Экстракционно-фотометрическое определение ванадия (V) с реагентами класса БФГА
1. Введение
Определение ванадия в природных и сточных водах
актуально в связи с токсичностью его соединений. В воду ванадий попадает из
полиметаллических и железных руд, а также из отходов производства: предприятий
тяжелой промышленности, переработки нефти и т.д. Ванадий долго остается в
почве, однако в растениях его мало - он не может накапливаться в их тканях.
Ванадий токсичен как в виде катионов, так и в виде анионов. Степень токсичности
зависит от валентности ванадия, дисперсности частиц аэрозоля и растворимости
соединений в биологических средах. Такие вещества как ионы хрома, хлорид-ион,
гидроксид алюминия, аскорбиновая кислота, белки и другие могут повышать
токсичность ванадия, повышая его абсорбцию. В связи с высокой токсичностью
соединений ванадия необходим надежный контроль за его содержанием в окружающей
среде, отходах производства, биологических объектах и т.д. Это особенно важно
из-за быстрого роста добычи и широкого потребления нефти с высоким содержанием
ванадия. Токсичность соединений ванадия обусловливает необходимость контроля
иногда очень малых содержаний ванадия. Для определения малых содержаний
ванадия(V) предложен ряд
реагентов и соответствующих экстракционно-фотометрических методик. Однако даже
лучшие из них характеризуются многооперационностью, трудоемкостью и
длительностью. Поэтому разработка новых методик экспрессного и надежного
аналитического контроля содержания ванадия в природных и сточных водах имеет
чрезвычайно важное значение.
Целью данной дипломной работы является
разработка простой и экспрессной методики экстракционно-фотометрического
определения ванадия, применимой для его определения в различных типах природных
вод.
2. Обзор литературы
.1 Ванадий в окружающей среде
Многообразие ионных форм и степеней окисления
ванадия определяют его высокую подвижность в земной коре и значительную
рассеянность в природе. Ванадий редко образует крупные скопления руд, но легко
концентрируется в почвах [1]. Обладая способностью к рассеиванию, он часто
встречается в качестве примеси в различных минералах и горных породах [2].
Миграция, рассеивание и концентрирование ванадия в биосфере осуществляется
путем извлечения его живыми организмами из воды, растительной и животной пищи,
из различных пород и путем высвобождения его в результате разложения и
минерализации органического вещества [3]. Содержание ванадия в почве
оценивается в 10−2 %, в растениях - 10−4 %, в организмах
млекопитающих и человека - 10−5 %. Ванадий в живых организмах играет роль
катализатора окислительно-восстановительных процессов. Но многие его соединения
являются ядами. Наибольшей токсичностью обладают соединения ванадия со степенью
окисления +5. Для человека и высших животных механизм токсичности соединений
ванадия недостаточно изучен. В тоже время известно, что ванадий является стимулятором
роста многих культурных растений и сельскохозяйственных животных. Концентрация
микроэлемента ванадия в природных водах составляет 0,01 - 20 мкг/л (в
незагрязненных пресных водах). В некоторых реках и озерах обнаружена более
высокая концентрация ванадия. В земной коре ванадия содержится 1,5∙10−2%
по массе [4].
Химия природных соединений ванадия чрезвычайно
разнообразна и порой настолько сложна, что до сих пор нет единого мнения о
природе и условиях существования отдельных форм этого элемента. Важной
особенностью пятивалентного ванадия является то, что его состояние в водных
растворах зависит от рН среды. В сильно щелочных растворах он существует в виде
моноядерной частицы - ортованадат-иона; с уменьшением концентрации щелочи
возникают полиядерные анионные частицы. В достаточно кислых растворах ванадий (V)
существует в виде моноядерного оксокатиона [5]. Ионы ванадия (V)
в сильнокислых средах (например, 3М H2SO4)
проявляют сильные окислительные свойства. Растворы ванадата применяются для
титриметрического определения многих восстановителей. При понижении
концентрации кислоты окислительные свойства пятивалентного ванадия ослабевают.
В слабокислых растворах ионы ванадата уже не окисляют многих восстановителей.
Соли ванадила (VO2+)
окисляются в умереннокислых растворах сильными окислителями. Так, они
количественно окисляются раствором перманганата в среде 5%-ной H2SO4
или раствором бромата калия в разбавленных солянокислых растворах, содержащих
соли аммония. При повышении рН тенденция к окислению V4+
усиливается. В щелочной среде он окисляется даже кислородом воздуха [6].
Заряд катионов ванадия в растворе не превышает
трех; в степенях окисления 4+ и 5+ он образует устойчивые оксикатионы VO2+,
VO23+ и VO3+.
Ванадий во всех степенях окисления образует комплексные соединения. По
способности преимущественно координироваться в комплексных соединениях с
донорными атомам лигандов N,
O и S
катионы ванадия(IV)
и ванадия(V) относятся к ряду
металлов, которые образуют с кислородом более прочную связь, чем с азотом, а
катионы ванадия(III)
- к ряду металлов, у которых связь с азотом прочнее, чем с кислородом. По
такому типу и протекает комплексообразование ванадия с природными лигандами
типа фульво- и гуминовых кислот [1]. Далее абзацы переставлялись и нумерацию
ссылок надо пересмотреть.
2.2 Методы определения ванадия
.2.1 Титриметрические методы
Титриметрическое определение ванадия основано на
его реакциях с окислителями (восстановителями) и комплексонами. Наибольшее
распространение получили оксидиметрические методы.
Оксидиметрическое титрование.
Ванадий в водных растворах может быть получен в
различных степенях окисления (II,
III, IV,
V), поэтому в
качестве титрантов применяют как окислители, так и восстановители.
Титрование восстановителями. Ванадий(V)
является сильным окислителем и в кислых растворах взаимодействует с
большинством восстановителей, переходя в ванадий(IV),
поэтому для титрования рекомендованы растворы солей железа(II),
олова(II), титана(III),
аскорбиновой кислоты и др. Для получения точных результатов ванадий
предварительно полностью переводят в пятивалентное состояние действием
различных окислителей. Для этой цели рекомендованы перманганат калия, хлорная
кислота, озон и бромат калия.
Широко распространенным и наиболее точным
методом определения ванадия является титрование его раствором соли железа(II)[7].
В кислой среде железо(II)
восстанавливает ванадий(V)
до ванадия(IV). Методом
амперометрического титрования показано, что эта реакция в 1-4М H2SO4
протекает стехиометрично и не зависит от форм существования ионов ванадия(V)
в растворе [8]. Для определения точки эквивалентности в зависимости от
кислотности среды используют ox-red
индикаторы, например фенилантраниловую кислоту.
Титрование окислителями. В водных растворах
ванадий(V) предварительно
восстанавливается до низших степеней окисления (II,
III, IV)
под действием различных восстановителей и далее титруют растворами окислителей.
Чаще всего используют восстановление ванадия(V)
до ванадия(IV) сернистым
ангидридом, сульфитом или бисульфитом натрия.
· Перманганатометрия. Соли ванадия(IV)
окисляются перманганатом калия с заметной скоростью только в слабокислой среде
(~0,05 М H2SO4)
при нагревании до 50 - 80 0С. При меньшей кислотности раствора (pH
2) ванадий(IV) начинает
окисляться кислородом воздуха, а при увеличении концентрации кислоты реакция
замедляется [9]. Титрование обычно проводят без индикатора до появления
слабо-розовой окраски раствора, устойчивой в течение 1 - 2 мин.
· Бихроматометрия. В сернокислой среде
бихромат калия взаимодействует только с ванадием(III),
но в присутствии следов марганца(II)
[10], а также в 3 - 12 М растворе фосфорной кислоты [11] он может окислять и
ванадий(IV).
Определение ванадия(III)
рекомендуют проводить в присутствии катализаторов: ионов меди(II)
в сернокислой среде и железа(III)
в фосфорнокислой среде . В качестве индикатора используют фенилантраниловую
кислоту или дифениламин [12].
· Иодометрия. В кислых растворах
ванадий(V) восстанавливается
иодидом калия до ванадия(IV).
В щелочных растворах иод окисляет ванадий(IV)
до ванадия(V). На
стехиометричность обеих реакций существенное влияние оказывают условия их
протекания, поэтому разработаны лишь косвенные методы иодометрического
определения ванадия.
Комплексонометрическое титрование.
Ванадий(V)
в водных растворах существует в виде сложных ионов, поэтому его взаимодействие
с комплексоном III
замедленно и в большой степени зависит от рН. Более распространены методы
титрования ванадия(IV),
который взаимодействует с комплексоном III
в широком интервале рН [13]. Для восстановления ванадия(V)
используют сульфит натрия или сернистую кислоту [14], аскорбиновую кислоту и
гидроксиламин [15]. Титрование ванадия(IV)
раствором комплексона III
можно проводить в присутствии различных индикаторов. При рН 3 - 3,5 используют N-бензоил-N-фенилгидроксиламин
(N-БФГА) [6,14].
Ванадий также может быть надежно определен
методами обратного титрования. К анализируемому раствору приливают комплексон III
и оттитровывают непрореагировавший избыток его растворами солей различных
металлов. Ванадий(V) определяют
при рН 3,5 - 6. Избыток комплексона III
титруют раствором соли цинка в присутствии индикатора дифенилкарбазона [16] или
растворами солей цинка, свинца или кадмия с пирокатехиновым фиолетовым [17].
Комплексонометрические методы не имеют
преимуществ перед методами оксидиметрического титрования. Их используют при
анализе ванадатов или сплавов на основе ванадия в том случае, если другие
компоненты определяют также комплексонометрически.
.2.2 Кинетические методы
Ванадий оказывает сильное каталитическое действие
на многие окислительно-восстановительные реакции. Скорость реакций в
большинстве случаев прямо пропорциональна концентрации ванадия . Каталитические
реакции высокочувствительны. Они позволяют определять до 10-4 - 10-5 мг/л
ванадия. В ряде случаев можно значительно повысить чувствительность реакций
введением очень небольших количеств активаторов (оксикислот, о-дифенолов,
8-оксихинолина и др.) [18]. Ванадий в минеральной, сточной, речной воде и
рассолах определяют [19] кинетическим методом по индикаторной реакции окисления
1,2-фенилендиаминаброматом калия, активатор - тайрон, предел обнаружения -
0,05-0,4 мкг/л в зависимости от состава воды .
Соединения ванадия ускоряют также реакцию
окисления анилина хлоратом калия при рН 6.5 - 7.5. Катализирующее действие
ионов ванадия усиливается в присутствии 8-оксихинолина [6].
2.2.3 Электрохимические методы
Потенциометрическое титрование.
Метод основан на резком изменении потенциала
индикаторного электрода вблизи точки эквивалентности при титровании. В методе
используют реакции окисления - восстановления, комплексообразования и
осаждения. С использованием потенциометрической регистрации ванадий(V)
титруют растворами железа(II)
в кислой среде , глицерина в щелочной среде [20], сульфата гидразина в
присутствии катализатора OsO4
, тиосульфата натрия в присутствии катализатора ионов Cu2+
[21], а также растворами молибдена(III),
хрома(II), титана(III)
и аскорбиновой кислоты [22]. Методом комплексонометрического титрования с
потенциометрической регистрацией определяют ванадий(IV)
обратным титрованием раствором Hg2+
при рН 8 (уротропиновый буфер) [23]. Осадительное аргентометрическое титрование
ванадия(V) до ортованадата
серебра проводят при рН 11.5, используя серебряный индикаторный электрод [24].
Полярографические методы.
Полярографическое поведение пятивалентного
ванадия сильно зависит от рН среды. В кислых растворах он дает две волны,
первая соответствует восстановлению ионов Hg22+,
получаемых в результате окисления металлической ртути ионами пятивалентного
ванадия, а вторая - восстановлению четырехвалентного ванадия до двухвалентного
(E1/2=-0.83В отн.
НКЭ) [25]. На последнюю волну дополнительно накладывается волна водорода, и
поэтому количественное определение ванадия затруднено.
Амперометрическое титрование.
Для косвенного амперометрического определения
ванадия применяют реакции окисления-восстановления, осаждения или
комплексообразования. За процессом титрования следят по изменению тока
восстановления или окисления одного из участников реакций. В качестве
индикаторных электродов используют ртутный капельный либо платиновый
микроэлектрод. Определение точки эквивалентности довольно просто и надежно, а
методически амперометрическое титрование не отличается от обычных
титрометрических методов.[1]
Кулонометрические методы.
Определение ванадия методом потенциостатической
кулонометрии применяют крайне редко из-за необходимости отделения его от многих
мешающих элементов и длительности подготовки проб при анализе сложных объектов
[26]. С другой стороны ванадий можно успешно определить титрованием
окислителями или восстановителями, генерированными в электролитической ячейке
(т.е. методом кулонометрического титрования). Точку эквивалентности
устанавливают спектрофотометрическим, потенциометрическим или амперометрическим
методом, а также визуально - по изменению окраски индикаторов. Содержание
ванадия находят по закону Фарадея, пересчитывая количество электричества,
пошедшего на получение титранта, необходимого для окисления (восстановления)
ванадия.
Микрограммовые количества ванадия титруют
электрогенерируемыми ионами Fe(II)
с амперометрическим установлением точки эквивалентности [27]. Наряду с ионами Fe(II)
в качестве титрантов-восстановителей применяют ионы Mo(V)
, W(V)
, Ti(III)
, Cu(I)
и V(III)
[28], а в качестве титрантов-окислителей - Cr(VI)
, Fe(III)
, хинон [29].
.2.4 Методы атомной спектроскопии
Методы эмиссионной спектроскопии.
В низкотемпературных пламенах определение
ванадия возможно лишь по измерению интенсивности молекулярных полос моноокиси
ванадия (VO) с
максимумами при 528,0; 550,0; 573,7 и 578,0 нм. Это обусловлено высокой
энергией диссоциацией VO,
равной 5,5 эв. Использование высокотемпера-турных пламен таких, как
кислород-ацетилен и закись азота- ацетилен, дает возможность определять до 3
мкг/мл ванадия по атомным линиям VI
318,5 и VI 437,9 нм
при существенном уменьшений различного рода влияний.
После экстракционного выделения
(концентрирования) ванадия в виде оксихинолината и распыления экстракта
непосредственно в водородно-кислородном пламени можно определить до 50 мкг/мл
ванадия по молекулярной полосе 528,0 нм. Повышение восстановительных свойств
пламени (за счет применения органических растворителей, в частности, этанола)
снижает предел обнаружения ванадия до 2 - 8 мкг/мл [1].
Предложен рентгенофлуоресцентный метод
определения ванадия в морской воде [30] (предел обнаружения 0,02 мкг/л) с
предварительным соосаждением ванадия с пирролидандитиокарбаматом кобальта(III).
Методы атомно-абсорбционной спектроскопии.
Собственной чувствительности
атомно-абсорбционного метода определения ванадия недостаточно для его
непосредственного определения в природных водах. Поэтому использование операций
концентрирования представляется неизбежным. Опробованы [31] различные методы
концентрирования ванадия из морской воды перед ААС определением: анионообменная
экстракция, метод хелатного обмена, соосаждение с гидратированной TiO2.
Лучшим оказался метод соосаждения . Исследована [32] возможность определения
ванадия в морской воде методом непламенной атомно-абсорбционной спектроскопии
после предварительного экстракционного концентрирования с помощью следующих
реагентов: ПАР+ хлорид тетрафениларсония; ПАР+хлорид тетрадецилдиметилбензил
аммония; N-бензоилфенилгидроксиамина,
5,7-дихлор-8-гидроксиламина, тетраметилендитиокарбамата натрия. Для ААС
определения ванадия на уровне мкг/л лучшими реагентами оказались ПАР с хлоридом
тетрадецилдиметилбензиламмония или тетраметилендитиокарбамат натрия. Для
определения ванадия в морской воде на уровне мкг/л разработана методика [33]
экстракционного концентрирования в виде роданидного комплекса триоктиламмония;
реэкстракты анализируют ААС с графитовой кюветой.
2.2.5 Методы молекулярной спектроскопии
Люминесцентные методы.
Известно всего несколько люминесцентных реакций
с участием ванадия. Ванадий дает желтую флуоресценцию с церулеином после
восстановления его цинком до V(II)
и добавления спирта. Метод позволяет обнаружить до 16 мкг/мл ванадия. С
резорцином ванадий дает в 10 М H2SO4
или конц. Н3PO4 красную
флуоресценцию, которая исчезает через 5 мин. Предел обнаружения ванадия
составляет 2,5 мкг/мл. Описан метод люминесцентного определения 0,1 - 0,8 мкг
ванадия(V) в водно-спиртовом
растворе с 1,4-диамино-5-нитроантрахиноном [34].
Фотометрические методы.
Для определения ванадия применяют методы,
основанные на измерении поглощения электромагнитного излучениями растворами
непосредственно ионов ванадия (V),
ванадия (IV), ванадия (III),
так и окрашенными продуктами взаимодействия ванадия с неорганическими и
органическими реагентами.
Методы определения, основанные на измерении
оптической плотности растворов, содержащих ионы ванадия (V),
ванадия (IV), ванадия (III),
малочувствительны, но позволяют быстро определять ванадий в простых по составу
объектах: растворах, окислах, ванадатах [1]. Ионы ванадия в степенях окисления II,
III, IV
и V различно окрашены
и имеют характерные абсорбционные спектры [35]. Ванадий (V)
в растворе в зависимости от его концентрации и кислотности среды может
находиться в различных ионных состояниях, которые характеризуются определенными
полосами поглощения [36]. Однако сложные ионные равновесия затрудняют
использование абсорбционных спектров ванадия (V).
Для его определения рекомендуют только две полосы поглощения с максимумами при
330 нм в конц. H2SO4
и при 270 нм в 1М NaOH.
Молярные коэффициенты погашения соответственно равны 4,6·103 и 8,4·103 [37].
Определению мешают многие элементы. Окрашенные
ионы Co(II),
Ni(II),
Cu(II)
и др. предварительно осаждают в виде гидроокисей. Хром отделяют в виде
оксихинолината или хромата свинца [38] большие количества железа(III)
отделяют электролизом на ртутном катоде. Титан маскируют фторид-ионами.
Определению ванадия при 270 нм в щелочной среде не мешают W,
Ti, Zr,
а также равные количества Re,
Mo и Nb
[37]. Для повышения избирательности определения ванадия в кислой среде
оптические плотности растворов измеряют при 450 - 480 нм.
Определение ванадия по образованию окрашенных
соединений с неорганическими реагентами.
Перекись водорода взаимодействует с ванадием в
кислой среде с образованием устойчивого комплексного иона [V(O2)]3+
красно-оранжевого цвета [39]. Его молярный коэффициент погашения при 460 нм
равен ~200 [40]. Перекисное соединение образуется в широком интервале
концентраций кислоты (0.3 - 3 М H2SO4)
и при содержании перекиси водорода 0,03 - 0,09 % [39]. Допустима замена серной
кислоты на соляную или азотную. Окраска растворов устойчива в течение 1 - 2
суток.
Возможно определение ванадия по окраске
фосфорновольфрамо-ванадиевой кислоты. Это гетерополисоединение (е400нм = 1400)
образуется при нагревании соединений ванадия в растворе минеральной кислоты (до
2.4 М) в присутствии 0.5 М фосфорной кислоты и 0.025 М вольфрамата натрия..
Определение ванадия по образованию окрашенных
соединений с органическими реагентами.
Вопрос о состоянии ионов ванадия(V)
при образовании комплексных соединений с органическими лигандами является
чрезвычайно сложным и по большей части не выясненным. В диапазоне рН 2ч5
ванадий(V) может входить в
состав комплексов либо в форме катионов VO3+,
VO(OH)2+,
VO2+, либо в форме анионов V14O114−,
V6O174−.
Поэтому найденные при изучении комплексообразования соотношения металл-лиганд и
константы устойчивости не объясняют химизма процесса и носят условный характер.
Это еще более справедливо, если речь идет об экстракции комплексных соединений
ванадия с органическими лигандами, поскольку в этом случае дополнительным
фактором, усложнившим протекание процесса, является наличие гетерогенного
равновесия между двумя несмешивающимися фазами. Наличие катионных форм ванадия(V)
в растворе подтверждено в работах по его экстракции трибутилфосфатом (ТБФ) из
солянокислых растворов в виде соединений HVO2Cl2∙2ТБФ,
VOCl3∙ТБФ [41].
Существование катионных форм ванадия в водных растворах минеральных и
органических кислот и их солей было также показано методом электрофореза на
бумаге [42].
Ванадий(V)
образует несколько окрашенных комплексов с пирокатехином (о-диоксибензолом)
[43]. Комплекс состава 1:1 розового цвета образуется при рН 1 - 3 за счет
вытеснения водорода одной спиртовой группы.


В интервале рН 3 - 4,7 и 4,7 - 7,0
устойчивы комплексы состава 1:1 и 1:2 соответственно. Для последнего комплекса
е590нм = 6800 [44]. Определению не мешают Ce, U, La, As, Pb, Sn, Th, Zr, Cd, Zn (20%), Sb (<10%), Ga (<10%),
а также щелочные и щелочно-земельные металлы. Мешают окрашенные ионы Cu(II), Ni(II), Co(II), Cr(III) и ионы,
взаимодействующие с реагентом: Fe(III), Al(III), Ti(IV), W(VI), Mo(VI), Nb(V), Bi(III). Большие
количества мешающих элементов отделяют в виде гидроокисей или хроматографически
[45]. Менее чем 100-кратные количества Fe, Co, Ni, Cr, Mo, W и Cu маскируют
аскорбиновой кислотой, цитрат- и цианид-ионами и комплексоном III,
104-кратные количества алюминия - фторид-ионами [44]. Измерение оптической плотности
при 800 нм полностью исключает влияние Nb, W, Ti, Bi, образующих
желтые комплексы с реагентом.
Интенсивно окрашенные соединения
образуются также с другими ортофенолами и 2-гидроксибензойными кислотами [43].
Комплексообразование в этих системах в наибольшей степени протекает в
слабокислых и нейтральных средах. Предполагается, что в системе образуются
устойчивые хелатные соединения за счет диссоциации гидроксо-групп реагентов.
3,4-диоксибензойная кислота [46] также образует синие комплексы состава 1:1 и
1:2 анионного характера.


Образование окрашенного в синий цвет
тройного комплекса ванадий(V)-трибромпирогаллол(ТБП)-дифенилгуанидин(ДФГ)
состава 1:2:1 в смеси хлороформа и изоамилового спирта (1:2) лежит в основе
методики экстракционно-фотометрического определения ванадия. Экстракт имеет
максимум светопоглощения при 600 - 650 нм. Определению не мешают ионы Ni, Mn, Cu, Cr, Al, а в
присутствии также Fe(III) и Mo маскируют
тиогликолевой кислотой, W - тартратом, Zr - фторидом
[6].
-оксихинолин (о-оксихинолин, оксин)
образует с ванадием (IV) и ванадием (V) окрашенные
в малиновый цвет комплексные соединения, хорошо экстрагирующиеся из слабокислых
растворов различными органическими растворителями. Окраска комплекса
развивается быстро и устойчива во времени (7 дней и больше) [47]. Помимо
ванадия окрашенные комплексы с оксихинолином в этих же условиях дают железо,
алюминий, медь, которые также экстрагируются хлороформом из слабо кислого раствора.
Отделение ванадия от этих элементов осуществляется реэкстракцией ванадия из
органической фазы содовым раствором при рН=9.4 [48]. Ванадий при этом переходит
в содовый раствор, а железо, медь и большая часть алюминия остаются в
хлороформе. Частично переходящий в щелочной реэкстракт алюминий дополнительно
комплексуется малоновой кислотой, раствор доводится до рН=4.5 и ванадий вновь
экстрагируется хлороформом. Методика экстракционно-фотометрического определения
ванадия с оксихинолином позволяет определить ванадий вплоть до содержаний 1
мкг/л в присутствии 0.5 мг алюминия, висмута, кобальта, хрома, меди, железа,
марганца, молибдена, никеля, олова, титана и урана, 0,1 мг вольфрама и 5 мг
цинка [49].
Исследовано взаимодействие ванадия с
5,7-дибром- и 5,7-дииод-производным 8-оксихинолина [50]. Образуемые комплексные
соединения экстрагируют высокомолекулярными спиртами. Молярные коэффициенты
погашения равны (6 - 7)·103. Основным мешающим элементом является железо (III), малые
количества которого могут быть замаскированы ионами PO43- и F-.
Фталексоны - красители
трифенилметанового ряда, включающие в свой состав группы - CH2N(CH2COOH)2 в
орто-положении к карбокси- группе. В кислой среде они взаимодействуют с
ванадием (V) с
образованием интенсивно окрашенных хелатных соединений различного состава
(табл. 1).
Таблица 1. Определение ванадия(V) с
фталексонами.
|
Реагент
|
рН
|
Состав
комплекса V:R
|
лmax,
нм
|
вуст
|
Интервал
определяемых содержаний V,
мг/л
|
|
|
|
комплекса
|
R
|
|
|
|
|
Ксиленоловый
оранжевый
|
4
|
2:1
1:1 1:2
|
580
590 520
|
440
|
20,0
13,0
|
|
0,1 - 4,5
|
|
Фталексон
S
|
3
|
1:1
|
540
|
434
|
21,0
|
4.7·105
|
0,25
- 1,25
|
|
Тимол-
фталексон
|
4
|
2:1
|
580
|
420
|
25,3
|
4.2·1011
|
1
- 3,5
|
|
Крезол-
фталексон
|
3
|
1:1
|
580
|
|
6,0
|
|
5,0
- 20
|
|
Метил-
тимоловый синий
|
3
- 4 5 - 7
|
1:2
1:1
|
580
510
|
440
|
19,0
9,8
|
2.3·1012
1.6·106
|
0,15
- 1,5
|
Ксиленоловый оранжевый (КО; 3,3′-бис(N,N'-ди(карбоксиметил)-аминометил)-о-крезолсульфофталеин)
- наиболее перспективный реагент для определения ванадия. Высокий молярный
коэффициент светопоглощения красителя сочетается с достаточной контрастностью
реакции с ванадием (V) и доступностью реагента. В интервале рН = 3ч5 КО
преимущественно находится в форме Н3RO33-, которая характеризуется максимумом
поглощения при длине волны 430 нм. При рН > 5 вследствие частичного перехода
реагента в форму Н2RO43- появляется второй максимум поглощения при 580 нм [51].
При избытке реагента и оптимальном значении рН 3 - 5 окраска комплекса КО с
ванадием развивается через 30 с и устойчива более суток. Определению мешают все
элементы, взаимодействующие с реагентом в этих условиях, а также дающие
окрашенные ионы в растворах (Fe(III),
Al, Cu,
Zn, Cd,
Ga, Bi(III),
Zr, Th,
Ti(IV),
Ni, Co,
Mn, Sc(III)).
Их предварительно отделяют от ванадия различными способами. Описан, например,
метод ионообменного разделения . Выбор маскирующих средств ограничен, так как
тартрат-, цитрат-ионы, а также перекись водорода и комплексон III
мешают определению ванадия [52]. В качестве маскирующих реагентов рекомендованы
фторид калия и комплексон IV
(1,2-диаминциклогексан-N,N,N’,N’-тетрауксусная
кислота) [53]. В их присутствии окраска развивается медленно (через 10 мин.) и
необходим достаточный избыток реактива.
Для определения ванадия(V)
рекомендован также метилтимоловый синий . Окрашенное соединение с ванадием
образуется при рН 3-4 и при достаточном избытке реагента. Оптическую плотность
раствора измеряют при 580 нм. Мешают многие элементы. Но выбор маскирующих
средств шире, чем при использовании ксиленолового оранжевого. Определению не
мешает 20-кратный избыток фтор-, тартрат_, пирофосфат-ионов [54]. Основную
массу мешающих элементов отделяют при вскрытии образцов щелочами.
Большой аналитический интерес представляет
комплексообразование ванадия с пирогаллоловым красным (ПК) и бромпирогаллоловым
красным (БПК). Взаимодействие ванадия(V) с ПК и БПК без третьего компонента не
представляет аналитического интереса для фотометрического определения [55]. В
качестве третьего компонента могут выступать катионные поверхностно-активные
вещества (ПАВ) или амины , сдвигающие реакцию комплексообразования в кислую
область и увеличивающие ее контрастность и чувствительность. Ванадий(V)
реагирует с ПК и БПК в присутствии ПАВ, образуя комплексы при рН 1,5-3,8 и
1,3-2,2 соответственно. С ПК в присутствии цетилпиридиния (ЦП) ванадий образует
комплекс с максимумом при 575 нм, а с БПК в присутствии ЦП - при 610 нм, при
этом контрастность составляет от 100 до 170 нм для комплексов разного состава
[56].
Определение ванадия с N-БФГА
и родственными реагентами.
Экстракционно-фотометрический метод определения ванадия
(V) с использованием N-бензоил-N-фенилгидроксиламина
получил наибольшее распространение среди методов, основанных на цветных
реакциях ванадия с различными реагентами.
Катионы ванадия(V)
преимущественно координируются в комплексных соединениях с донорными атомами
лигандами N, O,
S, но они относятся
к ряду металлов, которые образуют с кислородом более прочную связь, чем с
азотом. С этих позиций для аналитической химии ванадия(V)
интерес представляют реагенты, в молекулах которых содержаться как те, так и
другие донорные атомы. Среди органических реагентов давно находят широкое
применение производные фенилгидраксиламина и, в первую очередь, купферрона
(аммонийная соль N-нитрозофенилгидроксиламина)
[57]:
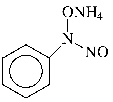
Купферронаты металлов в аналитической химии
применяют для различных целей и, главным образом, для разделения ионов и
концентрирования следовых количеств веществ в сочетании с экстракцией их
органическими растворителями. Как аналитический реагент купферрон обладает рядом
недостатков: неустойчив к воздействию света и тепла, легко окисляется
кислородом воздуха, при осаждении из горячих растворов претерпевает разложение
и осмоление. Купферронаты элементов нельзя использовать в качестве весовой
формы.
Значительное внимание в литературе уделено
изучению взаимодействия ванадия(V)
с гидроксамовыми кислотами (производными гидроксиламина с одним органическим
радикалом), приводящему к образованию интенсивно окрашенных соединений. В
работах [41,42] синтезировали многочисленные комплексы ванадия(V)
с различными замещенными гидроксамовыми кислотами. Было найдено [41], что
бензгидроксамовая кислота C7H6NO2
в водной среде при рН 3 - 4 реагирует с метаванадатом аммония с образованием
нерастворимого в воде комплекса темно-фиолетового цвета. На основании
химического анализа ему приписана формула [VO(OH)(C7H6NO2)2].формула
Бенз-, салицил- и никотингидроксамовые кислоты образуют соединения, растворимые
в водно-спиртовых растворах. Остальные дают осадки, растворимые в
высокомолекулярных спиртах, кетонах, альдегидах и эфирах. Для фотометрического
определения используют водно-спиртовые растворы и экстракты хелатов.
Экстрагируемость комплексных соединений зависит от кислотности среды, природы
растворителя, а также характера органического радикала [58]. Реагенты
малоизбирательны. При различных условиях с ними образуют комплексные соединения
ионы многих элементов.
Чаще всего для определения ванадия применяют
бензгидроксамовую кислоту, которая в слабокислой среде образует с ванадием (V)
окрашенный осадок, растворимый в водно-спиртовом растворе. Окраска раствора
устойчива в течение дня [40]. Определению мешают Fe(III),
Ti(IV),
Mo(VI),
Nb(V),
Hg(II),
Co(II).
Железо удаляют электролизом или предварительно экстрагируют совместно с титаном
в виде бензгидроксамата при рН 8,5 бутанолом [59].
Большинство из синтезированных в последнее время
гидроксамовых кислот не имеют особых преимуществ перед бензгидроксамовой, за
исключением капро- и метоксибензтиогидроксамовой, которые образуют соединения с
ванадием в более кислых средах (0,15 - 0,4 М H2SO4
и 6 M
HCl соответственно)
[60]. Это существенно снижает число мешающих элементов.
Производные гидроксиламина с двумя органическими
радикалами (двузамещенные гидроксамовые кислоты) - высоко избирательные
реагенты на ванадий (V).
Наиболее изучен и широко используется N-бензоил-N-фенилгидроксиламин
(N-бензоил-N-фенилгидроксамовая
кислота) [40].

-бензоил-N-фенилгидроксиламин
(БФГА) - бесцветное кристаллическое вещество с Тпл 121 - 122 оС. БФГА плохо
растворим в воде, умеренно - в этаноле, бензоле и диэтиловом эфире. Значительно
устойчивее к действию окислителей и света, чем купферрон. БФГА взаимодействует
с ванадием(V) в кислой среде (9
М - pH 6) с
образованием в водных растворах окрашенных хелатных соединений состава (C13H10O2N)2·VO2H,
(C13H10O2N)2·VO2CH3
и (C13H10O2N)2*VO2H·HCl
. В хлороформе комплекс имеет состав V2O3(C13H10O2N)4
[61]. Подобные комплексы образуют и салицилальдоксим, ацетилацетон,
бензоилацетон, изохинолин, гидразид и салицилизен-1,3-диаминопропан [5].
Образующийся комплекс ванадий-N-БФГА
можно экстрагировать хлороформом при рН 5,1 - 6,0. Это дает возможность
применять реагент для экстракционного разделения компонентов раствора сложного
состава и, что особенно важно, для экстракционно-фотометрического определения
следов веществ. При исследовании пригодности различных растворителей для
экстракции комплекса ванадия с БФГА был получен следующий ряд: бутанол >
пентанол > хлороформ > амилацетат > бутилацетат > бензол >
гексанол > толуол > метилизобутилкетон > четыреххлористый углерод .
Для работы рекомендован хлороформ [62]. Молярный коэффициент погашения
хлороформенного раствора равен 3700, максимум полосы поглощения - 445 нм.
Наибольшая избирательность определения ванадия
достигается, если проводить экстракцию хлороформом из очень кислых растворов
(9-11М по H2SO4
или 3-9 М по HCl) при
10-кратном избытке реагента [40]. При этом экстрагируется пурпурно-красное
соединение с широкой полосой поглощения (490-630 нм) и более высоким молярным
коэффициентом погашения (е = 4500 - 5100). Найдено, что в этом соединении
отношение ванадия к N-БФГА равно
1:2 [40,62]. В зависимости от кислотности раствора имеет место образование
различных комплексов ванадия(V)
с N-БФГА: в растворах
< 0.1М по HCl -
оранжево-красного цвета, > 2М по HCl
- фиолетового цвета. В 0.1 - 2 М солянокислых растворах существует смесь двух
комплексов, имеющая аметистовую окраску [63].
Из сернокислых растворов соединение ванадия(V)
с N-БФГА тоже
экстрагируется, но окраска хлороформенного слоя приобретает оранжево-желтый
цвет (л =530 нм). При подкислении раствора соляной кислотой и повторном
встряхивании с хлороформом оранжево-желтая окраска экстракта переходит в
пурпурную. Экстракции соединения ванадия(V)
с N-БФГА из солянокислых
сред отдается предпочтение, так как интенсивность светопоглощения экстракта по
сравнению с другими средами является максимальной. Однако экстракты, получаемые
из солянокислых растворов, обладают несколько меньшей устойчивостью. Было
показано, что это объясняется восстановлением ванадия(V)
слабым восстановителем N-БФГА,
протекающим в солянокислых растворах [64]. В этих условиях ванадий,
восстанавливаясь до степени окисления 4+, не взаимодействует с реагентом и не
экстрагируется хлороформом. По сравнению с солянокислыми растворами экстракция
соединения из сернокислых обеспечивает большую устойчивость окраски системы и
соответственно большую воспроизводимость получаемых результатов [65].
Чувствительность определения в сернокислых растворах несколько ниже, чем в
солянокислых средах, но может быть повышена использованием в качестве
растворителя смеси этилового спирта и хлороформа (2:8). Экстракт имеет максимум
светопоглощения при л = 440 нм. Железо,
титан, молибден(VI)
и вольфрам(VI)
маскируются с помощью фосфорной кислоты. Экстракция соединения ванадия с N-БФГА
из сернокислых растворов высокой кислотности перспективна с той точки зрения,
что разложение ванадийсодержащих сплавов и объектов минерального сырья чаще
всего производят с помощью серной кислоты.
Экстракция бензоилфенилгидроксиламинатов
представляет особый интерес при определении следовых содержаний ванадия.
Имеется ряд работ [66] по определению ничтожно малых количеств ванадия с
помощью N-БФГА. Закон Бера
для методики экстракционно-фотометрического определения ванадия с N-БФГА
соблюдается в пределах концентрации ванадия 0.7-12 мкг/л при л=510 нм . При
концентрации соляной кислоты 2.8-4 М образуется комплекс, имеющий максимум
поглощения при 510 нм и молярный коэффициент погашения 4650. Метод позволяет
определять ванадий при содержаниях более 40 мкг/л. Ванадий(IV)
в сильнокислых растворах с N-БФГА
не взаимодействует. Таким образом, можно считать, что в сильнокислых растворах N-БФГА
является специфическим и высокочувствительным реагентом для определения ванадия(V).
Обычно встречающиеся катионы определению ванадия
с N-БФГА не мешают.
Мешают окислители, разрушающие реагенты, и восстановители, изменяющие
валентность ванадия. Особенность взаимодействия ванадия(V)
с N-БФГА состоит в
том, что комплекс с ванадием избирательно экстрагируется из сильно кислых (5-9
М по HCl) растворов
хлороформом. В аналогичных условиях экстрагируются цирконий, титан, олово(IV),
ниобий(V), тантал, церий(IV),
плутоний(IV), молибден(VI),
вольфрам(VI) [67].
Однако либо содержание в пробах природных вод соответствующих металлов ничтожно
(ниобий, тантал, цирконий, плутоний) либо образуемые ими комплексы с N-БФГА
не поглощают излучение в видимой области (титан, олово(IV),
церий, молибден(VI),
вольфрам(VI)). Кроме
того, в сильно кислых растворах комплексы большинства ионов с этим реагентом
неустойчивы, тогда как комплекс ванадий-N-БФГА
вполне устойчив, а его окраска развивается мгновенно. Избирательная экстракция
ванадия(V) из сильно
солянокислых сред в широком интервале кислотности позволяет определять весьма
малые его количества без введения дополнительных маскирующих агентов.
В последние годы синтезировано большое число
новых органических производных гидроксиламина. Проведено сравнительное изучение
цветных реакций ванадия(V)
с реагентами, содержащими ароматические или гетероциклические радикалы
(бензоил-, метоксифенил-, фенилацетил-, о-, м-, п-толил-, 2-фуроил-, 2-теноил-,
циннамоил- и др.) в различных комбинациях [68,69], а также с ненасыщенными
гидроксамовыми кислотами.
В результате для определения ванадия (V)
рекомендованы N-2-теноил-N-п-толил-
и N-2-теноил-N-фенил-
[70], N-м-толил-N-о-метоксибензоил-
[68], N-толил-N-циннамоил-
[69], N-п-толил-N-2-фуроил-
[71], N-2-фуроил-N-фенил-
[72], N-о-метоксифенил-N-2-теноил-
[73], N-бензоил-N-о-толил-
[74], N-циннамоил-N-фенилгидроксиламины
[75]. Эти реагенты по чувствительности и избирательности превосходят БФГА. Их
сравнительная характеристика приведена в табл.2. Все реагенты в 2,5 - 8 М HCl
при достаточном избытке реагента образуют с ванадием хелатные соединения в
отношении 2:1. Соединения экстрагируются органическими растворителями,
преимущественно хлороформом. Молярные коэффициенты погашения при 510 - 555 нм
равны (3-7)·103. Они отличаются в основном по избирательности. Определению
ванадия с большинством реагентов этого типа мешают титан(IV),
молибден(VI), ниобий(V),
цирконий(IV) и гафний(IV).
Одним из наиболее избирательных реагентов
является N-м-толил-N-о-метоксибензоилгидроксамовая
кислота. Определению ванадия с ним не мешают щелочные и щелочно-земельные
металлы, Ti(IV),
Zr(IV),
Cr(III),
Mn(II),
Fe(III),
Co, Ni,
Cu(II),
Zn, Cd,
Hg(II),
Pb, Al,
As(III),
РЗЭ и анионы Cl-F-,
PO43-, C2O42-,
цитраты и тартраты [71].
Таблица 2. Определение ванадия(V)
с производными гидроксиламина.
|
Реагент
|
CHCl, M
|
лmax,нм
|
е*10-3
|
Кратные
количества ионов, мешающих определению ванадия
|
Сс
|
5
- 9
|
530
|
4,5
- 5,1
|
Zr(IV) 200; Mo(VI) 400; Ti(IV), Cr(VI) 10;
Fe(III) 103; Hf
|
[98]
|
|
N-2-Фуроил-N-фенилгидроксиламин
|
3
- 7
|
530
|
5,6
|
Ti(IV), Nb(V), Mo(VI), W(VI) > 100 - 1000
|
[106]
|
|
N-Циннамоил-N-фенилгидроксиламин
|
2
- 10
|
540
|
6,3
|
Ti(IV) 10; Mo(VI) 20; Zr(IV) 200; NO2-, Cr(VI)
|
[109]
|
|
N-2-Теноил-N-фенилгидроксиламин
|
5
|
530
|
5,7
|
Mo(VI), Ti(IV), Zr(IV)
|
[103]
|
|
N-о-Метоксифенил-N-2-теноил-гидроксиламин
|
3
- 6
|
545
|
7,2
|
30 - 100 мгTi(IV),
Zr(IV), Fe(III), Mo(VI), U(VI), PO43-, F-
|
[107]
|
|
N-Бензоил-N-о-толилгидроксиламин
|
4
- 8
|
510
|
|
Ti(IV),
который маскируютNaF
|
[108]
|
|
N-п-Толил-N-2-фуроилгидроксиламин
|
3
- 4
|
540
|
3,0
|
Ti(IV) иZr(IV)
300
|
[105]
|
|
N-м-Толил-N-о-метоксибензоил-гидроксиламин
|
6
- 8
|
550
|
6,5
|
|
[104]
|
. Экспериментальная часть
.1 Реактивы и аппаратура
. п-нитробензойная кислота, ч
. м-нитробензойная кислота
. 4-метилбензойная кислота
. п-бромбензойная кислота
. Нитробензол, ч
. Цинковая пыль
. Хлорид натрия, хч
. Метаванадат аммония, чда
. Соль Мора, 0,02 М
. Фенилантраниловая кислота, ч
. Фосфорная кислота, хч
. Хлороформ, ч, перегнан при 64оС
. Ионообменная смола КУ-2 в Н+ форме
. Ионообменная смола Амберлит в Cl-
форме
. Фотометрические измерения производили
на спектрофотометре модели UV
mini - 1240 фирмы “Shimadzu”
в кюветах с толщиной слоя 10 мм в области длин волн 400 - 700 нм. Раствор
сравнения хлороформ.
. Упаривание проб природных вод
производили на электрической плитке.
. Взятие навесок осуществляли с помощью
весов аналитических ВЛР-200.
3.2 Использованные растворы
. Стандартный раствор ванадия (V),
Cv = 0,829 г/л
Раствор готовили растворением навески (m
= 2,3934 г) ванадата аммония в 60 мл 0.5 М H2SO4
и доведением объема раствора до 1 л дистиллированной водой. Стандартизацию
раствора осуществляли титрованием стандартным раствором соли Мора с индикатором
фенилантраниловой кислотой. Рабочие растворы с меньшими концентрациями V(V)
готовили разбавлением исходного раствора дистиллированной водой непосредственно
перед использованием.
. Растворы реагентов в хлороформе, С =
2,00 г/л.
Растворы готовили растворением навесок сухих
реагентов (m = 0,1000 г) в 50
мл хлороформа.
3.3 Синтез
N-фенилгидроксиламина
(N-БФГА)
К раствору 5г хлористого аммония в 160 мл воды
добавляют при перемешивании 10 мл нитробензола, а затем маленькими порциями 13
г цинковой пыли в течении 15 - 20 минут. Реакция восстановления сопровождается
повышением температуры реакционной массы до 60 - 65°С. Реакционную массу
выдерживают при этой температуре в течение 10 - 15 минут, а затем фильтруют для
удаления гидроокиси цинка, которую затем промывают небольшим количеством
горячей воды. Фильтрат и промывные воды насыщают хлористым натрием (50г; необходимо
следить за тем, чтобы хлористый натрий целиком растворился), охлаждают до 0°С и
выдерживают при этой температуре не менее 0.5 часа. Выпавший фенилгидроксиламин
быстро отфильтровывают, хорошо отжимают и используют для конденсации с
хлористым бензоилом. Вес сырого продукта 11,8 г (93% теории, принимая во
внимание, что влажность его составляет 30%).
.3.2 Получение хлористого бензоила
Кипятят 0,1 моль карбоновой кислоты и 0,15 моля
тионилхлорида (на каждую карбоксильную группу) с обратным холодильником до
прекращения выделения газа. Затем избыток тионилхлорида отгоняют на водяной
бане. Образовавшийся хлорангидрид перегоняют под вакуумом.
.3.3 Получение N-бензоилфенилгидроксиламина
В колбе растворяют 6,7 г сырого, хорошо отжатого
фенилгидроксиламина в 200 мл теплой воды (40°С); полученный раствор
отфильтровывают. Фильтрат, охлажденный до 10°С, подщелачивают бикарбонатом
натрия до слабощелочной реакции и при энергичном размешивании к нему по каплям
добавляют 7,8 г бензоилхлорида. После добавления всего количества хлористого
бензоила реакционную массу выдерживают при перемешивании в течение 1,5 часов.
Далее, выпавшую смесь моно- и дибензоильных производных отфильтровывают,
выделенный осадок тщательно растирают с бикарбонатом натрия и небольшим
количеством воды, фильтруют и промывают водой до удаления запаха хлористого
бензоила. Монобензоильное производное отделяют от дибензоильного обработкой
смеси раствором аммиака, в котором растворяется только
монобензоилфенилгидроксиламин. Массу отфильтровывают, а фильтрат приливают при
размешивании к избытку раствора серной кислоты (1:1). Полученные белые
кристаллы бензоилфенилгидроксиламина отфильтровывают, промывают холодной водой
до отсутствия кислой реакции в промывных водах и хорошо отжимают. Полученный
продукт перекристаллизовывают из этилового спирта. Вес продукта 0.84 г (16%
теории).
.4 Синтез реагентов
По приведенной выше методике было получено
несколько различных реагентов для определения ванадия (V):
1. N-4-метилбензоилфенилгидроксиламин
(N-МБФГА)
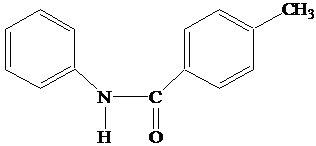
В синтезе N-МБФГА в
качестве карбоновой кислоты использовалась 4-метилбензойная кислота.
2. N-п-бромбензоилфенилгидроксиламин
(N-ББФГА)
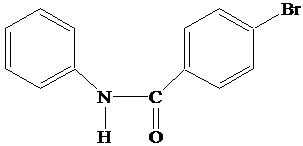
Для получения N-ББФГА
использовалась п-бромбензойная кислота.
3. N-п-нитробензоидфенилгидроксиламин
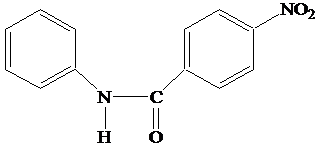
Для получения
п-нитробензоидфенилгидроксиламина использовалась п-нитробензойная кислота.
4. N-м-нитробензоилфенилгидроксиламин
Для синтеза
м-нитробензоилфенилгидроксиламина использовалась м-нитробензойная кислота.
5. N-циннамоилфенилгидроксиламин
(N-ЦФГА)

Для получения N-ЦФГА
использовалась коричная кислота.
Все полученные реагенты были
перекристаллизованы из этанола и высушены. Для идентификации синтезированных
веществ были получены их ЯМР - спектры. спектры
.5 Методика эксперимента
Экстракционно-фотометрический метод
определения ванадия(V) с N-БФГА основан
на образовании окрашенного в фиолетовый цвет комплексного соединения ванадия(V) с N-бензоилфенилгидроксиламином
в растворе соляной кислоты, экстрагировании его хлороформом и измерении
оптической плотности окрашенного органического раствора при длине волны 530 нм.
Порядок прибавления реактивов следующий: к раствору ванадия(V)
последовательно добавляли: 12 мл 6М раствора соляной кислоты, дистиллированную
воду до 30 мл и 3 мл хлороформенного раствора реагента. После тщательного
перемешивания и отстаивания хлороформенный экстракт сливали и измеряли его
оптическую плотность относительно хлороформенного раствора реагента на
спектрофотометре при 530 нм и толщине слоя 10 мм. Спектры хлороформенных
экстрактов комплексов ванадия (V) с реагентами представлены ниже.

Рис.1. Спектр хлороформенного
экстракта комплекса ванадий(V) - N-4-метилбензоилфенилгидроксиламин,
Cv = 0,825
мг/л.

Рис.2. Спектр хлороформенного
экстракта комплекса ванадий(V) - N-п-бромбензоилфенилгидроксиламин,
Cv = 0,825
мг/л.

Рис.3. Спектр хлороформенного
экстракта комплекса ванадий(V) - N-п-нитробензоилфенилгидроксиламин,
Cv = 0,825
мг/л.

Рис.4. Спектр хлороформенного
экстракта комплекса ванадий(V) - N-м-нитробензоилфенилгидроксиламин,
Cv = 8,25
мг/л.
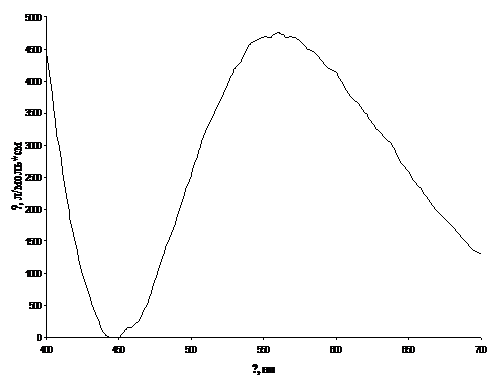
Рис.5. Спектр хлороформенного
экстракта комплекса ванадий(V) - N-циннамоилфенилгидроксиламин,
Cv = 0,825
мг/л.
Из данных спектров видно, что
хлороформенные экстракты комплексных соединений ванадия(V) поглощают
свет в видимой области спектра. Максимум поглощения для комплекса с N-4-метилбензоилфенилгидроксиламином
имеет место в области 530 - 540 нм, при этом коэффициент экстинкции ε ~ 5500
л/моль*см; для комплекса с N-п-бромбензоилфенилгидроксиламинов -
в области 520 - 530 нм, е ~ 3500 л/моль*см; для комплекса с N-п-нитробензоилфенилгидроксиламинов
- в области 520 - 530 нм, е ~ 2200 л/моль*см; для комплекса с N-м-нитробензоилфенилгидроксиламинов
- в области 510 - 520 нм, е ~ 60 л/моль*см; для комплекса с N-циннамолфенилгидроксиламином
- в области 550 - 560 нм, е ~ 4700 л/моль*см. Таким образом, наименьшее
значение коэффициента экстинкции имеет комплекс ванадий (V) - N-м-нитробензоилфенилгидроксиламин,
и поэтому данный реагент не представляет дальнейшего интереса.
Для получения более подробной
информации о значениях коэффициентов экстинкции комплексов ванадия (V) с
реагентами, были построены градуировочные графики.ПОЧЕМУ ТОЛЬКО ТРИ ТОЧКИ?

Рис.6. График зависимости оптической
плотности хлороформенного экстракта комплекса ванадия(V) с N-4-метилбензоилфенил-гидроксиламином
от концентрации ванадия(V) в водном растворе.
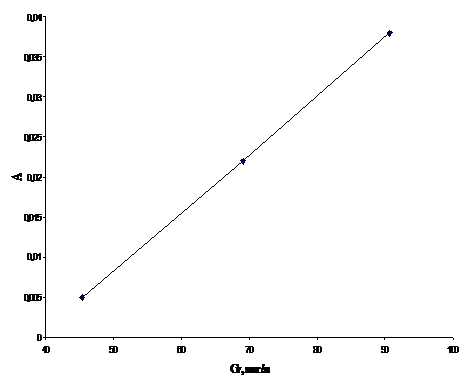
Рис.7. График зависимости оптической
плотности хлороформенного экстракта комплекса ванадия(V) с N-п-бромбензоилфенил-гидроксиламином
от концентрации ванадия(V).

Рис.8. График зависимости оптической
плотности хлороформенного экстракта комплекса ванадия(V) с N-п-бромбензоилфенил-гидроксиламином
от концентрации ванадия (V).
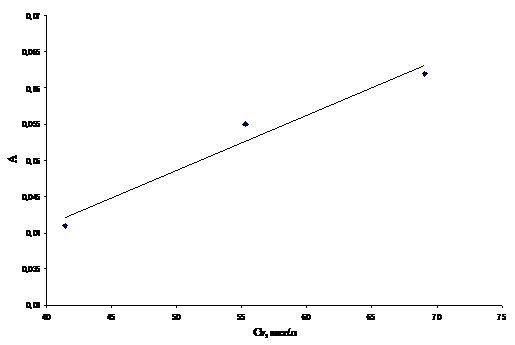
Рис.9. График зависимости оптической
плотности хлороформенного экстракта комплекса ванадия(V) с N-циннамоилфенил-гидроксиламином
от концентрации ванадия (V).
По представленным графикам из
наклона прямых были рассчитаны коэффициенты экстинкции комплексов ванадия(V) с
реагентами. Полученные данные представлены в таблице 3.
Табл.3 Значения коэффициентов
экстинкции комплексов ванадия (V) с различными реагентами.
|
Реагент
|
е,
л/моль*см
|
|
N-4-метилбензоилфенилгидроксиламин
|
5800
|
|
N-п-бромбензоилфенилгидроксиламин
|
3700
|
|
N-п-нитробензоилфенилгидроксиламин
|
2260
|
|
N-циннамоилфенилгидроксиламин
|
3870
|
Из представленных данных видно, что наибольшее
значение коэффициента экстинкции имеет комплекс ванадия(V)
с N-4-метилбензоилфенилгидроксиламином.
Таким образом, данный реагент представляет наибольший интерес.
ванадий титриметрический
спектроскопия реагент
3.6 Построение градуировочного графика методики
экстракционно-фотометрического определения ванадия(V)
с N-4-метилбензоилфенилгидроксиламином
Была построена зависимость оптической плотности
экстракта ванадия с N-4-метилбензоилфенилгидроксиламином
от концентрации ванадия в водных растворах. Для этого была приготовлена серия
растворов с содержанием ванадия(V)
4 - 250 мкг/л. Полученные экспериментальные данные приведены в таблице 4.
Табл.4 Значения оптических плотностей
хлороформенных экстрактов комплекс ванадий(V)
- N-4-метилбензоилфенилгидроксиламин
при различных концентрациях ванадия в водном растворе.
|
Cv, мкг/л
|
А±∆А
(n=3, P=0.95)
|
|
4,15
|
0,0083±0,0014
|
|
5,53
|
0,0127±0,0063
|
|
6.91
|
0,0167±0,0052
|
0,0203±0,0080
|
|
11,1
|
0,0223±0,0038
|
|
13,8
|
0,0243±0,0080
|
|
27,6
|
0,0447±0,0080
|
|
69,1
|
0,0907±0,0076
|
|
96,7
|
0,124±0,008
|
|
138,2
|
0,181±0,007
|
|
165,8
|
0,219±0,009
|
|
207,3
|
0,272±0,008
|
|
248,7
|
0,322±0,009
|
На основании данных таблицы 4 был построен
градуировочный график зависимости оптической плотности хлороформенных
экстрактов от концентрации ванадия(V)
в водном растворе (рис.10).

Рис.10. Градуировочный график методики
экстракционно-фотометрического опредления ванадия(V)
с N-4-метилбензоилфенилгидроксиламином.
Градуировочный график был обсчитан по методу
наименьших квадратов. Прямолинейная градуировочная зависимость описывается
уравнением:
= a
+ b·Cv
Где: a
= 0,00648
b = 0,00127
Sa = 0,00119
Sb = 1,27·10-5
При P
= 0,95 и n = 3 коэффициент
Стьюдента t = 2.2,
следовательно:
∆a
= 7,3·10-4
∆b
= 7,8·10-6
Наклон градуировочного графика соответствует
значению коэффициента экстинкции комплекса ванадий - N-4-метилбензоилфенилгидроксиламин
в хлороформе и равен 6470.ПОЧЕМУ НЕ СОВПАДАЕТ С НАЙДЕННЫМ РАНЕЕ?
.7 Экстракционно-фотометрическое определение
содержания ванадия(V) в
природных водах
Сравнительное изучение вновь предложенного
реагента на ванадий….. и традиционного …. было проведено с использованием проб
реальных вод. Содержание ванадия в природных водах чрезвычайно мало и его
концентрирование неизбежно. Также большая часть ванадия в природной воде
находится в неактивной закомплексованной форме, поэтому его определение
возможно лишь после полного кислотного разложения проб с окислением имеющихся в
них органических веществ. Выбранная нами методика пробоподготовки включает в
себя упаривание анализируемых проб с хлорной кислотой до появления ее паров,
т.е. до полного разрушения имеющихся органических веществ.
Стандартная методика пробоподготовки выглядела
следующим образом:
1. Фильтрование пробы воды (немногим больше 1
л) через бумажный фильтр с использованием системы колба Бунзена - воронка
Бюхнера.
2. Точное измерение объема забираемой пробы
воды с помощью мерной колбы на 1 л.
. Добавление к анализируемой пробе 1 мл
хлорной кислоты.
. Упаривание 1 л воды на электроплитке до
появления паров хлорной кислоты.
. Добавление к упаренной пробе 1 мл
фосфорной кислоты для маскирования сопутствующих ванадию элементов.
. Доведение объема пробы до 25 мл
дистиллированной водой.
Вопрос о знаке заряда присутствующих в природных
водах форм ванадия(V) может быть
разрешен предварительным пропусканием анализируемых проб через ионообменные
смолы. В таком случае часть пробы, которая прошла через катионообменную смолу,
не будет содержать катионные формы ванадия(V),
а только его анионные формы. Часть пробы, которая прошла через анионообменную
смолу, не содержит анионные формы ванадия(V),
а только его катионные формы. Для соответствующих целей использовали 10 см слои
ионообменных смол: катионообменной КУ-2 в H+
форме и анионообменной АМБЕРЛИТ в Cl-
форме. Через катионообменную смолу проба пропускалась со скоростью ~ 11,3
мл/мин, а через анионообменную - со скоростью ~ 4,8 мл/мин.
Суммарное содержание ванадия(V)
определялось с помощью N-БФГА
и N-4-метилбензоилфенилгидроксиламин,
а содержание катионных и анионных форм определялось с помощью только N-4-метилбензоилфенилгидроксиламин.
Результаты представлены в таблице 5.
Табл.5. Содержание ванадия(V)
в образцах анализируемых вод (n=3,
P=0.95).
|
Проба
воды
|
N-БФГА
|
N-4-метилбензоилфенилгидроксиламин
|
|
Суммарное
содержание ванадия (V),
мкг/л
|
Суммарное
содержание ванадия (V),
мкг/л
|
Содержание
катионных форм, мкг/л
|
Содержание
анионных форм, мкг/л
|
|
Водопровод,
Ст.Петергоф
|
7,5±1,3
|
7,3±1,5
|
1,7±0,9
|
4,5±0,2
|
|
Р.
Нарова, пос. Переволок, Лен. обл.
|
8,0±1,6
|
7,6±0,9
|
1,8±0,7
|
5,3±0,9
|
|
Троицкий
ручей, Ст. Петергоф
|
4,7±1,4
|
4,6±0,7
|
2,5±0,8
|
2,7±0,6
|
|
Пруд,
парк Сергиевка, Ст. Петергоф.
|
4,5±1,1
|
4,9±1,5
|
1,3±0,6
|
|
Ольгин
пруд, Н. Петергоф
|
4,1±1,4
|
4,6±0,8
|
0,8±0,4
|
3,4±0,5
|
3.8 Изучение влияние концентрации соляной
кислоты на экстракционно-фотометрическое определение ванадия(V)
При экстракционно-фотометрическом определении
ванадия(V) затрачивается
большой объем соляной кислоты. В связи с этим встает вопрос: возможно ли
проведение экстракционно-фотометрического определения ванадия(V)
в солянокислой среде меньшей концентрации? Для ответа на этот вопрос экстракция
комплексов ванадия(V) с N-БФГА
и N-4-метилбензоилфенилгидроксиламином
проводилась при различных значениях концентрации соляной кислоты. Результаты
этого эксперимента приведены в таблице 6.
Табл.6. Значения оптических плотностей
хлороформенных экстрактов комлексов ванадия(V)
с N-БФГА и N-4-метилбензоилфенил-гидроксиламином
при различных концентрациях соляной кислоты, Cv
= 0,138 мг/л.
|
CHCl, моль/л
|
А,
комплекс ванадий(V)
- N-БФГА
|
А,
комплекс ванадий (V)
- N-4-метилбензоилфенилгидроксиламин
|
|
0
|
0,035
|
0,043
|
|
0,4
|
0,060
|
0,059
|
|
0,8
|
0,065
|
0,082
|
|
1,2
|
0,102
|
0,118
|
|
1,6
|
0,120
|
0,121
|
|
2,0
|
0,126
|
0,134
|
|
2,4
|
0,152
|
0,174
|
По результатам, представленным в таблице 6, были
построены графики зависимости оптической плотности хлороформенных экстрактов
комлексов ванадия (V) с N-БФГА
и N-4-метилбензоилфенилгидроксиламином
от концентрации соляной кислоты.

Рис.11. График зависимости оптической плотности
хлороформенного экстракта комлекса ванадия(V)
с N-БФГА от
концентрации соляной кислоты.

Рис.12. График зависимости оптической плотности
хлороформенного экстракта комлекса ванадия(V)
с N-4-метилбензоилфенил-гидроксиламином
от концентрации соляной кислоты.
Из графиков видно, что концентрация соляной
кислоты оказывает значительное влияние на протекание реакции образования
комплекса ванадия(V) как с N-БФГА,
так и с N-4-метилбензоилфенилгидроксиламином.
Таким образом, … (не знаю, как выразить мысль о том, что не стоит уменьшать
количество добавляемой кислоты) Вы сначала должны показать, что исп. Кол-во не
мало!
Список литературы
1. Музгин
В.Н., Хамзина Л.Б., Золотавин В.Л., Безруков И.Я. Аналитическая химия ванадия,
М.: Наука, 1981
. Сонгина
О.А. Редкие металлы, М: Металлургиздат, 1951
. Рощин
А.В. Ванадий и его соединения, М: Медицина, 1968
4. Schiller
K., Thilo E. SpektrophotometrischeUntersuchung von Vanadatgleichgewichten in
verdunntenwassrigenLosungen. // Z. Enorg. Chem. - 1961. - Bd.
310. - HF 4 - 6. - s.
261 - 285
. Ивакина
А.А., Фотиева А.А. Химия пятивалентного ванадия в водных растворах, Свердловск,
Академия наук СССР, Уральский научный центр,1971
. Бусев
А.И., Типцова В.Г., Иванов В.М. Практическое руководство по аналитической химии
редких элементов, М: Химия, 1966
. Поляк
Э.А., Городенцева Т.Б. - Тр. Всесоюз. н.-и. ин-та станд. обр. и спектр.этал.,
6, 41, 1970
. Сонгина
О.А., Студенская Л.С., Бектурова Г.Б., Маслова П.И., Городенцева Т.Б. - Тр.
ВНИИ станд. обр. и спектр.этал., 4, 112, (1968)
. Кольтгоф
И.М., Белчер Р., Стенгер В.А., Матсуяма Дж., Объемный анализ. М.: Госхимиздат,
т.3, с. 719, 730, (1961)
. Хитаров
П.И. - Зав. лаб., 4, 491, (1935)
11. RaoG.G.,
RaoP.K. - Ibid., 14, 33, (1967)
. ChawlaK.L.,
TandonJ.P. - Ibid., 6 , 332, (1968)
13. ШварценбахГ.,
ФлашкаГ., Комплексонометрическоетитрование. М.: Химия, 1970
14. Kaimal
W.R.M., Shome .C. - Anal. chim. Acta, 27,594, (1962)
15. МариновВ.
Химияииндустрия (НРБ), 43, 353, (1971)
. КусакинаН.П.,
ЯкимецЕ.М. - Тр. Уральск. Политехн.
ин-та,
№ 121, 91, (1962)
17. SajoJ.
- Z. anal. Chem., 188, 168, (1962)
18. ЯцимирскийК.Б.
Кинетическиеметодыанализа. М.: Химия, 1967
. КрейнгольдС.У.,
ЮжальЕ.М., Покровская И.Е., Иванов Ю.А. Определение ванадия в веществах особой
чистоты и природных водах кинетическим методом. // Заводская лаборатория. 1981.
47Т. - №5. - с.17 - 19
20. Степин
В.В., Силаева Е.В., Плисс А.М., Курбатова В.И., Федорова Н.Д., Поносов В.И.
Анализ черных металлов, сплавов и марганцевых руд. М.:
Металлургия,
1971
. Rao
V.P.R., Sarma B.V.S. - Chemist-Analyst, 54, 107, (1965)
. Singh
V.B., Prasad B.N. - J. Sci. Res. Banaras Hindu Univ., 12, 116, (1961-1962)
. Khalifa
H., El-Sirafy A. - Z. anal. Chem., 227, 109, (1967)
. Khalifa
H., Ismail I.A. - Microchim. J., 14, 464, (1969)
25. КрюковаТ.А.,
СиняковаС.И., АрефьеваТ.В. Полярографическийанализ. М.: Госхимиздат, 1959
26. ЗозуляА.П.
Кулонометрический анализ. Л.: Химия, 1968
. Гончаренко
А.С. Электрохимия ванадия и его соединений. М.: Металлургия, 1969
28. АгасянП.К.,
БасовВ.Н., КостроминА.И.Журнал аналитической химии, с. 1933, (1970)
. Костромин
А.И., Ахметов А.А., Орлова Л.Н. Журнал аналитической химии, 25, 195, (1970)
30. Cole
P.C., Eckirt F.M., Williams K.Z. Determination of dissolved and particulate
vanadium in sea water by x-ray fluorescence spectrometry. // Anal. Chim. Acta.
1983. - Vol. 153. - p. 61 - 67
. СимузиТ.,Ксида
Ю., Шийо Ю. Определение ванадия в речной воде сочетанием метода соосаждения в
атомно-абсорбционной спектрометрии с использованием графитовой кюветы. //
БунсэкиКашаку. 1981. - Т.30. - №2. - с. 113 - 117 Рж. Химия. 1981.
- 18Г.
199
33. Yoshio
S., Yokio K., Yokuo S. Determination of vanadium in sea water by graphite
furnace atomic absorption spectrometry. // Bunseki Kagaku 1983. - Vol. 32. - №
9. - p. 285 - 291
. RomanM.,
Garcia-SanchesF., GomezH.A. - Quim. anal. (purayapl.), 28, 191,
(1974)
. Martin
E.L., Bentley K.E. - Ibid., 34, 354, (1962)
36. Золотавин
В.Л., Безруков И.Я., Санников Ю.И.Журнал неорганической химии, 6, 584, (1961)
. Харламов
И.П. Спектрофотометрический анализ сплавов. М.:
Металлургия,
с.
71, 170,
(1969)
38. Sarma
P.L. - Anal. Chem., 36, 1076, (1964)
39. Долгорев
А.В. - В кн.: Химия и химическая технология ванадиевых соединений. Пермь: Кн.
Изд-во, с.447, (1974)
40. Марченко
З. Фотометрическое определение элементов. М.:
Мир,
1971, с.128
- 137
. R.L.
Dutta, S. Lahiry. J.Indian Chem. Soc., 39, 860, (1962)
. R.L.
Dutta, S. Lahiry. J.Indian Chem. Soc., 40, 53, (1963)
43. ШнайдерманС.Я.,
КлименкоЕ.П., КнязеваЕ.Н., Черная Н.В. Журнал неорганической химии, 14, 1238,
(1969)
44. Patrowsky
V. - Chem. listy, 49, 854 (1955); Coll. Czech. Chem. Communs, 31, 3392,
(1966)
. Huart
A., Chaussabel L., Corgier J., Deron S., Detwiller J., Disant C. - Note CEA, N
1341, 55, (1970)
46. ШнайдерманС.Я.,
КлименкоЕ.П., Прокопьева Г.П. Журнал неорганической химии, 14, 1552, (1969)
. Смирнов
А.Н. Изучение фталексонов как реагентов для спектрофотометрического определения
ванадия. Саратов, 1983
48. Муликовская
Е.П. Определения ванадия в природных водах // В кн. Методы анализа минерального
сырья. Тр. Всесозн. н.-и. ин-та 1964. Т.117. - с. 79 - 84
49. Новиков
Ю.В., Ласточкина К.О., Болдина З.Н. Методы определения вредных веществ в воде
водоемов. М.: Медицина 1981. - с. 376
. Heitner-WirguinC.,
GanezM. - Talanta, 14, 671, (1967)
51. Калугин
А.А., Нипрук О.В., Егорова О.А. Определение микроколичеств ванадия (V)
в водных растворах труднорастворимых уранованадатов различных металлов.
Нижегородский ГУ им. Н.И. Лобачевского, 1999
. Janousek
J.J. - Coll. Czech. Chem. Communs, 27, 2972, (1962)
. Otomo
M. - Bull. Chem. Soc. Japan, 36, 137, (1963)
54. Тихонов
В.Н. Журнал аналитической химии, 21, 1172, (1966)
. Назаренко
В.А., Вещикова Н.А., Новоселова М.М. Журнал аналитической химии, 1984, 39.№12,
с. 2151
. Мамедова
АМ., Иванов В.М., Ахмедов С.А. Взаимодействие W(VI)
и V(V)
с пирогаллоловым красным и бромпирогаллоловым красным в присутствии ПАВ, Вести
Московского ун-та сер.2, Химия, 2004. Т.45.№2
. Попова
В.Ф. Спектрофотометрический метод определения ванадия путем сочетания его с
капрогидроксамовой кислотой: Автореф. Дисс.
Канд,хим.наук
/ ТашПи.
- Ташкент.
- 1970. - с. 18
58. Dutta
R.L. - J. Indian Chem. Soc., 35, 243 (1958); 36, 285, 339 (1959)
. Hulcher
F.H. - Anal. Chem., 32, 1183, (1960)
60. ПоповаВ.Ф.,
МаркманА.Л.
- Узб.
хим.
ж.,
№ 4, 18 (1966); Тр.
Ташкентск.
политехн. ин-та, вып. 64, 3, (1970)
. ЖаровскийФ.Г.,
ПилипенкоА.Т. Украинский химический журнал, 25, 230, (1964)
. Алимарин
И.П., Судаков Ф.П., Головкин Б.Г. - Усп. хим., 31, 989 (1962)
. Жаровский
Ф.Г., Пилипенко А.Т. Украинский химический журнал, 25, вып. 2, 230, (1959)
. Чмутова
М.К., Петрухин О.М., Золотов Ю.А. Журнал аналитической химии, 18, 588, (1963)
. ТомиокоХидэо
«БунсэкиКагаку» JapanAnalyst
12, 3, 271, (1963); РЖХ 21, 21 Г85,(1963); Repts.
Covt. Chem.
Industr. Res.
// Tokyo 58, 387,(1963);
РЖХ, 13, 13 Г78, (1964)
. Пограничная
Р.М., Нерубащенко В.В., Цевина А.В. Экстракция ванадия в виде комплекса с
4-(2-пиридилазо)-резорцином. // Ж. аналит. химии, 1972. - Т.24. Вып.9, с. 1845
- 1847
. Алимарин
И.П., ЦзеЮнь-сян. Вестник Московского университета, серия химия, 2, 53, (1960)
68. Gupta
V.K., Tandon S.G. - Anal. chim. acta, 66, 39 (1973)
. Tandon
S.G., Bhattacharya S.C. - J. Indian Chem. Soc., 47, 583 (1970)
. Tandon
S.G., Bhattacharya S.C. - Anal. Chem., 33, 1267 (1961); 36, 1378 (1964)
. Agrawal
Y.K. - Ibid., 7, 729 (1974)
72. ПилипенкоА.Т.,
ШпакЭ.А., РубанП.П. - Украинскийхимическийжурнал, 29, 1209 (1963)
73. Abbasi
S.A. - Anal. Lett., 9, 113 (1976)
. Majumdar
A.K., Das G. - Ibid., 31, 147 (1964)
75. Гусакова
Н.Н., Еременко С.Н., Муштакова С.П., Фрумина Н.С.- Журнал аналитической химии,
30, 721 (1975)