Разработка универсального набора функций, позволяющих описывать свойства молекул
1. Литературный обзор
.1 Методология исследования количественных
соотношений «cтруктура-свойство» / «cтруктура-активность»
Методы компьютерного прогнозирования свойств
химических соединений на основе поиска количественных (QSAR, Quantitative
Structure Activity Relationships, количественные соотношения
«структура-активность»; QSPR, Quantitative Structure Property Relationships,
количественные соотношения «структура-свойство») соотношений «структура -
активность» / «структура-свойство» применяются при конструировании веществ с
заранее заданными свойствами и прогнозировании свойств новых соединений.
Применение этих методов позволяет сократить время и затраты экспериментальных
процедур по синтезу и тестированию свойств новых соединений. В последние 20 лет
в связи ростом числа новых материалов, разрабатываемых для нужд
микроэлектроники, экологии, медицины, фармацевтики и др., востребованность
методов QSAR/QSPR в самых разных областях науки и техники значительно возросла.
Эти методы активно развиваются, и спектр объектов их применения расширяется,
особенно в области прогнозирования свойств материалов и супрамолекулярных
систем. Так, например, на сегодняшний день 29% публикаций в области QSPR
материалов посвящено исследованию полимеров, 19% - катализаторам, 16% -
наноматериалам, 12%, 10% и 2%, соответственно, суперкритическому СО2,
ионным жидкостям и керамике [1].
Методологии QSAR/QSPR и применению методов на её основе
посвящено значительное число монографий, обзоров и работ [1-8]. Методология
QSAR/QSPR основана на предположении, что изменения структуры молекулы влияет на
изменение макроскопических свойств вещества. Связь между молекулярной
структурой и макроскопическими свойствами (например, биологической активностью
или физико-химическими свойствами) рассчитывают в виде функциональной
зависимости с помощью статистического аппарата методов машинного обучения.
Простейшая схема методологии QSAR/QSPR представлена на Рис. 1.

Рис. 1. Схема методологии QSAR/QSPR
На основе набора структур соединений с известными значениями
исследуемого свойства / активности формируют базу данных. Далее с
использованием соединений обучающей выборки строят функциональную зависимость
(модель) между значениями свойства и набором молекулярных дескрипторов,
кодирующих определённую информацию о структуре молекулы. В QSPR материалов в
качестве входной информации также могут использовать характеристики,
описывающие внешние условия и исходные компоненты для синтеза [1].
Прогнозирующую способность модели оценивают с помощью соединений контрольной
(тестовой) выборки. После того, как модель построена и доказана её
прогнозирующая способность, она может быть использована для прогноза свойств
(активности) новых соединений, для которых свойства (активность) не известны из
экспериментальных данных. Способы разбиения базы данных на обучающую и
контрольную выборки описаны в обзоре [8].
Классификация методов QSAR/QSPR. В обзоре [3] методы
QSAR/QSPR классифицируют исходя из размерности описания структуры молекул или
способа представления дескрипторов:
• 1D QSAR - поиск
соотношений между активностью / свойством и общими молекулярными
характеристиками: pKa, logP и др. [10-14];
• 2D QSAR - построение
корреляций на основе 2D (топологических) характеристик структуры, например,
индексов связности, 2D фармакофоров и др. [15-18]. Эти методы имеют ограничения
при конструировании новых молекул, т. к. не учитывают характеристик их
пространственной структуры;
• 3D QSAR - построение
корреляций на основе 3D геометрии молекул с использованием потенциалов
молекулярных полей в качестве дескрипторов [1-7,19];
• 4D QSAR - в рамках 3D
QSAR используют дополнительную информацию о конформациях ансамбля лигандов
[20];
• 5D QSAR - в рамках 4D
QSAR используют дополнительную информацию об изменении конформации лиганда при
связывании с белком (induced-fit model) [21];
• 6D QSAR - в рамках 5D
QSAR используют дополнительную информацию об эффекте растворителя [22].
Методы статистического анализа данных. Для построения QSAR/QSPR
моделей используются различные методы машинного обучения. Их целью является
нахождение зависимости между входными значениями (дескрипторами) и зависимыми
параметрами (свойство, активность). Многие из этих методов рассмотрены в обзоре
[8].
Авторы [3] дифференцируют методы QSAR/QSPR по типам методов
статистического анализа, используемых для построения моделей - линейные и
нелинейные.
Классическими линейными методами статистического анализа,
традиционно используемым для целей QSAR/QSPR, являются множественная линейная
регрессия (Multiple Linear Regression, MLR), метод частичных наименьших
квадратов (Partial Least Squares, PLS), регрессия на главных компонентах
(Principal Components Regression, PCR), гребневая регрессия (Ridge Regression,
RR) [23].
К нелинейным методам относят искусственные нейронные сети
(ANN) [24-25], метод ближайших соседей (kNN) [26] и ряд других.
В последнее время в области QSAR/QSPR активно применяются
ядерные методы машинного обучения [27], которые имеют большие перспективы в
прогнозировании свойств химических соединений и материалов. Эти методы
приспособлены для работы с математическим аппаратом ядер и позволяют
представить произвольную нелинейную функцию в виде линейной комбинации
нелинейных ядер. К ним относят машину опорных векторов (Support Vector Machine,
SVM) [28], ядерную гребневую регрессию (Kernel Ridge Regression, KRR) [29],
ядерные частичные наименьшие квадраты (Kernel Partial Least Squares, K-PLS)
[30] и ряд других методов.
Дескрипторы. На сегодняшний день разработано и
описано более 5000 различных дескрипторов [8,31,32] для представления тех или
иных свойств молекул и, соответственно программное обеспечение для их расчёта
(DRAGON [33], CODESSA [34] и др.). В обзоре [1] приведена следующая
классификация типов дескрипторов:
· Конституционные (характеризуют
относительное число атомов различного типа);
· Топологические [35]. В их основе лежит
представление молекулы в виде молекулярного графа. Топологические дескрипторы
подразделяют на топоструктурные, содержащие информацию о смежности и
топологических расстояниях между атомами, и топохимические, которые, кроме
этого, указывают на элементную принадлежность атомов и гибридизацию. Среди
топологических дескрипторов особую роль играют фрагментные дескрипторы, которые
показывают наличие или отсутствие тех или иных фрагментов в структуре молекулы
[36]).
· Физико-химические (характеризующие
растворимость, дипольные моменты, формальный заряд, липофильность и т.д.);
· Квантово-химические (характеризуют
частичный заряд на атомах, поляризуемость, энергии орбиталей, и др. параметры,
рассчитываемые с помощью полуэмпирических методов, методом функционала
плотности и другими квантово-химическими методами);
· Структурно-геометрические (характеризующие
пространственную геометрию, форму и площадь поверхности молекулы, расстояние
между функциональными группами);
· Дескрипторы молекулярных полей, которые
рассчитывают как энергии взаимодействия между атомами молекулы и пробными
атомами, находящимися в узлах воображаемой трехмерной решетки, построенной
вокруг молекулы.
Важным элементом методов QSAR/QSPR является представление
химических структур в стандартных обменных форматах, которые обеспечивают
возможность их хранения в базах данных и работы с ними с помощью широкого
набора существующих компьютерных программ [2,4]. Для преобразования файловых
форматов разработаны специальные программы (ChemAxon, OpenBabel, Avogadro и
др.).
1.2
Методы 3D QSAR/QSPR
Несмотря на то, что по сравнению с подходами 3D QSАR/QSPR
классические методы 2D QSАR/QSPR более просты и лучше приспособлены для анализа
больших массивов данных, они имеют ограничения при конструировании новых
соединений и, особенно, супрамолекулярных систем. В частности, они 1) не
позволяют учитывать особенности пространственного строения молекул, и как
следствие, различать стереоизомеры, 2) не позволяют детально описывать
межмолекулярные взаимодействия лиганд-мишень, 3) в рамках этих методов
невозможна наглядная интерпретация результатов путем рассмотрения
пространственного строения комплексов лиганд-мишень.
Поскольку практически все свойства химических соединений,
обусловленные образованием межмолекулярных комплексов, зависят от их
пространственного строения, в настоящее время методы 3D QSAR являются ведущими
при поиске новых биологически активных соединений, в частности, при создании
лекарственных препаратов, а методы 3D QSPR представляют перспективный
инструментарий для компьютерного прогнозирования свойств соединений и
супрамолекулярных комплексов при конструировании новых материалов. Методам 3D
QSAR и их применению для прогнозирования биологической активности соединений
посвящено множество монографий и обзоров [1-7,38].
1.2.1
Методы 3D QSAR
К стандартными методам 3D QSAR можно отнести подходы, в
основе которых лежит предположение о том, что биологическая активность лигандов
обусловлена нековалентным взаимодействием с биологическими мишенями посредством
молекулярных полей. В рамках этих методов для описания таких полей вычисляют
энергию взаимодействия между атомами совмещенных в пространстве (выравненных)
молекул и пробными атомами, помещенными в узлы воображаемой трёхмерной решётки.
Такие энергии взаимодействия рассматривают как потенциалы молекулярных полей.
На основе результатов расчёта формируют матрицу, каждая строка которой отвечает
молекуле лиганда, а каждая колонка - энергии взаимодействия, рассчитанной на
определенном узле решётки. Количественные соотношения между значениями энергий
взаимодействия и значениями биологической активности получают с помощью
статистического анализа на базе методов машинных обучения.
CoMFA. Исторически первым и до сих пор одним из
наиболее распространённых методов 3D QSAR является CoMFA (Comparative Molecular
Field Analysis, метод сравнительного анализа молекулярных полей), разработанный
в 1988 г. Крамером [39]. В рамках этого метода в качестве дескрипторов
используются потенциалы электростатического и стерического полей, рассчитанные
на узлах гипотетической трёхмерной решётки, по умолчанию имеющей шаг 2Å и распространённая на 4Å в каждом направлении от
всех молекул (см. Рис. 2).

Рис. 2. Выравненная база соединений и гипотетическая
трёхмерная решётка, используемая в методе CoMFA
Электростатические и стерические поля обычно считаются
достаточными для описания нековалентных взаимодействий между лигандом и
биологической мишенью. Для расчёта потенциалов электростатического поля в узлы
решётки помещают пробные атомы водорода с зарядом +1 (протон), а для расчёта
потенциалов стерического поля - атомы углерода в sp3-гибридизации.
Электростатические потенциалы рассчитывается по закону Кулона, а стерические -
с использованием потенциала Леннард-Джонса 6-12. В качестве стандартного метода
статистического анализа используется метод частичных наименьших квадратов PLS
(Partial Least Squares). Метод CoMFA реализован в коммерческом программном
продукте SYBYL [Sybyl]. Метод CoMFA описан в ряде монографий [5-7] и обзорных
статей [40,41].
GRID. Программа GRID (Graphic Retrieval and
Information Display) [42] применяется в качестве альтернативы методу CoMFA
[43]. Программа GRID также рассчитывает взаимодействие между молекулой и
пробными атомами, расположенными в узлах трёхмерной решётки, но имеет ряд
преимуществ перед CoMFA: во-первых, вместо потенциалов Леннард-Джонса 6-12
используются более гладкие функции типа 6-4; во-вторых, для описания большого
разнообразия типов межмолекулярного взаимодействия в методе GRID используют
значительно большее число различных пробных атомов и даже групп атомов. В
частности, в дополнение к электростатическим и стерическим потенциалам, программа
вычисляет потенциалы водородной связи и гидрофобный потенциал. В работе [44]
силовое поле GRID использовали в сочетании с программой GOLPE (General Optimal
Linear PLS Estimation) для исследования ингибиторов гликогенфосфорилазы b и
получили хорошие статистические результаты.
CoMSIA. Другой метод, широко используемый в 3D QSAR,
Метод сравнительного анализа индексов молекулярного подобия (Comparative
Molecular Similarity Indices Analysis, CoMSIA) [45] был разработан как развитие
метода CoMFA. Подробное описание этого метода и его модификаций приведено в
работах [46,47]. В рамках этого подхода рассчитываются индексы молекулярного
подобия, которые используются в качестве дескрипторов. Расчёт проводят путём
сравнения каждой молекулы базы с пробными атомами радиуса 1Å с зарядом +1 и гидрофобностью +1, помещёнными в узлах решётки.
Наиболее часто с помощью индексов молекулярного подобия описывают
электростатические, стерические, гидрофобные поля, а также поля водородных
связей. В отличие от CoMFA, для описания потенциалов в этом методе используются
функции Гауссова типа, что позволяет избежать резких изменений при переходе из
одной ячейки к другой и не требует введения ограничительных значений для
потенциалов сверху. Кроме того, модели, полученные методом CoMSIA, легче
интерпретировать визуально.
Описанные методы 3D QSAR предполагают выполнение
определённого числа основных операций [3,19]:
Формирование базы лигандов. На первом этапе формируется база
данных, содержащая структурные формулы соединений и экспериментально
определённые свойства (активности). Для получения QSAR-модели с хорошей
предсказательной способностью важно, чтобы: (а) все лиганды имели одинаковый
механизм связывания с мишенью; (б) значения активностей были получены одним
методом; (в) активности должны быть приведены в одинаковых единицах измерения;
(г) диапазон активностей должен быть насколько возможно более широким,
желательно не меньше трёх логарифмических единиц; (д) желательно, чтобы
значения активностей были разбросаны симметрично относительно среднего
значения.
Генерация 3D-геометрии. Для построения пространственной
геометрии структур используют следующие подходы
· На основе экспериментальных данных. (Для
многих молекул трёхмерная структура определена методом рентгеноструктурного
анализа (РСА) и хранится в базах данных, доступных в электронном виде,
например, Кембриджская кристаллографическая база структурных данных (Cambridge
Crystallographic Structural Database) для малых молекул до 500 атомов или Банк
данных белковых молекул (Protein Data Bank) для полипептидов и полисахаридов).
· Библиотеки фрагментов. 3D-структуру можно
построить на основе фрагментов, собранных в специальные библиотеки. Длины и
углы связей фрагментов предварительно оптимизированы, таким образом, требуется
привести в соответствие лишь значения торсионных углов между фрагментами.
· Автоматическая конвертацией из 2D в 3D.
Часто информация о строении молекулы хранится в одно- или двумерном
представлении, которое необходимо перевести в трёхмерную систему координат.
Такие программы как CONCORD [48] и CORINA[49] генерируют 3D-геометрию с учётом
табулированных значений длин и углов связей.
Оптимизация геометрии. Для оптимизации геометрии применяют
три подхода: (а) методы молекулярной механики; (б) методы квантовой механики
(применяют для молекул с необычным распределением электронной плотности либо в
случае отсутствия необходимых параметров силового поля для конкретной молекулы;
квантово-механические методы являются очень точными, однако их недостатком
является сложность вычисления, не позволяющая применять их для больших
молекул.); (в) гибридные методы, сочетающие эти подходы (применяются для
больших молекул, которые невозможно рассчитать на основе квантовой механики;
часто точное квантово-механическое описание требуется лишь для небольшого
фрагмента молекулы, например, активного центра в ферменте, тогда как остальная
часть молекулы описывается при помощи молекулярной механики.).
Конформационный анализ. Молекулы не являются жёсткими
структурами, и их геометрия находится в процессе постоянного изменения. За счёт
кинетической энергии происходит вращение вокруг одинарных связей, благодаря
чему молекула в разные моменты времени находится в виде разных конформаций,
т.е. пространственных структур, отличающихся значениями торсионных углов. Для
построения модели необходимо привести все лиганды из базы в конформации, в
которых они предположительно связываются с биологической мишенью, - такая
конформация называется биологически активной. Если пространственная структура
мишени известна, найти биологически активную конформацию можно, выполняя докинг
лигандов в мишень. При неизвестной структуре мишени перебирают
низкоэнергетические конформации лигандов. Поскольку не всегда конформация с
наименьшей энергией является биологически активной, отбор последних из
предварительно найденного набора часто ведут путем построения фармакофорных
моделей для рассматриваемого типа биологической активности [50]. Необходимый
для этого перебор низкоэнергетических конформеров может, например, быть
выполнен при помощи систематического поиска, при котором систематически
изменяются значения торсионных углов с получением всех возможных конформаций.
Такая процедура даёт возможность найти все минимумы потенциальной энергии,
однако её недостатком является трудоёмкость, так как с увеличением числа связей
и уменьшением шага вращения число сгенерированных конформаций быстро
возрастает. По этой причине для исследования конформационного пространства
больших и гибких молекул применяется метод Монте-Карло, или метод случайного
поиска, который основан на случайном изменении торсионных углов на каждом шаге.
Также для конформационного анализа гибких молекул часто применяют методы
молекулярной динамики, которая воспроизводит движение молекулы в зависимости от
времени. Применяются также генетические, или эволюционные алгоритмы, которые
основаны на имитации биологической эволюции. На начальном этапе создаётся
популяция решений (конформеров), которые затем подвергаются мутациям, на каждом
шаге определяется энергия, и в случае уменьшения энергии характеристики
конформера, обеспечившие улучшение решения, его характеристики передаются
последующим поколениям конформеров.
Выравнивание базы структур. Для корректного расчета
потенциалов молекулярных полей в узлах решетки необходимо, чтобы все структуры
лигандов были расположены в пространстве единообразно, и группировки атомов
разных лигандов, обладающие сходной функциональностью, совмещались. Выбор
способа выравнивания зависит от структурной гомогенности базы данных.
Совмещение «атом-на-атом» проводится в случае, если все лиганды из базы
обладают общим фрагментом (шаблоном). Каждая молекула совмещается с заранее
заданным шаблоном путём минимизации среднеквадратичного отклонения расстояний
между атомами молекулы и шаблона. В случае отсутствия общего фрагмента у
лигандов можно проводить совмещение не на основе атомного скелета, а на уровне
молекулярных полей. В этом случае изменением пространственной ориентации
молекул добиваются максимального совмещения их молекулярных полей. Связывание
лигандов происходит в полости биологической мишени за счёт наличия в лигандах
структурных элементов со сходной функциональностью. Эти элементы, отвечающие за
наличие у соединения определенного типа биологической активности, называются
фармакофорами. Для выравнивания лигандов также проводят поиск фармакофоров и
изменение пространственной ориентации молекул таким образом, чтобы фармакофоры
накладывались друг на друга. Для того, чтобы избежать проблем, связанных с
выравниванием, разработан ряд методов 3D QSAR, не требующих пространственного
совмещения молекул [51].
1.2.2
Методы 3D QSPR
В различных областях науки и технологий (энергетики,
микроэлектроники, фармацевтики и др.) важной научной и практической задачей
является прогнозирования свойств, связанных с образованием супрамолекулярных
комплексов. В последние 10 лет методы в литературе появился ряд публикаций,
посвященных применению методов 3D QSAR, основанным на использовании
гипотетической решетки, к объектам, отличным от традиционных для этих методов -
межмолекулярных комплексов белок-лиганд. В этих работах исследованы возможности
3D QSPR прогнозирования свойств супрамолекулярных комплексов различной природы.
Например, среди этих публикаций можно отметить работы по прогнозированию
абсорбции красителей на целлюлозном волокне [51,52], и работы по
прогнозированию каталитических свойств металлокомплексных катализаторов,
которые обусловлены свойствами комплексов металлов с органическими лигандами
[53,54].
Следует заметить, что в соответствии с терминологией,
общепринятой в литературе [4], стандартным методами 3D QSAR принято считать
методы, в рамках которых при построении 3D моделей дескрипторы рассчитывают на
основе потенциалов молекулярных полей. Поэтому к методам 3D QSPR мы относим
методы, в которых используют подход аналогичный стандартными методами 3D QSAR,
в отличие от методов QSPR, в которых для расчёта 3D дескрипторов исследователи
применяли геометрические или энергетические характеристики. Публикации,
посвященные последним, подробно описаны в обзорах [9,32].
Полученные результаты демонстрируют перспективность
применения методологии 3D QSAR/QSPR для целей прогнозирования свойств
супрамолекулярных комплексов различной природы.
1.3
Концепция непрерывных молекулярных полей
Как уже отмечено выше, стандартные методы 3D QSAR основаны на
аппроксимации молекулярных полей потенциалами, которые рассчитываются в узлах
трёхмерной пространственной решётки и используются в последующем статистическом
анализе в качестве дескрипторов [5-7,39]. Такой подход обладает рядом
существенных недостатков. Во-первых, статистические модели оказываются
чувствительным к пространственной ориентации и шагу решётки. Во-вторых,
дискретные наборы дескрипторов не могут с достаточной точностью описать
молекулярные поля, которые являются непрерывными физическими объектами.
Уменьшения шага решётки возможно лишь до определённого предела, ниже которого
статистические параметры модели вновь ухудшаются из-за появления очень большого
числа дескрипторов, что ведёт к оверфиттингу (переподгонке). Увеличение шага
решётки ведёт снижению числа дескрипторов, но при этом большой объём важной
информации теряется.
Альтернативный подход на основе непрерывных молекулярных
полей был предложен в работах [56,57]. Суть его заключается в проведении
статистического анализа молекулярных объектов, представленных не в виде набора
дискетных дескрипторов, а в виде непрерывных гладких функций от
пространственных координат (т.н. непрерывных молекулярных полей). Авторами было
показано, что это может быть достигнуто путем конструирования специальных
ядерных функций в Гильбертовом пространстве и их использования для построения
регрессионных и классификационных моделей в рамках таких методов машинного
обучения как регрессионный метод опорных векторов, ядерная гребневая регрессия
и одноклассовая машина опорных векторов. Описание на основе непрерывных
молекулярных полей точнее соответствует физической природе взаимодействий между
мишенью и лигандом. Применение концепции непрерывных молекулярных полей на
практике стало возможным благодаря развитию методов машинного обучения с
использованием ядер (kernels) вместо векторов дескрипторов фиксированного
размера. Такая статистическая модель содержит не дискретный набор конечного
числа параметров, а непрерывное поле регрессионных коэффициентов [58].
Предложенный подход можно считать первым в мировой практике
примером применения статистических методов анализа функциональных данных в
хемоинформатике. В настоящее время в области хемоинформатики имеется очень
ограниченное число публикаций эвристического характера, касающихся методов
работы с непрерывными молекулярными полями. Так, в работах [59,60] описаны
индексы молекулярного сходства Карбо, которые вычисляются как интеграл
произведения функций электронной плотности для пары молекул. Их использование
для целей QSAR можно рассматривать как частный случай применения непрерывных
молекулярных полей без использования, однако, возможностей дуальных ядерных
методов машинного обучения. В работе [61] на основе концепции, внешне сходной
со способом сравнения молекул при помощи непрерывных молекулярных полей,
предложен метод проведения стерического и электронного совмещения структур
молекул. Абсолютное же большинство работ, касающихся прогнозирования свойств
соединений с использованием ядерных методов машинного обучения, например,
мaшины опорных векторов [62] основано на описании молекул с помощью векторов
признаков ограниченного и фиксированного размера и не используют способность
этих методов оперировать с функциональными данными, т.е. с фактически
бесконечным числом переменных.
В группе хемоинформатики физического факультета МГУ под
руководством И.И. Баскина предложенный подход реализован в рамках пакета
программ CMF (Continuous Molecular Fields, метод непрерывных молекулярных
полей). Математическое обоснование методологии непрерывных молекулярных полей
описано в работе [56].
1.3.1
Методология непрерывных молекулярных полей
В основе метода непрерывных молекулярных полей лежит расчёт
ядер (kernels) молекулярных полей. Ядро K(Mi, Mj)
описывает сходство молекулярных полей молекул Mi и Mj и
рассчитывается как линейная комбинация ядер, отвечающих каждому типу полей:
 (1)
(1)
где hf - коэффициент смешения для f-го типа поля, Kf(Mi,
Mj) - ядро, описывающее сходство между полем f-го типа молекул i и
j. Оно рассчитывается как сумма ядер всех пар атомов молекул i и j:
 (2)
(2)
где ядро kf(Ail, Ajm) описывает
сходство между полями f-го типа l-го атома i-й молекулы и m-го атома j-й
молекулы. Его значение вычисляют путём интегрирования произведения полей f-го
типа для l-го атома i-й молекулы и m-го атома j-й молекулы по всему трёхмерному
пространству:
 (3)
(3)
где  - значение поля f-го типа для l-го атома
i-й молекулы в точке с радиус-вектором r;
- значение поля f-го типа для l-го атома
i-й молекулы в точке с радиус-вектором r;  - значение для m-го атома j-й молекулы. Любое молекулярное поле
может быть представлено с помощью одной функции Гаусса:
- значение для m-го атома j-й молекулы. Любое молекулярное поле
может быть представлено с помощью одной функции Гаусса:
 (4)
(4)
где wfil - вес вклада l-го атома i-й молекулы в поле
f-го типа; αf - фактор аттенуации для поля f-го типа,
показывает ширину кривой Гаусса; ril - радиус-вектор l-го
атома i-й молекулы. В методе CMF задаётся различная параметризация wfil
для разных типов полей, например, для электростатического поля wfil
представляет собой частичный заряд l-го атома на i-й молекуле, для стерического
поля - величину:
 (4.1)
(4.1)
где  - ван-дер-ваальсова энергия;
- ван-дер-ваальсова энергия;  - ван-дер-ваальсов радиус. Эти параметры
берутся из силового поля Tripos. Гидрофобное молекулярное поле задаётся
коэффициентами wfil, представляющими собой вклады атомов данного
типа в величину гидрофобности молекулы. Поля кислотности и основности по
отношению к образованию водородной связи задаются коэффициентами wfil,
представляющими собой вклады атомов данного типа в величины констант Абрахама A
и B [63].
- ван-дер-ваальсов радиус. Эти параметры
берутся из силового поля Tripos. Гидрофобное молекулярное поле задаётся
коэффициентами wfil, представляющими собой вклады атомов данного
типа в величину гидрофобности молекулы. Поля кислотности и основности по
отношению к образованию водородной связи задаются коэффициентами wfil,
представляющими собой вклады атомов данного типа в величины констант Абрахама A
и B [63].
Аппроксимация молекулярного поля функцией Гаусса даёт возможность
вычислить ядро kf(Ail, Ajm) аналитически:
 =
=  (5)
(5)
В случае стерического поля выражение (5) несколько видоизменяется:
=
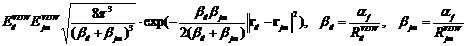 (5.1)
(5.1)
После вычисления ядра K(Mi, Mj) его можно
использовать для построения модели при помощи одного из методов машинного
обучения, приспособленных для работы с бесконечным числом переменных, таким как
метод опорных векторов (Support Vector Machine, SVM), ядерная гребневая
регрессия (Kernel Ridge Regression, KRR), ядерный вариант метода частичных
наименьших квадратов (Kernel Partial Least Squares, KPLS) и др. В общем виде
уравнение регрессии для свойства yt может быть записано в виде:
 (6)
(6)
Схема графической визуализации моделей CMF показана на Рис.
3.

Рис. 3. Визуализация молекулярных полей и полей регрессионных
коэффициентов
Теоретические аспекты рассматриваемой методологии построения
моделей SAR/QSAR/QSPR, в том числе методы построения ядер на основе
молекулярных полей, визуализации молекулярных полей и соответствующих полей
регрессионных коэффициентов, возможности применения методов статистического
анализа функциональных данных для прогнозирования свойств химических соединений
с использованием непрерывных полей при построении двух- и многоклассовых
классификационных моделей, а также перспективы применения этой методологии
построения моделей для виртуального скрининга биологически активных соединений
изложены также в работах [64-67].
1.3.2
Исследование биологической активности с использованием метода непрерывных
молекулярных полей
Применению методологии непрерывных молекулярных полей для
решения задач прогнозирования биологической активности соединений посвящен ряд
работ. В работах [56,57] метод непрерывных молекулярных полей с использованием
статистического аппарата регрессии на опорных векторах применяли для
прогнозирования биологической активности ингибиторов ферментов. Показано
преимущество этой методологии по сравнению со стандартными методами 3D QSAR в
отношении прогнозирования ингибирующей активности производных
3-амидинофенилаланина по отношению к трём ферментам подгруппы сериновых
протеаз: трипсину, тромбину и фактору Ха. Модели, построенные с помощью
методологии CMF, показали более высокую прогнозирующую способность на
скользящем контроле, чем модели, полученные стандартными методами CoMFA и
CoMSIA.
В работах [68,69] исследована возможность применения
одноклассовой классификации в сочетании с методом непрерывных молекулярных
полей для проведения виртуального скрининга химических соединений. Апробация
метода проводилась на базе DUD [70], для построения одноклассовых моделей по
методу 1-SVM [71] использовалась программа LIBSVM [72]. Значения
электростатических, гидрофобных и стерических ядер рассчитывали в соответствии
с изложенной выше методологией. В качестве статистического ядра использовали
суперпозицию электростатического, гидрофобного и стерического ядер. Оптимальные
значения параметров одноклассового классификатора как для комбинации ядер, так
и каждого ядра в отдельности находили путем максимизации площади под
ROC-кривой. Неактивные аналоги применяли для оценки прогнозирующей способности
моделей. Высокие значения площадей под ROC-кривыми построенных моделей
свидетельствуют в пользу работоспособности предлагаемого метода. Сделан вывод,
что использование непрерывных полей для сравнения структур позволяет применять
построенные модели для поиска принципиально новых лекарственных препаратов с
новыми мотивами расположения фармакофоров. Достоинством подхода являются его
малая чувствительность к выбору контр-примеров, возможность объединения в
рамках одной модели соединений, относящихся к разным структурным классам и
поиска потенциально активных лигандов с различным расположением фармакофорных
групп.
В работах [73,74] впервые в рамках одноклассового подхода с
использованием метода опорных векторов получены серии регрессионных и
одноклассовых SVM-моделей, демонстрирующие перспективность данного подхода для
виртуального скринига потенциальных ингбиторов обратной транскриптазы ВИЧ-1.
Для построения одноклассовой модели в этих работах использовали одноклассовый
метод опорных векторов (1-SVM) [71]. База данных включала сведения о lg(EC50)
для 1045, 330, 330 и 128 ненуклеозидных ингибиторов обратной транскриптазы
вируса ВИЧ-1 относительно 4-х штаммов - дикого и мутантных К103N, Y188L и
IRLL98 - соответственно. Показано, что представление молекулярной структуры
органических лигандов на основе непрерывных молекулярных полей позволяет
получать классификационные модели более высокого качества по сравнению с
подходами, базирующимися на использовании «молекулярных отпечатков»,
спектрофоров [75] и фрагментных дескрипторов Кархарта [76]. Наилучшие модели,
построенные на основе непрерывных молекулярных полей, имеют статистические показатели,
близкие к идеальному классификатору. Эти модели авторы рекомендовали для
проведения широкомасштабного виртуального скрининга. Разработанное авторами
сочетание методологии непрерывных молекулярных полей с методами одноклассовой
классификации является совершенно уникальным инструментом, не имеющим близкого
аналога в мировой практике. Примером дальних аналогов являются методы
виртуального скрининга, осуществляемые при помощи рассмотрения близости
молекулярных форм и фармакофорного описания [77]. Недостатками подобных методов
являются: использование только одного представителя активных соединений в виде
шаблона, отсутствие стадии обучения и невозможность оптимизации метрики,
определяющей близость молекул в химическом пространстве. Подход, сочетающий метод
непрерывных молекулярных полей и метод одноклассовой классификации свободен от
этих недостатков, что и объясняет значительно более высокие значения площади
под ROC-кривой для этого сочетания. Ещё одним дальним аналогом является метод
виртуального скрининга химических соединений на основе нейросетей Кохонена,
решающих задачу одноклассовой классификации [78]. Данная методология, однако,
не позволяет использовать описание молекул при помощи молекулярных полей, не
говоря уже о визуализации и интерпретации моделей.
1.4
Поиск количественных соотношений «структура-свойство» для комплексов
органических лигандов с Am+3 и Eu+3
В настоящее время одной из практически значимых экологических
проблем является проблема переработки ядерных отходов [79]. На сегодняшний день
в мире функционирует около 440 ядерных энергетических реакторов, в результате
работы которых выделяется отработанное ядерное топливо с высокой остаточной
радиоактивностью, обусловленной, в том числе, содержанием в нём минорных
актинидов. Для их удаления наиболее перспективными являются экстракционные
методы. Выделение минорных актинидов (например, Np, Am, Сm), однако,
осложняется присутствием в топливе редкоземельных элементов, обладающих
сходными химическими свойствами (Eu и др). Решение проблемы состоит в поиске
высокоселективных лигандов, образующих устойчивые комплексы с разделяемыми
элементами. Недостатками существующих лигандов являются их низкая
селективность, неустойчивость к окислению и радиационному воздействию и т.д.
[80]. Трудности при разработке высокоселективных лигандов связаны с наличием в
растворе сложных внутри- и межмолекулярных взаимодействий в комплексах металл -
лиганд [81,82]. Поэтому особое значение для направленного синтеза лигандов
имеет их конструирование с использованием теоретических методов.
В настоящее время теоретический анализ комплексов
лиганд-металл осуществляется почти исключительно методами молекулярного
моделирования, основанными на квантово-химических вычислениях [83] и / или на
применении метода молекулярной динамики с использованием эмпирических силовых
полей (см., например, [84]). Оба подхода являются, однако, чрезвычайно
трудоёмкими и поэтому не дают возможность осуществлять направленный дизайн
таких супрамолекулярных систем с использованием методов виртуального скрининга
либо de novo дизайна, требующих перебора очень большого числа возможностей.
Этого недостатка лишены методы QSPR с применением современных методов машинного
обучения [85,86]. В этом направлении в последние годы были достигнуты
определённые успехи.
Следует заметить, моделирование экстрагирующей способности
лигандов проводиться, как правило, раздельно для процессов комплексообразования
и экстракции. Поскольку константа стабильности комплекса в воде (logK) и
константа экстракции (logKex) могут быть связаны через коэффициенты
распределения свободных лигандов и их комплексов в водной и органической фазах
[87], на практике коэффициенты распределения часто бывают недоступны, поэтому
связь между logK и logKex редко используется.
Первыми попытками прогнозирования способности лигандов к
образованию комплексов с металлами было исследование эмпирических
корреляционных соотношений [88]. Были получены линейные корреляции способности
к комплексообразованию со свободной энергией (Linear free energy relationships,
LFER) и параметрические уравнения. LFER для серии металлов Mi при
одинаковых лигандах L1 и L2 имело следующий вид:
(MiL1) = a * logK(MiL2)
+ b (7)
тогда как для серии лигандов Li при одинаковых
металлах M1 и M2:
(M1Li) = a * logK(M2Li)
+ b (8)
Для построения параметрических уравнений использовали
различные функциональные зависимости способности к комплексообразованию от
свойств металлов, включая заряд, ионный радиус, электроотрицательность и
ионизационный потенциал [89].
В работе [90] предложено более сложное уравнение,
представляющее logK (ML) как функцию от характеристик металла и лиганда:
(K (M1L) / K (M2L)) = αEn + βH (9)
где параметры α и En -
мягкость, β и H - жёсткость для металла и лиганда
соответственно.
Попытки прогнозирования устойчивости комплексов лигандов Ca+2
с помощью 2D QSAR были предприняты в работе [91], в которой, наряду с
топологическими и физико-химическими дескрипторами, применяли молекулярные
фрагменты. Однако полученные модели не имели высокой прогнозирующей способности
(R2pred = 0.4).
Варнеком с соавторами был построен ряд 2D QSPR моделей,
связывающих комплексообразующую способность комплексонов некоторых классов
относительно металлов из групп лантанидов и актинидов со структурой
органического лиганда [92]. Все эти модели основаны на применении метода
множественной линейной регрессии с отбором переменных в сочетании с
использованием фрагментных дескрипторов [93]. Построенные модели позволили
осуществить направленный дизайн молекул комплексонов на основе процедуры
виртуального скрининга сгенерированной виртуальной комбинаторной библиотеки
молекул органического лиганда [94,95]. При этом, однако, были выявлены
существенные недостатки этого подхода. Из-за чисто топологического характера
используемых дескрипторов электронное влияние атомов и пространственные факторы
оказываются учтенными лишь косвенно, что делает область применимость
построенных моделей очень узкой и препятствует дизайну комплексонов с
принципиально новой структурой.
В работах [96,97] методами 2D QSPR моделировали фактор
разделения (Separation Factor, SF) америция и европия для набора, включающего
47 полиазагетероциклических лигандов. В работе [96] получены модели на основе
топологических дескрипторов с учётом некоторых характеристик атомов
(электроотрицательность, поляризуемость).
В другой работе [97] при построении 2D QSPR моделей
использовали набор линейных и нелинейных методов в рамках трех программ -
Cerius2, ISIDA (In SIlico Design and Data Analysis) и CODESSA-PRO
(COmprehensive DEscriptors for Structural and Statistical Analysis). Для
описания структур авторы использовали два вида дескрипторов: подструктурные
молекулярные фрагменты (substructural molecular fragments, SMF), рассчитанные
программой ISIDA, и молекулярные дескрипторы, рассчитанные программой CODESSA-PRO.
В качестве фрагментных дескрипторов (ISIDA) использовали «последовательности»,
которые могли содержать атомы и связи, только атомы или только связи, и
«расширенные атомы», представляющие отдельный атом с окружением. С помощью
программы CODESSA-PRO были вычислены различные классы молекулярных
дескрипторов: структурные, геометрические, топологические, электростатические,
квантовые, химические и термодинамические. При построении статистически
значимой модели проводился отбор дескрипторов. В программе ISIDA для этой цели
применяли процедура t-test, а в CODESSA-PRO - «best multi-linear regression».
Для нелинейного анализа моделей использовали алгоритмы Radial Basis Function
Neural Networks (RBFNN) и Associated Neural Networks (ASNN).
Статистичеcкие характеристики 2D QSPR моделей для
прогнозирования фактора разделения комплексов полиазагетероциклических лигандов
с Eu+3 и Am+3 представлены в Табл. 1.

Рис. 4. Фактор разделения (logSF) америция и европия для
t-Bu-hemi-BTP, рассчитанный: (1) по моделям [96] (0.75 - 5.13); (2) по моделям
[97] (1.07 - 1.46). Экспериментальное значение logSF=1.0
Несмотря на хорошее качество некоторых моделей, они не
показали удовлетворительную прогнозирующую способность в отношении синтезированного
лиганда t-Bu-hemi-BTP (logSF = 1.0). В первом случае был получен разброс
значений logSF в диапазоне 0.75 - 5.13, во втором более близкие значения 1.07 -
1.46.
Преодолеть недостатки подходов 2D QSPR можно только переходя
к построению т.н. 3D QSPR (3D Quantitative Structure Property Relationships)
моделей, основанных на явном рассмотрении геометрического строения молекул и
полей, описывающих электронное влияние атомов. Эти методы, однако, до сих пор
ни разу не применялись для прогнозирования свойств низкомолекулярных
супрамолекулярных систем, таких как комплексы металлов с органическими
лигандами. Более того, без существенных модификаций они и не могут быть
использованы для этой цели.
Таким образом, в настоящее время при конструировании
лекарственных препаратов и количественном прогнозировании биологической
активности органических соединений большое значение имеют методы 3D QSAR. Эти
методы основаны на анализе и сопоставлении пространственных структур молекул и
поиске количественных соотношений между пространственными структурами молекул и
проявляемой ими биологической активностью. Кроме того, аналогичные им методы 3D
QSPR в последние годы начали использоваться для прогнозирования каталитических
свойств в металлокомплексном катализе. Кроме того, имеются отдельные
публикации, свидетельствующие о возможности применения методов 3D QSPR для
прогнозирования адсорбционной способности красителей на целлюлозном волокне.
Отметим, что во всех вышеупомянутых случаях речь идет о прогнозировании
свойств, связанных с образованием и свойствами супрамолекулярных комплексов. В
частности, биологическая активность молекул лекарств в большинстве случаев
обусловлена образованием межмолекулярных комплексов белок-лиганд,
каталитическая активность в металлокомплексном катализе связана со свойствами
комплексом металлов с органическими лигандами, а абсорбция красителей на
целлюлозном волокне также обусловлена образованием сложных межмолекулярных
комплексов. Мы предполагаем, что это связано с тем, что при образовании
супрамолекулярных комплексов большую роль играют не только электронные
характеристики молекул, но и особенности их пространственного строения. Отсюда
естественным образом вытекает идея о том, что методы 3D QSAR/QSPR с успехом
могли бы быть применены при прогнозировании и других свойств, связанных с
образованием комплексов. В частности, для нас представлял особый практический
интерес распространение разрабатываемого метода непрерывных молекулярных полей,
реализующего 3D QSAR/QSPR-анализ, на прогнозирование способности органических
комплексонов проводить разделение ионов Am+3 и Eu+3, чему
посвящена заключительная часть настоящей дипломной работы. Осуществление этого
потребовало от нас разработки нового класса молекулярных полей как альтернативу
либо возможное дополнение существующему набору физико-химических полей.
Действительно, в существующих методах 3D QSAR для описания
молекулярных объектов традиционно используют молекулярные поля
физико-химической природы, в частности, электростатическое, стерическое и
гидрофобное поля, а также поля, описывающие образование водородных связей.
Отметим, однако, три существенных недостатка у такого подхода.
Во-первых, предложено множество разнообразных способов
аппроксимации таких полей, и все они приводят к построению различных моделей 3D
QSAR и, как следствие, к их неоднозначности и плохой воспроизводимости.
Например, известно множество способов расчета значений частичных зарядов на
атомах, имеется ряд методов для расчета липофильности, используемой при
описании гидрофобного поля, и т.д.
Во-вторых, стандартный 3D QSAR набор физико-химических полей
наилучшим образом приспособлен для описания взаимодействий лигандов с
биологическими мишенями, и, естественно, не является оптимальным выбором для
прогнозирования других типов свойств, например, связанных с образованием
комплексов органических лигандов с ионами металлов.
В третьих, модели, построенные на основе физико-химических
полей, не обеспечивают их структурно-химической интерпретации. Поэтому
представляла интерес замена набора молекулярных полей физико-химической природы
более универсальным набором функций («молекулярных полей»), позволяющих
описывать произвольные свойства молекул, зависящие от различных типов
взаимодействий, и позволяющих интерпретировать QSAR/QSPR модели на структурном
уровне.
В задачу дипломной работы входила разработка такого набора
функций и программного обеспечения на его основе в рамках метода непрерывных
молекулярных полей (CMF). Представляла также интерес оценка применимости
разработанного подхода для прогнозирования различных свойств, обусловленных
образованием супрамолекулярных комплексов - биологической активности
органических лигандов в отношении фармакологических мишеней, а также фактора
разделения комплексов органических лигандов с катионами Am+3 и Eu+3.
2.
Обсуждение результатов
.1
Построение моделей 3D QSAR/QSPR на основе функций принадлежности точки атомным
типам (непрерывных индикаторных полей)
В рамках выполнения задач дипломной работы в качестве
универсального набора функций для описания свойств молекул нами были предложены
и исследованы функции, определяющие меру принадлежности точки с заданными
координатами атому определённого типа, или функции принадлежности атому. Эти
функции представляют собой принципиально новый тип непрерывных молекулярных
полей, для которого мы предложили название - непрерывные индикаторные поля
(НИП, CIF - continuous indicator fields). Их дальним прототипом являются
атомные индикаторные переменные, значения которых на узле решётки равно
единице, если узел находится внутри какого-либо атома в молекуле, и нулю в
противоположном случае [98]. При этом рассматривается принадлежность
расположенной на узле решетки точки любому атому независимо от его типа.
Введённые таким способом индикаторные переменные были использованы в качестве
дескрипторов при построении моделей 3D QSAR/QSPR. Замена такими индикаторными
переменными 6-12 потенциалов стерического поля, рассчитанных с использованием
потенциала Леннард-Джонса, позволила авторам повысить качество 3D QSAR моделей,
полученных для 256 ингибиторов дигидрофолат - редуктазы, относительно
стандартных моделей CoMFA.
В основе упомянутого подхода лежит
предположение о том, что радиусы атомов имеют конечные размеры. В реальности,
однако, атомы чётких границ не имеют, поэтому вместо бинарных (0 или 1) индикаторных
переменных мы предлагаем использовать непрерывную функцию принадлежности точки
с координатами r атому с номером i, которая может принимать значения в
интервале от 0 до 1:
 (10)
(10)
Впервые такие непрерывные функции принадлежности атомам были
введены Хиршфельдом в рамках разработанного им подхода к анализу функции
электронной плотности молекул, что позволило разбить общую электронную
плотность молекулы на вклады атомов и тем самым оценить на них частичные заряды
[99]:
 , (11)
, (11)
где  - функция электронной плотности i-ого атома в «свободном»
состоянии (т.е. когда все остальные атомы в молекуле от него удалены на
бесконечное расстояние),
- функция электронной плотности i-ого атома в «свободном»
состоянии (т.е. когда все остальные атомы в молекуле от него удалены на
бесконечное расстояние),  - функция электронной плотности «промолекулы», которая
представляет собой сумму электронных плотностей всех входящих в молекулу атомов
в «свободном» состоянии. Поскольку форма функции принадлежности атому похожа на
Гауссову, в наших расчетах мы ее аппроксимируем одной Гауссовой функцией:
- функция электронной плотности «промолекулы», которая
представляет собой сумму электронных плотностей всех входящих в молекулу атомов
в «свободном» состоянии. Поскольку форма функции принадлежности атому похожа на
Гауссову, в наших расчетах мы ее аппроксимируем одной Гауссовой функцией:
 (12)
(12)
В рамках метода непрерывных молекулярных полей поле типа f
для молекулы аппроксимируется следующим образом:
 , (13)
, (13)
где wfi - взвешивающий коэффициент, определяющий
индивидуальность поля. Например, для электростатического поля он равен
частичному заряду на атоме i, а для гидрофобного поля - вкладу атома i в
гидрофобность молекулы. Таким образом, вклад атома i в молекулярное поле f
определяется набором коэффициентов wfi для разных типов молекулярных
полей.
Значения коэффициентов wfi могут быть табулированы
для определённого молекулярно-механического типа атома:
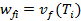 (14)
(14)
где Ti - молекулярно-механический тип атома i.
Подставляя (5) в (4), получаем:
 , (15)
, (15)
где внешнее суммирование ведётся по всем молекулярно-механическим
типам атомов в молекуле, а внутреннее - по атомом в молекуле, относящихся к
данному молекулярно-механическому типу, 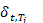 - дельта-функция, равная единице, если молекулярно-механический
тип атома i равен t, и нулю в противном случае. Функция принадлежности атома i
молекулярно-механическому атомному типу t может быть определена следующим
образом:
- дельта-функция, равная единице, если молекулярно-механический
тип атома i равен t, и нулю в противном случае. Функция принадлежности атома i
молекулярно-механическому атомному типу t может быть определена следующим
образом:
 (16)
(16)
Тогда:
 . (17)
. (17)
Таким образом, любое молекулярное поле Xf(r)
может быть представлено как линейная комбинация функций принадлежности атомным
типам 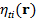 , и введённые нами поля - функции принадлежности атомным типам -
определяют универсальный набор полей, который может быть использован вместо
стандартных физико-химических полей для построения моделей 3D QSAR/QSPR.
, и введённые нами поля - функции принадлежности атомным типам -
определяют универсальный набор полей, который может быть использован вместо
стандартных физико-химических полей для построения моделей 3D QSAR/QSPR.
Иными словами, вместо бинарных индикаторных переменных мы
предлагаем использовать непрерывную функцию, показывающую меру принадлежности
(в интервале от 0 до 1) произвольной точки пространства атому определённого
молекулярно-механического типа. Именно это и есть «функция принадлежности
атомным типам» или НИП. Это функция от пространственных координат точки. Можно
предположить, что преимущество использования НИП при построении моделей 3D QSPR
будет проявляться тогда, когда стандартного набора полей недостаточно для
адекватного описания межмолекулярных взаимодействий, ответственных за
проявление моделируемого свойства. Кроме того, с помощью НИП можно описать
моделируемую зависимость со структурной точки зрения. В частности, поля
регрессионных коэффициентов таких моделей должны показывать, какие изменения на
структурном уровне надо ввести для модификации проявляемого молекулой свойства.
Под молекулярно-механическим типом атома понимается тип,
присвоенный атому в рамках определенного силового поля, например Tripos, Amber,
MM3 и др [19]. В наших расчётах мы использовали силовое поле, разработанное
фирмой Tripos (параметризация силовых полей Tripos приведена в Приложении, п.
1). Например, в силовом поле Tripos sp2-гибридизованному углероду
соответствует тип C.2, а sp3-гибридизованному углероду - тип C.3.
Для этих типов атомов в рамках силовых полей имеется набор табулированных
значений: радиусы Ван-дер-Ваальса, длины связей с их участием, жёсткость связей
с их участием, валентные углы, жёсткость валентных углов и др.
В рамках дипломной работы было создано программное
обеспечение для построения 3D QSAR/QSPR моделей на основе НИП и ядерной
гребневой регрессии. Описание программного обеспечения приведено в
Экспериментальной части.
2.1.1
3D QSAR моделирование биологической активности органических лигандов в
отношении фармакологических мишеней с использованием непрерывных индикаторных
полей
С использованием созданного программного обеспечения были
исследована активность органических лигандов супрамолекулярных комплексов по
отношению к различным фармакологическим мишеням. Исследовали 8 выборок
лигандов, традиционно используемых в исследованиях для сравнения возможностей
различных методов 3D QSAR [100-102]. Наборы содержали данные о структурах органических
лигандов, принадлежащих различным группам, а также сведения об их биологической
активности. Данные о структурах и активностях соединений выборок были взяты из
работы [102]. Наборы включали:
) 114 ингибиторов ангиотензинпревращающего фермента (ACE),
принадлежащих к 28 различным структурным типам [103];
) 111 ингибиторов ацетилхолинэстеразы (AСhE) [104];
) 163 лиганда бензодиазепинового рецептора (BZR) -
производных 1,4 - бензодиазепин-2-она [105];
) 322 ингибитора циклооксигеназы-2 (COX2) - производных 9
классов соединений: пирролы, имидазолы, циклопентены, бензолы, пиразолы,
спирогептены, спирогептадиены, изоксазолы и тиофены [106];
) 397 ингибиторов дигидрофолат-редуктазы (DHFR), содержащих
2,4 - диаминопиримидиновое кольцо[107];
) 66 ингибиторов гликогенфосфорилазы b (GPB) [108];
) 76 ингибиторов термолизина (THER) [109];
) 88 ингибиторов тромбина (THR) [47].
Структуры характерных представителей соединений различных
классов для каждого из исследованных наборов показаны на Рис. 5.

Рис. 5. Структуры характерных органических лигандов для
каждой из 8 баз: (А) эналаприл (ACE); (B) E2020 (AChE); (C) Ro14-5974 (BZR);
(D) целекоксиб (COX2); (E) метотрексат (DHFR); (F) спирогидантоин глюкопираноза
(GPB); (G) ZPLA (THER); (H) нафто-производное 4-TAPAP (THR)
При построении моделей на основе НИП для каждой из 8 баз
формировали соответствующую обучающую выборку, которую использовали для
построения моделей, и контрольную выборку, которая служила для независимой
оценки их прогнозирующей способности. Разбиение на выборки было сделано
согласно данными работы [102], в которой примерно 33% соединений были отобраны
в качестве контрольных выборок, остальные соединения были включены в обучающие
выборки. В Табл. 2 представлено разбиение лигандов на обучающие и контрольные
выборки для восьми баз данных.
Данные о пространственной геометрии и выравненных
пространственных структурах соединений были взяты из работы [102]. В этой
работе структуры лигандов были построены с помощью программы SYBYL на основе
фрагментов, минимизацию проводили с использованием силового поля TRIPOS.
В качестве моделируемых активностей для баз ACE, AChE, BZR,
COX2 и DHFR исследовали отрицательный логарифм концентрации ингибитора,
соответствующей 50% активности лиганда - log(IC50), для баз GPB,
THERM и THR - отрицательный логарифм константы связывания лиганда с
биологической мишенью - log(Ki).
Характеристики 3D QSAR моделей, полученных для восьми наборов
лигандов с помощью методов KRR1 и KRR2 и непрерывных индикаторных полей.
Формулы расчёта статистических параметров представлены в Экспериментальной
части.
Как видно из Табл. 4, для пяти из восьми исследованных баз
(AChE, BZR, COX2, GPB и THR) модели, полученные методом KRR1 (с оптимизацией
коэффициентов смешения hf), имеют сравнимые или более высокие
значения параметра q2, описывающем прогнозирующую способность в
условиях скользящего контроля, чем модели, полученные методом KRR2 (с
оптимизацией и без оптимизации коэффициентов смешения hf) и KRR1
(без оптимизации hf), соответственно - 0.67, 0.48, 0.56, 0.85, 0.77.
Эти модели имеют более низкие или равные значения среднеквадратичной ошибки
прогнозирования при скользящем контроле RMSEcv, соответственно -
0.70, 0.48, 0.67, 0.42, 0.46. Для баз АCE, DHFR и THERM значения q2 моделей,
полученные методами KRR1 (с оптимизацией и без оптимизации hf),
немного ниже, чем для моделей, полученных методом KRR2 (с оптимизацией hf),
и, соответственно, равны 0.78 и 0.79, 0.71. и 0.72, а также 0.77 и 0.79.
Соответственно, RMSEcv для этих моделей - 1.10 и 1.26, 0.68 и 0.70,
0.91 и 1.14.
Наиболее высокой прогнозирующей способностью на независимой
контрольной выборке характеризуются модели, полученные методом KRR2 (без
оптимизации hf) для 7 баз (ACE, BZR, COX2, GPB, DHFR, THERM и THR).
Для этих баз значения R2pred равны, соответственно -
0.56, 0.12, 0.13, 0.63, 0.58, 0.35, 0.35. Значения тех же параметров моделей,
полученных методами KRR2 (с оптимизацией hf) и KRR1 (как с
оптимизацией hf, так и без неё), значительно хуже. Только в случае
базы AChE значения R2pred модели (0.52), полученной
методом KRR1 (с оптимизацией hf и без нее), выше по сравнению с
моделью, построенной методом KRR2 (с оптимизацией hf). В целом же
можно сделать вывод о том, что метод KRR2 без оптимизации коэффициентов
смешения приводит к построению моделей с наилучшей прогнозирующей способностью
на независимой выборке.
Полученные результаты можно интерпретировать следующим
образом. При оптимизации коэффициентов смешения (hf) индикаторных
полей, общее число настраиваемых параметров модели значительно превышает число
параметров модели, построенной без их оптимизации, когда значения коэффициентов
смешения полей приняты постоянными. Эта разница равна числу типов атомов в параметризации
Tripos, присутствующих в молекулах обучающей выборки. Поскольку эта разница в
числе параметров весьма существенна, дополнительная ошибка прогнозирования,
возникающая при оптимизации hf вследствие оферфиттинга, в
большинстве случаев может превышать ошибку прогнозирования, возникающую при
отсутствии минимизации вследствие использования менее гибкой функции. Поэтому в
большинстве случаев оптимизация параметров смешения, хотя и приводит к
улучшению статистических характеристик, характеризующих прогнозирующую
способность в условиях скользящего контроля, тем не менее, ведёт к ухудшению
показателей, описывающих прогнозирующую способность на независимой выборке. С
другой стороны, наличие лишь одного дополнительного настраиваемого параметра в
KRR2 (свободного члена b) по сравнению с KRR1 ведёт лишь к незначительному
увеличению возможности проявления оверфиттинга, что в большинстве случаев не
может перевесить неблагоприятного эффекта, возникающего в KRR1 вследствие
отсутствия настраиваемого свободного члена. Поэтому в среднем прогнозирующая
способность моделей на контрольной выборке для моделей, построенных с помощью
KRR2 выше, чем при использовании KRR1.
Решение проблемы оверфиттинга является первоочередной задачей
будущего продолжения исследований.
Показатели моделей, полученных методами CoMFA, CoMSIA1 и
CoMSIA2, были взяты из работы [102]. В методе CoMFA было использовано 2 типа
молекулярных полей - электростатическое и стерическое. В CoMSIA1 использовали
электростатическое и стерическое молекулярные поля, в CoMSIA2 к ним добавлены
гидрофобное и поля водородных связей.лучшие модели, построенные на основе НИП
для 3 баз данных (ACE AChE и DHFR) имеют значения статистических характеристик,
описывающих прогнозирующую способность, как в условиях скользящего контроля,
так и на независимой выборке, превышающие соответствующие показатели лучших
моделей, полученных для этих же баз с помощью стандартных методов 3D-QSAR:
CoMFA, CoMSIA1 и CoMSIA2 [102].
В случае наборов BZR и GPB CMF-модели имеют более высокие
показатели на скользящем контроле по сравнению с вышеупомянутыми стандартными
методами 3D QSAR, a прогнозирующая способность этих моделей выше, чем у
соответствующих моделей CoMFA и CoMSIA1, и сравнима с аналогичными показателями
для моделей CoMSIA2. Так, для BZR значения R2pred моделей
CMF и CoMSIA2 равны 0.12, а для GPB аналогичные показатели составляют,
соответственно, 0.58 и 0.59.
Для базы COX2 при оценке предсказательной способности моделей
метод CMF показал лучшие результаты (R2pred = 0.13, RMSEpred
= 1.23) по сравнению с методом CoMSIA1 (R2pred = 0.03,
RMSEpred = 1.44), но худшие по сравнению с методами CoMFA (R2pred
= 0.29, RMSEpred = 1.24) и CoMSIA2 (R2pred =
0.37, RMSEpred = 1.17).
Для выборки THR модели, полученные с использованием метода
CMF, имеют более низкие статистические показатели, описывающие прогнозирующую
способность на независимой контрольной выборке, по сравнению с моделями
CoMSIA2.
Как видно из Табл. 5 и 7, модели, построенные для
исследованных баз с помощью метода CMF, практически во всех случаях имеют
лучшую прогнозирующую способность на скользящем контроле (более высокие
значения q2), чем соответствующие модели, полученные методами CoMFA,
CoMSIA1, и CoMSIA2. Исключение составляют модели для COX2 и THR, полученные
методом CoMSIA2. Особенно велика разность между значениями q2 между
моделями CMF и CoMFA для баз GPB (0.32) и DHFR (0.20). Существенная разница в
значениях q2 между моделями CMF и CoMSIA1 достигается для базы GPB -
0.31. Разность в значениях q2 между моделями CMF и CoMSIA2 наиболее
высока для баз GPB и THER (0.13) и DHFR (0.12).
Модели, построенные для исследованных баз с помощью метода
CMF, имеют лучшую прогнозирующую способность на независимой контрольной выборке
(более высокие значения R2pred) для баз ACE, AChE, BZR и
DHFR, чем соответствующие модели, полученные методами CoMFA, CoMSIA1, и
CoMSIA2. Для базы GPB значения R2pred CMF-моделей лучше,
чем для моделей, полученных методами CoMFA и CoMSIA1, но хуже, чем для моделей,
полученных методом CoMSIA2.
Таким образом, результаты анализа 3D QSAR моделей, полученных
при помощи метода CMF с использованием НИП, свидетельствуют о том, что их
прогнозирующая способность вполне сопоставима, а в ряде случаев и превышает
прогнозирующую способность лучших моделей, построенных с помощью стандартных методов
3D QSAR и физико-химических полей. Это подтверждает выдвинутое выше утверждение
о том, что НИП - это универсальный набор полей, который может быть использован
вместо стандартных физико-химических полей для построения моделей 3D QSAR/QSPR.
Это также свидетельствует о перспективности предлагаемого подхода и, как мы
считаем, необходимости его дальнейшего развития.
Использование НИП в рамках метода CMF позволяют оценивать
вклады атомов определенного типа в моделируемое свойство (активность) молекулы.
Кроме того, в рамках разрабатываемого подхода возможно проводить интерпретацию
моделей 3D QSAR путём рассмотрения перекрывания полей регрессионных
коэффициентов, характеризующих модель, и молекулярных полей, описывающих
молекулу: чем выше такое перекрывание, тем большей активностью должна обладать
молекула. Поля регрессионных коэффициентов 3D QSAR модели, полученные с помощью
НИП в рамках метода CMF, показывают расположение в пространстве вокруг молекулы
областей, присутствие в которых атомов определённого типа благоприятно для
проявления активности. Такого рода анализ удобно проводить путем визуализации
перекрывания полей регрессионных коэффициентов 3D QSAR модели и непрерывных
индикаторных полей, наложенных на трёхмерную модель молекулы. Для осуществления
такой схемы визуализации полей нами разработан специальный набор скриптов на
языке R, описание которого представлено в Экспериментальной части.
Представлен пример визуализации перекрывания полей
регрессионных коэффициентов CMF модели и непрерывных индикаторных полей,
рассчитанных для двух типов атомов (ароматического углерода, C.ar и азота
амидной группы, N.am) для наиболее и наименее активных соединений базы ACE,
соответственно, соединения №54 (THIOL_9) и соединения №76 (SQ29852_2Q).
Изо-поверхности полей регрессионных коэффициентов показаны как сплошные
окрашенные поверхности. Красным цветом отмечены области предпочтительного для
проявления активности расположения атомов данного типа, синим показаны области,
в которых присутствие атомов данного типа нежелательно. Молекулярные поля для
определённого типа атомов индивидуальных молекул представлены сеткой.
В случае наиболее активного соединения области непрерывных
индикаторных полей атомов обоих типов перекрываются с областями регрессионных
коэффициентов модели. В случае же наименее активного соединении перекрывание
полей регрессионных коэффициентов модели с НИП атомов обоих типов отсутствует,
так как структура этой молекулы не содержит ароматических колец, а атом азота
расположен далеко от позиции, в которой, согласно построенной модели, его
наличие благоприятно для проявления активности.
2.1.2
3D QSPR моделирование фактора разделения комплексов полиазагетероциклических
лигандов с Am+3/Eu+3 с использованием непрерывных
индикаторных полей
Для проверки возможности применения разработанного нами
универсального набора непрерывных индикаторных полей для моделирования свойств
комплексов органических лигандов с ионами металлов нами было предпринято
построение 3D QSPR модели на основе методологии непрерывных молекулярных полей
для прогнозирования фактора разделения Am+3/Eu+3 при
образовании комплексов этих катионов с полиазагетероциклическими лигандам.
Построение такой модели представляло для нас большой интерес как в
теоретическом, так и в практическом плане. Теоретический интерес обусловлен
тем, что из-за того, что строение внешней электронной оболочки атома Am (5f77s2)
аналогично строению внешней электронной оболочки атома Eu (4f76s2),
а значения ионных радиусов ионов Am+3 и Eu+3 различаются
всего лишь на 0.012 нм, существование селективных лигандов к ним с трудом может
быть объяснено «классическими» представлениями о геометрическом строении
комплексов и «чистым» электростатическим взаимодействием иона металла с
молекулами лиганда, лежащим в основе стандартного набора физико-химических
полей. Поэтому можно было ожидать, что непрерывные индикаторные поля, благодаря
своему универсальному характеру, окажутся способными в неявном виде
аппроксимировать другие типы взаимодействия, недоступные для описания
стандартным набором физико-химических полей, и, следовательно, привести к
построению моделей 3D QSPR с более высокой прогнозирующей способностью. Также
можно было надеяться, что в случае успеха дальнейший подробный анализ
построенных моделей в сопряжении с высокоуровневыми квантово-химическими
расчётами позволил бы определить ключевые факторы, необходимые для дизайна
новых комплексонов с рекордно высокой разделяющей способностью по отношению к
Am+3/Eu+3. Практический же интерес в построении такой
модели состоит в том, что такая модель в дальнейшем могла бы быть использована
для решения важнейшей задачи, связанной с утилизацией отходов атомной
промышленности.
Для построения модели нами была использована база данных, на
основе которой авторами работы [97] ранее были получены количественные модели
«структура-свойство» с помощью фрагментных 2D дескрипторов. Исходная база
включала сведения о 48 производных полиазагетероциклических соединений,
принадлежащих к 6-ти структурным группам: L1: 5,6 - диалкил-3 - (4,6 -
диалкилпиридин-2-ил) - 1,2,4 - триазины; L2: 5,6 - диалкил-3 - (пиразин-2-ил) -
1,2,4 - триазины; L3: 5,6 - диалкил-3 - (5,6 - диалкил - 1,2,4 - триазин-3-ил)
- 1,2,4 - триазины; L4: 3 - (4,6 - диалкилпиридин-2-ил) - 5,6 - ди
(пиридин-2-ил) - 1,2,4 - триазины; L5: 2,6 - бис (5,6 - диалкил - 1,2,4 -
триазин-3-ил) пиридины; L6: 5,6 - диалкил-3 - (4-алкил-6 -
(4-алкилпиридин-2-ил) пиридин-2-ил) - 1,2,4 - триазины.
В качестве моделируемого свойства нами был взят фактор
разделения (SF, separation factor) ионов Am+3/Eu+3,
представляющий собой отношение коэффициентов распределения между органической и
водной фазами для этих металлов (Dam/Deu). Значения SF
были получены путём экстракции катионов металлов (Am+3, Eu+3)
из водного раствора азотной кислоты в 1,1,2,2 - тетрахлороэтановую фазу, содержащую
альфа-бромкаприновую кислоту в качестве соэстрагента [110]. Концентрации
европия (152Eu) и америция (241Am) в жидких фазах были
измерены методом гамма-спектрометрии. Исходная концентрация азотной кислоты для
разных соединений варьировалась в пределах ([HNO3] = 0.012 - 0.045 M).
База 2D-структур лигандов была сформирована нами с помощью
программы ChemAxon Instant JChem версии 5.12.0 и экспортирована в файл в
формате Tripos Mol2. Так как структура соединения №47 ((5,6 - ди (пиридин-2-ил)
- 1,2,4 - триазин) - (5,6 - ди (пиридин-2-ил) - 1,2,4 - триазин)) в
значительной степени отличалась от скелета шаблона, выбранного нами для
процедуры выравнивания при построении трехмерной геометрии лигандов, мы
исключили его из выборки. Таким образом, исследованная нами база производных
полиазагетероциклических соединений включала 47 соединений. База данных
приведена в Приложении, п. 4.
Для понимания того, какие атомы лиганда участвуют в
образовании координационных связей с ионом металла, мы использовали взятые из работы
[111] данные рентгеноструктурного анализа комплекса 5,6 - диэтил-3-пиридин -
1,2,4 - триазина с Eu+3, имеющего код FIYFID в Кембриджской базе
данных. Структура этих лигандов наиболее близка к структурам соединений
исследуемой нами выборки. На основе анализа структуры комплекса мы уточнили,
что связывание лиганда с катионом Eu+3 происходит при участии 2-го
атома азота 1,2,4 - триазинового кольца.
Одна из проблем, с которой мы столкнулись в ходе выполнения
данного исследования, состояла в том, что фактор разделения Am+3/Eu+3
определяется строением комплексов с двумя разными металлами. В то же время
применение методологии 3D QSAR/QSPR предполагает, что для моделируемой системы
задано одно пространственное строение. В данном случае мы воспользовались тем обстоятельством,
что химические свойства Am и Eu, равно как и значения соответствующих атомных
радиусов, очень близки. Вследствие этого можно ожидать, что органические
лиганды в составе комплексов с ионами Eu+3 и Am+3 имеют
очень близкое пространственное строение, для нахождения которого достаточно
построить пространственную модель комплекса только с одним металлом, а также
воспользоваться средствами моделирования, не делающими различия между Eu и Am.
Поэтому для построения 3D геометрии мы воспользовались программным комплексом
ChemAxon 5.11.3, который не предусматривает различий между Eu+3 и Am+3
и для обоих металлов приводит к построению одной и той же 3D геометрии
связанных с ними органических лигандов.
Для построения 3D геометрии лигандов в комплексе с ионами Am+3/Eu+3
мы воспользовались входящей в состав комплекса ChemAxon программой
MarvinSketch 5.11.3. С её помощью мы в интерактивном режиме строили и
редактировали 3D cтруктуры комплексов органических лигандов с Eu+3
при соотношении 2:1. Конформацию лигандов в связанном с металлом состоянии
определяли на основе реализованного в комплексе ChemAxon подхода, который
включает в себя методы многомерной метрической геометрии и использование
библиотеки фрагментов [112]. Далее, чтобы упростить процедуру пространственного
выравнивания структур, один из лигандов удаляли и результаты сохраняли в файл.
Далее базу лигандов, связанных с Eu+3, выравнивали
с использованием структурного шаблона, представленного на Рис. 11. Он является
наибольшим общим фрагментом, содержащимся во всех соединениях базы, и поэтому
он был использован для ее выравнивания. Поиск наибольшего общего фрагмента
проводили с помощью специального приложения Library MCS, входящего в состав
программного комплекса ChemAxon 5.11.3.
Выравнивание проводили с использованием алгоритма совмещения
структур с использованием шаблона по методу наименьших квадратов [113].
Алгоритм был реализован нами в виде скрипта на языке R (mcmf-do-alignment.R) в
рамках комплекса программ CMF https://sites.google.com/site/conmolfields. Для
создания шаблона использовали скрипт mcmf-do-make-template.R.
После выравнивания структур лигандов, связанных с Eu+3,
из каждой из структур атом металла был удален. Тем самым была сформирована база
лигандов, пространственная структура которых обеспечивает связывание с Eu+3
и Am+3.
С использованием этой базы выравненных пространственных
структур лигандов в рамках дипломной работы с помощью метода CMF с
использованием предложенных нами непрерывных индикаторных полей нами были
построены 3D QSPR модели для фактора разделения для комплексов набора
полиазагетероциклических лигандов с Am+3 и Eu+3. Число
индикаторных полей определяли по числу присутствующих в базе данных
молекулярно-механических типов атомов (в рамках параметризации силового поля
Tripos).
Статистические характеристики 3D QSPR моделей, полученных
методом CMF на основе непрерывных индикаторных полей.
Статистические показатели построенных 3D QSPR моделей
практически не зависят от выбора модификации метода KRR. Лучший результат по
прогнозированию на независимой контрольной выборке (R2pred=0.76,
RMSEpred=0.20) был получен для трёх модификаций метода: с гребневой
регрессией KRR2 без оптимизации коэффициентов смешения hf и в случае
KRR1 вне зависимости от того, оптимизируются ли коэффициенты смешения.
Следует отметить, что 3D QSPR модели, полученные на основе
НИП и метода CMF, дают лучшие результаты прогноза, чем 2D QSPR модели,
полученные, согласно данным из литературных источников, с использованием
программы Cerius2, а также молекулярных дескрипторов, рассчитанных программой
CODESSA-PRO, при построении статистической модели методом множественной
линейной регрессии [97]. Параметры 2D QSPR моделей, полученных вышеупомянутыми
методами: R2pred = 0.44, RMSEpred = 0.34 и R2pred
= 0.73, RMSEpred = 0.24 [97]. Кроме того, прогнозирующая способность
построенных нами 3D QSPR моделей сопоставима с таковой для 2D QSPR модели,
полученной с помощью программы ISIDA с использованием подструктурных
молекулярных фрагментов (substructural molecular fragments, SMF), и статистического
метода множественной линейной регрессии MLR - R2pred =
0.76, RMSEpred = 0.22 [97].
Как уже было отмечено выше, использование непрерывных
индикаторных полей в рамках метода CMF и для построения 3D QSPR моделей
позволяют оценивать вклады атомов определенного типа в активность (свойства)
молекул. Представлена визуализация перекрывания полей регрессионных
коэффициентов 3D QSPR модели (KRR2, без оптимизации hf) и
молекулярных индикаторных полей для атомов, принадлежащих типам C.2 (углерод
гибридизации sp2) и C.3 (углерод гибридизации sp3), для
наиболее и наименее активных соединений. Красным цветом отмечены области
предпочтительного для проявления активности расположения атомов данного типа,
синим показаны области, в которых присутствие атомов данного типа нежелательно.
Молекулярные поля для соответствующих типов атомов представлены сеткой.
В случае наиболее активного соединения области непрерывных
индикаторных полей атомов обоих типов перекрываются с областями положительных
значений (красного цвета) регрессионных коэффициентов модели. В частности, из
рассмотрения перекрывания полей регрессионных коэффициентов, соответствующих
типу C.2 (sp2-гибридизованному углероду) с соответствующими
индикаторными полями, следует важное значение, которое имеет для высокого
значения коэффициента разделения Am+3/Eu+3 наличие
дополнительного пиридинового кольца, атом азота которого способен
координироваться с атомом металла. Это кольцо присутствует в наиболее активном
соединение (слева), но отсутствует в наименее активном (справа). С другой
стороны, в случае наименее активного соединения (справа) перекрывание полей
положительного значения (красного цвета) регрессионных коэффициентов модели с
соответствующими непрерывными индикаторными полями атомов обоих типов либо вообще
отсутствует (как в случае типа C.2), либо, наоборот, присутствует перекрывание
непрерывных индикаторных полей с областями отрицательного значения (синего
цвета) соответствующих полей регрессионных коэффициентов (как в случае типа
C.3). В частности, в наименее активном соединении присутствует отрицательное
перекрывание непрерывного индикаторного поля sp3-гибридизованного
атома углерода, входящего в состав присоединённой к пиридиновому кольцу
метильной группы, с областью отрицательных регрессионных коэффициентов (синего
цвета), свидетельствующее о нежелательности присутствия в ней атомов такого
типа. Подобное отрицательное перекрывание присутствует в наименее активной
структуре, но отсутствует в наиболее активной. Следует также отметить, что
рассмотренная выше область, неблагоприятная для sp3-гибридизованного
атома углерода, совпадает в пространстве с областью, благоприятной для наличия
sp2-гибридизованного атома углерода, которая, как следует из
приведенного до этого анализа, свидетельствует о благоприятном для повышения
коэффициента разделения присутствии дополнительного пиридинового кольца,
образующего координационную связь с атомом металла. Отсюда следует, что для
повышения активности метильная группа в соединении с низкой активностью,
находящаяся внутри соответствующей зоны отрицательных значений поля
регрессионных коэффициентов для типа C.3 должна быть заменена на присоединённое
через орто-положение пиридиновое кольцо. Также из рассмотрения приведённых на
Рис. 10 полей регрессионных коэффициентов для типа C.3 следует, что одним из
возможных способов повышения активности самого активного соединения могла бы
стать замена метоксифенильной группы на метильную.
Таким образом, в ходе выполнения дипломной работы были
впервые получены 3D QSPR модели для прогнозирования свойств супрамолекулярных
комплексов с редкоземельными элементами на примере моделирования фактора
разделения Am+3/Eu+3 для комплексов
полиазагетероциклических лигандов. Модели, полученные с помощью метода CMF и
непрерывных индикаторных полей, обладают хорошей прогнозирующей способностью и
позволяют интерпретировать результаты путем визуализации перекрывающихся полей
регрессионных коэффициентов и непрерывных индикаторных полей. Подобный анализ
позволяет на структурном уровне интерпретировать соотношения
«структура-свойства» и подсказать структурные модификации, приводящие к
желаемой модификации моделируемого свойства.
2.2
3D QSAR/QSPR моделирование на основе непрерывных молекулярных полей
физико-химической природы
Для того чтобы оценить сравнительные преимущества и
недостатки использования в рамках метода CMF непрерывных индикаторных и
физико-химических полей, мы осуществили построение 3D QSAR/QSPR моделей для
всех рассмотренных выше баз данных (т.е. для связывания с 8 фармакологическими
мишенями и для разделения Am+3/Eu+3) тем же методом, но c
заменой индикаторных полей на физико-химические.
2.2.1
3D QSAR моделирование биологической активности органических лигандов в
отношении фармакологических мишеней с использованием непрерывных молекулярных
полей
Все данные, использованные для моделирования биологической
активности в отношении 8 фармакологических мишеней, включая наборы структур и
активностей, разделение на обучающие и контрольные выборки, пространственную
геометрию и выравненные структуры описаны в разделе III.1.1. Значения частичных
зарядов на атомах были взяты из приложения к статье [102]. При построении
моделей CMF мы использовали 5 типов физико-химических полей -
электростатическое, стерическое, гидрофобное, а также поля кислотности и
основности по отношению к образованию водородных связей. Параметризация
молекулярных полей описана в Экспериментальной части.
Статистические характеристики 3D QSAR моделей, полученных с
использованием обучающих и контрольных выборок для восьми наборов лигандов с помощью
метода CMF и физико-химических непрерывных молекулярных полей, приведены в
Табл. 9. В таблице представлены четыре статистических показателя: q2
и RMSEcv, характеризующие прогнозирующую способность моделей,
определённую на основе 10-кратного скользящего контроля, и R2pred
и RMSEpred, характеризующие прогнозирующую способность моделей на
независимой контрольной выборке.
Как видно из таблицы, для баз ACE и THERM модели с наилучшей
прогнозирующей способностью на независимой выборке получены методом KRR1 (без
оптимизации hf), для баз AChE, GPB и DHFR - методом KRR2 (без
оптимизации hf), для базы BZR - KRR1 (с оптимизацией hf)
и для баз COX2 и THR - KRR2 (с оптимизацией hf).
Разности в значениях q2 (характеризующих
прогнозирующую способность моделей, рассчитанную на основе скользящего
контроля) и R2pred (характеризующих прогнозирующую
способность моделей, рассчитанную на основе независимой контрольной выборки)
для CMF и моделей, описанных в литературе на основе стандартных методов 3D
QSAR, представлены в Табл. 12.
Модели, построенные для исследованных баз с помощью метода
CMF, практически во всех случаях имеют лучшую прогнозирующую способность на
скользящем контроле (более высокие значения q2), чем соответствующие
модели, полученные методами CoMFA, CoMSIA1 и CoMSIA2. Исключение составляют
модели для BZR, полученные методами CoMSIA1 и CoMSIA2 [102].
Особенно велика разность между значениями q2 между моделями
CMF и CoMFA для баз GPB (0.27). Существенная разница в значениях q2
между моделями CMF и CoMSIA1 достигается для баз GPB (0.26) и DHFR (0.24).
Разность между моделями CMF и CoMSIA2 лучшая для базы THER (0.12).
Модели, построенные для исследованных баз с помощью метода
CMF, имеют лучшую прогнозирующую способность на независимой контрольной выборке
(более высокие значения R2pred) для баз ACE, AChE, BZR,
DHFR и THR, чем соответствующие модели, полученные методами CoMFA, CoMSIA1, и
CoMSIA2. Для базы GPB значения параметров R2pred
CMF-моделей лучше, чем для моделей, полученных методами CoMFA и CoMSIA1, но
хуже, чем для моделей, полученных методом CoMSIA2.
Следует отметить небольшое преимущество в прогнозирующей
способности (параметры R2pred и RMSEpred,
рассчитанные на основе контрольных выборок) для CMF моделей относительно CoMFA
и CoMSIA1 моделей для всех выборок, также CoMSIA2 моделей для 6 выборок (ACE,
AСhE, BZR, DHFR, GPB и THR).
Представляет интерес сопоставить результаты, полученные
методом СMF на основе НИП и непрерывных физико-химических молекулярных полей.
Результаты сравнения значений q2 и R2pred
лучших моделей, полученных для 8 выборок лигандов с использованием НИП и
непрерывных полей физико-химического типа.
Параметр q2, описывающий прогнозирующую
способность моделей в условиях внутреннего скользящего контроля, для моделей,
полученных на основе НИП для 4-х выборок (BZR, DHFR, GPB и THERM), выше по
сравнению с q2 соответствующих моделей, полученных на основе
непрерывных полей физико-химической природы. В то же время, для 3-х выборок
(AСhE, COX2 и THR) значения q2 моделей на основе физико-химических
полей несколько выше. Прогнозирующая способность моделей на независимой
контрольной выборке, полученных с использованием физико-химических полей,
несколько выше, чем для моделей на основе непрерывных индикаторных полей для 7
выборок (АСЕ, AСhE, BZR, COX2 DHFR, THERM и THR), за исключением GPB.
Таким образом, прогнозирующая способность 3D QSAR моделей,
полученных с помощью метода CMF с использованием непрерывных индикаторных
полей, вполне сопоставима с прогнозирующей способностью моделей, построенных с
помощью традиционных физико-химических полей. Это ещё раз подтверждает
выдвинутое ранее утверждение о том, что НИП - это универсальный набор полей,
который может быть использован вместо стандартных физико-химических полей для
построения моделей 3D QSAR/QSPR. Следовательно, в рамках метода CMF НИП могут
рассматриваться как разумная альтернатива и полезное дополнение
физико-химическим полям.
2.2.2
3D QSPR моделирование фактора разделения комплексов полиазагетероциклических
лигандов с Am+3/Eu+3 с использованием непрерывных
молекулярных полей
Представляло интерес провести сравнение прогнозирующей
способности 3D QSPR моделей, построенных с помощью метода CMF на основе
непрерывных индикаторных полей и полей физико-химической природы для базы
полиазагетероциклических комплексонов для разделения Am+3 и Eu+3.
Для построения 3D QSPR моделей на основе физико-химических полей мы
использовали те же самые наборы соединений и схемы выравнивания, как и в случае
непрерывных индикаторных полей (см. раздел III.1.2). Для описания молекулярных
объектов мы использовали 2 типа физико-химических непрерывных полей -
электростатическое и стерическое. При построении моделей поля гидрофобности и
водородных связей мы не учитывали, поскольку в образовании комплексов
соответствующие им типы взаимодействий участия не принимают.
Прогнозирующая способность 3D QSPR моделей, полученных с
использованием НИП, существенно превышает прогнозирующую способность моделей,
полученных на основе непрерывных молекулярных полей физико-химической природы.
Так, в случае НИП (KRR2 с оптимизацией и без оптимизации коэффициента смешения
полей) значение R2pred равно 0.76, а в случае полей
физико-химической природы (KRR2 с оптимизацией коэффициентов смешения hf)
- 0.50. Подобную разницу можно объяснить универсальным характером непрерывных
индикаторных полей, которые способны в неявном виде аппроксимировать такие типы
взаимодействия, которые недостаточно хорошо могут быть описаны с помощью
стандартного набора физико-химических полей, оптимизированного для описания
связывания органических лигандов с фармакологическими мишенями.
Таким образом, при 3D QSPR моделировании фактора разделения
комплексов полиазагетероциклических лигандов с Am+3 и Eu+3
можно отметить явное преимущество применения универсального набора непрерывных
индикаторных полей по сравнению с набором непрерывных стерических и
электростатических молекулярных полей.
3.
Экспериментальная часть
3.1
Построение регрессионных 3D QSAR/QSPR моделей методом CMF с использованием
разработанных в дипломной работе наборов скриптов
Для построения регрессионных 3D QSAR/QSPR моделей методом СMF
был использован набор скриптов, работающих в рамках среды статистического
анализа данных R, разработанный ранее на химическом факультете МГУ в
лаборатории физико-химических методов анализа строения вещества в группе,
руководимой д.ф.-м.н., в.н.с. И.И. Баскиным. В ходе выполнения дипломной работы
первоначальный набор скриптов был в существенной мере модифицирован и дополнен
возможностью работать с индикаторными полями, а также проводить анализ моделей
путём рассмотрения перекрывания полей регрессионных коэффициентов с
молекулярными полями при помощи средств интерактивной компьютерной графики.
Наборы скриптов были созданы в ходе дипломной работы с использованием
программой среды R #"661368.files/image031.gif">
где PRESScv - сумма квадратов разностей между
экспериментальными (yi) и предсказанными (ypredi)
значениями активности по всем n соединениям, принимающим участие в процедуре
скользящего контроля; SS - сумма квадратичных отклонений экспериментальных
значений (yi) от их среднего значения (ymean),

где yi - экспериментальное значение активности для
i-го соединения, ypredi - предсказанная активность этого
соединения, n - число соединений в базе данных. В методе непрерывных
молекулярных полей (CMF) происходит оптимизация гипер-параметров γ, α и, если указано,
коэффициентов смешения hf с целью минимизации значения RMSEcv.
Для определения прогнозирующей способности модели на
независимой контрольной выборке использовали параметры R2pred
и RMSEpred, рассчитанные на основе спрогнозированных значений
активностей для соединений, не вошедших в обучающую выборку, по следующим
формулам:
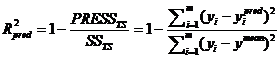
где PRESSTS - сумма квадратов разности между
экспериментальными (yi) и предсказанными (ypredi)
значениями активности для всех m соединений из внешней контрольной выборки; SSTS
- сумма квадратов отклонений экспериментальных значений (yi) от их
среднего значения (ymean) для соединений контрольной выборки,

где yi - экспериментальное значение активности для
i-го соединения из контрольной выборки, ypredi -
спрогнозированная активность того же соединения, m - число соединений в
контрольной выборке.
Выводы
. Впервые для описания свойств молекул в рамках методологии
3D QSAR/QSPR предложены и исследованы новые функции, определяющие меру
принадлежности точки с заданными координатами атому определенного типа -
«непрерывные индикаторные поля». Они представляют универсальный набор функций,
который является альтернативой молекулярным полям физико-химической природы при
построении моделей 3D QSAR/QSPR.
. На основе непрерывных индикаторных полей и метода ядерной
гребневой регрессии разработано программное обеспечение для построения моделей
3D QSAR/QSPR, позволяющих прогнозировать свойства, связанные с образованием
супрамолекулярных комплексов. Программа также позволяет проводить анализ
моделей путем визуализации перекрывания полей регрессионных коэффициентов
модели и непрерывных индикаторных полей атомов различного
молекулярно-механического типа.
. С использованием созданного программного обеспечения
построены 3D QSAR модели для прогнозирования биологической активности наборов
органических лигандов в отношении 8 фармакологически важных мишеней. Модели
обладают высокой прогнозирующей способностью, которая сопоставима, а для 4
выборок превышает прогнозирующую способность моделей, построенных с помощью
стандартных методов 3D QSAR CoMFA и CoMSIA на основе молекулярных полей
физико-химической природы.
Список
литературы
1. Le
T., Epa V. Ch., Burden F.R., Winkler D.A. Quantative Structure - Property
Relationship Modeling of Diverse Materials Properties. // Chemical Reviews.
2012. V. 112. P. 2889-2919.
2. Leach
A.R., Gillet V.J. An Introduction to Chemoinformatics. Springer. 2007. P.
1-255.
. Verma
J., Khedkar V.M., Coutinho E.C. 3D-QSAR in Drug Design - A Review. // Current
Topics in Medicinal Chemistry. 2010. V.10. P. 95-115.
. Gasteiger
J., Engel T. (Eds.). Chemoinformatics. Wiley-VCH. 2006.
. 3D
QSAR in Drug Design: Vol. 1. Theory, Methods and Applications
(Three-Dimensional Quantitative Structure Activity Relationships). (Ed. by H.
Kubinyi). Kluwer/Escom, Dordrecht. 2000.
. 3D
QSAR in Drug Design. Vol. 2. Ligand-Protein Complexes and Molecular Similarity.
(Ed. by H. Kubinyi, G. Folkers, and Y.C. Martin). Kluwer Academic Publishers,
Dordrecht. 2002.
. 3D
QSAR in Drug Design. Vol. 3. Recent Advances. (Ed. by H. Kubinyi, G. Folkers,
and Y.C. Martin). Kluver Academic Publishers, Dordrecht. 2002.
. Katritzky
A.R., Kuanar M., Slavov S., Hall C.D. Quantitative Correlation of Physical and
Chemical Properties with Chemical Structure: Utility for Prediction // Chem.
Rev. 2010. V.110, P.5714-5789.
. Thomas
L.J., Roy K. On Selection of Training and Test Sets for the Development of
Predictive QSAR models // QSAR & Combinatorial Science. 2006. V.25. I.3.
P.235-251.
. Richardson
B.J. Physiological research on alcohols. // Med. Times. Gaz. 1868. V.2.
P.703-706.
. Overton
E. Osmotic properties of cells in the bearing on toxicology and pharmacy. // Z.
Physik. Chem. 1897. V.22. P.189-209.
. Hammet
L.P. Some relations between reaction rates and equilibrium constants. // Chem.
Rev. 1935. V.17. P.125-136.
. Ferguson
J. The Use of Chemical Potentials as Indices of Toxicity. // Proc. R. Soc.
Lond. B. 1939. V.127. P.387-404.
. Taft
R.W. Polar and steric substituents constants for aliphatic and o-Benzoate
groups from rates of esterification and hydrolysis of esters. // J. Am. Chem.
Soc. 1952. V.74. P.3120-3128.
. Hansch
C., Maloney P.P., Fujita T., Muir R.M. Correlation of biological activity of phenoxyacetic
acids with hammett substituent constants and partition coefficients. // Nature.
1962. V.194. P.178-180.
. Free
S.M., Wilson J.W. A Mathematical contribution to structure-activity studies. //
J. Med. Chem. 1964. V.7. 395-399.
. Fujita
T., Ban T. Structure-activity study of phenethylamines as substrates of
biosynthetic enzymes of sympathetic transmitters. // J. Med. Chem. 1971. V.14.
P.148-152.
. Хёльтье
Х.-Д., Зиппль В., Роньян Д., Фолькерс Г. Молекулярное моделирование: теория и
практика. Бином. Лаборатория знаний. 2013.
. Hopfinger
A.J., Wang S., Tokarski J.S., Jin B., Albuquerque M., Madhav P.J., Duraiswami
C. Construction of 3D-QSAR models using the 4D-QSAR analysis formalism. // J.
Am. Chem. Soc. 1997. V.119 (43). P.10509-10524.
. Vedani
A, Dobler M. 5D-QSAR: The key for simulating induced fit? // J. Med. Chem.
2002. V.45. P.2139-2149.
. Vedani
A, Dobler M, Lill M.A. Combining protein modeling and 6D-QSAR. Simulating the
binding of structurally diverse ligands to the estrogen receptor. // J. Med.
Chem. 2005. V.48. P.3700-3703.
. Hoerl
A.E., Kennard R.W. (1970). Ridge regression: Biased estimation for
nonorthogonal problems. // Technometrics. 1970. V.12 (1). P.55-67.
. Гальберштам
Н.М., Баскин И.И., Палюлин В.А., Зефиров Н.С. Нейронные сети как метод поиска
зависимостей структура - свойство органических соединений // Успехи химии,
2003, Т. 72, №7, 706-727.
. Baskin
I.I., Palyulin V.A., Zefirov N.S. Neural networks in building QSAR models. //
Methods Mol. Biol. 2008. V.458. P.137-158.
. Cover
T.M., Hart P.E. Nearest neighbor pattern classification. // IEEE Transactions
on Information Theory. 1967. V.13 (1). P.21-27.
27. Hofmann T., Schölkopf B.,
Smola A.J. Kernel Methods in Machine Learning // The Annals of Statistics. 2008. V.36
(3). P.1171-1220.
. Cortes
C., Vapnik V. Support‐vector networks. // Mach.
Learn. 1995. V.20. P.273-297.
. An
S., Liu W., Venkatesh S. Fast cross-validation algorithms for least squares
support vector machine and kernel ridge regression. // Pattern Recognition.
2007. V.40 (8), P.2154-2162.
. Rosipal
R., Trejo L.J. Kernel partial least squares regression in reproducing kernel
hilbert space. // The Journal of Machine Learning Research. 2002. V.2.
P.97-123.
. Todeschini
R., Consonni V. Handbook of molecular descriptors. Wiley-VCH, Weinheim. 2000.
. Karelson
M. Molecular Descriptors in QSAR/QSPR. John Wiley & Sons: New York. 2000.
. Mauri
A., Consonni V., Pavan M., Todeschini R. // MATCH-Commun. Math. Co. 2006. V.56.
P.237-248.
. Karelson
M., Maran U., Wang Y., Katritzky A.R. QSAR and QSPR models derived using large
molecular descriptors spaces. A review of CODESSA application. // Collect.
Czech. Chem. Commun. 1999. V.64. P.1551-1571.
. Basak
S.C., Gute B.D., Grunwald G.D. Use of topostructural, topochemical, and
geometric parameters in the prediction of vapor pressure: A hierarchical QSAR
approach. // J. Chem. Inf. Comput. Sci. 1997. V.37. P.651-655.
. Baskin
I., Varnek A. Fragment Descriptors in SAR/QSAR/QSPR Studies, Molecular
Similarity Analysis and in Virtual Screening. In: Chemoinformatic Approaches to
Virtual Screening. (Ed. by A. Varnek, A. Tropsha). RCS Publishing. 2008.
. Rogers
D., Hopfinger A.J. Application of genetic function approximation to
quantitative structure-activity relationships and quantitative
structure-property relationships. // J. Chem. Inf. Comput. Sci. 1994. V.34.
P.854-866.
38. Molecular
Interaction Fields: Applications in Drug Discovery and ADME Prediction. (Ed. by
G. Cruciani, R. Mannhold, H. Kubinyi, G. Folkers). WILEY-VCH Verlag GmbH &
Co. KGaA, Weinheim. 2006.
. Cramer
R.D., Patterson D.E., Bunce J.D. Comparative molecular field analysis (CoMFA).
1. Effect of shape on binding of steroids to carrier proteins. // J. Am. Chem.
Soc. 1988. V.110. P.5959-5967.
. Green
S., Marshall G.R. 3D-QSAR: A current perspective. // Trends Pharmacol. Sci.
1995. V.16. P. 285-291.
41. Tominaga
Y., Fujiwara I. Prediction-weighted Partial Least-Squares regression method
(PWPLS) 2: Application to CoMFA. //J. Chem. Inf. Comput. Sci. 1997. V. 37 (6).
P. 1152-1157.
42. Goodford
P.J. A computational procedure for determining energetically favorable binding
sites on biologically important macromolecules. // J. Med. Chem. 1985. V.28.
P.849-857.
43. Davis
A.M., Gensmantel N.P., Johansson E., Marriott D.P. The use of the grid program
in the 3-D QSAR analysis of a series of calcium channel agonists. // J. Med.
Chem. 1994. V.37. P.963-972.
. Cruciani
G., Watson K.A. Comparative molecular field analysis using grid force field and
golpe variable selection methods in a study of inhibitors of
glycogen-phosphorylase-B. // Journal of Computer-Aided Molecular Design. 1994.
V.7. P.263-280.
. Klebe
G., Abraham U., Mietzner T. Molecular similarity indexes in a comparative
analysis (CoMSIA) of drug molecules to correlate and predict their biological
activity. // J. Med. Chem. 1994. V.37. P.4130-4146.
. Klebe
G., Abraham U. Comparative molecular similarity index analysis (CoMSIA) to
study hydrogen-bonding properties and to score combinatorial libraries. //
Journal of Computer-Aided Molecular Design. 1999. V.13. P.1-10.
. Bohm
M., Sturzebecher J., Klebe G. Three-dimensional quantitative structure-activity
relationship analyses using comparative molecular field analysis and
comparative molecular similarity indices analysis to elucidate selectivity
differences of inhibitors binding to trypsin, thrombin, and factor Xa. // J.
Med. Chem. 1999. V.42. P.458-477.
. Pearlman
R.S. Rapid generation of high quality approximate 3D molecular structures. //
Chemical Design Automation News. 1987. V.2. P.1-7.
. Gasteiger
J., Rudolph C., Sadowski J. Automatic generation of 3D-atomic coordinates for
organic molecules. // Tetrahedron Computer Methods. 1990. V.3. P.537-547.
. Langer
T., Hoffman R.D. Pharmacophores and Pharmacophore Searches. Wiley-VCH Publishers,
Weinheim. 2000.
. Pastor
M., Cruciani G., McLay I. Pickett S., Clementi S. GRid-INdependent descriptors
(GRIND): a novel class of alignment-independent three-dimensional molecular
descriptors. // J. Med. Chem. 2000. V.43 (17). P.3233-3243.
. Fabian
W.M.F., Timofei S., Kurunczi L. Comparative molecular field analysis (CoMFA),
semiempirical (AM1) molecular orbital and multiconformational minimal steric
difference (MTD) calculations of anthraquinone dye-fibre affinities. // J. Mol.
Struct.:THEOCHEM. 1995. V.340 (1). P.73-81.
. Fabian
W.M.F., Timofei S. Comparative molecular field analysis (CoMFA) of dye-fibre
affinities. Part 2. Symmetrical bisazo dyes. // J. Mol. Struct.: THEOCHEM.
1996. V.362. P.155-162.
. Cruz
V., Ramos J., Munoz-Escalona A., Lafuente P., Pena B., Martinez-Salazar J.
3D-QSAR analysis of metallocene-based catalysts used in ethylene
polymerisation. // Polymer. 2004. V.45 (6). P.2061-2072.
. Cruz
V.L., Ramos J., Martinez S., Munoz-Escalona A., Martinez-Salazar J.
Structure-Activity Relationship Study of the Metallocene Catalyst Activity in
Ethylene Polymerization. // Organometallics. 2005. V.24 (21). P.5095-5102.
. Жохова
Н.И., Баскин И.И., Бахронов Д.К., Палюлин В.А., Зефиров Н.С. Метод непрерывных
молекулярных полей в поиске количественных соотношений «структура - активность»
// Докл. РАН. 2009. Т.429. №2.