Рак легень в поєднанні з туберкульозом
Вступ
Питання про зв'язок між
двома етіологічно різними захворюваннями - раком і туберкульозом легені виник
більше 100 років тому і до теперішнього часу залишається актуальним. В Україні,
як і в багатьох країнах світу, рак легені стабільно займає провідне місце серед
інших злоякісних новоутворень в цілому. Оскільки епідемічна ситуація з
туберкульозу в нашій країні за останні роки погіршилась, а система протиракових
і протитуберкульозних профілактичних заходів недостатньо ефективна і потребує
перегляду, стає зрозумілим, чому поєднання туберкульозного процесу і раку
легені набуло найважливішу клінічну значимість Незважаючи на велику кількість
робіт, присвячених вивченню патогенетичної зв'язку раку з туберкульозом,
фахівці до цих пір не прийшли до загальної точки зору. За даними літератури,
існують три основних поглядів на розвиток в легенях поєднаної патології
туберкульозу і раку. [7]
Одні автори вважають, що
це два незалежні паралельно протікаючі процеси, інші відзначають наявність
якогось антагонізму і роз'єднання в перебігу цих захворювань, треті, що
становлять більшість, вважають, що всі хронічні запальні процеси в легенях,
ускладнені склерозом і цирозом, у тому числі туберкульоз, є сприятливим фоном
для розвитку злоякісних пухлин Загальновизнано, що хронічні запальні процеси в
легенях стимулюють метаплазію епітелію бронхів або грають роль канцерогенного
фактора, готуючи сприятливий грунт для розвитку злоякісного захворювання. У
розвитку раку при туберкульозі важлива роль відводиться циротичній перебудові органу,
порушень метаболізму та активності ферментативних процесів, що, як правило,
спостерігається у пацієнтів з хронічним неактивним фіброзно-кавернозним
туберкульозом. Велике значення мають рубцево-склеротичні зміни формування
сполучної тканини на місці активного специфічного запалення або такі явні
передракові зміни тканин легенів, як вогнища метаплазії епітелію слизової
оболонки бронхів або ділянки каверн. За даними Е.А. Коган і співавторів (1992),
біохімічний склад екстрацелюлярного матриксу рубців периферичного раку легенів,
метатуберкульозних і метапневмонічних ділянок вважається ідентичним. Багато
дослідників вказують на те, що рак формується в ураженій туберкульозом легені,
хоча є й інші думки. Відзначають вплив тривалої лікарської терапії на ініціацію
неоплазії в легенях, а також двосторонній зв'язок між раком і туберкульозом: в
одних випадках можливий розвиток раку легенів на тлі метатуберкульозних
рубцевих змін, в інших загострення специфічного процесу під впливом ракової
кахексії. Вказується, що рак легені може виникнути на основі хронічного
туберкульозу легень у хворих III і VII груп в старих рубцях, в фістулах
лімфатичних вузлів, з петрифіковані організованих ділянок старих каверн,
дренажних бронхів, бронхоектазів. Цьому сприяє також резистентність до терапії,
зниження імунітету і проведення променевої терапії, порушення регуляції
тканинного гомеостазу, в тому числі міграція клітин. За даними Є.І. Суслова і
співавторів, неопластичній трансформації передують зміни в легеневій паренхімі,
що призводять до порушення детермінації диференціювання клітин. Дані
рентгенологічного дослідження, бронхоскопії, бронхографії, реакцій Манту, Коха,
функціональних і цитологічних досліджень достовірні у 60 - 65% випадків, тому
альтеративні діагностичні тести в даний час досить актуальні. [2,4] Метою
нашого дослідження було встановлення впливу протитуберкульозних препаратів на
процеси проліферації та апоптозу високопроліферуючих клітин для подальшого
вивчення їх дії на трансформовані онкогенні клітини.
Актуальність. Більша кількість науковців вважають, що туберкульоз є сприятливим
фоном для виникнення пухлин у легенях, при цьому дія протитуберкульозних
препаратів на високопроліферуючі клітини в процесі онтогенезу недостатньо
вивчена. При вивченні впливу діючих речовин таких препаратів на процеси
проліферації та апоптозу клітин надасть можливість визначити потенційний
побічний онкогенний вплив препаратів при протитуберкульозній терапії.
1. Рак легенів і
туберкульоз
1.1 Рак легенів:
етіологія, класифікація
рак
легеня туберкульоз пухлина
Рак легені - злоякісне
новоутворення епітеліального походження, що розвивається зі слизових оболонок
бронхіального дерева, бронхіальних залоз (бронхогенний рак) або альвеолярної
тканини (легеневий або пневмогенний рак). Рак легені лідирує в структурі
смертності населення від злоякісних пухлин. Летальність при раку легені
становить 85% від загального числа хворих, незважаючи на успіхи сучасної
медицини.
Розвиток раку легені
неоднаковий при пухлинах різної гістологічної структури. Для диференційованого
плоскоклітинного раку характерно повільний плин, недиференційований рак
розвивається швидко і дає великі метастази. Самим злоякісним перебігом
характеризується дрібноклітинний рак легені: розвивається приховано і швидко,
рано метастазує, має поганий прогноз. [5]
Найчастіше пухлина
виникає в правій легені - в 52%, в лівій легені - в 48% випадків.
Ракова пухлина переважно
локалізується у верхній частці легені (60%), рідше в нижній або середній (30% і
10% відповідно). Це пояснюється більш потужним повітрообміном у верхніх
частках, а також особливостями анатомічної будови бронхіального дерева, в якому
головний бронх правої легені прямо продовжує трахею, а лівий у зоні біфуркації
утворює з трахеєю гострий кут. Тому канцерогенні речовини, чужорідні тіла,
частинки диму, прямуючи в добре аеруєми зони і тривало затримуючись у них,
викликають ріст пухлин.
Метастазування раку
легенів можливо за трьома шляхами: лімфогенним, гематогенним та імплантаційним.
[18]
Найбільш частим є
лімфогенні метастазування раку легенів у бронхопульмональні, пульмональні,
паратрахеальні, трахеобронхіальні, біфуркаційні, навколостравоходні лімфовузли.
Першими при лімфогенному метастазуванні уражаються пульмональні лімфовузли в
зоні поділу часткового бронха на сегментарні гілки. Потім в метастатичний
процес втягуються бронхопульмональні лімфатичні вузли вздовж часткового бронха.
Надалі виникають метастази в лімфовузлах кореня легені і непарної вени,
трахеобронхіальних лімфовузлах. Наступними залучаються до процесу
перикардіальний, паратрахеальні і навколостравоходні лімфатичні вузли.
Віддалені метастази виникають в лімфовузлах печінки, середостіння, надключичні
області. [11]
Метастазування раку
легені гематогенним шляхом відбувається при вростанні пухлини в кровоносні
судини, при цьому найбільш часто вражаються інша легеня, нирки, печінка,
надниркові залози, мозок, хребет.
Імплантаційне
метастазування раку легенів можливо по плеврі в разі проростання в неї пухлини.
За гістологічною структурою виділяють 4 типи раку легені: плоскоклітинний,
великоклітинний, дрібноклітинний і залозистий (аденокарцинома). Знання
гістологічної форми раку легені важливо в плані вибору лікування та прогнозу
захворювання. Відомо, що плоскоклітинний рак легені розвивається відносно
повільно і зазвичай не дає ранніх метастазів. Аденокарцинома також
характеризується порівняно повільним розвитком, але їй властива рання
гематогенна дисемінація. Дрібноклітинний та інші недиференційовані форми раку
легені швидкоплинні, з раннім обширним лімфогенним і гематогенним метастазуванням.
Помічено, що чим нижче
ступінь диференціювання пухлини, тим злоякісних її перебіг. [14]
За локалізацією щодо
бронхів рак легенів може бути центральним, що виникають у великих бронхах
(головному, пайову, сегментарному), і периферичним, котрий з субсегментарних
бронхів і їх гілок, а також з альвеолярної тканини. Центральний рак легенів
зустрічається частіше (в 70%), периферичний - набагато рідше (у 30%).
Форма центрального раку
легенів буває ендобронхіальною, перибронхіальною, вузловою і перибронхіально
розгалуженою. Периферична ракова пухлина може розвиватися у формі «кулястого»
раку (круглої пухлини), пневмоніє подібній, раку верхівки легені (Панкоста). [4]
1.2 Туберкульоз як
причина раку легенів
Встановлено, що
туберкульоз є причиною виникнення пухлин, статистика показує, що у хворих на
туберкульоз рак легені зустрічається частіше. Однак деякі вчені спростовують цю
теорію.
Проблема взаємозв'язку
між раком і туберкульозом легень вже давно привертає увагу, однак, і досі вона
не може вважатися повністю вирішеною. Вже понад двісті років точаться дискусії
про схильність туберкульозу викликати рак, та чи відбувається останній частіше
в осіб з туберкульозом легень в порівнянні з населенням в цілому. [25]
Таким чином,
Рокітанський (1881), Пеарле і співавт. (1929), В. І. Брауде (1968) встановили
існування антагонізму між цими захворюваннями. В. Добринін (I960), Цігельнік та
ін (1968) та інші заперечують зв'язок між ними. Шварца (1956), Барс (1962),
Бельке (1965), Хакл (1967), А. Є. Рабухін (1958, 1962), Хейфец (1962),
Плопський(1968), Раданов (1974) та інші вважають, що наявність туберкульозних
змін бронхогенний рак викликається частіше, в порівнянні з тими, у кого немає
такого ураження дихальних шляхів. Це протиріччя думок пояснюється насамперед
значною неоднорідністю клінічних і патологічних даних.
Частота поєднання
захворювань туберкульозу та раку легені змінюються в залежності від того де
проводилося дослідження та від статевого складу досліджуваних груп. Так у
туберкульозних лікарнях набагато частіше зустрічається рак на фоні
туберкульозу, у чоловіків зустрічається частіше, ніж у жінок.
Крім того, відповідаючи
на питання про частоту поєднання раку легенів і туберкульозу неможливо дати
вичерпну відповідь, якщо брати за основу тільки данні хворих в стаціонарних
лікарнях. Найбільш точну відповідь на це питання можна отримати тільки шляхом
порівняння диспансерного матеріалу з числом пацієнтів з раком легенів в
загальній популяції одного і того ж регіону, міста або країни, з урахуванням
тих, хто помер від хвороб.
При порівняні
інтенсивних показників, встановлено, що серед хворих на туберкульоз легень, в
осіб 40-49 років первинний рак легенів спостерігалася в 4,9 рази частіше, 50-59
років - у 7,5 рази, 60 років і старше - в 6,6 рази частіше. [17]
Епідеміологія поєднаного
туберкульозу і раку легенів
Важливу та досить
складну для лікарів задачу становить диференціювання процесів раку та
туберкульозу. За своїми ознаками ці захворювання дуже схожі. Нерідко рак легені
приймають за туберкульоз. Це пов’язано з неуважним вивченням
клініко-рентгенологічної картини та не використанням сучасних методів
діагностики.
Досить часто ракові
пухлини утворюються на тлі затихаючих туберкульозних процесів. Виникати пухлини
можуть також після втрати туберкульозної активності.
Туберкульозні та
онкологічні процеси в легенях можуть бути поєднані. В цьому випадку, очевидно,
туберкульоз стає благоприємним ґрунтом для утворення пухлини.
Локалізація ракових
утворень відбувається як в зонах специфічних змін, так і поодаль.
Рак легенів, як і туберкульоз
частіше розташований у верхніх відділах легенів, рідше - у середніх та нижніх.
Це зумовлено тим, що при диханні пил, бактерії, канцерогені речовини
потрапляють та осідають саме у верхній частині легені. Одним з найсильніших
чинників, викликаючи і рак, і туберкульоз є куріння.
Встановлено, що хворі на
туберкульоз - це зазвичай чоловіки 25-34 років. Рак легені частіше
зустрічається у чоловіків після 60 років. Поєднанні процеси найчастіше
виникають у чоловіків 50-60 років.
Озираючись на
статистичні данні, ми можемо ще раз засвідчити, що ймовірно, туберкульоз сприяє
розвиненню раку легені. [5]
Види туберкульозу, що
переходять в рак
Найчастіше рак
утворюються на тлі вогнищевого, фіброзно-кавернозного та циротичного
туберкульозу.
Вогнищевий туберкульоз -
це така форма захворювання, яка характеризується обмеженою протяжністю
запального процесу в легенях з переважанням продуктивного характеру запалення в
організмі. Протяжність зазвичай визначається 1-2 сегментом. Якщо брати
рентгенологічні ознаки, то зазвичай береться просторове поле не нижче 2 ребра,
тобто коли вогнищеві зміни в легенях знаходяться у верхніх сегментах. Якщо
вогнищеві зміни поширилися нижче другого ребра-то такий процес називається
дисемінований туберкульоз. [13,15]
Фіброзно-кавернозний
туберкульоз легень - хронічне захворювання, що протікає тривало і хвилеподібно,
з інтервалами затихання запальних явищ. Для нього характерна наявність однієї
або декількох каверн великої давності, з різко вираженим склерозом навколишніх
тканин, фіброзних перероджень легень і плеври. Розрізняють два клінічних
варіанти перебігу фіброзно-кавернозного туберкульозу легень:
) обмежений і відносно
стабільний фіброзно-кавернозний туберкульоз, коли завдяки хіміотерапії наступає
певна стабілізація процесу і загострення може бути відсутнім протягом декількох
років;
) прогресуючий
фіброзно-кавернозний туберкульоз, що характеризується зміною загострень і
ремісій, з різними періодами між ними. [8, 19]
Циротичний туберкульоз -
клінічна форма, яка характеризується розвитком виражених фіброзних змін у
легенях, наявністю емфіземи і бронхоектазів при збереженні
клініко-рентгенологічних проявів активного туберкульозного процесу. Циротичний
туберкульоз розвивається при тривалому перебігу фіброзно-кавернозного,
дисемінованого процесу. Зазначена форма може виникнути в результаті інволюції
поширеного інфільтративного процесу типу Лобіту, через розсмоктування
інфільтративних змін і розростання сполучної тканини, а також на ґрунті
ателектазу при наявності в легеневої тканини туберкульозних змін. 21]
Патогенез взаємодії
туберкульозу і раку легені
Проблема раннього
виявлення раку легені, що розвивається на тлі туберкульозного процесу, в даний
час є актуальною і складною. Це обумовлено як надзвичайною різноманітністю
клінічних варіантів перебігу цих захворювань, так і схожістю ряду клінічних
симптомних даних інструментальних методів обстеження. А.Є. Рабухін виділяв 4
типи проявів даної поєднаної патології:тип: поява в зоні стаціонарних, або
регресуючих, туберкульозних змін нової одиночної ізольованою великовогнищевої
або фокусної тіні неправильної округлої форми;тип: виникнення в зоні активного
туберкульозного процесу або поза ним на незмінній ділянці легені тіні округлої
форми або ділянки апневматозу, яка, незважаючи на протитуберкульозну хіміотерапію,
не зменшується в розмірах, хоча туберкульозні зміни при цьому регресують;тип:
приєднання до неактивних, метатуберкульозних змін або активного туберкульозу
пневмоніту, гіпопневматозу або ателектазу сегмента, частки або всього легені;
наростаючих перибронхіальних, інтерстиціальних ущільнень, що виходять з кореня
легені; одностороннього збільшення і ущільнення кореня легені, головним чином,
за рахунок внутрішньогрудних лімфатичних вузлів;тип: поява вираженого
асиметричного потовщення стінки туберкульозної каверни з поліпоподібні
горбистими розростаннями в просвіті або в перикавернозній зоні в відсутність
вираженого перифокального запалення і дисемінації. [3]
Аналіз спеціальної
літератури показав, що зазначена у більшості хворих клініко-рентгенологічна симптоматологія
легеневого процесу часто витлумачується неправильно [4,6,8]. За даними медичної
літератури, поєднання раку легень з туберкульозом має значну кількість «Масок»
[2,6]. На думку деяких авторів, найпоширенішими з них є пневмонічні. Плевритична
«маска» поєднання туберкульозу з раком легенів нерідко служить перегородою для
постановки правильного діагнозу. У хворого на туберкульоз похилого віку
розвиток ексудативного плевриту повинно викликати підозру на бронхокарциному.
«Маскою» бронхогенного раку у хворого на туберкульоз легень може бути
адгезивний процес в плевральній порожнині.
Діагностика раку при
туберкульозі
Рентгенологічні
дослідження часто не дають можливість встановити наявність пухлинного процесу,
і таким пацієнтам ставлять діагноз - туберкульоз. При гістологічному
дослідженні важливо взяти тканини з декількох ділянок легені, так як не завжди
вдається взяти пухлинні клітини, залежно від локалізації пухлинного процесу.
[11]
Далеко не завжди
вдається швидко встановити діагноз на підставі ознак, отриманих за допомогою
загальних методів, якщо не виявлено специфічні ознаки якогось певного
захворювання. У таких випадках необхідна диференціальна діагностика, яку
проводять в наступній послідовності.
. Оцінка виявлених ознак
захворювання і виділення найбільш важливої інформації про хворого з точки зору
її достовірності, інформативності і специфічності.
. Виділення
симптомокомплексу, що складається з достовірних, інформативних і, по
можливості, специфічних ознак. Симптомокомплекс може бути розширеним, якщо
включає велику кількість ознак (частіше спостерігається при недостатній
специфічності ознак), і звуженим при наявності кількості ознак, з яких один або
кілька високоспецифічні для певного захворювання.
. Складання переліку
захворювань, які мають подібні симптоми, з якими необхідно диференціювати
наявне у хворого захворювання, і побудова «моделі» альтернативних, тобто
взаємовиключних, симптомокомплексом,
. Зіставлення
симптомокомплексу, виявленого у хворого, з альтернативними симптомокомплексами
шляхом порівняння наявних і відсутніх ознак, що входять у симптомокомплекс. При
цьому вирішальне значення мають ознаки, найбільш специфічні для певного
захворювання: наявність одного або декількох таких ознак в симптомокомплекс,
характерному для того чи іншого захворювання, дозволяє встановити діагноз.
Для проведення
диференційної діагностики туберкульозу та інших захворювань легенів в клінічній
практиці використовують методику цитологічного дослідження мокротиння.
У хворих (особливо при
активному процесі) мокрота гнійно-слизиста або слизово-гнійна.
При цитологічному
дослідженні мазків вельми невелика кількість нейтрофілів виявляється в стадії
вираженої дегенерації на тлі детриту типу казеозного, що має вигляд крупинок і
пофарбованого в темно-фіолетовий колір. Характерні також скупчення
моноцитоїдного мононуклеарів, крупних клітин з ядром неправильної форми і
блідо-блакитний протоплазмою. Серед них відзначаються форми, перехідні до
епітеліоїдних. [23]
Елементи специфічного
запалення в мокроті виявляються в 14-32% спостережень. Нерідко при розпаді
звапніння вогнищ у легені можна виявити аморфні фосфати у вигляді кристалів
жовтуватого кольору різної величини. Враховуючи диференційно-діагностичне
значення аналізу мокроти, слід зазначити, що цитологія мокроти має істотне
значення в діагностиці бластоматозних процесів, деяких дисемінованих уражень
легенів, симулюють туберкульоз (як канцероматоз), медіастинальну легеневу формі
лімфогранулематозу, гемосидероз та ін.
Бронхологічне
дослідження - бронхоскопія є обов'язковим методом у діагностиці раку легені.
Бронхоскопія дозволяє виявити навіть початкові форми ендобронхіального раку і
верифікувати діагноз за допомогою щіткової або щипцевої біопсії та дослідження
промивних вод бронхів. Ендоскопічне дослідження необхідно виконувати усім
хворим з підозрою на рак легені, при наявності кашлю, ознак бронхіальної
обструкції, а тим більше - при кровохарканні. Флуоресцентна бронхоскопія з
використанням гематопорфірину, який вибірково накопичується в пухлині, сприяє
підвищенню ефективності ранньої діагностики ендобронхіального раку.
Ендоскопічна стравохідна
та ендобронхіальна сонографія застосовується для оцінки стану різних груп
медіастинальних лімфатичних вузлів, точність методу складає 93% і специфічність
- 94 - 100%.
Трансторакальна пункційна
біопсія застосовується для діагностики периферичних новоутворень, розташованих
у периферичних відділах легень. При аспіраційній біопсії матеріал досліджується
цитологічно, при трепан-біопсії - гістологічно. З найбільш серйозних ускладнень
методу треба відзначити кровохаркання, пневмоторакс і повітряну емболію. [14]
Плевральна пункція з
подальшим цитологічним дослідженням застосовується при наявності гідротораксу і
є обов'язковою навіть за мінімальної кількості вільної або осумкованої рідини у
плевральній порожнині. Одержана рідина досліджується на наявність пухлинних
клітин, мікобактерій туберкульозу, дріжджових грибків, визначається наявність
формених елементів крові і кількість білка. Після дослідження необхідне
спостереження за пацієнтом протягом 2-х годин з метою своєчасної діагностики
пневмотораксу.
Медіастиноскопія
виконується в умовах операційної під загальним знеболенням. Дослідження
показане для верифікації метастатичного ураження медіастинальних,
паратрахеальних і біфуркаційних лімфовузлів
Торакоскопія
(відеоторакоскопія) також виконується в умовах операційної під загальним
знеболенням. Через уведений в плевральну порожнину торакоскоп ретельно
оглядається легеня і плевра, виконується біопсія ділянок плеври, легені і
лімфовузлів, що викликають підозру.
З інших інструментальних
методів дослідження в діагностиці раку легені застосовуються трансбронхіальна
пункція лімфатичних вузлів (під час бронхоскопії), біопсія надключичних
лімфовузлів, радіоізотопне сканування скелета (для виявлення кісткових метастазів),
цитологічне дослідження мокроти.
Дослідження існуючих на
сьогоднішній день пухлинних маркерів щодо раку легені не є ефективним і
діагностичної цінності поки ще не становить.
2.
Молекулярно-генетичний механізм утворення пухлин легенів
2.1 Генетичні порушення
у клітинах
Рак легені і, зокрема,
його недрібноклітинний варіант, є основною причиною смерті від злоякісних
новоутворень. «Епідемія» цього захворювання обумовлена повсюдним поширенням
куріння. У переважної більшості пацієнтів хвороба діагностується в пізніх
стадіях, коли виконання радикальної операції вже неможливо, а сучасні променева
терапія і хіміотерапія мають лише паліативне значення. [16]
Таблиця 2.1. Генетичні
порушення в клітинах НДРЛ
|
Ген
|
Білок, що кодується, та його
функція
|
Характер генетичних порушень
|
Частота порушень %
|
Тирозинкіназний рецептор ЕGF
|
Гіперекспресія
|
>50
|
|
ЕRВВ2 (НЕR2/neu)
|
р 185 тирозинкіназний рецептор
|
Ампліфікація
|
25-30
|
|
RAS
|
Ras переносник сигналу
|
Мутація
|
20-30
|
|
МYC
|
Мус транскрипції, регулює
клітинний цикл та активність теломерази
|
Гіперекспресія Ампліфікація
|
50
|
|
РRAD1
|
Циклін Д1 в комплексі 3
циклінзалежною кіназою 4 стимулює перехід клітини із стану G0 у фазу G1
клітинного розподілу
|
Гіперекспресія Ампліфікація
|
47
|
|
RB
|
рRb - контролює вхід у 8-фазу,
регулюючи активність фактора транскрипції Е2F
|
Мутація
|
25
|
|
CDKN2
|
P16nk4 - інгібітор комплексу
циклін Д1 і циклінзалежної кінази 4, попереджає початок клітинної
проліфарації
|
Мутація
|
10-40
|
|
р53
|
р53 - транскрипційний фактор,
регулює клітинний цикл і апоптоз, контролює цілісність геному
|
Мутація
|
50
|
2.2 Передача сигналу
В кінці 70-х років M.B.
Sporn і G.J. Todaro визначили, що в патогенезі та прогресії багатьох злоякісних
пухлин, у тому числі і раку легені, важливу роль відіграють механізми
паракринної та аутокринної стимуляції [21]. При паракринній стимуляції
нормальні клітини, що оточують пухлину, або самі пухлинні клітини продукують
фактори росту або пептиди, стимулюючі проліферацію сусідніх пухлинних клітин.
При аутокринному механізмі самі пухлинні клітини продукують фактори росту, які
змушують їх безперервно ділитися. При НДРЛ такими чинниками є епідермальний
фактор росту (EGF), нейрорегулін, інсуліноподібний фактор росту 1 (IGF1) і
фактор росту гепатоцитів (HGF) [20].
Всі ці фактори росту
взаємодіють зі специфічно рецепторами, експресованими на поверхні пухлинної
клітини. Рецептори більшості факторів зростання є трансмембранними,
екстрацелюлярна їх частина безпосередньо пов'язує фактор росту, а
цитоплазматична частина представлена ферментом тірозінкіназою. Після зв'язування
ліганду відбувається димеризація рецептора, активація внутрішньої
тирозінкінази, її аутофосфорилювання і подальше фосфорилювання.
Молекули-переносники сигналу передають проліферативний сигнал по сигнальній
ланцюга факторів транскрипції, які ініціюють транскрипцію окремих генів з
подальшою продукцією відповідного білка (трансляція), стимулюючого клітинну
проліферацію.
Всі гени пухлинної
клітини можна розділити на дві категорії: онкогени, активація яких призводить
або сприяє проліферації клітини і метастазування пухлини; гени-супресори,
інгібуючі процес проліферації, стимулюючі диференціацію і апоптоз. У геномі
пухлинної клітини міститься пошкоджена генетична інформація (мутації,
неправильний обмін хромосомами, генетичною інформацією), в результаті якої відбувається
вимикання роботи або надмірна активація того чи іншого гена. В результаті таких
ушкоджень у пухлинних клітинах спостерігається активація онкогенів, що
стимулюють процес клітинного поділу, освіти пухлини та її метастазів. І,
навпаки, часто відбувається інгібування або повне випадання функції
генів-супресорів, які могли б перешкодить безконтрольній клітинній
проліферації. Відповідні білки, що кодуються визначеними генами, діляться на
онкобілки, що запускають процес клітинного поділу, стимулюючі розплавління
матриксу та утворення нових судин (ангіогенез), і білки-супресори, які
відповідають за інгібування клітинного поділу, запуск процесу диференціювання
та апоптозу. Ці білки можуть бути факторами росту або рецепторами до них,
передатчиками сигналів, факторами транскрипції, ферментами, відповідальними за
підтримання просторової структури ДНК, і т.д.
При НДРЛ виявлені різні
генетичні порушення на всіх етапах передачі внутрішньоклітинного сигналу.
По-перше, клітини НДРЛ експресують значно більше число рецепторів EGF, ніж
нормальні клітини епітелію бронхів, особливо при плоскоклітинному варіанті [5].
При аденокарциномі спостерігається підвищений вміст на мембрані пухлинної
клітини іншого рецептора з сімейства рецепторов EGF - p185. У цих клітинах відзначається
підвищення ефективна експресія гена erbB2 (HER2/neu), кодуючого цей рецептор,
що корелює з короткою тривалістю життя і є маркером лікарської резистентності
[24]. Після зв'язування фактора росту (або ліганду) відбувається димеризація
рецептора і активація плазматичної частини рецептора, представленої
тирозинкінази.
Сигнал від рецепторів
EGF і р185 передається на молекулу-переносник сигналу сімейства Ras. При НДРЛ у
20 - 30% хворих є точкова мутація гена RAS, в результаті якої білок Ras втрачає
здатність втрачати фосфатну групу й постійно перебуває в активованому стані,
імітуючи і передаючи стимулюючі до проліферації сигнали [20]. Вважається, що
мутація гена Ras виникає внаслідок впливу нітрозоамінів тютюнового диму.
Проліферативний сигнал від білка Ras проходить через ланцюг
молекул-передатчиків сигналів (МАР - кіназний каскад), щоб досягнути фактора
транскрипції Myc, кодованого геном MYC. У 50% хворих НДРЛ відзначається
гіперекспресія або ампліфікація гена MYC, що призводить до транскрипціі генів,
що стимулюють проліферацію пухлинних клітин [20].
Таким чином, сигнальний
ланцюг рецепторів EGF і erbB2, який передає проліферативний сигнал з
зовнішнього середовища до ядра пухлин клітини НДРЛ, має цілий ряд генетичних
порушень, які можуть бути мішенями протипухлинної терапії.
2.3 Клітинний цикл
Підсумком передачі
проліферативного сигналу є вступ пухлинної клітини в процес клітинного циклу,
що закінчується поділом і утворенням двох дочірніх клітин. Клітинний цикл є
строго регульованим процесом. Він стимулюється на різних етапах комплексом
циклінів і циклінзалежних кіназ [1]. Виявилося, що в клітинах НДРЛ концентрація
циклін Д1 (відповідального за входження клітини в клітинний цикл) підвищена в 5
- 100 разів в порівнянні з нормальними клітинами [19]. Це відбувається за
рахунок ампліфікації або гіперекспресії гена PRAD1, що кодує цикліну Д1, у 25%
і 47% хворих НДРЛ відповідно [3]. Підвищена продукція циклін Д1 сприяє
ініціації клітинного поділу. Найважливішим білком, який гальмує входження
клітини в процес розподілу, є білок ретінобластоми (Rb). Білок Rb пов'язує
фактор транскрипції E2F. Комплекс циклін Д1 і ціклінзалежної кінази 4
фосфорилюється білок Rb, в результаті чого відбувається розрив зв'язку з
чинником транскрипції E2F. E2F, будучи активним, запускає транскрипцію різних
генів і наступний синтез білків, ферментів і їх інгібіторів, необхідних для
переходу клітини з фази G1 у фазу S [1]. Втрата однієї копії і точкова мутація
залишилася копії гена-супресора RB, кодуючого білок Rb, зафіксована у 15 - 30%
хворих НДРЛ [15]. Білок p16INK4 пригнічує активність комплексу циклін Д1 і
ціклінзалежної кінази 4, запобігаючи початок клітинної проліферації. У 10 - 40%
хворих НДРЛ відзначаються делеція і точкова мутація залишилася копії гена
CDKN2, що кодує p16INK4, а у 70% хворих - відсутність експресії гена [20].
Ключовим регулятором
клітинного циклу і процесу природної загибелі клітини (апоптозу) є білок р53.
Ген р53 отримав назву «страж генома» завдяки своєї центральної ролі в підтримці
стабільності геному багатоклітинного організму і запобігання подвоєння
пошкодженої ДНК. р53 блокує процес розподілу клітки в разі пошкодження її ДНК
тимчасово для репарації або повністю, якщо ці пошкодження неможливо усунути. В
останньому випадку p53 запускає механізм апоптозу [2]. Білок р53 є чинником
транскрипції і регулює активність великої кількості генів, які беруть участь у
гальмуванні клітинного циклу, активації апоптозу та пригніченні ангіогенезу.
Зокрема, білок р53 є фактором транскрипції гена р21, найважливішого інгібітору
ціклінзалежних кіназ, і запускає виробництво білка р21 для зупинки клітинного
поділу в разі пошкодження ДНК. р53 є центральним компонентом механізму,
забезпечує видалення з організму патологічно змінених клітин. Цей механізм
повністю відповідає у 50% хворих НДРЛ у зв'язку з делецією однієї та мутацією
інший копії гена р53 [9]. Таким чином, генетичні зміни в клітинах пухлини НДРЛ
призводять до істотної втрати контролю за ключовими етапами клітинного циклу і,
як наслідок, за контролем проліферації пухлинної клітини. [28]
.4 Канцерогенні фактори
Канцерогенами можуть
виступати різні чинники. За своїм походженням вони розділяються на хімічні,
біологічні та фізичні.
Хімічні канцерогени. У
природі існує кілька мільйонів природних і синтезованих людиною хімічних речовин
і з'єднань. Людина активно контактує з десятками тисяч. Серед безлічі хімічних
агентів безсумнівно канцерогенними визнані кілька десятків. Вони присутні в
навколишньому середовищі, виділяються в процесі промислового виробництва або є
продуктами життєдіяльності живих організмів. Хімічні канцерогени можуть
надавати свою дію самі по собі (так звані прямі канцерогени) або потребують для
цього в активації (це відбувається в процесі обміну речовин в організмі
людини). [17]
Фізичні канцерогени. Це
агенти фізичної природи. Найбільш широка група їх відноситься до різних видів
іонізуючого випромінювання - рентгенівські промені, гамма промені, різні
елементарні частинки атома - протони, нейтрони, альфа і бета частинки). Фізичні
канцерогени так само є компонентом природного середовища або є продуктом
життєдіяльності людини. У деяких випадках постійне механічне травмування тканин
людини теж може сприяти розвитку злоякісної пухлини.
Біологічні канцерогени.
На початку 20 століття активно розвивалася і пропагувалася інфекційна теорія
розвитку злоякісних новоутворень, яка в той час була відкинута. У другій
половині 20 століття з розвитком медичної та мікробіологічної науки до цієї
проблеми повернулися знову. Результатом досліджень було відкриття декількох
вірусів, здатних прямо чи опосередковано викликати виникнення злоякісних пухлин
як у тварин, так і у людини. Далеко не всі ракові захворювання викликані
вірусами. Але зв'язок їх з деякими формами пухлин безсумнівна. [18,19]
Розрізняють 2 основні
групи причин, що сприяють розвитку раку легені. Перша - це ті канцерогенні
(викликають рак) речовини, які потрапляють в тканини бронхів та легенів з
зовнішнього середовища з диханням. Друга - ця велика група хронічних запальних
захворювань легенів.
Найважливіша причина
раку легені - це куріння. Приблизно 90 відсотків усіх випадків цього
захворювання розвивається у курців. Постійне багаторічне вдихання продуктів
тління тютюну (а вони містять цілий букет канцерогенних смол, газів і інших
речовин) викликає порушення у функціонуванні клітин слизової оболонки бронхів і
альвеол і веде до метаплазії (перебудові) цих клітин. Крім того, висока
температура вдихуваного тютюнового диму також веде до постійного подразнення
бронхів і тканини легень. У результаті порушуються процеси нормального ділення
клітин і у багато разів підвищується ймовірність захворіти на рак. Вірогідність
захворіти безпосередньо залежить від стажу куріння, кількості викурених сигарет
на добу і якості тютюну. Особи, що викурюють дві і більше пачок сигарет на
день, занедужують в 15-25 разів частіше некурців. Найбільшому ризику піддаються
курці, що вживають дешеві сорти тютюну без фільтру. Стаж куріння теж відіграє
величезну роль і чим він вищий, тим шанси отримати рак легені більше. Але деякі
дослідження показували, що навіть невеликий стаж уже підвищує ці шанси -
захворюваність серед курців і некурців була однаковою всього лише до 26-28
років, після чого курці починали хворіти частіше. Тютюновий дим небезпечний не
тільки для кращого, але і для його оточення. Регулярне «пасивне паління» має
абсолютно таку ж дію, як і безпосереднє тютюнопаління та підвищує ризик
захворіти на рак легені в 1,5 - 2 рази.
Підвищують ризик
захворіти раком легені і професійні шкідливості. В першу чергу це виробництва,
пов'язані з використання миш'яку, хрому, нікелю і азбесту (особливо азбестовий
пил), важких металів і хлорметіловим ефіром.
У виникненні пухлини
відіграють роль і хронічний запальні процеси в тканині легенів і бронхів, а
також захворювання, що супроводжуються ущільненням і перебудовою нормальної
тканини. Рубцеві зміни після перенесеного раніше туберкульозу, вогнища
пневмосклерозу (розростання сполучної тканини), бронхоектатична хвороба
(розширення стінок бронхів у вигляді мішечків) також підвищують вірогідність
захворювання раком легені. [22]
2.5 Стадії формування
пухлини
Перетворення нормальної
клітини в трансформовану - процес багатостадійний.
. Ініціація. Майже кожна
пухлина починається зі зміни ДНК в окремій клітці. Цей генетичний дефект може
бути викликаний канцерогенами, наприклад канцерогенними речовинами (зокрема,
компонентами тютюнового диму), фізичними факторами (УФ-випромінювання,
рентгенівські промені) або онкогенними вірусами. По-видимому, протягом
людського життя чимале число клітин організму із загального їх числа зазнає
пошкодження ДНК. Однак для ініціації пухлини важлива лише активація
протоонкогенів. Ці ушкодження є найбільш важливим фактором, що визначає
трансформацію соматичної клітини в пухлинну. До ініціації пухлини може
призвести і пошкодження антіонкогена (гена-онкосупресора). [25]
2. Промоція пухлини - це
переважне розмноження генетично змінених клітин. Такий процес може тривати
роками.
. Прогресія пухлини - це
процеси розмноження малігнізованих клітин, інвазії та метастазування, провідні
до прояву злоякісної пухлини.
Вивчення злоякісних
пухлин показує, що на початкових етапах вони ростуть і розвиваються повільно.
Найбільша зустрічальність різних форм раку відзначається після 40 років. Можна
припустити, що подібне явище не залежить від збільшення з віком частоти мутацій,
так як у міру старіння швидкість ділення клітин зменшується, ймовірність
подібних помилок падає. Є припущення, що з віком знижується ефективність
механізмів, що руйнують мутаційно змінені клітини. При цьому треба також
враховувати і супутнє зменшення кількості стовбурових клітин, зниження
ефективності фізіологічного поновлення - регенерації тканин, роль якого в
процесі виникнення пухлин не ясна. [26,27]
Отже, існує «прихований
період» розвитку пухлини, який охоплює проміжок часу між ініціюванням
канцерогенного процесу і клінічним виявленням пухлини. Послідовні «багаторазові
поштовхи», які є ступенями канцерогенезу, відбуваються протягом першої частини
«прихованого періоду». Він може тривати від декількох років до 3 або більше
десятиріч, в результаті чого утворюється перша неопластична - злоякісна
клітина. Другий період триває ще декілька місяців або років, складаючи останній
етап прихованого періоду. У цей час відбуваються мітози нащадків цієї клітини і
утвориться клінічно виявлене новоутворення (злоякісний клон, що складається
приблизно з 109 клітин).
Встановлено, що пухлина,
що розвивається (імовірно) з однієї первинної клітини діаметром 10 мікрон за
час, необхідний для тридцяти подвоєнь (розподілів), досягає умовно
диагностуємого розміру, рівного 1 см (злоякісний клон, що складається приблизно
з 109 клітин), - це той розмір, коли пухлина доступна для клінічної
діагностики. Прийнято вважати, що при вазі злоякісної пухлини 1,5 кг
відбувається загибель хворого. До цього часу здійснюється 40 подвоєнь (зазвичай
максимальний час існування пухлини). Тобто до появи перших ознак пухлина
проживає більшу частину часу - ѕ періоду існування системи людина - злоякісна
пухлина.
Темпи подвоєння різних
видів пухлини різні і залежать від цілого ряду факторів, індивідуальних особливостей
пухлини і організму, в тому числі: локалізації, виду пухлини, стану імунітету
та психіки (стресові ситуації), гормонального статусу (наприклад, в період
вагітності), споживання необхідних харчових і протипухлинних інгредієнтів. [30]
2.6 Імунологічні
особливості
Важливим науковим
досягненням стало розуміння того, що пухлинні клітини можуть індукувати
імунітет. Введені в здоровий організм, вони є вакциною - але тільки проти тієї
самої пухлини, з якої вони були виділені. Можна сказати, що практично неможливо
підібрати з величезної безлічі пухлин дві з однаковими антигенів. Передбачити,
яка саме пухлина може розвинутися у людини, і передбачити її антигенні
характеристики неможливо. Протективний (захищає) імунітет розвивається тільки
після профілактичної імунізації (до виникнення пухлини, і така імунізація поки
можлива тільки в умовах експерименту, коли здоровій тварині пересаджують
злоякісну пухлину від хворого); якщо імунізувати після виникнення пухлини
(терапевтична вакцинація), використовуючи клітини, отримані з цієї ж пухлини,
перешкодити її подальшому зростанню, на жаль, не вдасться. [29]
Незважаючи на чітко
встановлені дані про існування пухлинноспецифічних антигенів і викликаний ними
імунної відповіді, пухлини, як правило, продовжують своє зростання.
У 1908 р. Пауль Ерліх
знайшов наступний парадоксальний факт. Тварини, у яких вже існувала пухлина,
виявилися дивно стійкими до імплантації (пересадці) другий пухлини - навіть у
тому випадку, якщо перша продовжувала зростати. Цей феномен отримав назву
«супутній імунітет» і до теперішнього часу не знайшов повного пояснення. [11]
Супутній імунітет
відображає два важливих аспекти протипухлинного імунітету:
) зростаюча первинна
пухлина викликає захисну протипухлинну імунну відповідь;
) ця відповідь достатня
для елімінації вторинної пухлини, але впливу на пухлину, яка її індукує, не
надає.
Був проведений простий
експеримент: досвідченої миші від миші, ураженої злоякісною пухлиною,
пересадили клапоть шкіри і фрагмент пухлини. Незважаючи на те що в тому і іншому
випадку був ллогенних (від іншої тварини) трансплантат, шкірний клапоть
відірвав, а пухлина почала своє зростання. Це дозволило зробити найважливіший
висновок: пухлина володіє якимись особливими властивостями, які допомагають їй
уникнути нормальної алоспеціфічної реакції відторгнення. Був також зроблений
найважливіший висновок про існування кардинальних відмінностей імунного статусу
організму до і після розвитку пухлини..A. Janeway і P. Trovers показали, що
варіанти пухлинних клітин відбираються в організмі, перш за все, по безлічі
пов'язаних з виживанням in vivo (всередині людини) властивостей, у тому числі
найважливішої властивості пухлинних клітин - їх імунодепресивної здібності,
здійснюваної шляхом захисту себе від ефекторних систем господаря, неспецифічно
розпізнаючих чуже за допомогою макрофагів, нейтрофілів і натуральних кілерів
(NK), шляхом секреції антигенів, які пригнічують специфічний імунна відповідь.
Такі пухлинні клітини не експресують антигени тканинної сумісності 1-го класу,
необхідні для розпізнавання їх цитотоксичними CD8 Т-лімфоцитами. Відомий
феномен «вислизання» (escape) полягає в постійній мутації пухлинних антигенів,
коли в результаті відбору зберігаються тільки ті пухлинні клітини, які здатні
змінюватися постійно з великою швидкістю, випереджаючи реакції системи
імунітету. Генетична нестабільність пухлинних клітин, їх гетерогенність
забезпечують раку надзвичайну життєстійкість. Частота генетичних змін до швидко
метастазують пухлинах в 5-7 разів вище, ніж у неметастазуючих. [18]
Крім того, J. Bradley і
J. McClaskey до феномену «вислизання» відносять:
• маскування пухлинних
антигенів надлишком антитіл або імунних комплексів, що виробляються імунною
системою хазяїна пухлини;
• зниження «рівня
презентації» пухлинних антигенів макрофагами, що призводить до пригнічення
продукції протипухлинних цитокінів (ФНП, інтерферони і т.д.) макрофагами.
Ще в 50-і роки був
описаний феномен посилення пухлинного росту (enhancing), який полягає у захисті
антитілами господаря пухлинних клітин від дії імунних Т-лімфоцитів. В
результаті такого відбору вижили варіанти пухлинних клітин, як правило, більш
резистентні до цитотоксичної дії активованих макрофагів, NK-клітин і
Т-лімфоцитів. Пухлинні клітини набувають селективні переваги in vivo,
розвиваючи і використовуючи для свого захисту проти цих ефектів цілком
адекватні і специфічні механізми (наприклад, посилений катаболізм перекису
водню у відповідь на його продукцію активованими макрофагами і нейтрофілами, а
також викид імуносупресорів ПГЕ2 при контакті з NK-клітинами і макрофагами).
Сьогодні ні в кого не
викликає сумнівів той факт, що імунні механізми відіграють в організмі важливу
роль задовго до появи клінічно визначеної пухлини.
З'ясовано, що пухлина
формується і зростає під впливом протилежно спрямованих, але не взаємовиключних
імунних реакцій. Динаміка пухлинного росту визначається рівновагою між
факторами імунного нагляду, з одного боку, і пробластомними факторами тієї ж
власної імунної системи, що сприяють росту пухлини, - з іншого.
В даний час розрізняють
три групи факторів, що беруть участь у розвитку пухлини:
) антибластомні
(протипухлинні - вроджені і набуті імунологічні механізми знищення пухлинних
клітин в організмі людини);
) імунорезстентності
(стійкість пухлини, її здатність уникати або відбивати атаки імунної системи
людини-господаря пухлини);
) пробластомні
(здатності імунної системи підсилювати зростання пухлини за рахунок: а) оберігання
зростання злоякісної пухлини, при виборчому придушенні протипухлинного
імунітету; б) механізмів стимуляції росту пухлини).
Імунна система реагує на
появу пухлинної тканини і розвиває нормальний імунну відповідь, спрямований на
знищення пухлини за рахунок роботи компонентів (факторів) протипухлинної
захисту.
Але розвивається пухлина
постійно вислизає від імунного нагляду за рахунок факторів імунорезстентності
пухлини. [4]
Імунорезистентність
пухлини забезпечується:
) слабкою імуногенністю
пухлинних антигенів;
) постійної модифікацією
антигенів пухлини;
) селекцією імунологічно
стійких клітин;
) втратою експресії
антигенів системи HLA класу I;
) виділенням розчинних
пухлинних антигенів;
) експресією на поверхні
пухлинних клітин і викидом в міжклітинний простір рецепторів до різних
цитокінам;
) придбанням стійкості
до запрограмованої клітинної загибелі за рахунок: втрати рецептора до ФНП,
появи на мембрані молекули FasL;
) продукцією пухлинними
клітинами ІЛ-6, ІЛ-10, ФНП і т.д.
Більш того, на певному
етапі розвитку пухлини імунна система господаря починає виділяти пробластомні
(сприяють росту пухлини) фактори, які: а) пригнічують імунітет, б) сприяють
посиленню росту пухлини. [24]
Зазвичай пухлина виникає
в тих ділянках тканин і органів, де в ході регенерації найбільш інтенсивно йде
розмноження клітин - в так званих проліферативних центрах зростання. Тут
зустрічаються менш диференційовані клітини (клітини-попередники) і частіше
з'являються умови для розвитку клітинної дисплазії з подальшою трансформацією в
пухлину. Такі центри спостерігаються в периваскулярній тканини, в базальної
зоні багатошарового плоского епітелію, в криптах слизових оболонок.
Формування пухлини, або
перехід передпухлинних змін в пухлину, вивчено недостатньо. На підставі
експериментальних даних можна припустити наступну схему розвитку пухлини:
• порушення
регенераторного процесу;
• передпухлинні зміни,
що характеризуються гіперплазією і дисплазією;
• виникаюча стадійно
малігнізація проліферуючих клітин;
• виникнення пухлинного
зачатка;
• прогресія пухлини. [7]
3. Вплив
протитуберкульозних препаратів на життєдіяльність проліферуючих клітин
3.1 Проліферація клітин
і апоптоз
Кількість клітин в
тканини регулюється двома процесами - проліферацією клітин і «програмованою,
або фізіологічною, загибеллю клітин» (апоптозом). Обидва процеси в організмі
перебувають під контролем стимулюючих або інгібуючих факторів, які присутні в
розчинній формі або експресуються на поверхні сусідніх клітин.
Апоптоз - генетично
запрограмована загибель клітин, яка призводить до «акуратного» розбирання та
видалення клітин. Морфологічними ознаками цього активного процесу є зміни
клітинної мембрани («відшнурування» бульбашок, так званих апоптотичних тілець),
розпад клітинного ядра, ущільнення хроматину та фрагментація ДНК. Клітини, які
зазнали апоптозу, розпізнаються макрофагами і іншими фагоцитуючими клітинами і
швидко елімінуються Дуже важливо те, що при апоптозі не розвивається запальний
процес. Інший вид загибелі клітин, некроз, відрізняється від апоптозу тим, що
він розвивається в результаті пошкодження клітинної мембрани хімічними агентами
чи фізичними факторами. При некрозі пошкоджені клітини набухають, а потім
лізуються; при цьому часто розвивається запальний процес. За допомогою апоптозу
здійснюється регуляція об'єму або, точніше, кількості клітин в тій чи іншій
тканині. Особливо це стосується швидко проліферуючих клітин, таких, як клітини
кровотворної системи або гепатоцити печінки. За допомогою апоптозу організм
позбавляється від непотрібних, або «відпрацьованих», клітин, наприклад під час
ембріонального розвитку, при формуванні нервової системи і при імунній
відповіді. Шляхом апоптозу елімінуються трансформовані клітини, наприклад при
канцерогенної дегенерації, вірусної інфекції або необоротному пошкодженні ДНК в
разі опромінення. Прикладом апоптозу є лущення шкіри при сонячній засмазі.
[1,5]
Регуляція апоптозу
Апоптоз запускається
зовнішніми сигналами, які використовують різні сигнальні шляхи, більшість цих
шляхів дійсно запускають апоптоз, однак деякі шляхи його блокують.
Фактор некрозу пухлин
(б-ФНП (TNFб)) зв'язується з ФНП-рецептором першого типу і запускає апоптоз.
Центральне місце в регуляції апоптозу належить цистеїновихм протеїназам,
спорідненим інтерлейкін-1в-конвертазі (ІК). Припускають, що активація цих протеїназ
через ФНП-рецептор відбувається як багатоступеневий процес білок-білкового
взаємодії. Протеїнази специфічним чином розщеплюють полі - (АДФ-рибоза) -
полімеразу (ПАРП), білки sn-рібонуклеопротеідних комплексів, ламін (білок
ядерної мембрани) і інші білки. Ці змінені за рахунок протеолізу білки
запускають процес апоптозу.
Аналогічним шляхом
реалізується сигнал від Fas-ліганда, білка клітинної мембрани сусідніх клітин.
Fas-ліганд у вигляді тримера зв'язується з Fas-рецептором. Потім, по аналогії з
ФНП-рецептором, сигнал передається на цистеінові протеінази. Для ФНП-та
Fas-специфічних рецепторів характерно, що вони активуються шляхом утворення
олігомерів.
Джерелом сигналу може
бути і клітинне ядро. Так, білок р53, продукт онкосупресорного гена, який теж
активує цистеінові протеінази, може бути активований за допомогою
нерепарабельного розриву ДНК (DNA). Втрата клітиною білка p53 веде до
підвищеної швидкості росту пухлини.
Сигналам, які активують
апоптоз, протистоять інші сигнали, що блокують апоптоз. Таким сигналом може
бути білок bcl-2 або споріднені білки. Ген цього білка присутній в геномі
деяких вірусів. За допомогою продукту цього гена віруси перешкоджають
передчасної загибелі клітини-хазяїна за допомогою апоптозу. [1,32]
Ультраструтурні ознаки
апоптозу
Клітини в стані апоптозу
мають ультраструктурні характеристики.
● Відсутні
спеціалізовані структури клітинної поверхні (мікроворсинок, міжклітинні
контакти). На відміну від некрозу йдеться завжди про зміни в окремих клітках.
● Зменшення
розмірів клітини у зв'язку з конденсацією цитоплазматичних органел, зміни форми
клітини. Часто клітка розщеплюється на кілька апоптозних тілець, кожне з яких
має свій фрагмент ядра, обмежений двоконтурною ядерною мембраною, і
індивідуальний набір органел.
● Збереження та
інтегративність органел, насамперед мітохондрій. При цьому є місце аггрегації
рибосом в навпілкристалоїдні структури, поява пучків мікрофіламентів під
цитолемой, розташованих паралельно мембрані. Майже завжди спостерігається
короткочасна дилатація гладкого ЕПР з формуванням пухирів, наповнених рідиною,
яка виводиться з клітини. При скануючий електронній мікросокпії поверхня клітки
здобуває кратерообразной впячування.
● Конденсація
ядерного хроматину під каріолемою у вигляді півсфер і грудочок. У ядрі виявляються
осміофільні тільця, сформовані транскрипційними комплексами, які надходять з
ядерець. Ядро змінює свої розміри, стає смугастим, фрагментується, ядерні пори
концентруються тільки в місці, де не відбувається маргінація хроматину.
● Клітина в стані
апоптозу стає об'єктом фагоцитозу для сусідніх парнехіматозних і стромальних
клітин і, перш за все, для макрофагів. Фагоцитоз відбувається настільки швидко,
що в умовах in vivo апоптозні клітини зберігаються лише протягом пари хвилин,
що ускладнює їх контроль і вивчення. [34]
Проліферація
Це новоутворення клітин
і внутрішньоклітинних структур (мітохондрій, ендоплазматичної сітки, рибосом та
ін.) Лежить в основі росту і диференціювання тканин, забезпечує безперервне
оновлення структур організму. Проліферація. різних клітин імунокомпетентних
систем є основою імуногенезу. За допомогою проліферації ліквідується утворений
при пошкодженні тканин дефект і нормалізується порушена функція. Проліферація.
може виникати і внаслідок порушення гормональних впливів, приводячи до
жахливого збільшення органа, наприклад при акромегалії. Проліферація. клітин,
які втратили здатність диференціюватися в клітини того чи іншого органу, веде
до виникнення пухлин. Одні органи і тканини володіють дуже високою здатністю до
проліферації клітин (сполучна, кровотворна, кісткова тканина, печінка,
епідерміс, епітелій слизових оболонок), інші - більш помірної (скелетні м'язи,
підшлункова залоза, слинні залози та ін), треті - зовсім або майже позбавлені
цієї здатності (ЦНС., міокард).
До основних властивостей
усіх злоякісних пухлин відносять підвищену здатність до проліферації, втрату
здатності до повного диференціювання і апоптичної загибелі, а також інвазивний
ріст і метастазування. Завдяки їм неоплазма має перевагу перед нормальними
клітинами під час росту і виживання. Якщо у здорової тканини існує баланс між
процесами проліферації і загибелі клітини, то в тканині пухлини має місце
автономна і необмежена проліферація клітин. При цьому в трансформованих
клітинах виникає стійкість до індукції апоптозу [9]. В результаті генетичних
мутацій знижується здатність трансформованих клітин активувати програму
апоптозу, що, з одного боку, сприяє прогресуванню пухлинного процесу, а з
іншого - може стати причиною множинної лікарської стійкості [1]. У зв'язку з
цим останнім часом приділяється велика увага вивченню молекулярних маркерів, що
характеризують апоптоз і проліферацію при різних злоякісних новоутвореннях [7,
8] Провідну роль у розвитку апоптозу та регуляції клітинної проліферації грає
природний (Wt) тип гена-онкосупресора p53 і кодуємий їм ядерний білок р53 [3].
Останній модулює експресію генів, які відповідають за репарацію ДНК, поділ
клітин і апоптоз [1, 19]. У значної частини злоякісних пухлин виявляють мутації
в хромосомі 17 в області локалізації гена-супресора р53 [4, 21]. Bcl-2 є одним
з основних компонентів системи захисту клітини проти факторів, що викликають
апоптоз. Відомо, що Bcl-2 інгібує р53-залежний і незалежний апоптозні
метаболічні шляхи. При цьому сприяє утворенню в мітохондріях іонних каналів,
стабілізуючи цим мітохондріальну цитохромоксидазу С і пов'язує білки, що беруть
участь в апоптозі. Цитохром-оксидаза С здатна ініціювати активацію каскаду
каспаз і, як наслідок, деградацію ДНК і апоптоз. Зі станом основних
регуляторних систем програмованої клітинної загибелі пов'язані неопластичні,
репаративні і проліферативні процеси в тканинах. Показано, що порушення
механізмів проліферації - одна з головних особливостей пухлинних клітин. А
рівень проліферативної активності визначає агресивність і злоякісність
пухлинного процесу [7].
3.2 Вплив
протитуберкульозних препаратів нам показники життєдіяльності клітин в культурі
Протитуберкульозні
препарати - препарати активні по відношенню до мікобактерії туберкульозу.
Деякі автори вважають,
що тривале лікування туберкульозу протитуберкульозними препаратами може
викликати розвиток пухлин. [28]
У сучасній класифікації
протитуберкульозні препарати прийнято розділяти на два ряди в залежності від
клінічної ефективності
Препарати першого
ряду
Основна група препаратів.
За хімічною природою належать до різних класів сполук (табл..3.1.). Препарати,
що входять в неї, справляють максимальний протитуберкульозний ефект при
мінімальній токсичності.
Таблиця 3.1.
Протитуберкульозні препарати першого ряду
|
Назва
|
Медичне скорочення
|
Код АТХ
|
Група
|
|
Ізоніазид
|
H
|
J04AC01
|
Гідразиди
|
|
Рифаміцин
|
R
|
J04AB02
|
Ансаміцини
|
|
Піразинамід
|
Z
|
J04AK01
|
Сінтетичні антибактеріальні
препарати
|
E
|
J04AK02
|
|
|
Стрептоміцин
|
S
|
A07AA04
|
Аміноглікозиди
|
Препарати другого
ряду
Препарати другого ряду
(табл..3.2) справляють більш слабкий вплив на збудника туберкульозу, ніж
препарати першого ряду, будучи в той же час більш токсичними для організму
людини. Тому вони застосовуються лише тоді, коли у хворих визначається
стійкість мікобактерій туберкульозу до препаратів першого ряду. Звичайно це має
місце після антибактеріальної терапії, але у частини вперше виявлених пацієнтів
виявляється первинна стійкість в результаті первинного зараження
лікарськостійкими штамами мікобактерій туберкульозу. [1]
Таблиця 3.2
Протитуберкульозні препарати другого ряду
|
Назва
|
Медичне скорочення
|
Код АТХ
|
Група
|
|
Циклосерин
|
C
|
J04AB01
|
Антибіотики
|
|
Офлоксацин
|
Of
|
J01MA01
|
Фторхінолоны
|
|
Ципрофлоксацин
|
Cf
|
J01MA02
|
Фторхінолоны
|
|
Амікацин
|
A
|
D06AX12
|
Аміногликозіди
|
|
Канаміцин
|
K
|
A07AA08
|
Аміногликозіди
|
|
Капреоміцин
|
Cp
|
J04AB30
|
Глікопептиди
|
|
Протіонамид
|
Pt
|
J04AD01
|
Сінтетичні антибактеріальні
препарати, похідні ізоникотинової кислоти
|
|
Этіонамид
|
Et
|
J04AD03
|
Сінтетичні антибактеріальні
препарати, похідні ізоникотинової кислоти
|
|
ПАСК - парааміносаліцилова кислота
|
PAS
|
J04AA01
|
Сінтетичні антибактеріальні
препарати
|
3.3 Матеріали та методи
Отримання,
культивування, оцінка проліферативної активності та типування мезенхімальних
стовбурових клітин щурів
Забір клітин кісткового
мозку проводили у білих безпородних статевозрілих щурів масою 250 г. У
експериментах використовувалися тварини після проходження 14-ти денного
карантину. Всі щури знаходилися на стандартних умовах віварію, згідно
Страсбурзької конвенції з захисту прав лабораторних тварин, що використовуються
для експериментальних та інших наукових цілей (Страсбург, 1986 р.) і «Загальних
етичних принципів дослідів на тваринах» (Україна, 2001 р.). Евтаназію тварин
здійснювали за допомогою передозування ефірного наркозу. Кістковий мозок
отримували з стегнових, великогомілкових і плечових кісток. Клітини кісткового
мозку вимивали з порожнини кістки 3 мл живильного середовища Ігла МЕМ в
стерильну пробірку, мезенхімальні стовбурові клітини строми (МСК)
відокремлювали від гемопоетичних стовбурових клітин шляхом ресуспензування,
фільтрували через капронову сіточку, підраховували кількість життєздатних
клітин за допомогою трипанового синього [33]. Крім того, поділ досягався і за
рахунок адгезії до культуральної пластики. Кінцеву концентрацію життєздатних
клітин доводили до 5 • 104 в мл і поміщали в пластикові одноразові
флакони, площею 25 см2. Культивування клітин проводили в середовищі
Ігла МЕМ, збагаченої L-глутамином, 10% ембріональної телячої сироваткою і з
додаванням антибіотиків протягом 12 днів зі зміною Ѕ середовища кожні 5 діб.
При досягненні 80 - 90% моношару в острівцях клітин первинної культури або
наступних субкультурах МСК проводили їх пассивування. При цьому використовували
20 мМ фосфатний буфер, розчин 0,05% трипсину. Щільність посадки клітин складала
2000 Кл/см2. Культивування в стандартних умовах проводили при
температурі 37° в атмосфері 5% СО2 в HF151UV СО2 -
інкубаторі в умовах насиченої вологості.
Аналіз проліферативної
активності клітин грунтувався на імунофлюоресцентному визначенні ядерного антигену
поділу клітин. Отримані клітини фіксували крижаним метанолом протягом 15
хвилин. В якості первинних антитіл використовували антитіла до Ki67 антигену
(Sigma), які наносили на препарати та інкубували 30 хвилин при 4° С. Потім
препарати відмивали фізіологічним розчином, забуферений фосфатно-сольовим
буфером (РВS), і для виявлення пов'язаних первинних антитіл вносили вторинні
FITC - кон'юговані антитіла до мишачих імуноглобулінів(Sigma) на 30 хвилин при
4° С. На промиті і висушені препарати наносили 50% гліцерин. Мікроскопію
проводили за допомогою люминисцентного мікроскопа МС300 (Micros Austria) при
400-кратному і 1000 - кратному збільшенні.
Для оцінки
життєздатності та проліферативної активності клітин в культурах досить широко
застосовується колориметричний метод з використанням МТТ - реактиву (3 - [4,5 -
діметілтіазол-2-іл] -2,5 - діфенілтетразоліум бромід). Метод заснований на
тому, що дегідрогенази мітохондрій в життєздатних клітинах, в яких висока
метаболічна активність, здатні перетворювати додану в культуру водорозчинну МТТ
- тетразоліеву сіль жовтого кольору в кристали нерозчинного у воді формазану
лілового кольору. Реакцію проводили в пробірках, в кожній з яких містилося по 2
Ч 106 клітин на 1 мл живильного середовища, причому МСК заздалегідь
знімали з культурального пластика сумішшю розчину версії і трипсину,
центрифугували 5 хвилин при 200g, знімали супернатант і заливали живильним
середовищем. У кожну пробірку додавали по 10 мкл розчину МТТ. Після інкубації
протягом 1,5 годин при 37° С клітини центрифугували при 400g 10 хвилин і
відмивали РВS в аналогічних умовах. До осаду додавали 150 мкл ДМСО
(диметилсульфоксид), вимірювали оптичну щільність при л = 570 нм проти контролю
(ДМСО) за допомогою спектрофотометра (СФ-46). За 100% приймали кількість
формазану, що утворився в аліквоті інтактних МСК [36].
Оцінка морфологічних
змін в культах клітин при апоптозі
Для прижиттєвого
виявлення клітин з конденсованим або фрагментованим ядром був використаний
флюоресцентний ядерний барвник Hoechst 33342. [31]. У експеримент брали
мононуклеарні клітини після 24 і 48 годин культивування, а мезенхімальні
стовбурові клітини попередньо відокремлювали від підкладки. Суспензію клітин
від живильного середовища відмивали в РВS і інкубували протягом 10 хвилин при
кімнатній температурі з Hoechst 33342 (1 мкг/мл). Клітини відмивали від
барвника і фіксували в 50% гліцерині.
Морфологічну оцінку
проводили за допомогою люмінісцентного мікроскопа МС300 (Micros Austria) при
400-кратному збільшенні, загальна кількість клітин оцінювали за допомогою
фазовоконтрастной мікроскопії.
Визначення рівня
фрагментованою ДНК в клітинах
Найбільш характерним
біохімічним ознакою апоптозу є розщеплення ендонуклеазами двоспіральної ядерної
ДНК, на виявленні якій засновані сучасні методи діагностики апоптозу. Так
одними з найбільш чутливих є гель-електрофорез поодиноких клітин [35],
пульс-електрофорез [18], Саузерн - гібридизація ДНК [21]. Нами було використано
метод спектрофотометричного визначення фрагментованою ДНК з використанням
діфеніламінового реагенту [19]. Після культивування клітини відмивали у фізіологічному розчині,
поміщали в гомогенізатор з тефлоновим товкачиком і гомогенізували в сахарозному
середовищі виділення (0,25 М сахарози, 1мМ ЕДТА, рН = 7,4). Гомогенат
центрифугували, супернатант, який може містити ДНК некротичних клітин,
відкидали. До осаду ядер додавали лізис-буфер (5 мМ Tris HCl, 20 мМ ЕДТА (рН =
8), 0,5% Triton X-100) в об'ємному співвідношенні 1: 9. Гомогенат готували при
t = 4оС. Аліквоту проби центрифугували протягом 15 хв при 13 000 g,
відокремлюючи інтактний хроматин (осад) від фрагментованою ДНК (супернатант).
Потім проби ресуспензували в 500 мл ТЕ-буфера (10 мМ Tris HCl, 1 мМ ЕДТА, рН =
8) і преципітували в 600 мл 12% трихлороцтової кислоти при t = 4оС.
Далі преципітати центрифугували при 4000 g 10 хв і супернатант відкидали.
У пробірки з осадом
додавали по 300 мл 5% три хлороцтової кислоти і проводили гідроліз на водяній
бані при 90оС протягом 10 хв, білок осаджували центрифугуванням.
Потім піпеткою обережно з пробірок відбирали супернатант. Після охолодження в
пробірки з супернатантом додавали дифеніламіновий реагент (100 мг дифеніламіну,
10 мл крижаної оцтової кислоти, 0,28 мл Н2SO4) в
об'ємному співвідношенні 1: 2. Максимальний розвиток забарвлення досягалося
через 17 - 18 годин при кімнатній температурі. Інтенсивність забарвлення прямо
пропорційна вмісту ДНК в пробі. Контролем служила проба, що містить
дифеніламіновий реагент і 5% трихлороцвої кислоти.
Після проведення
кольорової реакції аліквоту проб переносили в кювети спектрофотометра (СФ-46)
та вимірювали оптичну щільність при л = 570 нм проти контролю.
Кількість фрагментованою
ДНК (фДНК) розраховували у відсотках за формулою:
D (фДНК)
Х =
--------------------------------- • 100%, де
D (фДНК) + D (іДНК)
Х - кількість
фрагментованої ДНК у%;(фДНК) - оптична щільність фрагментованої ДНК;(іДНК) -
оптична щільність інтактної ДНК.
Для постановки даної
методики були використані реактиви вітчизняного виробництва кваліфікації х.ч.
або ч.д.а.
В експеримент були взяті
5 дослідних груп для протитуберкульозних препаратів 1 ряду: у першій групі
клітини культивували з ізоніазідом, у другій - з рифампіцином, у третій - з
піразинамідом, у четвертій - з етамбутолом, у п'ятій клітини культивували з
стрептоміцином. (табл. 3. 3,3.4) Для протитуберкульозних препаратів 2
ряду було взято 7 дослідних груп: у першій групі клітини культивували з
циклосерином, у другій - з офлоксацином, у третій - з ципрофлоксацином, у
четвертій - з амікацином, у п'ятій - з канаміцином, у шостій - з протіонамідом,
у сьомій - з етіонамідом. (табл. 3. 5,3.6) Контролем служила інтактна
культура пухлинних клітин з 24 і 48 годинною інкубацією.
Індекс апоптозу (ІА)
визначався як відношення апоптозних тілець до загального числа клітин помножена
на 100%.
Статистичну обробку результатів
дослідження проводили на персональному комп'ютері із застосуванням стандартних
пакетів прикладних програм. [5]
.4 Результати і їх
обговорення
Аналізуючи вплив
протитуберкульозних препаратів першого ряду на культуру МСК кісткового мозку
щурів, ми виявили, що через 24 години культивування індекс проліферації у
культурі з ізоніазидом складав 5,1, що на 11,8% більше, ніж у контрольній
групі, а індекс апоптозу - 4,9, що на 22, 4% більше, ніж у контрольній групі, у
культурі з рифаміцином індекс проліферації складав 3,6 (на 20% менше, ніж у
контрольній групі), а індекс апоптозу - 4,1 (на 7,3% більше, ніж у контрольній
групі), у культурі з піразинамідом індекс проліферації складав 4,8 (на 6,25%
більше, ніж у контрольній групі), а індекс апоптозу - 4,5 (на 15,6% більше, ніж
у контрольній групі), у культурі з етамбутолом індекс проліферації складав 5,9
(на 23,7% більше, ніж у контрольній групі), а індекс апоптозу - 5,7 (на 33,3%
більше, ніж у контрольній групі), у культурі з стрептоміцином індекс
проліферації складав 4,0 (на 22,2% меньше, ніж у контрольній групі), а індекс
апоптозу - 4,2 (на 9,5% більше, ніж у контрольній групі).
Таблиця 3.3. Індекс
проліферації (ІП) та індекс апоптозу (ІА) для клітин, культивованих з
препаратами першого ряду через 24 години культивування
|
Препарат
|
ІП, %
|
ІА, %
|
|
Контроль
|
4,5
|
3,8
|
|
Ізоніазид
|
5,1
|
4,9
|
|
Рифаміцин
|
3,6
|
4,1
|
|
Піразинамід
|
4,8
|
4,5
|
|
Етамбутол
|
5,9
|
5,7
|
|
Стрептоміцин
|
4,0
|
4,2
|
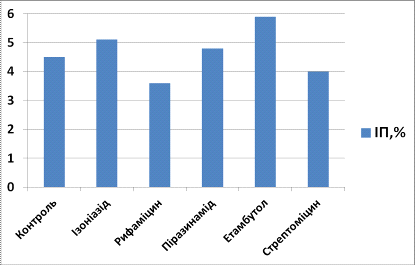
Рис. 3.1. Індекс
проліферації (ІП) для клітин, культивованих з препаратами першого ряду через 24
години культивування
1 - Контроль, 2 -
Ізоніазид, 3 - Рифаміцин. 4 - Піразинамід, 5 - Етамбутол, 6 - Стрептоміцин
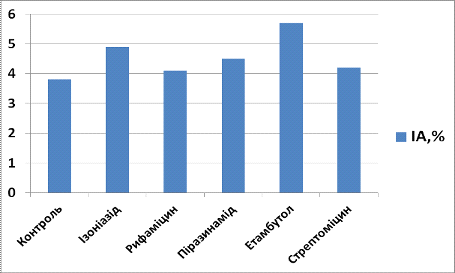
Рис. 3.2. Індекс
апоптозу (ІА) для клітин, культивованих з препаратами першого ряду через 24
години культивування
- Контроль, 2 -
Ізоніазид, 3 - Рифаміцин. 4 - Піразинамід, 5 - Етамбутол, 6 - Стрептоміцин
Таблиця 3.4. Індекс
проліферації (ІП) та індекс апоптозу (ІА) для клітин, культивованих з
препаратами першого ряду через 48 годин культивування
|
ПрепаратІПІА
|
|
|
|
Контроль
|
4,9
|
3,9
|
|
Ізоніазид
|
5,6
|
5,0
|
|
Рифаміцин
|
4,3
|
4,6
|
|
Піразинамід
|
5,0
|
4,7
|
|
Етамбутол
|
6,1
|
5,9
|
|
Стрептоміцин
|
4,7
|
4,4
|
Через 48 годин індекс
проліферації у культурі з ізоніазидом складав 5,6, що на 12,5% більше, ніж у
контрольній групі, а індекс апоптозу - 5,0, що на 22% більше, ніж у контрольній
групі, у культурі з рифаміцином індекс проліферації складав 4,3 (на 12,2%
менше, ніж у контрольній групі), а індекс апоптозу - 4,6 (на 15,2% більше, ніж
у контрольній групі), у культурі з піразинамідом індекс проліферації складав
5,0 (на 22% більше, ніж у контрольній групі), а індекс апоптозу - 4,7 (на 17% більше,
ніж у контрольній групі), у культурі з етамбутолом індекс проліферації складав
6,1 (на 19,7% більше, ніж у контрольній групі), а індекс апоптозу - 5,9 (на
33,9% більше, ніж у контрольній групі), у культурі з стрептоміцином індекс
проліферації складав 4,7 (на 4% меньше, ніж у контрольній групі), а індекс
апоптозу - 4,4 (на 11,4% більше, ніж у контрольній групі).

Рис. 3.3. Індекс
проліферації (ІП) для клітин, культивованих з препаратами першого ряду через 48
годин культивування
- Контроль, 2 -
Ізоніазид, 3 - Рифаміцин. 4 - Піразинамід, 5 - Етамбутол, 6 - Стрептоміцин
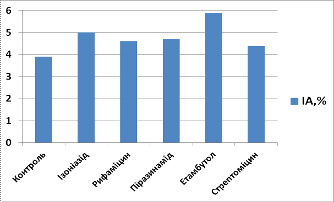
Рис. 3.4. Індекс
апоптозу (ІА) для клітин, культивованих з препаратами першого ряду через 48
годин культивування
- Контроль, 2 -
Ізоніазид, 3 - Рифаміцин. 4 - Піразинамід, 5 - Етамбутол, 6 - Стрептоміцин
Отримані нами данні, як після
24 годин культивування, так і після 48 годин культивування, свідчать про те, що
протитуберкульозні препарати першого ряду практично не стимулюють проліферацію
клітин, індекс проліферації у дослідних групах значно не відрізняється від
індексу проліферації у контрольній групі. Індекс апоптозу у культурах клітин з
дослідних груп також практично не відрізнявся від культури клітин з контрольної
групи. Отже можна сказати, що терапія протитуберкульозними препаратами першого
ряду не спричиняє вплив на життєздатність проліферуючих клітин.
Таблиця 3.5. Індекс
проліферації (ІП) та індекс апоптозу (ІА) для клітин, культивованих з
препаратами другого ряду через 24 години культивування
|
Препарат
|
ІП
|
ІА
|
|
Контроль
|
4,5
|
3,8
|
|
Циклосерин
|
4,2
|
3,9
|
|
Офлоксацин
|
3,8
|
|
Ципрофлоксацин
|
4,7
|
3,5
|
|
Амікацин
|
4,3
|
3,7
|
|
Канаміцин
|
4,5
|
3,8
|
|
Капреоміцин
|
4,0
|
3,6
|
|
Протіонамід
|
4,1
|
3,2
|
Аналізуючи вплив
протитуберкульозних препаратів другого ряду на культуру МСК кісткового мозку
щурів, ми виявили, що через 24 години індекс проліферації у культурі з
циклосерином складав 4,2, що на 6,7% меньше, ніж у контрольній групі, а індекс
апоптозу - 3,9, що на 2, 6% більше, ніж у контрольній групі, у культурі з
офлоксацином індекс проліферації складав 4,4 (на 2,2% менше, ніж у контрольній
групі), а індекс апоптозу - 3,8 (дорівнює контролю), у культурі з
ципрофлоксацином індекс проліферації складав 4,7 (на 4% більше, ніж у
контрольній групі), а індекс апоптозу - 3,5 (на 7,9% меньше, ніж у контрольній
групі), у культурі з амікацином індекс проліферації складав 4,3 (на 4,4% менше,
ніж у контрольній групі), а індекс апоптозу - 3,7 (на 5,5% менше, ніж у
контрольній групі), у культурі з канаміцином індекс проліферації складав 4,5
(дорівнює контролю), а індекс апоптозу - 3,8 (дорівнює контролю), у культурі з
капреоміцином індекс проліферації складав 4,0 (на 11,1% меньше, ніж у
контрольній групі), а індекс апоптозу - 3,6 (на 5,3% більше, ніж у контрольній
групі), у культурі з протіонамідом індекс проліферації складав 4,1 (на 8,9%
меньше, ніж у контрольній групі), а індекс апоптозу - 3,2 (на 15,8% меньше, ніж
у контрольній групі).
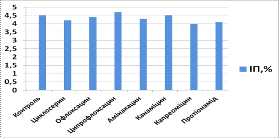
Рис. 3.5 Індекс
проліферації (ІП) для клітин, культивованих з препаратами другого ряду через 24
години культивування
- Контроль, 2 -
Циклосерин, 3 - Офлоксацин, 4 - Ципрофлоксацин, 5 - Амікацин, 6 - Канаміцин, 7
- Капреоміцин, 8 - Протіонамід

Рис. 3.6 Індекс апоптозу
(ІА) для клітин, культивованих з препаратами другого ряду через 24 години
культивування
- Контроль, 2 - Циклосерин,
3 - Офлоксацин, 4 - Ципрофлоксацин, 5 - Амікацин, 6 - Канаміцин, 7 -
Капреоміцин, 8 - Протіонамід
Таблиця 3.6. Індекс
проліферації (ІП) та індекс апоптозу (ІА) для клітин, культивованих з
препаратами другого ряду через 48 годин культивування
|
Препарат
|
ІП
|
ІА
|
|
Контроль
|
4,9
|
3,9
|
|
Циклосерин
|
4,4
|
4,1
|
|
Офлоксацин
|
4,7
|
4,1
|
|
Ципрофлоксацин
|
4,8
|
3,8
|
|
Амікацин
|
4,5
|
3,9
|
|
Канаміцин
|
4,6
|
4,0
|
|
Капреоміцин
|
4,2
|
3,9
|
|
Протіонамид
|
4,5
|
3,5
|
Аналізуючи вплив
протитуберкульозних препаратів другого ряду на культуру МСК кісткового мозку
щурів, ми виявили, що через 48 годин індекс проліферації у культурі з
циклосерином складав 4,4, що на 10,2% меньше, ніж у контрольній групі, а індекс
апоптозу - 4,1, що на 4,9% більше, ніж у контрольній групі, у культурі з
офлоксацином індекс проліферації складав 4,7 (на 4% менше, ніж у контрольній
групі), а індекс апоптозу - 4,1 (на 4,9% більше, ніж у контрольній групі), у
культурі з ципрофлоксацином індекс проліферації складав 4,8 (на 2% меньше, ніж
у контрольній групі), а індекс апоптозу - 3,8 (на 2,6% меньше, ніж у
контрольній групі), у культурі з амікацином індекс проліферації складав 4,5 (на
8% менше, ніж у контрольній групі), а індекс апоптозу - 3,9 (дорівнює
контролю), у культурі з канаміцином індекс проліферації складав 4,6 (на 6% менше,
ніж у контрольній групі), а індекс апоптозу - 4,0 (на 2,5% більше, ніж у
контрольній групі), у культурі з капреоміцином індекс проліферації складав 4,2
(на 14,3% меньше, ніж у контрольній групі), а індекс апоптозу - 3,9 (дорівнює
контролю), у культурі з протіонамідом індекс проліферації складав 4,5 (на 8,2%
меньше, ніж у контрольній групі), а індекс апоптозу - 3,5 (на 10,3% меньше, ніж
у контрольній групі).
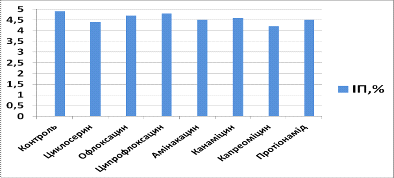
Рис. 3.7 Індекс
проліферації (ІП) для клітин, культивованих з препаратами другого ряду через 48
годин культивування
- Контроль, 2 -
Циклосерин, 3 - Офлоксацин, 4 - Ципрофлоксацин, 5 - Амікацин, 6 - Канаміцин, 7
- Капреоміцин, 8 - Протіонамід

Рис. 3.8 Індекс апоптозу
(ІА) для клітин, культивованих з препаратами другого ряду через 48 годин
культивування
- Контроль, 2 -
Циклосерин, 3 - Офлоксацин, 4 - Ципрофлоксацин, 5 - Амікацин, 6 - Канаміцин, 7
- Капреоміцин, 8 - Протіонамід
Отримані нами данні, як
після 24 годин культивування, так і після 48 годин культивування, свідчать про
те, що протитуберкульозні препарати другого ряду практично не стимулюють
проліферацію клітин, індекс проліферації у дослідних групах значно не
відрізняється від індексу проліферації у контрольній групі. Індекс апоптозу у
культурах клітин з дослідних груп також практично не відрізнявся від культури
клітин з контрольної групи.
Отже, можна сказати, що
терапія протитуберкульозними препаратами другого ряду суттєво не впливає на
життєздатність проліферуючих клітин.
Також ми можемо
зазначити, що ІП та ІА у культурах клітин з препаратами другого ряду менше
відрізнялися від контролю, ніж культури клітин з препаратами першого ряду.
Висновки
На основі проведених
досліджень показано, що протитуберкульозні препарати першого і другого ряду можуть
по-різному впливати на життєздатність клітин з високим проліферативним
потенціалом
• 1. Протитуберкульозні препарати першого ряду в більшому ступені
стимулюють процеси проліферації та процеси апоптозу, порівняно з препаратами
другого ряду. При цьому інтенсивність змін практично не залежить від часу
впливу препаратів (24-48 год)
• 2. Протитуберкульозні препарати другого ряду в 0,5-1,5 рази знижують
індекс проліферації у дослідних групах порівняно з показником контрольної
групи.
• 3. Індекс апоптозу у культурах клітин з дослідних груп під дією
препаратів другої групи практично не відрізняється від контрольного показника.
• 4. Висловлено припущення, що при протитуберкульозній терапії
препарати другого ряду можуть мати менший ризик виникнення побічних ефектів з
індукцією пухлинних процесів, порівняно з препаратами першого ряду.
Список літератури
1. Абраменко И.В.
Оценка параметров апоптоза в диагностике онкологических заболеваний, их
прогнозе и оптимизации схем терапии. // . Абраменко ИВ, Фильченков АА. - Вопр
онкол 2003; 49: 21-30.
. Кноринг Б.Е.
Оценка клинической значимости туберкулиновой сенсибилизации у больных раком
легких // Кноринг Б.Е., Ариэль Б.М Пробл. туберкулеза, - 1996, с. 26-30.
. Копнин Б.П.
Мишени действия онкогенов и опухолевых супрессоров: ключ к пониманию базовых
механизмов канцерогенеза // Копнин Б.П Биохимия. - 2000.-Т. 65.-С. 5 - 33.
. Лапач С.Н.
Статистические методы в медико-биологических исследованиях с использованием Excel. // Лапач С.Н., Чубенко
А.В., Бабич П.Н. Киев: Морион, - 2000. - 320 с.
. Національний
канцер-реєстр України (1997): Розповсюдженість злоякісних новоутворень в
популяціях України в 1991-1996 рр.
. Пятночка И.Т.
Пропаганда профилактики рака легкого среди контингентов противотуберкулезных
диспансеров. // Пятночка И.Т. - 1999, с. 71 - 72
. Раданов Р.
Туберкулез легких в сочетании с другими заболеваниями (Пер. с болгарського) //
Раданов Р., Тодоров С., 1974, 261 с.
. Хейфец С.Л. Рак
и туберкулез // Хейфец С.Л Медицина, Москва, - 1969, 167 с.
. Чумаков П.М.
Функция гена р53: выбор между жизнью и смертью // Чумаков П.М. Биохимия. -
2000.-Т. 65.-С. 34-47.
11. Betticher
D.C., Heighway J. Hasleton P.S. et al. Prognostic significance of CCND1 (cyclin
D1) overexpression in primary resected nonQ smallQcell lung cancer. // Brit. J.
Cancer. - 1996.-Vol.-, 73p. 294-300.
12. Bonner J.,
Ezekiel M., Robert F. et al. Continued
response following treatment with IMCQC225: an EGFr MoAB, combined with RT in
advanced head and neck cancer // Proc. ASCO. - 2000. - Vol.19. - Abstr. 5F.
13. Cerny T.,
Barnes D.M., Hasleton P. et al. Expression
of epidermal growth factor receptor (EGFQR) in human lung cancer // Brt. J.
Cancer. - 1986.-Vol.54. - 265-268.
14. DeVore R.F.,
Fenrenbacher L., Herbst R.S. et al. A
randomized phase II trial comparing Rhumab VEGF (recombinant humanized
monoclonal antibody to vascular endothelial cell groiwth factor) plus
carboplatin/paclitaxel (CP) to CP alone in patients with stage IIIB/IV NSCLC //
Proc. ASCO. - 2000. - Vol.19. - Abstr. 1896.
. Ferry D.,
Hammond L., Ranson M. et al. Intermitten oral Zd1839 (Iressa), a novel
Epidermal Growth Factor receptor tyrosine kinase inhibitor (EGFQTki), shows
evidence of good tolerability and activity: final results from a phase I study
// Ibid. - Vol.19. - Abstr. 5E.
. Folkman J.
Clinical application of research on angiogenesis // N. Engl. J. Med. -
1995.-Vol.333.-P.1757-1763.
. 16.
Greenblatt M.S., Bennett W.P., Hollstein M., Harris C.C. Mutation in the p53
tumor suppressor gene: clues to cancer etiology and molecular pathogenesis //
Cancer Res. -1994.-Vol. 54.-P.4855 - 4878.
. Kubba S.,
Adak S., Schiller J. et al. Phase I trial of adenovirus p53 in
bronchioloalveolar cell lung carcinoma administered by bronchoalveolar lavage
// Proc. ASCO. - 2000. - Vol.19 - Abstr. 1904.
. Kuss B.,
Cotter F. Antisesnse Q time to shoot the messenger // Ann. Oncol. - 1999. -
Vol.10.-P.495Q503.
. Mace PD,
Wallez Y, Egger MF, Dobaczewska MK, Robinson H, Structure of ERK2 bound to
PEA-15 reveals a mechanism for rapid release of activated MAPK. // Pasquale EB,
Riedl SJ. - 2012
. Meng F, Li
H, Shi H, Yang Q, Zhang F, Yang Y, Kang L, Zhen T, Dai S, Dong Y, Han A. MACC1
Down-Regulation Inhibits Proliferation and Tumourigenicity of Nasopharyngeal
Carcinoma Cells through Akt/в-Catenin Signaling Pathway. // PLoS One. 2013; 8
(4):e60821. doi: 10.1371/journal.pone.0060821. Epub - 2011
. Nelson
A.R., Fingleton B., Rothenberg M.L. et al. Matrix metalloproteinases: biologic
activity and clinical implications // J. Clin. Oncol. - 2000.-Vol.
18.-P.1135Q1149.
. Nemunaitis
J., Swisher S.G., TimmonsT. et al. AdenovirusQmediated p53 gene transfer in
sequence with cisplatin to tumors of patients with nonQsmallQcell lung cancer
// Ibid.-Vol.18.-P.609Q22.
24. Norton L.,
Slamon D., LeylandQJones B. et al. Overall
survival advantage to simultaneous chemotherapy plus the humanized antiQ HER2
monoclonal antibody Herceptin in HER2Qoverexpressing metastatic breast cancer
// Proc. ASCO. - 1999.-Vol. 18. - Abstr. 483.
25. Reissmann
P.T., Koga H., Takahashi R. et al. Inactivation
of the retinoblastoma susceptibility gene in nonQsmallQcell lung canver
//Oncogene. - 1993.-Vol.8.-P.1913-1919.
. Roth J.A.
Modification of tumor supressor gene expression in nonQsmallQcell lung cancer
with a retroviral vector expressing wildtype (normal) p53 // Human Gene Ther. -
1996.-Vol.7.-P.861-74.
. Rowinsky
E.K., Windle J.J., Von Hoff D.D. Ras protein farnesyltransferase: a strategic
target for anticancer therapeutic development // J. Clin. Oncol. -
1999.-Vol.17.-P.3631-52.
. Sausville
E.A. CyclinQdependent kinases: novel targets for cancer treatment // ASCO
Educational book. - 1999 (spring).-P.9-21.
. Schauer I.E.,
Siriwarnada S., Langan T.A., Sclafani R.A. Cyclin D1 overexpression vs.
Retinoblastoma inactivation:implicationfor growth control evasion in nonQsmall
cell and small cell lung cancer // Proc. Natl. Acad. Sci. USA. -
1994.-Vol.91.-P.7827-7831.
. Sekido Y.,
Fong K.M., Minna J.D. Progress in understanding the molecular pathogenesis of
human lung cancer // Biochim. Biophis. Acta. - 1998.-Vol.1378. - F21-F59.
. Sporn
M.B., Todaro G.J. Autocrine growth factors and cancer // Nature. -
1985.-Vol.313.-P.747-751.
. Taga S.,
Osaki T., Ohgami A. et al. Prognostic Impact of Telomerase Activity in
NonQSmall Cell Lung Cancers // Ann. of Surgery. - 1999.-Vol.230.-P.968-974.
. Wang J.,
Xie L., Allan S. et al. Myc activates telomerase // Genes Dev. -
1998.-Vol.12.-P.1769-1774.
. Wang L,
Zhou BB, Yu K, Su ZH, Gao S, Chu LL, Liu JR, Yang GY. Novel Antitumor Agent,
Trilacunary Keggin-Type Tungstobismuthate, Inhibits Proliferation and Induces
Apoptosis in Human Gastric Cancer SGC-7901 Cells. // Inorg Chem. - 2013
35. Weiner D.B.,
Nordberg J., Robinson R. et al. Expression
of the neu geneQencoded protein (p185neu) in human nonQsmall cell carcinomas of
the lung // Cancer Res. - 1990.-Vol.50.-P.421-425.
. Wong VK,
Zhang MM, Zhou H, Lam KY, Chan PL, Law CK, Yue PY, Liu L. Saikosaponin-d
Enhances the Anticancer Potency of TNF-б via Overcoming Its Undesirable
Response of Activating NF-Kappa B Signalling in Cancer Cells. // Evid Based
Complement Alternat Med. 2012; 2012:745295. doi: 10.1155/2012/745295. Epub -
2012