование вредных примесей
|
Время
|
|
- окислов азота
|
13 мин
|
- триэтиламина
|
6 мин
|
|
- окиси углерода
|
13 мин
|
- ацетона
|
3 мин
|
|
- окиси углерода высокой концентрации
|
7 мин
|
- сернистого ангидрида
|
15 мин
|
|
- сурьмянистого и мышьяковистого водорода
|
6 мин
|
- сероводорода
|
15 мин
|
|
- предельных углеводородов
|
6 мин
|
- гептила
|
20 мин
|
Принцип действия прибора основан на
изменении цвета наполнителя индикаторной трубки (ИТ) при наличии в просасываемом
через нее воздухе вредных примесей. Просос определяемого объема воздуха
осуществляется сильфонным насосом. Содержание вредной примеси определяется по
длине столбика наполнителя ИТ, изменившего окраску.
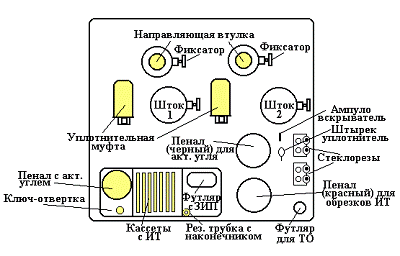
Рисунок 7 - Назначение органов
управления ПГА-ВПМ
Концентрацию измеряемой вредной
примеси определяют путем совмещения границы маркировки индикаторной трубки с
нулевым делением шкалы, находящейся в кассете для хранения трубок.
Общий порядок эксплуатации прибора
Для подготовки прибора и трубок к
работе необходимо:
открыть крышку прибора,
отвести вверх рычаг уплотнительной муфты, вставить шток в направляющую втулку
соответствующего сильфонного насоса (с одинаковой маркировкой), так, чтобы зуб
фиксатора вошел в продольный паз штока. Сжимать сильфонный насос с подключенной
ИТ запрещается;
оттянуть правой рукой
фиксатор и надавить левой рукой на головку штока. Фиксатор отпустить, а пружину
сильфона сжимать до тех пор, пока зуб фиксатора не западает в верхнее
углубление штока;
вынуть индикаторные трубки.
Работать осторожно: содержимое трубок при попадании в глаза и на кожу вызывает
болевые ощущения, а также разъедает одежду;
с помощью роликов
стеклореза на концах ИТ сделать надрезы и обломать концы трубок в отверстии
стеклореза. Концы трубок сложить в красный пенал, расположенный справа;
уплотнить с помощью штырька
ватный тампон со стороны конца трубки без перехвата;
подготовить к работе
окислительную трубку ТО-1 (для определения окислов азота). Для этого необходимо
отрезать сначала тонкий конец ТО-1, затем утолщенный. Взять ампуловскрыватель в
правую руку, а обрезанную окислительную трубку в левую так, чтобы из нее не
выпала ампула с окислителем, и вставить утолщенный конец ее в отверстие ампуловскрывателя,
чтобы игла вошла во внутреннюю полость трубки. Раздавить ампулу нажатием штыря.
Затем встряхивать ТО-1 до полного смачивания стекловаты окислительной
жидкостью.
Индикаторные трубки,
используемые в газоанализаторах для определения окислов азота
Индикаторная трубка на
окислы азота ИТМ-1Б представлена на рисунке 7.
Индикаторная трубка
заполнена силикагелем, обработанным реактивом сложного состава (иодид калия,
перманганат калия, тиосульфат натрия, хлорид натрия, крахмал). Индикаторная
трубка определяет только двуокись азота. Окись азота предварительно на
окислительной трубке окисляется до двуокиси азота.
Наполнитель окислительной трубки -
раствор марганцевокислого калия в ампуле. Индикаторная масса имеет исходный
желтый цвет. При взаимодействии с двуокисью азота в ней образуется свободный
йод, вызывающий посинение крахмала. Окрашенная шкала нелинейна и имеет три
ориентира: 0,5; 1,5; 5 мг\м3 (Рисунок 8).Объем прокачиваемого
воздуха 280 мл.
При использовании индикаторной
трубки ИТМ-1Б в приборе ПГА-ВПМ для соединения окислительной и индикаторной
трубок используется муфта малинового цвета. Окислительная трубка сохраняет свои
окисляющие свойства до момента сохранения цвета.
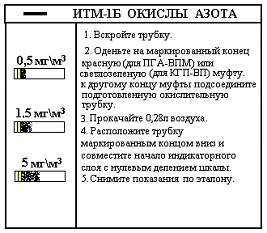
Рисунок 8 - Шкала для снятия
показаний ИТМ-1Б
Порядок определения:
обрезать с обоих концов
индикаторную трубку;
обрезать с обоих концов
окислительную трубку (обрезание со стороны ампулы нужно выполнить так, чтобы
образующееся отверстие было меньше диаметра ампулы. Если этого не сделать, то
при наклоне трубки ампула может выпасть);
ампуловскрывателем
раздавить ампулу окислительной трубки и встряхнуть ее в направлении
стекловолокна для смачивания;
через муфту соединить
окислительную и индикаторную трубки;
вставить трубки в
газоанализатор и прокачать объем 280 мл;
снять показания.
Все выше указанные действия
отображены на рисунке 9.
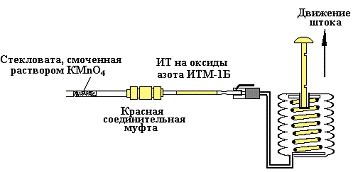
Рисунок 9 - Порядок
определения окислов азота
Главным недостатком данной
индикаторной трубки является ее малый диапазон измерения. В практике имели
место случаи с проливом азотного тетраоксида, который создавал исключительно
высокие концентрации. В процессе определения наблюдались случаи отрабатывания
ИТ по всей длине уже на этапе ее вскрытия. Дальнейшее измерение было
невозможно. Диапазон концентраций при проливе очень широк и угадать, в каком
поддиапазоне можно провести определения весьма затруднительно. По этой причине
любые работы в области исследования процессов определения азотного тетра оксида
являются важными и перспективными.
Колориметрическое
определение двуокиси азота по реакции с бензидином и b-нафтолом
При протягивании
воздуха, содержащего двуокись азота, через индикаторную трубку с силикагелем,
пропитанным растворами солянокислого бензидина и b-нафтола, цвет
индикаторного порошка изменяется. Интенсивность появившейся окраски измеряют по
стандартной шкале и определяют концентрацию двуокиси азота в воздухе.
Чувствительность метода
0,5 мг/м3 при отборе 100 мл воздуха.
Реактивы:
1) солянокислый бензидин,
насыщенный спиртовой раствор (0,5 мл, 1 мл, 5 мл;
) b-нафтол, 0,5%-ный спиртовой раствор;
) хлорид никеля NiCl2 - 6Н2О, 0,1%-ный спиртовой раствор.
Индикаторный порошок. Смешивают 0,5
г мелкого силикагеля с 0,2 мл насыщенного раствора солянокислого бензидина до
тех пор, пока порошок не станет сухим на вид. Затем прибавляют 0,1 мл 0,5%-ного
раствора b-нафтола, 0,1 мл
0,1%-ного раствора хлорида никеля, порошок перемешивают и высушивают при
комнатной температуре. Индикаторный порошок должен быть белого цвета.
Индикаторные трубки. В стеклянную
трубку диаметром 3 мм и длиной 100 мм насыпают слоем в 2 мм индикаторный
порошок. По обе стороны порошка помещают мелкий силикагель слоем 3 мм и вводят
по небольшому ватному тампону.
Приготовленные индикаторные трубки
запаивают.
Искусственная стандартная шкала.
Через индикаторные трубки протягивают по 100 мл воздуха, содержащего 0,5; 1,0;
10 мг/м3 двуокиси азота с интервалом 1,0 мг/м3. Оттенки
окраски индикаторного порошка зарисовывают на полосках бумаги акварельными
красками. Окраска в зависимости от концентрации NO2 изменяется от светло-розовой до вишнево-красной.
Ход определения.
Через индикаторную трубку
протягивают 100 мл воздуха в течение 30 сек. Через 1-2 мин. после отбора пробы
сравнивают интенсивность окраски индикаторного порошка со стандартной шкалой.
Другие способы
определения двуокиси азота и суммы ее с окисью азота
В.П. Федотов [2] применил
индикаторный порошок, состоящий из силикагеля, пропитанного раствором риванола.
Индикаторный порошок окрашен в лимонно-желтый цвет. Под действием двуокиси
азота окраска индикаторного порошка переходит в бледно-розовую до красной. Для
количественного определения использовал искусственную стандартную шкалу.
Чувствительность метода 0,004 мг/л
при протягивании 1 л воздуха.
Кобаяси и Китаева [3] рекомендуют
линейно-колористическое определение с использованием силикагеля, пропитанного
солянокислым о-толидином. При этом цвет слоя прореагировавшего индикаторного
порошка изменяется от серовато - белого до желто-зеленого.
Для количественного определения
авторы приводят кривые, выражающие зависимость между длиной окрашенного слоя
индикаторного порошка и концентрацией NO2. Метод позволяет
определять NO2 при концентрациях
(0,008-0,01) мг/л.
Киносян и Хаббард [4] применили для
пропитки силикагеля свежеприготовленный 1%-ный раствор сульфаниловой кислоты и
0,08%-ный раствор солянокислого N - (1-нафтил) - этилендиамина.
По длине окрашенного слоя
индикаторного порошка определяют содержание NO2 в воздухе. Чувствительность метода 0,002 мг/л при отборе 200 мл
воздуха.
Л.А. Мохов и сотр. [5] применили
колориметрический метод, основанный на получении азокрасителя при действии NO2 на индикаторный порошок, представляющий собой силикагель,
пропитанный Н-кислотой и n-аминобензойной кислотой. Полученную окраску колориметрируют по
искусственной стандартной шкале. Чувствительность метода 0,5 мг/м3.
К.Г. Парфенова [6] разработала метод
для суммарного определения в воздухе азотной и азотистой кислот, основанный на
восстановлении азотной кислоты цинковой пылью в уксусной среде до двуокиси
азота и последующего суммарного определения NO2 по реакции с реактивом Грисса-Илосвая.
Воздух протягивают через трубку,
наполненную силикагелем, смешанным с цинковой пылью, активированной сульфатом
марганца. После отбора пробы содержимое индикаторной трубки обрабатывают двумя
каплями реактива Грисса-Илосвая. Через 10 мин. интенсивность полученной окраски
сравнивают с искусственной стандартной шкалой. Чувствительность метода в
пересчете на N2O5 3 мг/м3 при
отборе 125 мл воздуха.
Предложен способ приготовления
наполнителя контрольных трубок для определения нитрозных газов, основанный на
пропитке силикагеля спиртовым раствором диэтиланилина и сульфаниловой кислоты.
Пропитывают 3- г предварительно промытого водой и высушенного при 105 0С
силикагеля 0,002 г. диэтиланилина и 0,1 г сульфаниловой кислоты, растворенными
в спирте. Затем силикагель просушивают при 30 0С до полного удаления
запаха спирта, заполняют реактивным порошком трубки и заплавляют их. Высушенный
белый порошок при соприкосновении с нитрозными газами окрашивается в
орнжево-желтый цвет.
Предложен наполнитель [7]
индикаторных трубок для определения двуокиси азота, основанной на реакции NO2 с N, N’ - дифенилбензидином. Для повышения чувствительности реакции и
срока хранения реактивного порошка в носитель добавляют кислые соли или
буферные растворы с рН 5,5. Для приготовления носителя 100 г. силикагеля с
величиной зерен (0,3-0,4) мм заливают 160 мл воды, в которой растворен 1 г
хлорида аммония. Равномерно пропитанный силикагель высушивают при 110 0С,
затем пропитывают 130 мл ацетона, в котором растворено 0,006 г. N, N’ - дифенилбензидина. После пропитки
для удаления ацетона порошок высушивают при 60 0С. Приготовленным
белым порошком заполняют индикаторные трубки. В присутствии NO2 порошок окрашивается в темно-голубой цвет. Реакция
высокочувствительная, минимальная определяемая концентрация NO2 равна 0,04, максимальная равна 200 мг/м3.
Предложен метод [8] основанный на
окислении азота до диоксида хромовой кислотой, нанесенной на твердый сорбент, и
дальнейшем определении диоксида азота по образованию азокрасителя с реактивом
Грисса-Илосвая.
Минимальная определяемая
концентрация 0,065 мкг в анализируемом объеме.
Определению мешает диоксид азота,
влияние которого устраняют в процессе отбора пробы. Влияние окислителей,
присутствующих обычно в атмосферном воздухе, незначительно.
Реактивы.
Сорбент. Инертный пористый носитель
типа ТС-М, промытый горячей водой и высушенный, с размером зерен (0,32-0,5) мм
смачивают 20%-ным водным раствором триэтаноламина, помещают в чашку и сушат
40-60 мин. при (90-95) 0С. Сухой сорбент должен рассыпаться.
Стабилизатор влажности. К 40 г.
безводного ацетата натрия медленно, по каплям, при постоянном перемешивании
добавляют 13 мл воды. Получают гранулированные кристаллические зерна.
Окислитель. Инертный пористый
носитель с размером зерен (0,32-0,5) мм смачивают раствором, содержащим 17 г.
триоксида хрома в 100 мл воды. Избыток раствора сливают, реагент высушивают при
105-115 0С и хранят в стеклянной банке с притертой пробкой.
Фильтр - стеклянная трубка
(внутренний диаметр 20 мм, длина 150 мм), заполненная 15 мл сорбента для
диоксида азота, 15 мл гранулированного порошка стабилизатора влажности, 10 мл
окислителя. Сорбенты разделены между собой тампонами стекловаты.
Поглотительный раствор. 40 г. иодида
калия растворяют в 500 мл воды. Полученный раствор должен быть бесцветным.
Сохраняют в банке из темного стекла. Срок хранения две недели.
Сульфит натрия. 0,06% раствор.
Готовят перед употреблением.
Уксусная кислота. 12% раствор. 64 мл
концентрированной кислоты наливают в мерную колбу на 500 мл и доводят водой до
метки.
Сульфаниловая кислота. 0,5 г
сульфаниловой кислоты растворяют в 150 мл 12% раствора уксусной кислоты.
Растворы сохраняют в банке из темного стекла.
a-Нафтиламин. 0,2 г
реактива растворяют в 20 мл воды при нагревании на водяной бане до образования
лиловых капель на дне колбы. Раствор осторожно сливают в темную склянку,
оставляя осадок в колбе, и приливают к раствору 150 мл 12% раствора уксусной
кислоты.
Составной реактив (реактив
Грисса-Илосвая). Перед анализом смешивают растворы a-нафтиламина и сульфаниловой кислоты в отношении 1:1.
Исходный стандартный раствор нитрита
натрия. (2-3) г NaNO2 растворяют и сушат при
50-60 0С в течение 2 ч. Навеску 0,15 г. растворяют в мерной колбе на
100 мл. 1 мл полученного раствора соответствует 1 мг NO2.
Рабочий стандартный раствор с
содержанием NO2 1 мкг/мл готовят
соответствующим последовательным разбавлением исходного стандартного раствора
поглотительным раствором иодида калия.
Отбор проб. Для определения разовой
концентрации исследуемый воздух со скоростью 0,25 л/мин протягивают в течение
20 мин. через систему, состоящую из стеклянной трубки-фильтра, заполненной
сорбентом для поглощения NO2, стабилизатором
влажности и окислителем, и U-образного поглотительного прибора, содержащего 6 мл 8% раствора
иодида калия.
Ход определения.
После отбора проб поглотительный
раствор доводят до первоначального объема. Отбирают 5 мл исследуемого раствора
в колориметрическую пробирку, добавляют 0,5 мл составного реактива и
встряхивают. Через 20 мин в пробирку вносят 5 капель 0,06% раствора сульфита
натрия и тщательно взбалтывают. Измеряют оптическую плотность раствора в кювете
с толщиной слоя 10 мм при длине волны 540 нм относительно воды. Одновременно
измеряют оптическую плотность контрольного раствора, для чего 5 мл
поглотительного раствора обрабатывают аналогично анализируемой пробе.
Выбор стандарта для замены
концентраций NxOy, в ГВС, моделирующего оксиды азота в зоне реакции.
Концентрацию диоксида азота находили
по калибровочному графику, для построения которого готовили серию стандартных
растворов:
номер стандарта;
рабочий стандартный
раствор, мл;
поглотительный раствор;
содержание диоксида азота,
мкг.
Затем из каждой колбы отбирали по 5
мл в пробирки и проводили все операции согласно ходу определения.

где:
V0 - объем исследуемого
воздуха, приведенный к нормальным условиям, л;
b - количество вещества, введенного в объем исследуемого раствора,
мкг.
Определение диоксида азота с N - (1-нафтил) - этилендиамином [8].
Принцип метода.
Диоксид азота реагирует с
сульфаниловой кислотой с образованием диазосоли, которая с N - (1-нафтил) - этилендиамином дает
красную окраску. Содержание диоксида азота определяют фотометрически.
Минимальная определяемая
концентрация 0,02 мкг/мл. Предельно допустимая концентрация диоксида азота в
атмосферном воздухе (максимальная разовая и среднесуточная) 0,085 мг/м3.
Гидропероксиды, сероводород, диоксид
серы, хлор и формальдегид мешают определению; оксид азота (I), триоксид азота и нитраты - не
мешают.
Реактивы.
Сульфаниловая кислота. Растворяли 4
г сульфаниловой кислоты в 800 мл дистиллированной воды, подкисляли 140 мл
ледяной уксусной кислоты и доводили объем водой до 1 л.
N - (1-нафтил) этилендиаминхлоргидрат, 0,1% водный раствор. Из-за
высокой стоимости не применяли. Взамен этого индикатора найден [8] реактив
в-нафтол.
3.3 Исследование и
разработка способа линейно - колористического и фотометрического определения
окислов азота по реакции с бензидином и Я-нафтолом
Данный метод был разработан в [1],
однако метод достаточно подробно исследован не был. По этой причине в качестве
дипломной работы была выбрана тема по исследованию колориметрических
возможностей метода.
Разработка установки для
проведения экспериментов
Для проведения экспериментов была
разработана установка, приведенная на рисунке 10 и на рисунке 12.

Рисунок 10 - Установка
для проведения экспериментов
Принципиально важным вопросом для
проведения анализа содержания окислов азота является наличие методики
приготовления стандартной парогазовой смеси окислов азота. В литературе ссылок
на методику такого типа найти не удалось. Сложность приготовления парогазовой
смеси заключается в высокой реакционной способности окислов азота практически
по отношению ко всем веществам. Кроме того, по современным представления окислы
азота представляют собой достаточно устойчивые во времени свободные радикалы.
АТ и его исходный окисел - двуокись азота представляют собой таким образом
представляет собой радикалы пятивалентного азота NO2•.
Проблема приготовления парогазовой
смеси была решена следующим образом. В герметичный сосуд объемом 5 литров
помещался нитрит натрия в навеске определенной массы (7,4; 29,6; 74) мг. Затем
к нему добавляли 0,2 мл воды дистиллированной и 0,5 мл концентрированной серной
кислоты. Добавление дистиллированной воды принципиально важно, т.к. реакция с
концентрированной серной кислотой с высоким выходом окислов азота не идет.
2NaNO2 + H2SO4
= Na2SO4 + NO + NO2 + H2O
2NO + O2 = 2NO2
После протекания реакции двуокись
азота вытеснялась в воздух, а окись азота в течение небольшого времени
окислялась до двуокиси. Данный процесс записан на цифровую фотокамеру и
демонстрирует образование окислов азота в 2 концентрациях (7,4; 74) мг. Внешний
вид образовавшихся окислов приведен на рисунке 10.
Объем прокачиваемой пробы сильфоном
ПГА-ВПМ равен 280 см3, а диапазон линейного определения окислов
азота лежит в пределах от 0 до 160 мкг в пробе. Тогда предельная концентрация,
которую можно определить при прокачивании 280 мл будет:
С = 160 мкг/280 мл = 0,57 мкг/мл
,57 мкг/мл*5000 мл = 2850 мкг - это
максимальное количество NO2 в воздухе, на объем 5
литров, которое определяется в пределах линейности данного способа. Для
создания этой концентрации необходима навекска нитрита натрия, которая
определяется из расчета:
138 у. е. NaNO2 →
46 у. е. NO2мкг NaNO2 →
2850 мкг NO2
Х = 8550 мкг NO2
Количество окиси азота, которое
будет образовано из нитрита натрия можно рассчитать по соотношению: 30 у. е. NO дают при протекании
реакции 92 у. е. NO2.
138 у. е. NaNO2 →
30 у. е. NO
,55 мг NaNO2→
Х мг NO
Х = 1,86 мг NO Из 30 у. е. NO образуется 92 у. е. NO2. Тогда из 1,86 мг NO будет образовано:
30 у. е. NO → 92 у. е. NO2
,86 мг NO → Х мг NO2
Х = 1,86*92/30 = 5,7 мг NO2
Навеска NaNO2 весом 8,55 мг дает: по реакции m (NO2) = 2850 мкг, NO2 путем окисления NO получается 5700 мкг.
Следовательно, для получения нужной концентрации в баллоне необходимо уменьшить
навеску в 3 раза. Т.о. масса навески равна 2,85 мг.
Навеска 2,85 мг NaNO2 дает концентрацию 2850 мкг NO2 в 5 литрах паровоздушной смеси. Соответственно:
мг NaNO2 → 2000 мкг NO2 в 5 литрах;
мг NaNO2 → 1000 мкг NO2 в 5 литрах;
,5 мг NaNO2 → 500 мкг NO2 в 5 литрах.
Для объема пробы в 10 мл - объем
шприца, посредством которого можно дозировать пробу, максимальная концентрация
в пределах диапазона линейности будет:
Смакс = 160 мкг/10 мл =
16 мкг/мл
Тогда масса NO2 в сосуде объемом 5 литров составит:
мкг/мл * 5000 мл = 80 мг NO2 Таким образом, из:
138 у. е. NaNO2 →
46 у. е. NO2
Х мг NaNO2→
80 мг NO2
Х = 240 мг NaNO2
240 мг NaNO2 дают 80 мг - NO2 и 160 мг по реакции NO → NO2. Следовательно: 80 мг NaNO2 образуют 80 мг NO2, включая реакцию NO→NO2. Соответственно:
мг NaNO2 → 60 мг NO2, включая реакцию NO→NO2
40 мг NaNO2 → 40 мг NO2, включая реакцию NO→NO2
20 мг NaNO2 → 20 мг NO2, включая реакцию NO→NO2
5 мг NaNO2 → 5 мг NO2, включая реакцию NO→NO2.
В герметичный сосуд объемом 5 литров
помещался нитрит натрия в навеске определенной массы (20; 40; 60; 80) мг. Затем
к нему добавляли 0,2 мл воды дистиллированной и 0,5 мл концентрированной серной
кислоты.

На фотографии представлены
парогазовые смеси с количеством NO2: 80, 60, 40, 20 мг.
В верхней части баллона для
обеспечения максимально полного реагирования компонентов и для равномерного
распределения паро-газовой смеси по всему объему была прикреплена кювета малого
объема. Для впрыскивания серной кислоты и воды через отверстие в крышке баллона
использовался медицинский щприц (V=2 мл).


Рис. Техника введения компонентов
для приготовления паровоздушной смеси окислов азота
Внутри сосуда установлены две
стеклянные трубки разной длины. Через длинную осуществлялся отбор проб, а через
короткую - приток свежего воздуха в сосуд в процессе отбора. В связи с тем, что
пары NO2• тяжелее воздуха при
отборе проб объемом 280 мл в течение 10 раз концентрация существенно не
изменялась. Отбор проб осуществлялся с помощью прибора ПГА-ВПМ. Объем
прокачиваемого воздуха - 280 мл.
Баллон с двуокисью азота был
подсоединен к двум последовательно соединенным поглотительным устройствам
Рыхтер, в которые был помещен спиртовой раствор солянокислого бензидина. Второе
поглотительное устройство подсоединялось с целью определения возможного
проскока. С помощью сильфона из баллона с паро-газовой смесью была прокачена
проба V=280 см3 (Рис.).

Рисунок - Лабораторная установка для
определения окислов азота и емкости для приготовления паровоздушной смеси
В ходе опыта было установлено, что
реакция оксида азота и солянокислого бензидина проходит через промежуточную
стадию окрашивания раствора в темно-зеленый цвет. С течением времени около 3-5
минут раствор приобретает характерный темно-красный цвет. Во втором поглотителе
имеет место небольшой проскок, ~5%.


Основные направления работы были
связаны с исследованием цветной реакции в поглотительном приборе. Разбавляя
полученный в ходе опыта темно-красный раствор оксидов азота и солянокислого
бензидина до состояния, приемлемого для измерения оптической плотности, можно
определить концентрацию данного разбавленного раствора по полученной ранее
шкале и с учетом коэффициента разбавления пересчитать концентрацию исходного
раствора (для получения различной интенсивности окрашивания и увеличения точности
определения разбавляли полученный раствор этиловым спиртом).
Была исследована возможность прямого
определения двуокиси азота по ее характерной коричневой окраске путем
фотометрирования. Для этого было взято три навески NaNO2 40 мг, 60 мг, 80 мг и приготовлено три парогазовых смеси.
Предварительно был снят спектр во всей возможной области для СФ-26: от 210 нм
до 760 нм. Областью наибольшего поглощения является УФ область 220-230 нм.
Таким образом данный диапазон может быть рекомендован для определения двуокиси
азота прямым методом. На приведенных рисунках видно наличие характерной окраски
в закрывающихся кварцевых кюветах. Техника приготовления определенной
концентрации двуокиси азота показана на рисунке…. Приготовленная стандартная
концентрация окислов азота прокачавалась через кювету, которая была закрыта
пробкой из резины протыкаемой двумя иглами. После прокачивания кювета
освобождалась от иголок и фотометрировалась.



Приготовленные смеси были прокачаны
через кварцевые кюветы и была измерена их оптическая плотность относительно
воздуха. Результаты представлены в таблице.
|
D
|
Масса навески, мг
|
Объем баллона, л
|
Концентрация, мг/л
|
|
0,007
|
40
|
5
|
8
|
|
0,03
|
60
|
5
|
12
|
|
0,04
|
80
|
6
|
13,3
|
По графику можно сделать вывод, что
зависимость D = f(C) линейная.
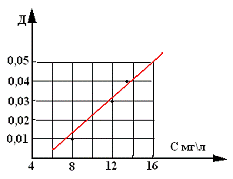
Зависимость D = f(C) для двуокиси азота определяемой
спектрофотометрическим методом по пару в воздухе
Приготовление
составляющих ингридиентов для проведения эксперимента
Приготовление индикаторного порошка
Смешивают 0,5 г мелкого силикагеля с
0,2 мл насыщенного раствора солянокислого бензидина до тех пор, пока порошок не
станет сухим на вид. Затем прибавляют 0,1 мл 0,5%-ного раствора b-нафтола, 0,1 мл 0,1%-ного раствора хлорида никеля, порошок
перемешивают и высушивают при комнатной температуре (Рисунок 13). Индикаторный
порошок должен быть белого цвета, как показано на рисунке 14.
 (а)
(а)
 (б)
(б)
 (в)
(в)
Рисунок 13 - Приготовление
индикаторного порошка:
а - добавление к силикагелю
солянокислого бензидина;
б - добавление спиртового раствора
хлорида никеля;
в - перемешивание полученной смеси
ингредиентов

Рисунок 14- Исходный индикаторный
порошок
Следует заметить, что приготовленный
индикаторный порошок с течением времени, приблизительно равного одному месяцу,
изменяет свою окраску с белого цвета на коричневый, как представлено на рисунке
15. Порошок коричневого цвета является не пригодным для проведения опытов. Для
повышении срока годности данного реактива, его хранили в инертной гелийной
атмосфере. Как показала практика в инертной атмосфере инкдикаторный порошок
сохраняет свои свойства и внешний вид до года.

Рисунок 15- Индикаторный порошок с
истекшим сроком годности
Приготовление индикаторных трубок
В стеклянную трубку диаметром 3 мм и
длиной 100 мм насыпают слоем в 2 мм индикаторный порошок. По обе стороны
порошка помещают мелкий силикагель слоем 3 мм и вводят по небольшому
стекловатному тампону. Полученная индикаторная трубка представлена на рисунке
16.

Рисунок 16 - Вид индикаторной трубки
Ход эксперимента
Для приготовления концентраций NO2•, равных (1, 4, 10) мг/м3, взвешивали три разные
навески нитрита натрия: 7,4 мг; 29,6 мг; 74 мг соответственно и смешивали их с
0,2 мл воды дистиллированной и 0,5 мл концентрированной серной кислоты.
При увеличении или уменьшении
величины навески возможно приготовление более и менее концентрированных
паровоздушных смесей. Смесь является нестойкой и спустя несколько часов ее
концентрация сильно меняется. По этой причине приготовленные смеси
использовались сразу после приготовления, а при постановке новой серии
экспериментов готовились заново.
В ходе пропускания через ИТ
парогазовой смеси 1 стандартной концентрации уже приблизительно при пропускании
50 мл воздуха, на ней появилось бирюзовое (сине-зеленое) окрашивание, которое
быстро переходило первоначально в коричневый цвет, а затем, спустя (15-20)
минут в розовый.
Таким образом по результату
эксперимента можно сделать вывод, что ИТ позволяет контролировать концентрацию
окислов азота по наличию всех типов окрашивания. При этом чувствительность
трубки может быть повышена.
Проведение эксперимента с хлоридом
никеля в изопропиловом спирте.
При проведении экспериментов, было
установлено, что на процесс определения сильно влияет растворитель. Замена
растворителя - этилового спирта на изопропанол приводит к изменению цвета
окрашивания в ИТ.
Для проведения опыта была взята
навеска хлорида никеля массой 200 мг и растворена в 127 мл изопропилового спирта.
Из полученного 0,25% раствора NiCl2*6H2O в изопропаноле, был приготовлен
новый индикаторный порошок. Далее была приготовлена ИТ, содержащая слой 7 мм
данного индикаторного порошка.
Для приготовления воздушной смеси
бралась наибольшая имеющаяся навеска нитрита натрия, равная 74 мг, которая
создает концентрацию двуокиси азота С=10 мг/м3 в 5 л объема. При пропускании
воздуха, через ИТ на изопропаноле было зафиксирована равномерное желтое
окрашивание, что соответствует рисунку 17.

Рисунок 17 - Окрашивание ИТ,
приготовленной на основе растворителя - изопропанола
3.4 Определение окислов
азота на основе спектрофотометрического метода
Спектрофотометрические методы
обладают значительными преимуществами по сравнению с ЛКМ, выражающиеся в
большем линейном диапазоне и повышенной чувствительностью определения. Поэтому
была исследована возможность построения шкалы для определения окислов азота по
данному способу определения.
Для проведения следующих
экспериментов были применены поглотительные приборы ТУ 9452-016-00480514-95. В
промышленности выпускаются поглотительные приборы следующих типов: простой,
поглотитель с пористой пластиной, Рыхтер (Рисунок 18), Полежаева, Петри,
Зайцева, Яворской. Поглотители имеют сравнительно небольшой коэффициент
поглощения, поэтому при анализе часто устанавливаются по 2-3 в связке. Часто
поглотители объединяются в системы для большего удобства в работе.
Рисунок 18 - Поглотитель Рыхтер
Основные направления работы были
связаны с исследованием цветной реакции в поглотительном приборе и ее изменения
при различных вариантах проведения:
) для получения различной
интенсивности окрашивания и увеличения точности определения разбавляли
полученный раствор этиловым спиртом в (1,5-2) раза;
) заменяли растворитель
хлорида никеля с этанола на изопропанол;
) изменяли концентрацию
каждого ингредиента на 50% в обе стороны:
· солянокислый
бензидин 0,5:1:1; 1,5:1:1;
· Я-нафтол 2:1,5:1;
2:0,5:1;
· NiCl2*6H2O 2:1:1,5; 2:1:0,5.
Реактивы для определения в жидкой
фазе брались в том же соотношении, в каком они используются при приготовлении
ИТ.
В поглотительный прибор помещали в
соотношении 2:1:1:
) солянокислый бензидин
(спиртовой раствор);
) Я-нафтол (спиртовой
раствор);
) спиртовой раствор хлорида
никеля.
Объем исходной смеси был выбран
равным 5 мл - оптимальным для поглотительного прибора Рыхтер. При пропускании
280 мл смеси двуокиси азота в воздухе с концентрацией 100 мг/м3
наблюдается изменение окраски от светло-желтой до бирюзово - зеленой, а затем
до вишнево красной в диапазоне возрастающего количества двуокиси азота от 1 до
28 мкг (Рисунок 19).

Рисунок 19 - Окрашивание раствора в
поглотительном приборе по окончании прокачки 280 мл
Данный процесс записан на цифровую
видеокамеру и представлен в приложении к дипломной работе (Файл.
«Покачивание»).
Диапазон концентраций при
определении может быть по чувствительности достаточно сильно изменен. Для этого
достаточно разбавить исходную смесь этиловым спиртом. В качестве примера брали
исходную смесь всех компонентов в соотношении (1:0,5:0,5) соответственно,
разбавляем ее 2 мл этилового спирта, затем пропускали через поглотительный
прибор паровоздушную смесь окислов азота. При пропускании наблюдается растягивание
окрашивания во времени - формирование зеленого окрашивания, переходящего в
красное, что позволяет более точно регистрировать величину концентрации окислов
азота а также итоговое окрашивание из-за разбавления становится менее
выраженным, что представлено на рисунке 20.

Рисунок 20 - Сравнение интенсивности
окрашивания при разбавлении реактивного раствора этанолом
Данное разбавление позволяет более
точно определить концентрации окислов азота в диапазоне от 1 до 5 мкг. (Файл
«Прокачивание при разбавлении»).
При замене спирта в хлориде никеля с
этилового на изопропиловый и, проводя эксперимент в таких же соотношениях,
существенного отличия не получали. Окраска изменяется аналогично этанолу.
При изменении соотношения в
реактивном растворе солянокислого бензидина в сторону увеличения до 1,5, в
сторону уменьшения 0,5 были получены следующие данные:
При увеличении объема полученный
реактивный раствор имел более длинный период зеленого окрашивания а при
развитии красного имел более темный оттенок.
При уменьшении объема полученный
реактивный раствор имел более короткий период зеленого окрашивания, а при
развитии красного имел более коричневый оттенок, как показано на рисунке 21.


Рисунок 21 - Сравнение влияния
количества солянокислого бензидина в реактивном растворе: справа исходный
вариант развития окраски при соотношении реактивов 2:1:1, в центре - соотношение
3:1:1, слева соотношение - 1:1:1
Изменение соотношений других
реактивов реактивного раствора на цвет окрашивания не повлияли.
Построение шкалы для
спектрофотометрического определения оксида азота
Для построения шкалы был приготовлен
растворы нитрита натрия с различными концентрациями мкг/мл в этиловом спирте.
1. C (NaNO2)
= 4 мкг/мл
В 100 мл раствора содержится 400 мкг
= 0,4 мг NaNO2. Было взято 0,4 мг NaNO2 и растворено в 100 мл этанола.
2. C (NaNO2)
= 20 мкг/мл
В 100 мл содержится 2000 мкг = 2 мг.
Было взято 2 мг NaNO2 и растворено в 100 мл
этанола.
3. C (NaNO2)
= 200 мкг/мл
В 100 мл содержится 20000 мкг или 20
мг. Было взято 100 мл этанола и растворено в них 20 мг NaNO2.
. Раствор солянокислого
бензидина должен быть объемом 150-200 мл.
. 0,5% спиртовой раствор
в-нафтола
,5 г в-нафтола в 100 г. этанола
с = 0,81 г./см3
V = 100г/0,81 = 123,45 мл
Т.о. 0,5 г в-нафтола заливаем 123 мл
этанола.
. NiCl2Ч6H2O 0,1 спиртовой раствор
М = 59+35,5*2+18*6 = 237
В 237 г. NiCl2Ч6H2O → 129 г. NiCl2
x ← 0,1 г NiCl2
x = 237*0,1/129 = 0,184 г.
Т.о. 0,184 г. NiCl2Ч6H2O растворяем в 123 мл этанола.
. Итого этанола необходимо взять 746
мл.
Шкала строилась при внесении
реактивов, представленных в таблице 6, и показана на рисунке 22.
Таблица 6 - Построение шкалы для
спектрофотометрического анализа
|
Наименование раствора
|
Объем раствора, мл
|
|
Спиртовой раствор NaNO2
(4 мкг/мл)
|
-
|
0,6
|
1,2
|
-
|
-
|
-
|
-
|
-
|
|
Спиртовой раствор NaNO2,
(20 мкг/мл)
|
-
|
-
|
-
|
0,75
|
1,5
|
-
|
-
|
-
|
|
Спиртовой раствор NaNO2,
(200 мкг/мл)
|
-
|
-
|
-
|
-
|
-
|
3
|
0,6
|
1,2
|
|
Спирт, мл
|
2
|
1,4
|
0,8
|
1,25
|
0,5
|
1,7
|
1,4
|
0,8
|
|
Солянокислый бензидин, мл
|
2
|
2
|
2
|
2
|
2
|
2
|
2
|
2
|
|
в-нафтол, 0,5%
|
1
|
1
|
1
|
1
|
1
|
1
|
1
|
1
|
|
NiCl2,
0,1%
|
1
|
1
|
1
|
1
|
1
|
1
|
1
|
1
|
|
Эквивалент NO2
|
0
|
2,4
|
8
|
15
|
30
|
60
|
120
|
240
|

Рисунок 22 - Шкала растворов нитрита
натрия
В результате фотометрирования
образовавшегося окрашивания было установлено, что шкала становится линейной и
воспроизводимой спустя 20-30 минут после ее приготовления. В более ранний
период зависимость нелинейна. Полученная зависимость спустя 20-30 минут
выражено линейна в диапазоне до 200 мкг иона NО2 в пробе, что является достаточно
высоким показателем для определения высоких концентраций двуокиси азота
(Рисунок 23).
Оптическая плотность растворов была
измерена с помощью СФ-26 относительно нулевого стандарта при длине волны л=400
нм.
Была получена зависимость:
|
№ стандарта
|
Оптическая плотность
|
Количество NO2
в пробе по шкале
|
|
1
|
0,01
|
2,4
|
|
2
|
-
|
-
|
|
3
|
0,105
|
15
|
|
4
|
0,145
|
30
|
|
5
|
0,300
|
60
|
|
6
|
0,560
|
120
|
|
7
|
1,200
|
240
|
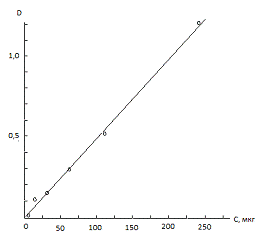
Рисунок 23 - Зависимость оптической
плотности от концентрации иона NO2 в пробе
Выпала точка №3.
Для определения длины волны, на
которой должно проводиться спектрофотометрическое определение был снят спектр
окрашенного продукта, который приведен на рисунке 24.

Рисунок 24 - Спектр окрашенного
продукта при определении на спектрофотометре СФ-26
Исходя из спектра целесообразной
длиной волны для определения является длина волны 400 нм. Данные спектра
представлены в таблице 7.
Таким образом, экспериментально
установлена возможность определения окислов азота спектрофотометрическим
методом по рассматриваемой реакции.
Таблица 7 - Спектр окрашенного
соединения
|
л, нм
|
D
|
|
400
|
0,74
|
|
420
|
0,69
|
|
440
|
0,6
|
|
460
|
0,57
|
|
480
|
0,54
|
|
500
|
0,5
|
|
520
|
0,48
|
|
540
|
0,44
|
|
560
|
0,37
|
|
580
|
0,32
|
|
600
|
0,27
|
|
620
|
0,25
|
|
640
|
0,23
|
|
660
|
0,22
|
|
680
|
0,22
|
Из растворов был исключен NiCl2. Шкала получилась, первые четыре пробы отливают зеленым цветом.
Для данных растворов также была измерена оптическая плотность.

|
№ стандарта
|
Оптическая плотность
|
Количество NO2
в пробе по шкале
|
|
1
|
0,006
|
2,4
|
|
2
|
0,015
|
8
|
|
3
|
0,075
|
15
|
|
4
|
0,175
|
30
|
|
5
|
0,4
|
60
|
|
6
|
0,863
|
120
|
|
7
|
1,55
|
240
|

NiCl2 на качество шкалы не
влияет. Он применяется для увеличения контрастности окрашивания.
Из растворов были убраны NiCl2 и в-нафтол →шкала получилась.

|
№ стандарта
|
Оптическая плотность
|
Количество NO2
в пробе по шкале
|
|
1
|
0,035
|
2,4
|
|
2
|
0,025
|
-
|
|
3
|
0,125
|
15
|
|
4
|
0,25
|
30
|
|
5
|
0,60
|
60
|
|
6
|
1,25
|
120
|
|
7
|
2,00
|
240
|
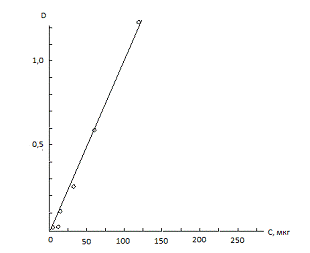
Было установлено, что в области
высоких концентраций наблюдается нарушение закон Будера - Ламберта, т.е.
линейная зависимость D от количества NO2 существует при
концентрациях NO2, не превышающих 155
мкг/мл.
В ходе разработки данного метода
получили колористическую шкалу на базе спиртовых растворов солянокислого
бензидина с NaNO2. Соотношения объемов
реагентов приведены в таблице.
Объем раствора, мл
|
|
Спиртовой раствор NaNO2
(4 мкг/мл)
|
-
|
0,6
|
1,2
|
-
|
-
|
-
|
-
|
-
|
|
Спиртовой раствор NaNO2,
(20 мкг/мл)
|
-
|
-
|
-
|
0,75
|
1,5
|
-
|
-
|
-
|
|
Спиртовой раствор NaNO2,
(200 мкг/мл)
|
-
|
-
|
-
|
-
|
-
|
3
|
0,6
|
1,2
|
|
Спирт, мл
|
2
|
1,4
|
0,8
|
1,25
|
0,5
|
1,7
|
1,4
|
0,8
|
|
Солянокислый бензидин, мл
|
2
|
2
|
2
|
2
|
2
|
2
|
2
|
2
|
|
Эквивалент NO2
|
0
|
2,4
|
8
|
15
|
30
|
60
|
120
|
240
|
NiCl2 можно добавить в
растворы для улучшения контрастности шкалы.
4. Результаты работы и
их обсуждение
В результате выполнения дипломной
работы были получены следющие основные результаты:
опробована методика
определения высоких концентраций окислов азота по реакции с солянокислым
бензидином;
отработан способ получения
стандартных парогазовых смесей окислов азота в воздухе статическим методом;
на базе реакции окислов
азота с солянокислым бензидином предложен спектрофотометрический метод
определения окислов азота в области высоких концентраций.
Главная проблема данного объекта
исследования заключается в точности приготовления стандартных парогазовых
смесей. В исходной методике и в литературе никаких сведений по приготовлению
калибровочных смесей обнаружить не удалось. В литературе приведены только
способы получения двуокиси азота, которые не позволяют по ряду причин
приготовить стандартную концентрацию. При использовании реакции
концентрированной азотной кислоты с медью весьма проблематично обеспечить ее протекание
внутри сосуда с воздухом из-за слишком долгого процесса взаимодействия от 20 до
30 минут. Результатом такого протекания реакции является длительный процесс
установления неизменной концентрации.
Реакция прокаливания нитрата свинца
обладает теми же недостатками с точки зрения длительности установления
концентрации и осуществить прокаливание в герметичном пластиковом сосуде, в
котором готовятся смеси, практически не возможно.
Предложенный мною способ так же не
является идеальным, хотя обладает главным достоинством - быстрым, одномоментным
выделением оксидов азота в герметичный объем, в котором осуществляется
перемешивание. Расчетное значение концентрации реально будет несколько выше
из-за потерь окислов азота в небольшом количестве разбавленной серной кислоты
(за счет их растворения). Проведенные эксперименты показывают, что установление
постоянной концентрации происходит в течение (5-6) минут. Однако, при стоянии
происходит падение концентрации окислов азота, что даже заметно, спустя сутки,
невооруженным глазом. В дальнейшем смесь устойчиво храниться около двух суток.
Для приготовления парогазовой смеси использовались инертные газы (гелий, азот),
которые способствуют устойчивости смеси во времени. Вытеснению воздуха из
сосудов емкостью 5-6 литров проводили продувкой гелием сосуда в течение (3-4)
минут. Навеска нитрита натрия заранее до продувки размещалась в пластиковом
контейнере, а серная кислота вводилась шприцом в емкость при проколе крышки.
Приготовление в инертном газе обеспечивает большую стабильность парогазовой
смеси. Вместе с тем оценка точности приготовления проведена не была.
Приготовленные парогазовые смеси в воздухе обеспечили описанный в исходной
методике аналитический результат. Идеальным вариантом приготовления смесей
является вариант их разбавления в газообразном виде из баллона с известной
стандартной концентрацией, приготовленной в заводских условиях. Такой вариант
реализован не был из-за высокой стоимости. Теоретически возможно достаточно
точное приготовление парогазовой смеси из баллона, путем отбора из него
определенного объема большим шприцом и внесение этого объема в емкость.
При отработке линейно -
колористического метода определения была достигнута хорошая воспроизводимость
результатов в соответствии с описанием метода. Однако, при разбавлении
реактивного раствора этиловым спиртом было получено зеленое окрашивание ИТ,
возникающее дополнительно перед розовым. Данный эффект может быть использован
как колориметрический в другом диапазоне определения, который к настоящему
моменту не выявлен и может быть реализован в последующих исследованиях.
Возможно использование данной ИТ со
слоем наполнителя не (4-5) мм, а (30-50) мм с реализацией ЛКМ. При
приготовлении ИТ такого типа наблюдалось значительное размывание реагирующего
слоя. При выдержке трубки в течение нескольких часов, наблюдалось достаточно
сильное изменение окраски. Это позволяет сделать вывод о невозможности
реализации метода ЛКМ при данной реакции определения. По этой причине была
предпринята попытка разработки спектрофотометрического метода определения.
Предложен новый способ
спектрофотометрического определения двуокиси азота в воздухе, обладающий
линейностью в диапазоне высоких концентраций окислов азота от 10 до 200 мкг в
пробе.
Были выполнены эксперименты с
использованием реагирующей системы - солянокислого бензидина, b-нафтола, хлористого никеля в виде спиртовых растворов при
использовании линейно колористического метода и спектрофотометрического
методов. Для образующегося окрашенного соединения сняты зависимости оптических
плотностей от длины волны видимого света и построен градуировочный график для
оксидов азота по нитрит иону. Установлено наличие нескольких окрашенных
продуктов, позволяющих выполнять определения в различных диапазонах
концентраций.
Выполнено исследование возможности
определения по окрашенному веществу красно-коричневого цвета. Установлено
наличие линейного диапазона, позволяющего проводить определение.
Установлен максимум поглощения
спектра для продукта красно-коричневого цвета с длиной волны 400 нм. Данная длина
волны и является рекомендуемой при определении окислов азота
спектрофотометрическим методом.
Были сняты спектры продуктов
окрашенной реакции в видимой области на спектрофотометре СФ-26, которые
показывают возможное наличие в реактивном растворе однотипных, но различно
окрашенных соединений, образующихся с увеличением концентрации двуокиси азота.
Для использования фотометрического
способа определения окислов азота возможно установление диапазона линейности
для каждого продукта при изменении:
соотношения исходных
компонентов - бензидина, бетта-нафтола, хлористого никеля;
разбавление их в
реагирующей смеси этиловым спиртом;
приготовление растворов на
изопропиловом спирте и других спиртах;
выбор оптимальной длины
волны для спектрофотометрического определения на каждом участке концентраций.
Воспроизведен способ контроля
концентраций двуокиси азота по реакции с солянокислым бензидином в присутствии
хлорида никеля и бетта нафтола.
ИТ реагируют с двуокисью азота с
образованием продукта красно-коричневого цвета в широком диапазоне
концентраций. Предел чувствительности составляет около 0,5 мг\м3. С
изменением концентрации цвет отработавшего слоя изменяется от розового до
красно-коричневого. В области высоких концентраций цвет становится желтым,
переходящим в красный.
ИТ должна храниться в герметично
закрытом состоянии, в атмосфере азота (гелия), без доступа света. При хранении
ИТ в открытом виде реагирующая способность сохраняется в течение (2-3) суток. С
увеличением срока хранения ИТ в открытом виде, реактивное вещество испаряется с
поверхности силикагеля и частично разлагается. Результатом этих процессов
является сильное снижение чувствительности ИТ за время около (5-10) дней.
Предпочтительным вариантом хранения является хранение реактивного порошка в
инертной атмосфере герметично закрытым. В этом случае ориентировочное время
сохранения индикаторного порошка в пригодном для использования виде составляет
около 0,5 года.
5. Технико-экономическое
обоснование работы
.1 Расчет затрат на
проведение исследовательской работы
Затраты на проведение
исследовательской работы могут состоять из текущих и капитальных. В состав
текущих входят затраты на израсходованные реактивы, сырье, материалы, различные
виды энергии, заработная плата исследователя, руководителя, консультанта, на
исследуемые в процессе приборы, амортизационные отчисления от стоимости этих
приборов, расходы, связанные с выполнением специальных анализов и
общеинститутские расходы.
При проведении данной
исследовательской работы имели место только текущие затраты, т.к. специальные
приборы и монтаж дополнительного оборудования не требовался.
Для определения общей суммы текущих
затрат составляется смета, представляющая собой свод всех текущих затрат на
выполнение исследовательской работы.
Затраты на сырье, материалы и
реактивы (Зм), израсходованные при проведении работы определяются по
формуле (2):
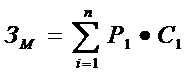 , (2)
, (2)
где Рi - расход i-го видов материальных ресурсов,
натур. ед.;
Сi - цена i-го вида материальных ресурсов,
руб.;
i = 1, 2, 3,… n - виды материальных ресурсов.
Результаты расчета этих затрат
отражены в таблице 8.
Таблица 8 - Затраты на сырье,
материалы и реактивы
|
№
|
Наименование сырья, материалов, реактивов
|
Ед. изм.
|
Кол-во израсходованного материала
|
Планово-заготовительная цена единицы ресурса, руб.
|
Стоимость ресурсов, руб.
|
|
1
|
Нитрит натрия (NaNO2) ч
|
кг
|
0,293
|
36,40
|
10,7
|
|
2
|
Серная кислота (H2SO4)
|
л
|
0,03
|
25
|
0,75
|
|
3
|
Вода дистиллированная
|
л
|
3
|
27
|
81
|
|
4
|
Я-нафтол (спиртовой раствор) (C10H7OH)
|
кг
|
0,05
|
320
|
16
|
|
5
|
Хлорид никеля (спиртовой раствор) (NiCl2*6H2O)
|
кг
|
0,05
|
200
|
10
|
|
6
|
Этиловый спирт (C2H5OH)
|
л
|
0,325
|
24
|
7,8
|
|
7
|
Изопропиловый спирт (CH3CH(OH) CH3)
|
л
|
0,250
|
420
|
105
|
|
8
|
Силикагель КС КГ
|
кг
|
0,25
|
56
|
14
|
|
9
|
Солянокислый бензидин
|
л
|
0,002
|
1200
|
2,4
|
|
Итого: 244,2 руб.
|
В состав энергетических затрат
входят затраты на электроэнергию, пар, холод и т.д., израсходованные при
выполнении исследования на технологические нужды. При выполнении данной
исследовательской работы использовалась только электроэнергия.
Затраты на электроэнергию (Зэ)
определяются по формуле (3):
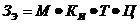 , (3)
, (3)
где М - паспортная
мощность электродвигателей электроприборов, кВт;
Ки -
коэффициент использования мощности электрооборудования (принимается Ки
= 0,7 - 0,9);
Кэх -
коэффициент, учитывающий затраты на содержание энергохозяйства предприятия
(принимаем Кэх =1,07-1,1);
Т - время работы
электрооборудования за весь период выполнения исследования, ч;
Ц - цена 1 кВт ч
электроэнергии, 2,55 руб./кВт.
ч
электроэнергии, 2,55 руб./кВт.
Затраты на
электроэнергию представлены в таблице 9.
Таблица 9 - Затраты на
электроэнергию
|
Наименование приборов
|
Номинальная паспортная мощность единицы электрооборудования кВт
|
Количество единиц электрооборудования, шт.
|
Номинальная мощность всего установленного электрооборудования,
кВт
|
Коэффициент спроса
|
Коэффициент увеличения заявленной мощности за счет потерь
энергии в сетях
|
Заявленная мощность электрооборудования, кВт ,
|
Эффективный годовой фонд времени работы электрооборудования в
году, час
|
Годовой расход электроэнергии, кВт*ч
|
|
Весы аналитические БЛР-200
|
0,10
|
1
|
0,1
|
0,9
|
1,1
|
0,099
|
5
|
0,495
|
|
Весы лабораторные WAS 220/C2
|
0.0083
|
1
|
0,0083
|
0,9
|
1,1
|
0,008
|
3
|
0,024
|
|
Вытяжной шкаф (вентилятор осевого типа общего назначения)
|
0,2
|
1
|
0,2
|
0,9
|
1,1
|
0,198
|
10
|
1,98
|
|
Спектрофотометр СФ-26
|
0,1
|
1
|
0,1
|
0,9
|
1,1
|
0,099
|
2
|
0,198
|
|
Газоанализатор ПГА-ВПМ
|
0,1
|
1
|
0,1
|
0,9
|
1,1
|
0,099
|
6
|
0,594
|
|
ИТОГО:
|
|
0,503
|
|
3,291
|
Ц=2,55 руб./кВт.
Зэ=3,29·2,55·1,1=9,23
руб.
Затраты на воду рассчитываются
исходя из ориентировочного расхода и цены по формуле (4):
Зв=Q·Ц, (4)
где Зв - затраты на воду, руб.;
Q - расход воды, л;
Ц - цена на воду за 1 л, руб.
Ц =19 руб./л.
Определение затрат на воду приведено
в таблице 10.
Таблица 10 - Затраты на воду
|
Наименование
|
Расход, л
|
Цена, руб./л
|
Затраты, руб.
|
|
Вода
|
2
|
19
|
38
|
При переходе научных подразделений
на хозрасчет, важным показателем оценки проводимого исследования является
договорная цена на НИР Цнир, которая рассчитывается по формуле (5):
 , (5)
, (5)
где Знир -
затраты на исследование, предусмотренные планом дипломной работы, руб.;
Р - уровень
рентабельности исследования, 20%;
К - коэффициент,
учитывающий поощрительную надбавку за качество разработки (К = 1).
Научно-технический
эффект НИР рассчитывается по формуле (6):
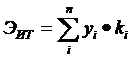 ,
(6)
,
(6)
где yi - величина частного показателя,
балл;
Исходя из степени
новизны исследования у1 = 50.
Исходя из
научно-технического уровня исследования у2 = 70.
Исходя из уровня глубины
воздействия в процессе использования
у3 = 30.
ki
- весовой коэффициент i-го
частного показателя;
k
= 0,33
i
= 1, 2, 3,… n - количество учитываемых частных показателей.
Эит =50·0,33
+ 70·0,33 + 30·0,33 = 49,5 следовательно К=1,5.
Затраты на стеклянную
посуду приведены в таблице 11.
Так как оборудование для
опытов не покупалось, а использовалось то, что было в лаборатории, то затраты
на посуду и приборы составят всего 30% от суммы полученной выше.
Таблица 11 - Затраты на
стеклянную посуду и приборы
|
№
|
Наименование прибора
|
Кол-во, шт.
|
Цена, руб.
|
Стоимость, руб.
|
|
1
|
Поглотительные приборы
|
2
|
235
|
470
|
|
2
|
Цилиндр с пришлифованной пробкой (емкость 25 мл.)
|
1
|
82
|
82
|
|
3
|
Цилиндр мерный 100 мл.
|
2
|
115,75
|
231,2
|
|
4
|
Пипетка градуированная
|
4
|
18,00
|
72
|
|
5
|
Пробирка с оттянутым концом
|
65 см. d=7
|
1
|
19
|
19
|
|
|
100 см d=7
|
1
|
25
|
25
|
|
6
|
Колба мерная
|
100 мл.
|
2
|
34
|
68
|
|
|
1 л.
|
1
|
316
|
316
|
|
7
|
Трубка гофрированная вертикальная
|
2
|
18
|
36
|
|
8
|
Палочка стеклянная
|
2
|
5,3
|
10,6
|
|
9
|
Пробирка стеклянная без делений с одной отметкой
|
12
|
5
|
60
|
|
10
|
Пробирка центрифужная с пробкой, ПС, Ш 16х100 мм, емкостью 10 мл
нестерильная
|
7
|
20
|
140
|
|
11
|
Кюветы кварцевые из оптического стекла
|
4
|
52
|
208
|
|
12
|
Стакан высокий
|
2
|
47,75
|
95,5
|
|
13
|
чашка Петри ЧБН-1-100
|
2
|
29,5
|
59
|
|
14
|
ИТ (индикаторные трубки) Наружный диаметр 6,9 мм
|
5
|
25
|
125
|
|
ИТОГО
|
|
2017,3
|
Таким образом затраты на стеклянную
посуду и приборы определяются по формуле:
Зпп=2017,3·0,3=605,19
руб.
Сумма амортизационных отчислений
определяется по формуле (7):
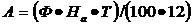 , (7)
, (7)
где Ф - стоимость
использованного в работе оборудования и приборов, руб.;
На - годовая
норма амортизации, %;
Т - время их
использования для данного исследования, мес.
А=(32800·15·1+53200·15·1+18600·15·1+22300·20·1+68023·20·1)/(100∙12)=
2812,92 руб.
Затраты на основную
заработную плату исполнителя определяются исходя из месячной стипендии по
формуле (8):
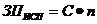 , (8)
, (8)
где n - число месяцев, отведенных для выполнения дипломной работы;
n
= 3;
С - размер месячной
стипендии исследователя, руб.
С = 1600 руб.
ЗПисп=1600∙3=4800
руб.
Таблица 12 - Расчет
амортизационных отчислений
|
№
|
Наименование прибора
|
На, %
|
Время использования, мес.
|
Стоимость, руб.
|
Амортизационные отчисления, руб.
|
|
1
|
Весы аналитические БЛР-200
|
15
|
1
|
32800,00
|
410,00
|
|
2
|
Весы лабораторные WAS 220/C2
|
15
|
1
|
53200,00
|
665,00
|
|
3
|
Вытяжной шкаф (вентилятор осевого типа общего назначения)
|
15
|
1
|
18600,00
|
232,5
|
|
4
|
Газоанализатор ПГА-ВПМ
|
20
|
1
|
22300
|
371,7
|
|
5
|
Спектрофотометр СФ-26
|
20
|
1
|
68023
|
1133,72
|
|
ИТОГО:
|
2812,92
|
Затраты на основную заработную плату
руководителя рассчитываются по формуле (9)
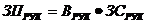 , (9)
, (9)
где ЗСрук -
часовая оплата доцента, руб.;
ЗСрук = 235
руб.;
Врук - время
руководителя, затраченное на работу с дипломником;
Врук = 60
час.
ЗПрук=60∙235=
14100 руб.
Итого затраты на
основную заработную плату:
ЗПосн = ЗПрук
+ ЗПисп = 14100 + 4800= 18900 руб.
Отчисления на страховые
взносы в размере 34% от основной заработной платы составляют:
ЗПсоц=14100 ∙
0,34= 4794 руб.
Накладные расходы,
включающие затраты на содержание учебно-вспомогательного и административно-управленческого
персонала, а также на отопление, освещение, на содержание в чистоте и ремонт
зданий, канцелярские и прочие хозяйственные расходы, составляют 70% от суммы
затрат по предыдущим статьям.
Смета затрат на
проведение научно-исследовательской работы приведена в таблице 13.
Цнир
=46586,02· (1 + 20/100) =83855 руб.
Анализ сметы затрат на
проведение работы показывает, что наибольшую долю затрат составляют затраты на
стеклянную посуду, основную заработную плату и накладные расходы.
Таблица 13 - Смета затрат
на проведение научно-исследовательской работы
|
№
|
Наименование статей затрат
|
Сумма затрат, руб.
|
Удельный вес статьи в общей сумме затрат, %
|
|
1
|
Сырье, материалы, реактивы
|
244,2
|
0,52
|
|
2
|
Электроэнергия
|
9,23
|
0,02
|
|
3
|
Водопотребление
|
38
|
0,08
|
|
4
|
Основная з/плата исследователя Ст. Преподавателя с уч. степенью
Итого
|
4800,00 14100,00 18900,00
|
40,57
|
|
5
|
Страховые взносы
|
4794
|
10,29
|
|
6
|
Стеклянная посуда
|
605,19
|
1,3
|
|
7
|
Амортизационные отчисления
|
2812,92
|
6,04
|
|
Итого прямых затрат
|
27403,54
|
58,82
|
|
Накладные расходы (70% от прямых затрат)
|
19182,48
|
41,17
|
|
ИТОГО
|
46586,02
|
100
|
Заключение
В ходе представленной работы на
высокие концентрации был произведен поиск и сделан анализ существующих средств
индикации оксидов азота в газовоздушной среде, в связи с отсутствием средств контроля
за оксидами азота. Так же для определения окислов азота в газовоздушной среде
были предложены и частично экспериментально доказаны, оптимальный и доступный
для использования спектрофотометрический и линейноколористический метод
определения окислов азота в воздушной среде.
Дальнейшие исследования будут
продолжены, а разработанная нами методика исследования процесса определения
окислов азота в воздухе будет использоваться студентами на кафедре ИЗОС для
дальнейших исследований.
Список использованных источников
1) Kobayashi S.,
Matsumoto A., Yamada T., Chem. Soc Japan, Ind. Chem. Soc, 61, 625 (1958); Хим. 1959, реф. №827958.
2) Мохов Л.А.,
Халтурин В.С., Лабор. Дело, №226 (1958).
3) Kobayashi
J., Kitagama T., цит. по Hobbs A.P., anal. Chem., 30, 788 (1959).
) Kinosian
J.R., Habbard B.R., Am. Ind. Hyg. Assoc J., 19, 453
(1958).
5) Мохов Л.А.,
Удалов Ю.Ф., Халтурин В.С., ЖПХ 32, 452 (1959).
) Парфенова К.Г.,
Сборник работ и авторефератов по вопросам гигиены труда и профпаталогии в
горнорудной, химической и машиностроительной промышленности, Харьков, 1958.
7) Ratzka E., Nowak G., нат ГДР 31567, 5/IX 1964.
8) Hesse B., нат ГДР 43296, 15/IX 1965.
) Новый
справочник химика и технолога. Радиактивные вещества. Вредные вещества.
Гигиенические нормативы. СПб: АНОНПО Профессионал, 2005 г., с. 1142.
) Пожаровзрывоопасность
веществ и материалов и средства тушения. Спр.изд. в 2 книгах. Баратов А.Н.,
Корольченко А.Я., М., Химия, 1990 - 384 с.
11) Сборник общих правил
и инструкций по технике безопасности при работе в химической лаборатории. - Л.:
ЛТИ им Ленсовета, 1991 - 336 с.
) ГОСТ 00000-00.
CCБТ. Опасные и вредные производственные факторы. Класификация. М.: Изд-во
стандартов, 1974.
13) СНиП 23-05-95
Естественное и искусственное освещение. Нормы проектирования. - М.: Стройиздат,
1996.
14) НПБ 105-03
Определение категорий помещений, зданий и наружных установок по взрывопожарной
и пожарной опасности.
15) Правила устройств
электроустановок. ПУЭ. - М.: Энергоатомиздат, 1985.
) ГОСТ 12.4.021 -
75 CCБТ. Системы вентиляционные. Общие требования. М.: Изд-во стандартов, 1975.
17) Техническое описание
и конструкция по эксплуатации СФ - 26. ЛОМО им. Ленина, 1981.
18) Кожухов С.Г.,
Калистратов Ю.К.: Комплекс многоцелевого контроля КМК-1, Пушкин, 1998.
) Перегуд Е.А.,
М.С. Быховская, Е.В Гернет, Быстрые методы опр. вред. вещ. в воздухе, «Химия»,
1970.
) Глинка Н.Л.
Общая химия. Л.: Химия, 1981.
) Ахметов Н.С.
Общая неорганическая химия. М.: Высшая школа, 1981.
22) Sentek A.,
Prace Centr. Inst. Ochrony Pracy, 15, 1966.
23) Thomas M.D.,
McCleod I.A, Roblins R.C., Anal. chem., 28, 1956.
24) Adley F.E.,
Skillern E.P., Am. Ind. Hyg. Assoc. J., 19, 1958.
25) Gummings W.G.,
Redflarn M.W. Inst. Fuel, 30, 1967.
26) Смирнов Н.Ф.
Исследование влияния: Дипломная работа. - СПбГТИ(ТУ), рукопись, 2009. - 0110 с.
) Е.А. Перегуд, Д.О.
Горелик, Инструментальные методы контроля загрязнения атмосферы. Л.: Химия,
1981.