Разработка методов синтеза нитроксильных радикалов ряда имидазолина, имидазолидина и пирролидина
Введение
Нитроксильные радикалы (нитроксиды, НР) - наиболее распространённый класс
стабильных органических радикалов. Небольшие молекулы нитроксидов с легко
варьируемой структурой обладают уникальными свойствами, обеспечивающими
многообразие и постоянное расширение их применений в различных областях науки и
техники. Наиболее быстро развиваются работы по применению НР в биофизике, в
медицине и в химии полимеров.
В биофизике НР используются для изучения строения и механизмов
взаимодействия сложных биомолекул и исследования процессов, обеспечивающих
различные аспекты жизнедеятельности клеток. Развитие новых физических методов
исследования на основе низкопольного ЭПР и эффекта Оверхаузера открывает новые
возможности для неинвазивного исследования живых объектов и диагностики при
помощи парамагнитных молекулярных зондов. Широкое применение НР в этой сфере
осложняется их восстановлением в диамагнитные продукты (гидроксиамины) под
действием биогенных антиоксидантов и ферментативных систем.
Применение НР в медицине связано с их свободно-радикальной природой и
способностью как к одноэлектронному окислению, так и восстановлению. Благодаря
этой особенности НР чрезвычайно легко взаимодействуют с активными радикалами,
образующимися в результате протекания различных окислительных процессов в живых
системах. Благодаря этому НР блокируют развитие цепных радикальных процессов с
участием активных форм кислорода (АФК), с которыми связывают развитие различных
патологий. Реагируя с активными радикалами, НР могут образовывать устойчивые
аддукты либо претерпевать одноэлектронное восстановление или окисление с
образованием гидроксиамина (ГА) или оксоаммониевого катиона соответственно.
Последний быстро превращается в гидроксиамин, реагируя с органическими
соединениями. Гидроксиамины в свою очередь способны реагировать с АФК, при этом
регенерируются НР. Совокупность этих процессов обусловливает высокую
антиоксидантную активность НР. Показано, что НР, подобно природному ферменту
супероксиддисмутазе, катализируют диспропорционирование супероксидного
радикала, являющегося основным источником всех АФК в организме. Антиоксидантная
активность обусловливает применения НР в фармакологии для защиты от
ионизирующего излучения, профилактики и лечения онкологических заболеваний,
нейродегенеративных болезней, гипертонии, а также предотвращения повреждения
тканей вследствие ишемии/реперфузии. Следует отметить, что биологическая
активность НР и соответствующих гидроксиаминов может различаться. Различие в
биологической активности часто наблюдают в экспериментах на клеточных
культурах, где небольшое количество клеток не может быстро изменить соотношение
НР и ГА. В экспериментах in vivo, напротив, эти различия сглажены.
Вследствие реакции гидроксиаминов с постоянно образующимися АФК, в живых тканях
наблюдается некое равновесие между НР и ГА, сдвинутое в сторону образования
гидроксиамина. Можно ожидать, что изменение положения этого равновесия может
существенно повлиять на биологическую активность.
В основе использования НР в химии полимеров лежит их способность обратимо
реагировать с короткоживущими С-центрированными радикалами. Проведение
радикальной полимеризации виниловых мономеров в присутствии НР приводит к
обратимому захвату радикалов растущих полимерных цепей с образованием
макроалкоксиаминов. В результате концентрация растущих цепей понижается до
необходимого минимума, что существенно снижает вклад процессов рекомбинации
(необратимого обрыва цепи) и позволяет получать полимеры более высокого
качества с узким распределением по молекулярной массе. Более того, полученные
полимеры, как правило, имеют структуру макроалкоксиамина, что позволяет
реинициировать полимеризацию в присутствии другого мономера и получать
блок-сополимеры регулярного строения. Уникальные свойства последних, например,
способность к самоорганизации в наноструктуры, делают их перспективной основой
для создания высокотехнологичных материалов.

Развитие
упомянутых выше применений порождает постоянный интерес к синтезу новых
производных. В связи с этим химия НР продолжает быстро развиваться. Одним из
активно развиваемых направлений является синтез НР, содержащих объёмные
заместители у α-атомов углерода нитроксильной группы. Пониженная
пространственная (топологическая) доступность нитроксильного фрагмента придаёт
этим радикалам ряд полезных свойств. В частности, значительно возрастает
устойчивость НР к восстановлению в диамагнитные соединения, протекающему в
биологических образцах, что открывает перспективы применения пространственно
затруднённых НР в биофизических исследованиях in vivo
и в медицине. Кроме того, меняется положение равновесия в обратимой реакции НР
с короткоживущими С-центрированными радикалами, что позволяет использовать
пространственно-затруднённые НР в контролируемой радикальной полимеризации
различных виниловых мономеров при более низкой температуре и улучшить
характеристики получаемых полимеров и блок-сополимеров.
В связи с этим разработка новых методов синтеза нитроксильных радикалов с
объёмными заместителями представляется весьма актуальной задачей.
Целью данной работы являлась разработка методов синтеза нитроксильных
радикалов ряда имидазолина, имидазолидина и пирролидина, содержащих объёмные
алкильные заместители у a-атомов
углерода нитроксильной группы.
В главе 1 (литературной обзор) рассмотрены различные подходы к синтезу
нитроксильных радикалов с пространственно-затруднённым нитроксильным
фрагментом. Литературные данные показывают, что объёмные заместители в
циклические НР могут быть введены как на стадии построения гетероцикла, так и
при последующих превращениях.
В данной работе в качестве основного метода достижения поставленной цели
выбраны реакции циклических нитронов производных пирролина, 2Н- и 4Н-имидазола
с металлоорганическими соединениями. Хотя этот метод имеет существенные ограничения
и в ряде случаев даёт невысокие выходы целевых нитроксильных радикалов, он
позволяет использовать единый подход для синтеза набора различных производных.
Кроме того, в НИОХ СО РАН разработаны удобные способы получения исходных
циклических нитронов и выбранный метод позволяет использовать это преимущество.
В результате исследования реакций 2Н-имидазол-1-оксидов с реактивами
Гриньяра (Глава 2) был синтезирован набор 4-фенил-3-имидазолин-1-оксилов с
различными объёмными заместителями в положениях 2 и 5. Однако, было
установлено, что использование этого метода для получения НР ряда 3-имидазолина
с метильной группой в положении 4 даёт низкие выходи и не рационально.
Основными побочными процессами при этом являются переметаллирование и
дезоксигенирование нитронной группы. Было обнаружено, что введение на начальном
этапе синтеза объёмного заместителя, не склонного к металлированию, к атому
углерода нитронной группы позволяет в дальнейшем использовать
высокореакционноспособные литийорганические соединения для введения второго
объёмного заместителя к тому же атому, в результате чего сильно затрудненные НР
могут быть получены с хорошим выходом.
Эта находка была была успешно использована для получения НР из нитронов
другого строения - производных 4Н-имидазол-3-оксида (Глава 4) и
1-пирролинон-4-1-оксида (Глава 5). В результате были синтезированы
пространственно затруднённый рН-чувствительный зонд с рК 6.49 и несколько
спиновых меток и зондов пирролидинового ряда. Последние показали чрезвычайно
высокую устойчивость к восстановлению в модельных системах, гомогенатах тканей
и изолированных органах.
НР 3-имидазолина превращали в радикалы имидазолидинового ряда через
алкилирование диметилсульфатом и восстановление полученных имидазолидиниевых
солей боргидридом натрия (Глава 3). Было установлено, что при наличии в
имидазолиниевой соли асимметрического центра соотношение образуюшихся при
восстановлении диастереомеров зависит от условий проведения реакции. Полученные
НР имидазолидинового ряда обладают рН-зависимыми спектрами ЭПР. Один из
полученных НР, содержащий в составе молекулы карбоксильную группу имеет
необычно высокий рК (6.2) и представляет интерес в качестве рН-чувствительного
спинового зонда.
Интересной особенностью НР 3-имидазолинового и 3-пирролидинонового рядов,
содержащих бутильный и трет-бутильный заместители у одного атома углерода
является их термическая неустойчивость. Исследование этого необычного
превращения (Глава 6) показало, что при нагревании происходит отщепление
трет-бутильного радикала с образованием соответствующего бутилнитрона (реакция
обратная спиновому захвату).
На основе полученных пространственно-затруднённых НР были синтезированы
алкоксиамины (Глава 7), которые в дальнейшем использовались как для определения
констант скорости гомолиза С-O связи,
необходимых для оптимизации условий контролируемой полимеризации с участием
полученных НР, так и в качестве инициаторов для получения полимеров.
Многие из полученных пространственно затруднённых НР и соответствующих
алкосиаминов были переданы для исследования полимеризации в МТЦ СО РАН и НИИ
химии Нижегородского государственного университета. Среди переданных соединений
обнаружены эффективные регуляторы полимеризации стирола, метилметакрилата и
акриламида (в водном растворе), позволяющие получить полимеры с низкой
полидисперсностью.
В настоящее время в Государственном университете штата Огайо (Коламбус,
США) продолжаются исследования с использованием синтезированных в ходе этой
работы спиновых меток и зондов, направленные на разработку новых способов диагностики
онкологических заболеваний.
Работа выполнена в Лаборатории азотистых соединений НИОХ СО РАН. Запись
ИК, УФ, ЯМР, ЭПР и масс-спектров, а также рентгеноструктурный анализ
осуществлены в Лаборатории физических методов исследования НИОХ. Элементный анализ
выполнен в Лаборатории микроанализа НИОХ. Определение рК (титрование) и
констант скорости восстановления полученных НР аскорбиновой кислотой
проводилось Комаровым Д.А и Глазачевым Ю.И. (ИХКиГ СО РАН).
Проведённые исследования были частично поддержаны РФФИ (проекты
04-03-32299-а и 08-03-00432-а).
Глава 1. Синтез пространственно затрудненных нитроксильных радикалов
В последние годы наблюдается большой рост интереса к нитроксильным
радикалам (НР), содержащим объёмные заместители в ближайшем окружении группы N-O●. Подавляющее большинство известных на сегодняшний
день НР содержат метильные группы у α-атомов углерода нитроксильной группы.
Установлено, что увеличение объема этих заместителей всего лишь на один атом
углерода приводит к существенному изменению свойств НР. Поэтому, здесь и далее
под термином «объёмные заместители» мы будем понимать любые группы большие, чем
метильная.
На сегодняшний день известно много подходов к синтезу стабильных НР
различной структуры. Однако для производных с объемными заместителями при
нитроксильной группе существующие методы синтеза имеют определённые
ограничения. Данный обзор представляет собой попытку систематизации известных
подходов к синтезу нитроксильных радикалов с пространственно затруднённым
нитроксильным фрагментом и анализа возможностей и ограничений различных
методов.
Образование нитроксильной группы может происходить следующими способами:
1) окисление пространственно-затруднённых аминов или гидроксиламинов,
которые в свою очередь могут быть получены различными методами, включая
разнообразные конденсации или реакции нитроной группы с нуклефильными агентами,
например, с металлоорганическими соединениями;
2) термолиз алкоксиаминов;
) присоединение радикальных агентов к нитронам или нитрозо
соединениям (спиновый захват);
) присоединение нуклеофила к катион-радикалу нитрона.
Способ 4 не рассматривается в данном обзоре, т.к. имеет очень узкое
применениев синтезе НР. Присоединение нуклеофила к катион-радикалу нитрона
используется только в синтезе радикалов со специфическим окружением (с алкокси-
или амино- группами у α-атома углерода).
Способы 2-4 также имеют весьма ограниченное значение, поэтому мы коротко
рассмотрим их в этом разделе.
Спиновый захват обычно не используется как прямой препаративный метод
синтеза НР. Это связано с тем, что скорость захвата С-центрированных радикалов
нитроксильными выше, чем скорость захвата их спиновыми ловушками (нитронами,
нитрозо соединениями или оксидами азота). В результате образующийся НР
претерпевает дальнейшие превращения, чаще всего, дает алкоксиамин. Например,
окисление замещенного гидразина 1 диоксидом свинца в присутствии
нитрозосоединения 2 описано в работе как метод синтеза алкоксиамина 3 (схема
1). Реакция, очевидно, идёт через образование 1-фенилизобутильного радикала, который
затем последовательно реагирует с 2 и образовавшимся нитроксильным спиновым
аддуктом.
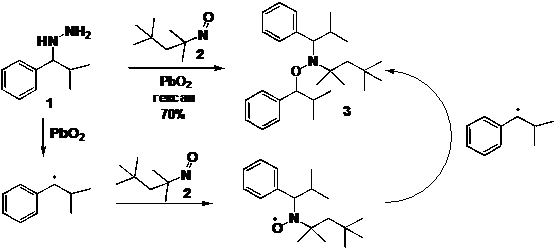
СХЕМА
1
Аналогичные превращения использованы в патентованных методах синтеза
алкоксиаминов 3a-3c через термолиз азо-бис-изобутиронитрила
в присутствии нитрозосоединений, нитронов или окиси азота.
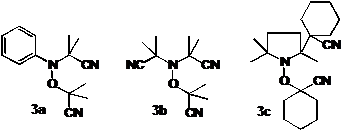
Рисунок
1
Некоторые алкоксиамины способны к обратимому гомолитическому распаду по
связи С-О, что позволяет использовать их в качестве источников инициирующих
радикалов и НР в контролируемой радикальной полимеризации. При нагревании таких
алкоксиаминов на воздухе образующийся алкильный радикал необратимо связывается
кислородом, освобождая НР. Эта реакция иногда используется для препаративного
получения НР, например, в тех случаях, когда для построения скелета требуется
использование сильно кислых сред, в которых НР неустойчивы (например, см. схему
2).
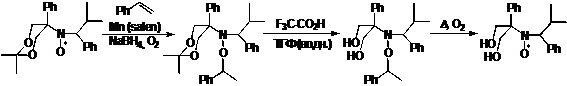
СХЕМА 2
Окислительные
методы
Далее будут рассмотрены методы синтеза НР, в которых последней стадией
является окисление. Важнейшими субстратами для получения стабильных НР являются
вторичные амины и гидроксиамины. Методики окисления этих групп носят достаточно
общий характер, поэтому ключевым этапом в синтезе пространственно-затрудненных
НР является получение прямых предшественников. Здесь и далее амины и
гидроксиамины, окисление которых приводит к образованию НР с объёмными
заместителями у α-атомов углерода к нитроксильной группе, мы будем
называть пространственно затруднёнными аминами (гидроксиаминами).
Пространственно
затрудненные амины
Препаративное значение приобрели лишь немногие окислители. Одна из
важнейших окислительных систем - пероксид водорода в присутствии солей
вольфрамовой кислоты (окислителем в данном случае является первольфрамат-ион)
приводит к образованию НР через промежуточный гидроксиамин (см. схему 3).
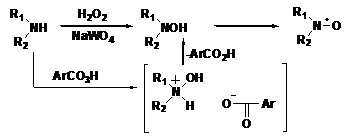
СХЕМА
3.
Метод окисления пероксидом водорода в присутствии вольфраматов позволяет
получать НР и из третичных аминов.
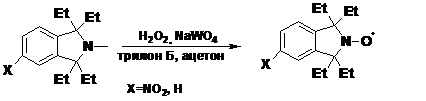
СХЕМА
4
Другие неорганические катализаторы окисления перекисью водорода не
приобрели существенного препаративного значения в химии НР.
Альтернативой системе Н2О2-Na2WO4 являются органические надкислоты,
чаще всего м-хлорнадбензойная (MCPBA)
и п-нитронадбензойная кислоты (схема 3). Этот метод позволяет проводить реакцию
в неводных средах (хлороформе, хлористом метилене).
Окисление пространственно затрудненных вторичных аминов надкислотами
обычно происходит более успешно, чем перекисью водорода в присутствии
вольфраматов. Например, для пространственно затрудненного пиперидинового
производного 4 образование радикала с выходом 62% наблюдается только под
действием м-хлорнадбензойной кислоты.
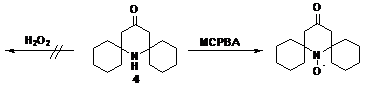
СХЕМА 5
Способы
получения циклических пространственно-затрудненных аминов
Синтезы с
использованием реакции конденсации
Синтезы на
основе 4-оксопиперидина (триацетонамина)
Триацетонамин (2,2,6,6-тетраметил-4-оксопиперидин, TAA), легко получаемый конденсацией
ацетона с аммиаком, является основой для синтеза наиболее распространенных
2,2,6,6-тетраметилзамещенных НР пиперидинового ряда. Кроме того, из ТАА также
получают важнейшие НР пирролинового, пирролидинового и диазепинового рядов.
Этим объясняются многочисленные попытки исследователей получить аналоги ТАА,
содержащие объёмные заместители в положениях 2 и 6.
В некоторых случаях стерически затрудненные пиперидины удается получить с
неплохим выходом, непосредственно конденсируя кетоны с аммиаком. Так при
взаимодействии аммиака с метилэтилкетоном образуется диметилтриэтилзамещенный
тетрагидропиримидин 5, который далее превращается в целевой пиперидон, с общим
выходом 55%. Окисление надуксусной кислотой позволяет получить НР 6 с выходом
89% (схема 6). Однако другие метилкетоны, (типа Me(CO)R, где R > Et)
дают в этой реакции очень низкий выход соответствующего пиперидона, а более
затрудненные кетоны, например 3-пентанон не реагируют совсем.
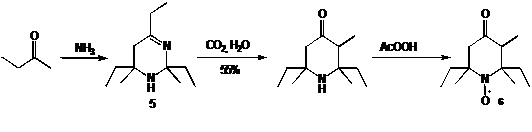
СХЕМА
6
Поскольку
ТАА образуется в результате последовательности обратимых реакций, неоднократно
предпринимались попытки получить из триацетонамина или его прямого
предшественника - ацетонина
(2,2,4,4,6-пентаметил-2,3,4,5-тетрагидропиримидина)- более затрудненные НР. При
взаимодействии ацетонина с кетонами в присутствие хлорида аммония наблюдается
образование трех основных типов продуктов, соотношение которых обусловлено,
главным образом, строением кетона (схема 7). Однако для большинства кетонов
выход целевого сильно затрудненного амина типа 7b незначителен.
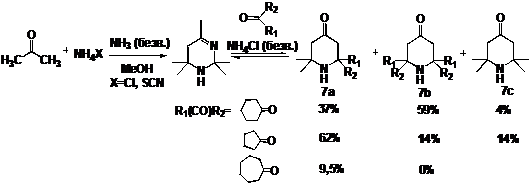
СХЕМА
7
Препаративное
значение этот подход имеет только для циклогексанона, а также некоторых
шестичленных гетероциклических кетонов, например тетрагидропиран-4-она (схема
8). Очевидно, в данном случае значение имеет не только циклическая структура
кетона, но, видимо, и размер цикла, так в реакции с циклогептаноном и
циклопентаноном преимущественно образуется несимметричный амин 7а (схема 7).
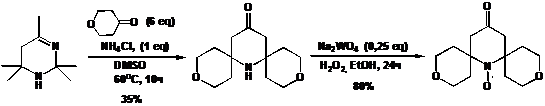
СХЕМА 8
Тем ни менее, 2,2,6,6-тетраэтилзамещенный пиперидиновый радикал может
быть получен несколькими способами.
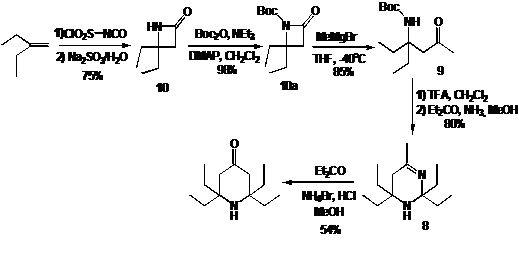
Ключевой стадией одного из вариантов синтеза также является
перегруппировка/гидролиз соответствующего тетрагидропиримидина 8 в присутствии
диэтилкетона и бромистого аммония. Соединение 8 удается получить с высоким
выходом конденсацией диэтилкетона и аммиака с солью бета-аминокетона 9 и
трифторуксусной кислоты (TFA).
Бета-аминокетон в свою очередь получают при раскрытии цикла β-лактама 10. Лактам 10 получают из
коммерчески доступного 2-этилбутена (схема 9).
СХЕМА
9
На
схеме 10 представлен другой способ формирования этильных заместителей в НР 11
посредством десульфирования пиперидинона 12, получаемого конденсацией ацетонина
с тетрагидротиопиран-4-оном.
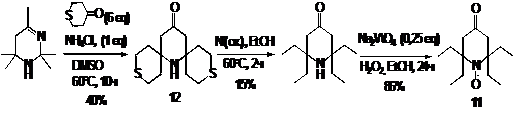
СХЕМА
10
Еще
один подход к синтезу пространственно затрудненных пиперидин-4-онов заключается
в формировании замещенного производного форона и последующей конденсации его с
аммиаком. Этим путем НР 11 был получен, исходя из коммерчески доступного
металлил дихлорида. Ключевым этапом синтеза является олефинирование по
Уодсворту-Хорнеру-Эммонсу (УХЭ) 3-пентаноном полученного по известной методике
бисфосфоната 13 (см. схему 11, II). Присоединение одновременно двух молекул
диэтилкетона осуществить не удается. Поэтому олефинирование проводится
последовательно и требует тщательного подбора условий и реагентов. Так, для
протекания первой реакции УХЭ, по-видимому, необходимо образование соответствующего
дианиона из бисфосфоната 13, для чего последний обрабатывается LDA,
а затем бутиллитием. Повторное олефинирование протекает с трудом, однако его
можно осуществить в достаточно мягких условиях в присутствии гидроксида цезия.
Соотношение образующихся таким образом двух изомерных продуктов 14 и 14а не
меняется при длительном нагревании в условиях реакции, однако разделение
изомеров не требуется, т.к. необходимая изомеризация происходит в условиях
последующей реакции циклизации.
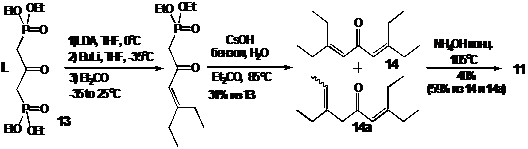

СХЕМА
11
Аналогичный
подход позволяет получить 7-аза-диспиро[5.1.5.3]гексадекан-15-он 1 из ацетона и
циклогексанона (схема 12). Интересно отметить, что поэтапное альдольное
присоединение 2-х эквивалентов циклогексанона к ацетону с последующим
элиминированием приводит к образованию 3-х продуктов, которые конденсируются с
аммиаком в единственный 4-оксо-пиперидин 1. Особенности окисления 1 в радикал
были рассмотрены выше.
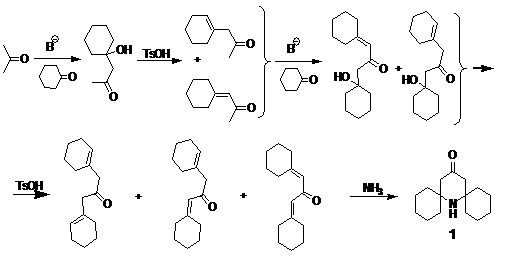
СХЕМА
12
Как и в случае ТАА, пространственно-затруднённые 4-оксопиперидины могут
быть использованы для синтеза НР других типов. Наибольшее значение имеют
реакции расширения цикла, приводящие через нитреновые интермедиаты или через
внедрение карбена к производным диазепанов или азепанов. Производные диазепана
получают по реакции Шмидта или при помощи перегруппировки Бекмана:
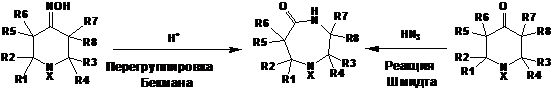
СХЕМА
13
Обе
упомянутые реакции требуют сильно кислой среды, поэтому преобразование цикла
проводят на предшественниках НР, пространственно затруднённых аминах, или
используют защитные группы. Например для синтеза триметилдиэтил замещенного, а
также тетраэтил замещенного радикалов диазепанового ряда может быть
использована следующая тактика. Используя методику Матьяшжевского из
пиперидиновых НР 15a,b получают соответствующие алкоксиамины. После
превращения карбонильной группы в оксимную и последующей перегруппировки по
Бекману с хорошим выходом образуются диазепановые алкоксиамины 16a,b,
которые снова дают НР 17a,b при нагревании в присутствии кислорода (схема14).
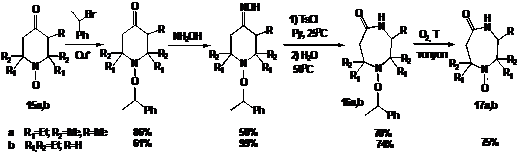
СХЕМА
14
Аналогичным
образом поступают при синтезе НР азепанового ряда. Под действием на пиперидоны
триметилсилилдиазометана происходит расширение цикла с внедрением метиленовой
компоненты. Так был получен, например, тетраэтилзамещенный НР 18

СХЕМА
15
Рассмотренная в этом разделе группа методов является модификацией
традиционных подходов к синтезу НР. Некоторые методики основаны на
использовании доступного сырья, хорошо проработаны. Однако, многостадийность,
умеренные выходы пространственно затруднённых НР, необходимость разделения
сложных смесей снижают привлекательность этих подходов.
Синтезы
оксазолидинов конденсацией аминоспиртов с кетонами
Большое количество нитроксильных радикалов было получено в
оксазолидиновом ряду. Оксазолидиновые циклы образуются при взаимодействии β-аминоспиртов с кетонами в присутствии
кислотного катализатора при азеотропной отгонке воды (т.к. продукт легко
гидролизуется).

СХЕМА
16
Этим методом были получены моно- и бирадикальные соединения, в том числе,
содержащие в положениях 2 и 5 гетероцикла объемные адамантановые заместители
(схема 17).

СХЕМА
17
В некоторых случаях вместо кетонов бывает удобнее использовать
соответствующие кетали. Поскольку вместо воды при этом образуется спирт, то
необходимость в азеотропной отгонке отпадает. Этот прием позволил, к примеру,
получить оксазолидины 19 и 20 (схема 18). Соединение 19 было успешно окислено в
радикал действием м-хлорнадбензойной кислоты (MCPBA), тогда как аминогруппа оксазолидина 20 оказалась
недоступной не только для MCPBA,
но и для диоксирана, молекула которого существенно меньше.

СХЕМА
18
К
классу оксазолидинов принадлежат также и одни из первых стероидных НР,
например, синтезированный на основе холестан-3-она, бирадикал (схема 19). Эти
соединения нашли широкое применение при изучении биологических мембран.

СХЕМА
19
Доступность
b-аминоспиртов и простота делают эти методы
привлекательными несмотря на низкий выход целевых продуктов. Основными
недостатками НР оксазолидинового ряда являются их низкая устойчивость и быстрый
гидролиз продуктов их восстановления. Кроме того, НР этого ряда не нашли
применения в контролируемой радикальной полимеризации.
Синтезы на
основе a-аминонитрилов
α-Аминонитрилы реагируют с кетонами,
давая замещенные 4-оксоимидазолидины с выходами до 90% (схема 20, I). В работе описан ряд интересных
соединений, в том числе бирадикалов, полученных по данной схеме (см. рис.2).
Исходные α-аминонитрилы получают из кетонов, аммиака и цианида.
Благодаря обратимости этого процесса, в отсутствие других кетонов α-аминонитрилы могут вступать в реакцию
самоконденсации(схема 20, II).
Образующиеся имидазолидиноны легко окисляются перекисью водорода с вольфраматом
в соответствующие НР типа 21. Это простой и удобный метод имеет большое значение,
поскольку полученные с его помощью НР 21а-с (рис.2) являются эффективными
регуляторами радикальной полимеризации.


СХЕМА
20
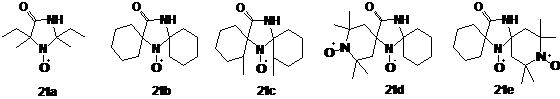
Рисунок
2
Интересно отметить, что автоконденсация α-аминонитрилов в жёстких условиях
может проходить по другому направлению, приводя к образованию
3,5-диоксопиперазинов 22 (схема 21), которые также могут быть окислены в
пространственно затруднённые НР.

СХЕМА
21
Синтезы с
участием трихлорметильного аниона
Разнообразные гетероциклические соединения, содержащие
пространственно-затруднённую вторичную аминогруппу в составе гетероцикла, могут
быть получены конденсацией диаминов или аминоспиртов с кетонами и хлороформом в
щелочной среде. Реакцию проводят в двухфазной системе в условиях фазового
переноса, в присутствии концентрированного водного раствора щёлочи. Согласно
данным работы, образующийся из хлороформа в щелочной среде трихлорметильный
анион реагирует с кетоном, образуя дихлороксиран, который в свою очередь
реагирует по аминогруппе. Замыкание цикла обычно происходит сразу через внутримолекулярное
ацилирование.
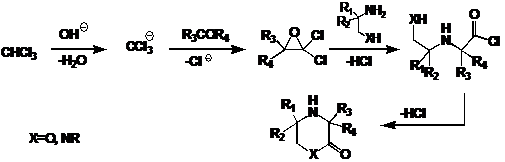
СХЕМА 22
С использованием этого метода были получены пространственно затруднённые
НР пиперазинового, диазепанового и морфолиновго рядов. Для синтеза НР
пиперазинового ряда исходные диамины могут быть получены по реакции Манниха:
вторичное нитросоединение обрабатывают смесью первичного амина с
формальдегидом, после чего нитрогруппу восстанавливают водородом на скелетном
никеле. Согласно этой схеме был получен, например, тетраэтилпиперазиновый НР 23
(схема 23, Ia). Невысокий выход радикала 23
очевидно связан с тем, что конденсация диаминов с кетонами протекает с
образованием двух продуктов 24 и 25 (схема 23, I). Селективность, а также скорость реакции существенно
повышаются в присутствии катализатора фазового переноса (PTC). Например, в реакции II (cхема 23), если PTC = BnNEt2Cl, то соотношение продуктов 24а : 25а = 86 : 14, а если PTC = BnNEt2Cl : NaCN = 2 : 6, то соотношение 24а : 25а=
95 : 5.

СХЕМА
23
Аналогичным
образом из 1,3-диаминов получают семичленные, диазепановые циклы. Известны
также соединения 26, содержащие два цикла (схема 24).

СХЕМА
24
3,3,5,5-Тетразамещенные-2-оксоморфолины
образуются подобно 3-оксопиперазинам - из β-аминоспиртов и кетонов в присутствии хлороформа в сильнощелочной среде.
В условиях реакции происходит гидролиз образующегося лактона, обратная
циклизация происходит при действии хлористого водорода. Восстановление
2-оксоморфолина 27 алюмогидридом лития также разрушает цикл, который, однако,
можно сформировать заново действием метансульфоновой кислоты при нагревании в
инертной атмосфере (схема 25).
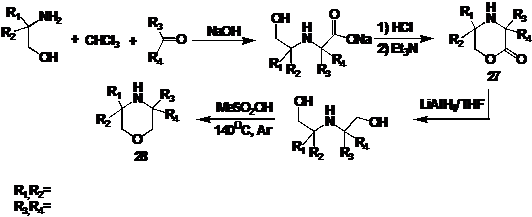
СХЕМА 25
Реакции
лактамов с металлорганическими соединениями
N-Замещённые
фталимиды и циклические имидаты отличаются способность реагировать с
несколькими эквивалентами металлорганического соединения, в результате чего в
одну стадию к α-атому(ам) углерода вводятся сразу два заместителя. Это
позволяет избежать постадийного введения заместителей и сильно упрощает синтез
пространственно затруднённых аминов.
На схеме 26 показан синтез НР изоиндолинового ряда. Все 4 этильных заместителя
вводятся в ходе одной процедуры. Именно на этих НР впервые был обнаружен эффект
объёмных заместителей у α-атома углерода нитроксильной группы, понижающих
склонность НР к восстановлению. НР этого ряда нашли широкое применение как в
биофизических исследованиях, так и в химии полимеров.
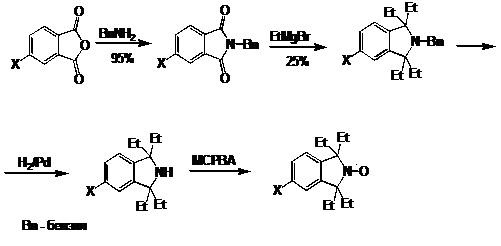
СХЕМА
26
Алкилирование лактамов по атому кислорода приводит к образованию
циклических имидатов (метоксииминов), обладающих более высокой реакционной
способностью по отношению к реактивам Гриньяра, чем лактамы. Взаимодействие
активированного метилированием имидата 29 с аллилмагнийбромидом позволяет сразу
ввести пару объемных заместителей к азометиновому атому углерода. Спиновая
метка в таком соединении встроена прямо в основной скелет молекулы, это
открывает путь к синтезу спин-меченных аналогов природных стероидов, которые
затем могут принимать участие в метаболических процессах организма.

СХЕМА 27
Это же метод был использован для синтеза азетидиновых НР 30 и 31.
Исходный метоксиимин может быть получен метилированием β-лактама, который получали в одну
стадию из изобутилена.

СХЕМА
28
Синтезы с
использованием реакции 1,3-диполярного циклоприсоединения нитронов
Несмотря на то, что реакция 1,3-диполярного циклоприсоединения широко
применяется в органическом синтезе, сведения об использовании её для синтеза НР
появились совсем недавно. Так ключевой стадией в синтезе НР 33 является
циклоприсоединение нитрона 32 (синтез см. стр.28) и метилакрилата (схема 29).
Важное достоинство этого метода заключается в том, что в результате раскрытия
изоксазолидинового цикла у α-атома углерода аминогруппы
оказывается заместитель, содержащий функциональную группу.
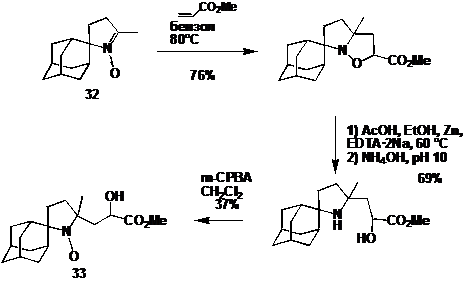
СХЕМА
29
Ещё
более привлекательным представляется использование внутримолекулярной реакции
1,3-диполярного циклоприсоединения. В этой модификации реакция становится менее
чувствительной к стерическим препятствиям и идёт даже с неактивированными
алкенами. Первый и весьма удачный пример такого синтеза представлен на схеме
30.

СХЕМА
30
Следует
отметить также удачный выбор исходного нитрона 34, благодаря особенностям
строения которого реакции на всех стадиях синтеза, кроме второй (окисления
гидроксиламина в нитрон), идут стерео- и региоспецифично и приводят к
единственному стереоизомеру.
Пространственно-затрудненные
гидроксиамины
В отличие от пространственно затруднённых аминов, окисление которых в НР
не всегда проходит легко, окисление пространственно-затруднённых гидроксиаминов
(ПГ), как правило, не вызывает затруднений. Нередко образующийся гидроксиламин
не удаётся выделить, поскольку он легко окисляется в соответствующий НР.
Гидроксиамины превращаются в радикалы под действием разнообразных
неорганических и органических окислителей, среди которых наиболее часто
используются PbO2, MnO2, периодаты, К3Fe(CN)6, Ag2O, или кислород воздуха.
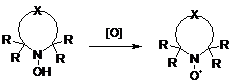
СХЕМА
31.
Однако по доступности ПГ сильно уступают аналогичным аминам. Это связано
как с относительной лабильностью гидроксиламина и его органических производных,
так и с особенностями реакционной способности NHOH-группы, более чувствительной к стерическим
ограничениям, чем аминогруппа, и способной реагировать по разным реакционным
центрам. Поэтому примеры синтезов ПГ через конденсации не отличаются
разнообразием. Основным методом их получения является реакция нитронов с
металлорганическими соединениями.
Способы
получения пространственно-затрудненных гидроксиаминов
Синтезы с
использованием реакции конденсации
Циклические N,N-дизамещённые гидроксиамины могут
быть получены конденсацией бифункциональных 1,2- и 1,3-гидроксиаминооксимов,
гидроксиаминоспиртов или бис-гидроксиаминов, уже содержащих объемные
заместители, с кетонами. К сожалению, известно мало соединений, содержащих
объёмные заместители при атоме углерода у гидроксиамино группы, а немногие
известные либо неохотно реагируют с кетонами, либо эти реакции не изучены. Среди
известных синтезов НР с более, чем одним объёмным заместителем у α-атомов углерода, можно упомянуть
синтез НР 3-имидазолин-3-оксида конденсацией 1,2-гидроксиаминооксимов с
циклическими кетонами.
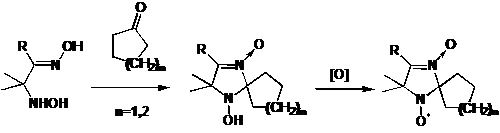
СХЕМА
32
Гораздо
легче проходит конденсация 1,2-гидроксиаминокетонов с кетонами и ацетатом
аммония.
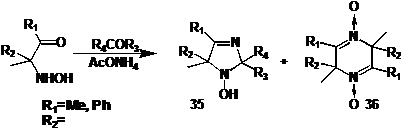
СХЕМА
33
Помимо
ПГ 35 наблюдается образование значительного количества продуктов
самоконденсации гидроксиаминокетонов - дигидропиразинов 36 (схема 33), особенно
если реакция не завершается в течение 5 часов. В случае если R1=Ph,
практически с любыми субстратами образуется только пиразин
Известные
примеры получения НР с четырьмя объёмными заместителями сводятся к реакциям
3-гидроксиамино-3-этилпентанона-2. Конденсации этого гидроксиаминокетона с
кетонами в присутствии ацетата аммония с хорошими выходами приводят к
образованию 1-гидрокси-2,5-дигидроимидазолов. На воздухе эта реакция
осложняется окислением образующихся ПГ в соответствующие НР и
гидроксиаминокетона в нитрозосоединение, поэтому конденсацию проводят в инертной
атмосфере.

СХЕМА 34
Пространственно затрудненные НР ряда имидазолина легко превратить в
имидазолидиновые производные алкилированием по азоту в третьем положении
гетероцикла и последующим восстановлением иминиевой соли боргидридом натрия.
Нитроксильный и другие фрагменты молекулы при этом не затрагиваются (схема 35).
Имидазолидиновые НР являются менее сильными окислителями из-за отсутствия
электроноакцепторного эффекта фрагмента С=N. В связи с этим, они более привлекательны для использования
в биофизике и в химии полимеров.
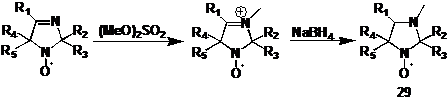
СХЕМА
35
Синтезы с
использованием металлорганических соединений
Реакция нитронов с металлорганическими соединениями весьма широко
используется в синтезе НР различного строения. Этот подход имеет огромное
синтетическое значение, т.к. позволяет ввести до четырех разных заместителей в
уже сформированный скелет молекулы. Разработано большое число методов синтеза
исходных нитронов.
Реакция нитронов с металлоорганическими соединениями чувствительна к
стерическим препятствиям. Это особенно характерно для реакций с реактивами
Гриньяра, которые обычно образуют объёмные комплексы с молекулами растворителя.
Например, реактив Гриньяра легко присоединяется альдонитронам (схема 36, I), но при взаимодействии с
кетонитронами образуются, как правило, имины (схема 36, II).

СХЕМА 36
Дезоксигенирование может происходить и в реакциях с цикличепскими
нитронами. Например, 3-имидазолин-3-оксиды реагируют с реактивом Гриньяра с
образованием продуктов 37 и 38. При этом в реакции с метилмагнийиодидом
образуется в основном имин 38, а с фенилмагнийбромидом - гидроксиамин 37.
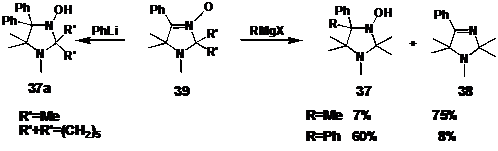
СХЕМА 37
Литийорганические соединения менее склонны к
окислительно-восстановительным реакциям. Например, в реакциях с нитронами 39
образуются только продукты 37a.
Недостатком литийорганических соединений является склонность к металлированию,
особенно, алкильных заместителей, в результате чего могут образовываться
продукты самоконденсации.
Несмотря на упомянутые ограничения, имеется масса примеров успешного
использования реакции нитронов с металлоорганическими соединениями для
получения ПГ и пространственно-затруднённых НР. Ниже приведены некоторые примеры.
Ациклические
нитроксильные радикалы
Реакция металлорганических соединений с альдонитронами является основным
способом получения пространственно-затруднённых НР с β-атомом водорода. Устойчивость таких
НР с атомом водорода у α-углеродного атома, очевидно, связана с тем, что
стерическое напряжение делает невыгодной конформацию, необходимую для
отщепления β-атома водорода. Поскольку торсионный угол O-N-C-H в радикале близок к 180о,
возможность диспропорционирования на нитрон и гидроксиамин отсутствует, и такие
соединения могут быть не только выделены в индивидуальном виде, но и храниться
длительное время при комнатной температуре без разложения (рис.3).
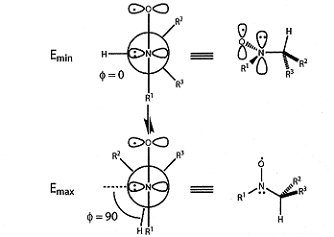
Рисунок
3
Исходные ациклические нитроны получают обычно конденсацией альдегидов с
трет-алкил гидроксиламинами. Последние часто получают in situ восстановлением нитросоединений. Полученный таким
образом альдонитрон обрабатывают реактивом Гриньяра, после чего образующийся
гидроксиамин окисляют воздухом в присутствии солей меди (II) - эта система обеспечивает
необходимые для окисления в радикал мягкие условия (схема 38).
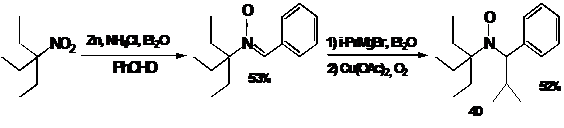
СХЕМА
38
Например,
по этой схеме, можно синтезировать НР 40. Исходный триэтилнитрометан получают
из соответствующего спирта с использованием реакции Риттера. НР 40 является
эффективным регулятором полимеризации стирола и n-бутилакрилата.
Синтез НР, содержащего адамантановый фрагмент, 41 можно осуществить двумя
путями. Первый метод аналогичен описанному выше. Нитроадамантан 42 получают
окислением адамантиламина диметилдиоксираном, который образуется in situ из ацетона и оксона. Интересно отметить, что побочный
нитрозоадамантан 42а, образующийся в реакции, не требует удаления из смеси,
т.к. в присутствии цинка также как и нитросоединение 42 превращается в
гидроксиламин.
Второй подход дает несколько более высокий выход НР 41. Имин 43,
полученный конденсацией адамантиламина с бензальдегидом, окисляют в оксазиридин
44 м-хлорнадбензойной кислотой. Далее оксазиридин превращается в нитрон 45 при
термической перегруппировке в кипящем ацетонитриле.
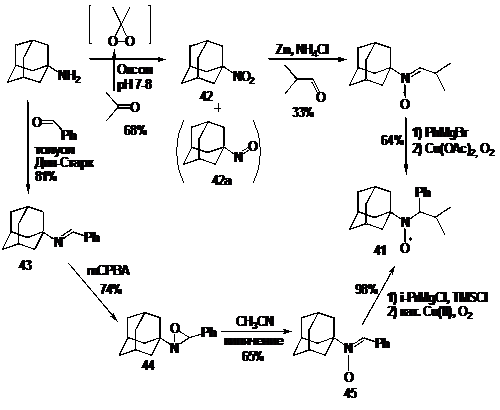
СХЕМА
39
Интересным
направлением в химии ациклических НР является синтез функциональных
производных, содержащих, к примеру, гидроксиметильные заместители в α-положении к нитроксильной группе. В отличие от
описанного выше, проводя реакции, приходится учитывать наличие в молекуле
высокореакционноспособных заместителей, поэтому особое внимание уделяется
защите гидроксильных и нитроксильной групп. Синтез НР 46 происходит по обычной
схеме, но после формирования 1,3-диоксанового цикла для защиты 2-х гидроксиметильных
заместителей. Однако для снятия защиты необходима сильно кислая среда, которой
нитроксильная группа может не выдержать. Поэтому НР 47 предварительно
превращают в алкоксиамин.

СХЕМА
40
Аналогичным
образом, исходя из 2-(гидроксиметил)-2-нитропропан-1,3-диола, можно получить НР
с тремя гидроксиметильными заместителями рядом с нитроксильной группой (схема
41).
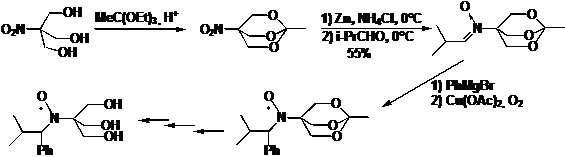
СХЕМА
41
Известно,
что 3-имидазолин-3-оксиды могут существовать ввиде 2-х таутомерных форм 48А и
48B. В случае, если соединение существует в циклической
форме 48А (R4=Alk), при взаимодействии с металлорганическими реагентами
образуются 1,3-дигидроксиимидазолидины, если же нитрон существует в
ациклической форме 48В (R4=Ar), то действие фениллития или фенилмагнийбромида
приводит к образованию ациклических гидроксиаминооксимов 49. Окисление
гидроксиаминов 49 двуокисью марганца или свинца приводит к стабильным
ациклическим НР 50.
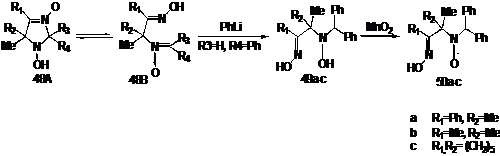
СХЕМА
42
Пиперидины
Оригинальный метод синтеза 2,6-дитрет-бутил замещенных пиперидиновых НР
предложен в работе. Такой подход тем более интересен, учитывая, что ввести 2
трет-бутильных заместителя в ближайшее окружение нитроксильной группы другим
способом чрезвычайно сложно. Гидрированием коммерчески доступного
2,6-дитретбутилпиридина получают циклический вторичный амин 51, содержащий при α-атомах углерода 2 объемных
заместителя. Последующее окисление пиперидина 51 различными окислителями
приводит к гидроксиамину 52, а затем нитрону 53. Интересно отметить, что
трет-бутильные группы настолько осложняют доступ к N-O-фрагменту молекулы, что
первично образующийся при окислении гидроксиамина 52 НР имеет время жизни ~
нескольких часов при комнатной температуре, и дальнейшее превращение его в
нитрон 53 происходит существенно медленнее, чем в случае менее затрудненных
производных. Нитрон 53 далее обрабатывают реактивом Гриньяра. Последний
заместитель вводят аналогичным образом.

СХЕМА
43
-Имидазолины
Для получения НР имидазолинового ряда с объёмными заместителями в
положения 2 и 5 реакция нитронов с металлорганическими соединениями является
гораздо более эффективным методом синтеза, чем конденсации (см. стр.23). В
качестве исходных нитронов могут быть использованы 2Н-имидазол-1-оксиды с двумя
объёмными заместителями в положении 2, легко образующиеся в конденсациях
изонитрозокетонов с дифенилметанимином или кетонами и ацетатом аммония.
Последовательное введение двух заместителей в положение 5 при взаимодействии с
металлоорганическими соединениями и окисление даёт НР 54,55a,b.
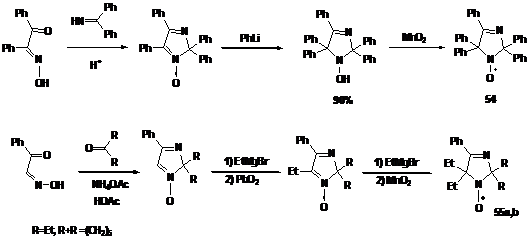
СХЕМА
44
Присоединение
металлорганических соединений к 4Н-имидазол-3-оксидам может происходить как по
атому углерода нитронной группы, так и по положению 5, причём направление
реакции зависит от электронного влияния заместителей в положениях 2 и 5. При
взаимодействии 5-диалкиламино-замещённых 4Н-имидазол-3-оксидов с
этилмагнийбромидом выделены только НР 56a,b,
что обусловлено электрондонорным эффектом аминогруппы. Однако, выход этих НР
невелик.

СХЕМА 45
Пирролидины
Благодаря высокой стабильности, НР ряда пирролидина получили широкое
распространение. Реакции пирролин-N-оксидов с реактивами Гриньяра являются основным методом получения этих
НР (схема 46).
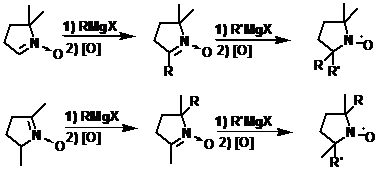
СХЕМА 46.
Следует отметить, что перечень заместителей, которые можно ввести в
ближайшее окружение нитроксильной группы посредством взаимодействия
металлорганических соединений с пирролин-N-оксидами, не ограничивается только алкильными и арильными
группами. Введение в α-положение к нитроксильному фрагменту сложных
заместителей с различными функциональными группами в своем составе позволяет
получать новые НР с необычными свойствами: от водорастворимых и амфифильных
радикалов до различных спиновых меток и веществ, способных к
комплексообразованию и самоорганизации.
В качестве примера можно привести синтез радикала 57 с гидроксифенильным
заместителем. Пирролин-N-оксид
58 получают в 2 стадии: при конденсации метилвинилкетона с нитроциклогексаном
образуется соединение 59, которое циклизуется при восстановлении цинком с
хлористым аммонием. Для сохранения гидроксильной группы используется
трет-бутилдиметилсилильная защита, которая снимается после получения радикала.

СХЕМА
47
-Адамантил-5-метилпирролин-N-оксид
32, получают из 2-нитроадамантана и метилвинилкетона аналогично нитрону 58.
Обработка нитрона 32 этинильным реактивом Гриньяра дает
спироадамантилпирролидиновый НР 60. Пирролидинпропаргиловый спирт 61 получают в
реакции нитрона 32 с BrMgCºCCH2OMgBr
- реактивом Гриньяра, образующимся при взаимодействии пропаргилового спирта и
2-х эквивалентов этилмагнийбромида. НР 61 - ключевое соединение в синтезе
липофильных спиновых меток. Так после замещения гидроксильной группы на бром
62, в молекулу можно ввести SSO2CH3 функцию с образованием тиол-специфического
метантиосульфонатного НР 63.

СХЕМА
48
Стерическая
доступность нитроксильного фрагмента в радикалах 64,65 понижена за счет
фрагмента краун-эфира, в который нитроксильная функция встроена подобно
спиновой метке. В некоторых конформациях нитроксильная группа находится внутри
полости молекулы. В комплексах таких краун-эфиров с подходящими ионами металлов
нитроксильный радикал, в силу пространственных ограничений будет
взаимодействовать с металлом напрямую. Синтез спин-меченных краун-эфиров 64, 65
пытались проводить различными путями, исходя из пирролинового метилнитрона 66.
Однако успешным оказался только один подход. Он заключается в следующем. После
последовательного присоединения к исходному нитрону 66 двух метоксифенильных
групп и снятия защиты соединение 67 алкилируют дигалогенидами 68 в присутствии
гидрида натрия и ДМФА с образованием краун-эфиров 69 и 70. Обработка нитронов
69 и 70 метиллитием и последующее окисление приводят к преимущественному
образованию цис-диметил замещенных НР 64 и 65, т.к. метиллитий подходит к
нитронной группе с более пространственно-досупной стороны.

СХЕМА
49
С
использованием того же принципа, что и для краун-эфиров, осуществлен синтез
нитроксил-меченого криптанда 71 (схема 50). Исходный нитроксильный радикал с
двумя карбоксилсодержащими заместителями превращают в крайне нестабильный
диацилхлорид действием оксалилхлорида на калиевую дисоль 72. Ацилирование
диамина диаза-18-краун-6 хлорангидридом дикислоты 72 является ключевой стадией
синтеза. Образующийся диамид далее восстанавливают в системе BH3-ТГФ.
Для полученных соединений исследована их способность к комплексообразованию.
Интересно, что криптанд 71 связывает ионы натрия и калия существенно лучше, чем
краун-эфиры 64,65.
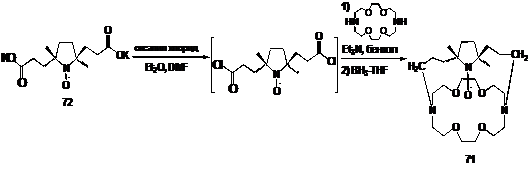
СХЕМА
50
Заключение
Таким образом, в рамках представленного обзора рассмотрены известные
методы синтеза пространственно затрудненных НР различной структуры. Для
получения прямых предшественников НР - вторичных аминов и гидроксиаминов как
правило используют два основных подхода: конденсации и реакции
металлорганических соединений с нитронами или активированными иминами.
Применение конденсации для синтеза затрудненных НР сильно ограничивается
необходимостью формирования исходных синтонов, обладающих довольно сложным
строением. Другим недостатком является неоднозначность протекания превращений в
реакционных условиях, что часто приводит к накоплению значительных количеств
побочных продуктов. Нередко протекание побочных процессов обусловлено именно
стерическими затруднениями, создаваемыми объемными заместителями.
Пространственная недоступность нитронной группы в реакциях с реактивами
Гриньяра иногда приводит к деоксигенированию субстрата и образованию иминов.
Также металлорганические соединения могут металлировать заместители при нитроне
по α-метиленовой компоненте, что
способствует протеканию разнообразных конденсаций.
Тем ни менее второй метод, по-видимому, обладает более широкими
синтетическими возможностями, т.к. позволяет ввести в α-положение к нитроксильной группе до
четырех различных заместителей, открывая путь к самым разным производным, в том
числе функциональным. Кроме того, развитие химии нитронов, металлорганических
соединений и метода защитных групп делает легко доступными все необходимые
синтоны.
Весьма перспективными представляются также методы, основанные на реакции
1,3-диполярного циклоприсоединения, отличающиеся пониженными стерическими
ограничениями. Благодаря наличию функциональных групп во вводимых заместителях
эти методы оставляют возможность для построения дополнительных циклических
систем, ограничивающих доступность нитроксильной группы.
Глава 2. Синтез нитроксильных радикалов на базе
2Н-имидазол-1-оксидов
Необычно высокая устойчивость к восстановлению, обнаруженная для
имидазолидиновых нитроксильных радикалов с четырьмя этильными группами в
положениях 2 и 5 имидазолидинового цикла, делает весьма актуальным исследование
влияния ближайшего окружения нитроксильной группы на
окислительно-восстановительные свойства нитроксильных радикалов. Первоочередной
задачей данного исследования было получение разнообразного набора нитроксильных
радикалов имидазолина и имидазолидина, различающихся по стерической
затруднённости нитроксильного фрагмента, для последующего исследования их
свойств. При этом наибольщий интерес вызывали НР с несколькими (четырьмя)
алкильными заместителями, большими чем метил, у α-атомов углерода нитроксильной группы,
для которых стерический эффект заместителей дополняется, а не компенсируется их
электронным эффектом. К началу наших исследований в литературе описан метод
синтеза НР имидазолина с четырьмя этильными заместителями или двумя этильными
группами и спиро-циклогексановым фрагментом у α-атомов углерода нитроксильной группы,
который включает 7 стадий (из диэтилкетона) с суммарным выходом НР менее 20%.
При этом использование подобного метода для получения НР с заместителями,
отличными от этильных групп, в положении 5 представляется весьма проблематичным
и требует разработки специальных методов синтеза гидроксиламинооксимов для
введения каждой новой комбинации заместителей.
Рациональным решением этой проблемы представлялось использование реакции
нитронов с реактивами Гриньяра (см. главу 1). Хотя этот метод имеет
существенные ограничения и в ряде случаев даёт невысокие выходы целевых
нитроксильных радикалов, он позволяет использовать единый подход для синтеза
набора различных производных.
Для синтеза имидазолиновых нитроксильных радикалов, содержащих объёмные
заместители в положениях 2 и 5 гетероцикла, нами был выбран подход, включающий
двукратное присоединение магнийорганического соединения по атому углерода
альдонитронной группы 2Н-имидазол-1-оксидов (схема 51).
СХЕМА
51.
Для
выбора именно этого пути имелись следующие предпосылки:
)
2Н-имидазол-1-оксиды получают в одну стадию из доступных изонитрозокетонов,
причём метод их синтеза позволяет легко вводить объёмные алкильные заместители
в положение 2 гетероцикла.
)
Присоединение металлоорганических соединений к 2Н-имидазол-1-оксидам ранее уже
было успешно использовано в синтезе нитроксильных радикалов.
)
В отличие от ранее разработанного метода, этот поход принципиально пригоден для
введения разнообразных заместителей в положение 5 и позволяет избежать
необходимости синтеза набора различных 1,2-гидроксиаминокетонов.
Исходные
2H-имидазол-1-оксиды 16b,d,f получали согласно известным литературным методикам
конденсацией соответствующих изонитрозокетонов 15 с кетонами и ацетатом
аммония. Синтез ранее не описанных 2Н-имидазол-1-оксидов 16a,с,e
осуществляли аналогичным образом.
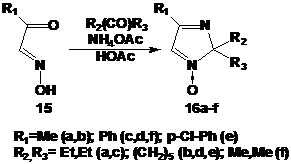
СХЕМА
52.
Следует
отметить, что диэтилкетон вступает в реакцию менее охотно, чем циклогексанон и,
тем более, ацетон, а конденсации с участием изонитрозоацетона дают меньшие
выходы, чем конденсации изонитрозоацетофенона и оксима
2-(4-хлорофенил)-2-оксоацетальдегида. Тем не менее, в большинстве случаев
получены приемлемые выходы 2Н-имидазол-1-оксидов (табл. 1). Спектральные
характеристики полученных соединений подобны характеристикам известных
2,2-диалкил 2Н-имидазол-1-оксидов. В ИК спектрах соединений 16a,с,e
имеются характерные полосы симметрических и асимметрических колебаний связей C=N при
1510-1520 и 1570-1590 см-1 и связи С-Н альдонитронной группы при 3015-3070
см-1, в УФ-спектрах - характерная полоса поглощения при 270-285 нм, положение
которой мало зависит от заместителя в положении 4, а в спектрах ПМР - сигнал
протона альдонитронной группы при 7.1-8.1 м.д. Сигналы альдонитронного атома
углерода в спектре ЯМР 13С находятся в обычной для альдонитронов области
125-130 м.д., а сигнал иминного атома углерода смещен на 5 м.д. в сильное поле
по сравнению с изолированной фенилиминовой группой. В ПМР-спектрах 16a,c
сигналы метиленовых протонов этильных групп в положении 2 гетероцикла
представляют собой АВ-систему в области 1.66-2.08 м.д. (JAB=14-20
Гц), каждый сигнал которой расщепляется в квартет на соседней метильной группе
(Jк=7.2-7.4 Гц). В спектрах соединений 16b,d,e
сигналы протонов спироциклогексанового фрагмента представляют собой три
мультиплета интенсивностью 3Н, 5Н и 2Н. Фенильные группы в спектрах ПМР
2H-имидазолов 16c,d,f выходят в виде двух характерных мультиплетов
интенсивностью 2H и 3H, а в спектре соединения 16e имеется AA’BB’-система
(J= 8.5 Гц), соответсвующая протонам п-хлорфенильной группы. Можно было
ожидать, что двукратная обработка 2Н-имидазол-1-оксидов 16a,b
этилмагнийбромидом с последущим разложением реакционной массы и окислением
позволит получить описанные ранее НР 19а,b в меньшее
число стадий, чем при использовании литературных методов. Однако при проведении
синтеза с самого начала возникли осложнения. При взаимодействии
2Н-имидазол-1-оксидов 16a,b, содержащих метильную группу в 4 положении цикла, с
этилмагнийбромидом количество продуктов и их соотношение существенно зависит от
порядка прибавления реагентов. Соединения 17a,b
образовывались лишь в случае, когда 2Н-имидазолы 16a,b
вводили в эфирный раствор реактива Гриньяра, но не наоборот. Вероятно, основной
причиной протекания множества побочных процессов является склонность соединений
16a,b к металлированию по метильной группе в основных
средах, свойственных растворам металлорганических соединений. Такая
металлированная частица может конденсироваться с исходным 2Н-имидазолом,
образуя множество продуктов разного строения. Кроме того, продукт присоединения
реактива Гриньяра - гидроксиамин, может подвергаться дегидратации в
сильноосновной реакционной среде. По-видимому, попадая в избыток
магнийорганческого соединения, 2Н-имидазолы 16a,b
сначала целиком металлируются, что частично подавляет побочные процессы самоконденсации
и делает присоединение реактива Гриньяра предпочтительным.

СХЕМА
53.
Действительно
при взаимодействии 2H-имидазола 16a с избытком этилмагнийбромида
образуется сложная смесь продуктов, из которой однако при растирании с гексаном
выпадает осадок ожидаемого имидазолина 17а. К сожалению, вещество 17а не
удается полностью очистить от примесей ни перекристаллизацией, ни
хроматографически, и выход его не превышает 25%. Спектральные характеристики
полученного соединения типичны для 1-гидрокси-3-имидазолинов. В ИК-спектре
присутствует сигнал ОН-группы при 3154, а также единственная полоса колебаний
связи C=N при 1652 см-1. В ПМР спектре сигнал метинового
протона при С5 выходит в виде триплета при 3.76 м.д. с константой СТВ 4.0 Гц, а
в спектре ЯМР 13С имеется сигнал атома углерода С5 при 74.76 м.д.. В отличие от
2Н-имидазол-1-оксида 16а этильные группы во 2-м положении неплоского
имидазолинового цикла 17a неэквивалентны, триплетные сигналы их СН3-фрагментов
выходят при 0.81 и 0.85 м.д., с константой СТВ 6.6 Гц. Также в спектре
присутствует триплет еще одного этильного заместителя при 0.74 м.д. с
константой 6.4 Гц для CH3-фрагмента. Сигналам СН2-фрагментов всех 3-х этильных
групп соответствует широкий мультиплет при 1.60 м.д.. 2Н-Имидазол-1-оксид 18а
получали окислением имидазолина 17а избытком диоксида свинца в хлороформе.
Реакция сопровождается образованием трудноотделимых побочных продуктов, что не
позволило выделить это соединение в чистом виде.
Синтез
другого этилнитрона 18b осуществляли без выделения гидроксиамина, окисляя
реакционную смесь сразу после разложения водой. Выход 2Н-имидазола 18b (40%)
несколько превышает выход его триэтильного аналога 18а (22%). Также как и в
предыдущем случае, очистка соединения 18b проблематична. Кроме того было
обнаружено, что при хранении этилнитроны 18а,b разлагаются с образованием
сложной смеси неидентифицированных продуктов. Вероятно, неустойчивость
кетонитронов 18а,b является одной из причин, по которым их не удается
выделить в чистом виде. Спектральные характеристики 2Н-имидазол-1-оксидов 18а,b
подобны приведённым для исходных соединений 16a,b, но в ПМР-спектрах
отсутствует сигнал протона при С5 имидазольного цикла, и появляются сигналы,
относящиеся к новому этильному заместителю.
Добавление
неочищенных кетонитронов 18a,b к избытку этилмагнийбромида приводит к
образованию продуктов присоединения реактива Гриньяра по нитронной группе. В
процессе обработки реакционной смеси образующиеся гидроксиамины частично
окисляются кислородом воздуха. Поэтому их доокисляли избытком диоксида марганца
без выделения. При этом образовывались 3-имидазолиновые НР 19a,b с
выходами ~ 40-50%. Характеристики соединений 19а,b полученных по
этому методу, совпадают с характеристиками заведомых образцов.

СХЕМА
54.
Таким
образом, присоединение реактивов Гриньяра к 2H-имидазол-1-оксидам мало подходит
для препаративного получения НР имидазолинового ряда с метильной группой в
положении 4. Этот подход, по-видимому, является существенно менее эффективным,
нежели описанный в работе метод получения имидазолиновых НР конденсацией α-гидроксиаминокетонов с кетонами.
В
отличие от 4-метил-2Н-имидазол-1-оксидов 16a,b
2H-имидазол-1-оксиды 16c-f не содержат кислых протонов и, следовательно, не
склонны к самоконденсации в присутствии оснований. Благодаря этому качеству
4-фенилзамещенные соединения 16c-f представляются гораздо более перспективной основой
для синтеза пространственно затрудненных НР.
Обработкой
2Н-имидазол-1-оксидов 16c,d,f различными реактивами Гриньяра синтезировали
набор 5-замещенных 1-гидрокси-2,5-дигидроимидазолов 20a-f (см. схему 55).
Выходы гидроксиаминов 20 в этой реакции зависят от природы используемого
магнийорганического соединения и уменьшаются в ряду PhMgBr, t-BuMgCl ≥
EtMgBr > AllMgBr. Пара этильных заместителей во 2-м положении гетероцикла
субстрата также затрудняет реакцию в большей степени, нежели гем-диметильный
или спироциклогексановый фрагменты. Порядок прибавления реактива Гриньяра,
напротив, не оказывает значительного влияния на выходы целевых продуктов.
Причиной понижения выхода имидазолинов 20a,b и
с, по-видимому, является их легкая дегидратация в щелочной среде, которая может
происходить при обработке реакционной массы.

СХЕМА
55.
Спектральные
характеристики полученных соединений сходны с описанными ранее для имидазолина
17а. Так в ИК-спектрах продуктов присутствуют широкая полоса колебаний
ОН-группы при 3165-3185, и сигнал связи C=N при
1620-1630 см-1. Однако в УФ-спектрах наблюдается поглощение фенилиминного
фрагмента при ~240 нм; в ПМР спектрах - сигнал метинового протона при С5
выходит в виде дублета дублетов при 4.4-4.6 м.д. с константами СТВ 6-10 и 3.6-4
Гц на диастереотопных протонах соседнего СН2-фрагмента. В спектре ПМР
имидазолина 20c фрагменту =СН2 принадлежат сигналы при 5.02 и 5.06
м.д., а при 5.93 м.д. выходит мультиплет =СН- - фрагмента. В ЯМР 13С сигналы
СН2= и =СН- проявляются при 116.77 и 135.23 м.д. соответственно. Отнесение
сигналов -СН2- групп в спектре ПМР этого соединениябыло сделано на основании
данных спектров с частичным подавлением спин-спинового взаимодействия (ССВ).
Метиленовым протонам ллильной группы в составе этого соединения соответствуют
два мультиплета при 2.35 и 2.59 м.д..
В
спектрах 13C имидазолинов 20d,e присутствуют два набора сигналов, принадлежащих
фенильным группам. При этом, о- ,м- и п-атомам углерода фенильного заместителя
в 4-м положении имидазолинового цикла соответствуют более слабопольные (128-130
м.д.) сигналы, а атомам фенила в 5-м положении - более сильнопольные (127-128
м.д.). Однако сигнал и-С заместителя в 5-м положении гетероцикла, напротив,
лежит в более слабом поле на 6-7 м.д.. Такое расположение сигналов, очевидно,
связано с индуктивным и мезомерным эффектами, оказываемыми на ароматические
системы донорным (-CH-N-OH) и акцепторным (-C=N-) фрагментами. Кроме того,
благодаря дезэкранирующему влиянию фенильной группы, сигнал протона при С5 в
спектрах ПМР имидазолинов 20d,e сдвинут на ~1 м.д. в слабое поле.
В
5-трет-бутил-замещенном имидазолине 20f π-система фенильного заместителя выведена из сопряжения с эндоциклической
связью C=N, что обусловлено стерическим напряжением, создаваемым трет-бутильной
группой, поэтому некоторые спектральные характеристики его отличаются от характеристик
соединений 20a-e. Так гидроксиамин 20f имеет сравнительно более
коротковолновый максимум поглощения (~230 нм) в УФ-спектре. Кроме того, сигнал
С5 в спектре ЯМР 13С лежит на ~10 м.д. в более слабом поле, чем у других
имидазолинов 20. Последнее характерно для трет-бутил-замещенных соединений и
полностью согласуется с литературными данными по зависимости химсдвигов атома С
от степени его α-замещенности в алканах.
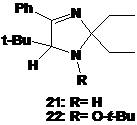
РИСУНОК
4
Помимо
имидазолина 20f из реакционной смеси были выделены минорные продукты
21 и 22 с выходом 4 и 3 % соответственно. Спектральные характеристики обоих
соединений весьма близки к характеристикам гидроксиамина 20f, что может
свидетельствовать об их имидазолиновой природе. Так УФ- и ЯМР-данные соединения
21 почти совпадают с таковыми имидазолина 20f. Однако в ПМР-спектре 21
отсутствует уширенный сигнал от протона N-OH-группы
при 4.98 м.д., а в спектре ЯМР 13С сигналы, принадлежащие С2 и С5 имидазольного
цикла, имеют химсдвиги, 92.83 и 77.44 м.д., т.е. находятся в более сильном
поле. В ИК-спектре 21 полоса при 3379 см-1 имеет характерную для колебаний
связи N-H-группы в 1-незамещённых имидазолинах узкую форму. Исходя из всего,
выше сказанного, а также данных элементного анализа, был сделан вывод, что
выделенному соединению соответствует структура
5-трет-бутил-4-фенил-2,2-диэтил-2,5-дигидро-имидазола (21) (см. рис. 4).
В
спектрах ЯМР соединения 22 помимо сингналов, близких к имеющимся в спектрах
имидазолина 21 присутствуют сигналы ещё одной трет-бутильной группы, причём,
все сигналы последней сдвинуты в слабое поле, что указывает на связь с
гетероатомом (кислородом). На основании приведенных данных, а также данных элементного
анализа выделенному соединению присвоена структура
5-трет-бутил-1-трет-бутокси-2,2-диэтил-4-фенил-2,5-дигидроимидазола (22) (см.
рис. 4) Интересно, что подобно имидазолинам 20a-e алкоксиамин 22 имеет максимум
поглощения в УФ-спектре при 238 нм, что характерно для сопряженного
фенилиминного фрагмента. Предположительно, стерическое напряжение, создаваемое
объемными трет-бутильной и трет-бутоксильной группами, искажает
гетероциклическую систему соединения таким образом, что наиболее энергетически
выгодной становится конформация, в которой фенильное кольцо вступает в
сопряжение.
Соединение
22, очевидно, образуется в результате алкилирования трет-бутилхлоридом,
непрореагировавшим при получении реактива Гриньяра, аниона - продукта
присоединения трет-бутилмагнийхлорида к нитрону 16с (Схема 56).
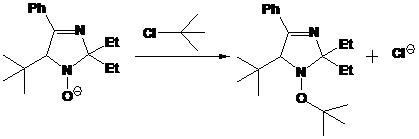
СХЕМА
56
Окисление
гидроксиаминов 20a-f избытком двуокиси свинца в хлороформе с хорошим
выходом приводит к образованию 2Н-имидазол-1-оксидов 23a-f
(схема 55).
Таблица
1: 4-фенил замещенные 2Н-имидазол-1-оксиды и гидроксиамины, их выходы.
|

|
|
№
|
R1
|
R2,R3
|
R4
|
Выход, %
|
|
16a
|
Me
|
Et,Et
|
H
|
17
|
|
16b
|
Me
|
(CH2)5
|
H
|
60см.
[63]
|
|
16c
|
Ph
|
Et,Et
|
H
|
65
|
|
16d
|
Ph
|
(CH2)5
|
H
|
90см.[63]
|
|
16e
|
p-Cl-C6H4
|
(CH2)5
|
H
|
47
|
|
16f
|
Ph
|
Me,Me
|
H
|
95 [63]
|
|
17a,18a
|
Me
|
Et,Et
|
Et
|
22, 90*
|
|
18b
|
Me
|
(CH2)5
|
Et
|
40
|
|
20a,23a
|
Ph
|
(CH2)5
|
Et
|
57, 53*
|
|
20b,23b
|
Ph
|
Et,Et
|
Et
|
45, 89*
|
|
20c,23c
|
Ph
|
(CH2)5
|
All
|
15, 80*
|
|
20d,23d
|
Ph
|
(CH2)5
|
Ph
|
86, 83.5*
|
|
20e,23e
|
Ph
|
Me,Me
|
Ph
|
65, 58.5*
|
|
20f,23f
|
Ph
|
Et,Et
|
t-Bu
|
45, 97*
|
|
23g
|
Ph
|
(CH2)5
|
Me
|
93
|
|
23h
|
Ph
|
Me,Me
|
Me
|
95.6
|
|
23i
|
p-Cl-C6H4-
|
(CH2)5
|
2-(1,3-диоксолан-2-ил)-этил
|
5.4
|
|
23j
|
Ph
|
(CH2)5
|
t-Bu
|
70
|
* Первая цифра относится к гидроксиамину, а вторая - к соответствующему
нитрону отдельно на каждую стадию (номера соединений указаны через запятую в
первой колонке таблицы)
Соединения 23g-j получали аналогично описанному выше, исходя из
2H-имидазолов 16d-f без выделения промежуточных гидроксиаминов. Избыток
диоксида свинца прибавляли к реакционной смеси непосредственно после ее
разложения водой (схема 56). Также как и при получении имидазолинов 20a-f
выходы 2H-имидазолов 23g-j существенно различаются в зависимости от
используемого реактива Гриньяра.
Взаимодействие альдонитронов с метилмагнийиодидом, как правило, протекает
легко, обеспечивая высокие выходы соответствующих гидроксиаминов.
Действительно, присоединение этого реактива Гриньяра к нитронам 16d,f
завершается в течение получаса при комнатной температуре. Выходы
соответствующих метилнитронов после окисления реакционных смесей превышают 90%,
и вещества не требуют дополнительной очистки для дальнейшего использования в
синтезе. Присоединение других реактивов Гриньяра происходит менее однозначно.
Кроме того, аналогично 4-метил-производным 18a,b
4-фенил-замещенные кетонитроны 23 с α-метиленовым фрагментом в заместителе
при С5 разлагаются при хранении.
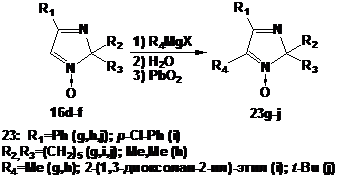
СХЕМА
56.
РИСУНОК
5.
Подобные
продукты (например, соеднинение х, см. схему 57) ранее наблюдали при
взаимодействии 2Н-имидазол-1-оксидов с диизопропиламидом лития, образование их
происходит при металлировании альдонитронной группы и нуклеофильном
присоединении образующегося аниона по нитронной группе неметаллированной
молекулы.
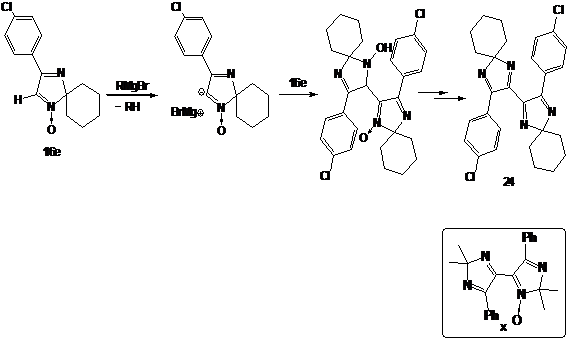
СХЕМА
57.
При
обработке 2Н-имидазол-1-оксида 16d трет-бутилмагнийхлоридом с последующим окислением с
выходом ~ 2% из реакционной смеси был также выделен
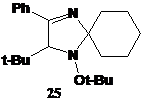
РИСУНОК
6
-трет-бутил-1-трет-бутокси-3-фенил-1,4-диаза-спиро[4.5]дека-3-ен
(25) (рис. 6). Спектральные характеристики этого соединения практически не
отличаются от соответствующих характеристик его 2,2-диэтильного аналога 22,
описанного выше. Однако вместо сигналов протонов этильных групп в спектре ПМР
25 присутствуют 3 мультиплета интенсивностью 1Н, 7Н и 2Н, относящиеся к
спироциклогексановому фрагменту.
Спектральные
характеристики полученных соединений 23 подобны приведённым для 2Н-имидазолов
16, но в ПМР-спектрах отсутствует сигнал протона при С5 имидазольного цикла, и
появляются сигналы, относящиеся к новому заместителю, а в спектрах ЯМР 13С
сигнал С5 имидазольного цикла смещается в область, характерную для кетонитронов
~140 м.д.. Особенностью соединений 23f,j является
отсутствие сопряжения π-системы имидазольного цикла с фенильным заместителем,
что обуславливает отличия в спектральных данных от остальных кетонитронов 23.
Так
в УФ-спектрах отсутствует промежуточный максимум при 240 нм, в спектрах ПМР
наблюдается сильнопольный сдвиг о-протонов фенильного цикла на ~ 0.4 м.д.
Нитроны
23а,b снова обрабатывали 4-х-6-ти кратным избытком реактива
Гриньяра. Присоединение этилмагнийбромида к кетонитронам 23a,b происходит труднее,
чем к соответствующим альдонитронам 16c,d, но без
образования такого большого числа побочных продуктов, какое наблюдалось для
реакций кетонитронов 18a,b. Также как и в случае 4-метил-замещенных производных
выделить соответствующие тетраалкил-замещенные гидроксиамины не удается, т.к.
они частично окисляются кислородом воздуха при обработке реакционной смеси.
После доокисления реакционной массы избытком диоксида марганца из нее
хроматографией выделяли образовавшиеся 3-имидазолиновые НР 26a,b. В
УФ-спектрах этих соединений снова наблюдаются полосы поглощения фенилиминной
группы соответственно 245 и 244 нм, в ИК-спектрах наблюдается полоса колебаний
С=N, характерная для имидазолинов, при 1599 см-1 для 26а
и 1601 см-1 для 26b.

СХЕМА
58.
Суммарный
выход НР 26а (9 % на 4 стадии) может быть существенно повышен (до 40 %), если
на первой стадии использовать уксусную кислоту для гашения реакционной массы и
проводить синтез без выделения промежуточных продуктов.
Таким
образом, нами было показано, что последовательная двукратная обработка
2,2-диалкил-4-фенил-2Н-имидазол-1-оксидов алкилмагнийгалогенидом с последующим
разложением и окислением позволяет получать
4-фенил-2,5-дигидроимидазол-1-оксилы, содержащие 4 этильных заместителя или 2
этильных заместителя и спироциклогексановый фрагмент в положениях 2 и 5, с
удовлетворительным выходом.
Аналогичным образом на базе кетонитронов 23d,e и g,h был осуществлен синтез имидазолиновых радикалов 27a,b, содержащих сильно отличающиеся по объемам метильный и
фенильный заместители в 5-м положении гетероцикла (схема 59). На примере этого
синтеза было изучено влияние порядка, в котором вводятся более и менее объемный
заместители, на выходы целевых радикалов. Однако суммарный выход во всех случаях
составил 30-40%. Существенной зависимости выхода от последовательности введения
фенильного и метильного заместителей не наблюдается.

27: R,R = (CH2)5 (a), CH3, CH3 (b)
СХЕМА
59.
При
взаимодействии наиболее затрудненного кетонитрона 23d с
метилмагнийиодидом помимо целевого НР 27a из реакционной
массы с выходом 20% было выделено бесцветное кристаллическое соединение, спектр
ПМР которого напоминает спектр исходного соединения. Спектры отличаются только
положением 2-х мультиплетов, относящихся к спироциклогексановому фрагменту:
мультиплет интенсивностью 3Н сдвинут в слабое поле, а мультиплет интенсивностью
2Н - в сильное на 0.3-0.5 м.д. С другой стороны в УФ спектре этого соединения
отсутствует характерный для 2Н-имидазол-1-оксидов длинноволновый максимум, а в
ИК-спектре полосы в области колебаний С=N
малоинтенсивны. В спектре ЯМР 13С присутствует всего один набор сигналов,
соответствующих фенильному заместителю, что указывает на симметричность
структуры, и нет сигнала при ~ 135 м.д., который можно было бы отнести к
фенилнитронному атому углерода. По данным элементного анализа в состав этого
вещества не входит кислород. На основании всего выше сказанного соединению
приписана структура 2,3-дифенил-1,4-диаза-спиро[4.5]дека-1,3-диена 28 (см.
схему 59). Важно отметить, что в реакции пространственно менее затруднённого
2,2-диметил-4,5-дифенил-2Н-имидазол-1-оксида 23e с MeMgI
дезоксигенирования не происходит.
Дезоксигенирование
нитронной группы при обработке реактивами Гриньяра свойственно стерически
затруднённым нитронам и обсуждается нами в разделе 2.2 Главы 1.
Действительно,
при дальнейшем увеличении размера заместителя при С-5 выход целевого НР заметно
снижается именно за счет возрастания роли процесса деоксигенирования. Так, в
реакции 2Н-имидазол-1-оксида 23j с этилмагнийбромидом образуется в основном
продукт дезоксигенирования 29, а выход целевого
2-трет-бутил-3-фенил-2-этил-1,4-диаза-спиро[4.5]дека-3-ен-1-оксила 30 не
превышает 37% (суммарный выход на 2 стадии - 11%) (схема 60).
Известно,
что литийорганические соединения более реакционноспособны и менее склонны к
реакциям восстановления, чем реактивы Гриньяра. Однако, они более склоны к
реакциям металлирования, чем магнийорганические соединения. Реакции метил- или a-н-алкилнитронов с литийорганическими соединениями, как правило,
приводят к металлированию. В то же время известны примеры успешного применения
литийорганических соединений в синтезе НР из a-фенилнитронов,
например синтез 2,2,4,5,5-тетрафенил-2,5-дигидроимидазол-1-оксила из
2,2,4,5-тетрафенил-2Н-имидазол-1-оксила. Кетонитроны 23d,j также не содержат
кислых протонов, что позволяет использовать для получения НР литийорганические
соединения. Действительно, реакция 2Н-имидазол-1-оксида 23d с метиллитием
проходит без образования заметных количеств побочных продуктов. После обработки
реакционной массы НР 27a был выделен с выходом 87% (суммарный выход - 72%)
(см. схему 59). Аналогичным образом при обработке трет-бутилнитрона 23j
бутиллититием с высоким выходом был получен единственный продукт -
2-бутил-2-трет-бутил-3-фенил-1,4-диаза-спиро[4.5]дека-3-ен-1-оксил (31),
спектральные характеристики которого очень близки к характеристикам НР 30,
синтезированного с использованием реактива Гриньяра (схема 60).

СХЕМА
60.
По
аналогии с синтезом НР 31 при взаимодействии трет-бутилнитрона 23f с
бутиллитием должен образовываться соответствующий имидазолиновый НР с двумя
этильными заместителями при одном α-углеродном атоме нитроксильной группы и бутильной и трет-бутильной
группой - при другом. Через час после прибавления бутиллития к 2Н-имидазолу 23f
мы наблюдали (методом ТСХ) практически полное отсутствие в реакционной смеси
исходного соединения и присутствие 2-х новых веществ. Однако после разложения реакционной
смеси водой этих веществ в составе смеси обнаружено не было. Вместо этого
удалось выделить два других продукта, не имеющих характерной для имидазолиновых
радикалов желтой окраски.
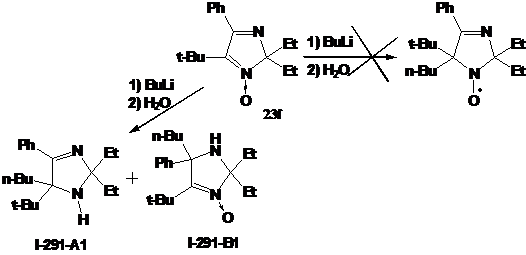
СХЕМА
61
Первое
из соединений - I-291-A1 представляет собой бесцветное масло, обладающее
спектральными характеристиками, похожими на характеристики имидазолина 21. В
ИК-спектре I-291-A1 также присутствует малоинтенсивная полоса колебаний
N-H-группы при 3369 см-1 и полоса колебаний C=N-фрагмента
при 1623 см-1. Отнесение сигналов в спектре ПМР соединения было сделано с
использованием Н-Н корреляционного спектра (H1 cosy)
(см. рис. 7). Интересно отметить, что все пары протонов в СН2-фрагментах n-бутильной
группы не являются эквивалентными. В спектре ЯМР 13С сигналы узловых атомов
углерода находятся при 91.46 и 78.19 м.д. соответственно, т.е. там же, где и у
имидазолина 21, а сигналы иминного атома углерода и и-С фенильной системы
сдвинуты на ~ 8 м.д. в слабое поле, сигнал п-С, напротив, сдвинут в сильное
поле на 3-4 м.д.. Кроме того, в УФ-спектре I-291-A1 не наблюдается поглощения.
Упомянутые спектральные особенности, вероятно, связаны с тем, что фенильная
группа в этом соединении полностью выведена из сопряжения с иминным фрагментом
имидазолинового цикла. На основании спектральных данных и данныx
элементного анализа.соединению I-291-A1 приписана структура
5-трет-бутил-5-бутил-4-фенил-2,2-диэтил-2,5-дигидро-имидазола.

РИСУНОК
7
Второй
выделенный продукт - I-291-B1, бесцветное кристаллическое вещество, в УФ-спектре
которого присутствует полоса поглощения при 240 нм. Как и в случае с первым
продуктом реакции, ИК-спектр I-291-B1 содержит узкую полосу колебаний N-H
группы при 3444 см-1, но сигнал, относящийся к колебаниям фрагмента C=N,
сдвинут в область, характерную, скорее, для нитронов - 1551 см-1. Набор
сигналов n-бутильной группы в спектре ПМР имеет почти тот же
вид, что и в спектре соединения I-291-A1. Однако ЯМР 13С-спектры выделенных
соединений I-291-A1 и I-291-B1 имеют принципиальные отличия. Так в спектре I-291-B1
самый слабопольный сигнал с химическим сдвигом 151.62 м.д. лежит в области,
типичной для нитронных атомов углерода, из чего можно предположить, что
соединение имеет структуру 3-имидазолин-3-оксида. Тогда сигналы при 71.06 и
92.25 м.д. принадлежат атомам С5 и С2 соответственно. Таким образом, I-291-B1
представляет собой
4-трет-бутил-5-бутил-5-фенил-2,2-диэтил-2,5-дигидроимидазол-3-оксид и,
вероятно, является продуктом нетипичного присоединения бутиллития по иминному
фрагменту 2Н-имидазол-1-оксида 23f.
Природа
происходящих в реакции кетонитрона 23f с бутиллитием превращений
остается пока не вполне ясной и требует проведения дополнительных исследований.
Нужно отметить, однако, что стерическое напряжение, создаваемое в молекуле
двумя этильными группами, больше напряжения от спироциклогексанового фрагмента.
Поэтому соответствующий НР, вероятно, должен быть еще менее стабилен
термически, нежели радикал 31 (см. главу 6). Разложение целевого радикала,
предположительно, могло произойти при добавлении в реакционную смесь воды,
которое сопровождалось сильным разогревом раствора, а также при стоянии смеси
при комнатной температуре.
Таким
образом, весьма перспективной представляется стратегия синтеза сильно
пространственно-затруднённых НР из циклических нитронов, включающая введение на
начальном этапе синтеза объёмной не склонной к металлированию группы к
нитронному атому углерода, что позволяет использовать высокореакционноспособные
литийорганические соединения для введения второго объёмного заместителя к тому
же атому углерода.
Глава 3. Синтез имидазолидиновых НР
Нитроксильные радикалы ряда имидазолидина с объемными заместителями в
ближайшем окружении нитроксильной группы представляют особый интерес для
некоторых прикладных областей научных исследований. Стерически затрудненные НР
имидазолидинового строения потенциально более привлекательны для использования
для регуляции псевдоживых радикальных процессов, чем имидазолиновые радикалы.
Отсутствие сильно акцепторных группировок в составе имидазолидинового цикла
изменяет константы прямой и обратной реакции рекомбинации НР с радикалом
растущей полимерной цепи таким образом, что диссоциация происходит легче, т.е.
полимеризация может происходить в контролируемом режиме при более низких
температурах. С другой стороны, имидазолидиновые нитроксильные радикалы - один
из немногих классов НР, обладающих рН-зависимым спектром ЭПР. Неподелённая пара
электронов у атома азота N-3 в
нитроксильных радикалах имидазолидина придаёт этим радикалам основный характер.
Обратимое протонирование по этому основному центру, находящемуся в
непосредственной близости к нитроксильной группе, вызывает заметные изменения в
спектре ЭПР. Это явление лежит в основе оригинального метода определения рН
среды по спектру ЭПР. Спектр ЭПР, записанный при рН близком к значению рК,
представляет собой суперпозицию протонированной и непротонированной форм
нитроксильного радикала. В общем случае для точного определения соотношения
протонированной и непротонированной форм требуется симуляция спектров. На
практике константа сверхтонкого расщепления на атоме азота нитроксильной
группы, измеряемая, как расстояние между неразрешёнными компонентами спектра,
может быть использована в качестве чувствительного к рН маркера. Кислотность
среды (рН) является одним из важнейших, часто измеряемых параметров в биологии,
биофизике, медицине. Изменения рН отражают развитие различных процессов в
организме, и могут служить признаком развития патологий, таких как ишемия, инфекции,
воспаления и др. Значение межклеточной кислотности играет существенную роль в
процессе возникновения опухолей, их росте и терапии. В настоящее время активно
изучается возможность применения таких рН-чувствительных НР в качестве спиновых
меток и зондов, позволяющих неинвазивно проводить мониторинг изменений
кислотности среды в органах т тканях зонах внутри живых организмов с помощью
новых физических методов исследования на основе низкопольного ЭПР и эффекта
Оверхаузера.
Известно, что взаимодействие 1-метил-3-имидазолин-3-оксидов с
металлорганическими соединениями может использоваться для получения НР
имидазолидинового ряда, например, НР, содержащего у a-атомов углерода два фенильных
заместителя и спироциклогексановый фрагмент [Резников, Tetrahedron], (см. Главу 1). К сожалению, нам не
удалось использовать этот метод для синтеза НР, содержащего только алкильные
заместители у a-атомов
углерода, т.к. аналогично литературным данным основным результатом этого
взаимодействия оказывается деоксигенирование исходного нитрона (схема 1).

СХЕМА
1
Другой
способ получения подобных соединений из соответствующих имидазолиновых НР
заключается в алкилировании по атому азота в положении 3 гетероцикла и
последующим восстановлением имидазолиниевых солей (схема 2).
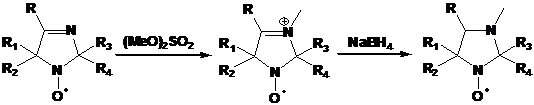
СХЕМА
2
Для
получения НР имидазолидинового ряда этим способом соединения 19a,b и
26a,b, синтез которых был рассмотрен в разделе 1 данной
главы, обрабатывались диметилсульфатом, по методу, описанному в, в результате
образовывались иминиевые соли 1. Алкилирование НР 19a,b
проходит значительно легче, чем алкилирование их фенил- замещенных аналогов (26a,b),
что обусловлено влиянием фенильного заместителя, понижающего нуклеофильность
иминного атома азота. Следует отметить, что алкилирование производных 19b и
26b, содержащих спиро-циклогексановый фрагмент, проходит
медленнее, чем алкилирование соответствующих 2,2-диэтильных производных 19a и
26а, что, по-видимому, вызвано пространственным затруднением, создаваемым
спироциклогексановым фрагментом, при этом в случае 26b довести процесс до конца
не удается, т.к. при длительном нагревании начинают накапливаться побочные
продукты. Провести алкилирование сильно затрудненного НР 31 не удается из-за
его термической нестабильности (см. раздел 5). Иминиевые соли без выделения
восстанавливали боргидридом натрия.
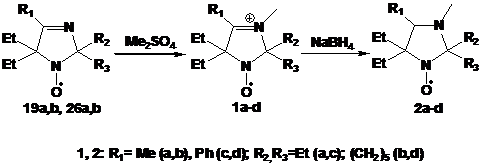
СХЕМА
3
Спектральные
характеристики полученных НР имидазолидинового ряда близки к характеристикам
известных описанных в литературе аналогов.
Для
получения имидазолидиновых НР с метильными группами в положении 4 использовали
5,5-диэтил-3-имидазолин-1-оксилы, полученные из
3-этил-3-(гидроксиамино)пентанона-2 по аналогии с литературной методикой
[ОВС1]. К примеру, диметил, диэтил-замещенный имидазолиновый НР 4 легко
получается конденсацией упомянутого 1,2-гидроксиаминокетона с ацетоном и
ацетатом аммония с последующим окислением (схема 4).

СХЕМА
4
Синтезированные
имидазолидиновые радикалы обладаюи рН-зависимыми спектрами ЭПР (табл.1), однако
их рК не превышают 5, что выходит за рамки физиологически-важных значений.
Кроме того, все они являются липофильными соединениями, трудно растворимыми в
воде, что препятствует их использованию в качестве спиновых зондов. Для
получения гидрофильного спинового зонда 3-этил-3-(гидроксиамино)пентанон-2
конденсировали с левулиновой кислотой, полученный 3-имидазолин алкилировали
диазометаном и окисляли (схема 8). Полученный НР 8 по аналогии с известными методиками
превращали в нитроксильный радикал имидазолидинового ряда 10.
Таблица
1 Значения рК для некоторых синтезированных имидазолидиновых НР
|
номер
|
Структура
|
рК
|
kred
|
|
2a
|
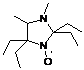 4.950.04±0.003 (0.020±0.005) 4.950.04±0.003 (0.020±0.005)
|
|
|
|
2b
|
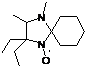 4.900.083±0.005 4.900.083±0.005
|
|
|
|
5
|
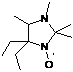 4.750.08±0.01 4.750.08±0.01
|
|
|
|
10A
|
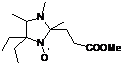 3.960.085±0.006 3.960.085±0.006
|
|
|
|
11A
|
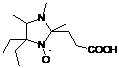 6.210.65±0.2 6.210.65±0.2
|
|
|
Интересно, что при постепенном добавлении к этанольному раствору
метилсульфата 9 небольших порций боргидрида натрия происходит образование двух
диастереомерных продуктов 10A и10B, которые могут быть разделены
хроматографически. В случае же добавления сразу большого избытка боргидрида в
реакции образуется только один диастереомер 10А. Нам удалось определить
конфигурацию этого продукта, проведя рентгеноструктурный анализ соответствующей
карбоновой кислоты 11А, полученной после щелочного гидролиза сложноэфирной
группы 10А и подкисления раствора соответствующей соли. Таким образом,
2-карбоксиметильный заместитель в положении 2 и метильная группа в положении 4
имидазолидинового цикла 10А цис-ориентированы друг относительно друга.
Соответствующие центры ассиметрии имеют конфигурации: 2R(S),4R(S) (рис.1).
Образование именно этого изомера при быстром восстановлении
имидазолиниевой соли можно объяснить следующим образом: в плоском цикле
имидазолиниевого катиона присоединение нуклеофила к атому углерода при связи С=N происходит со стороны,
противоположной объёмному 2-(2-метоксикарбонилэтильному) заместителю. В случае
же медленного прибавления боргидрида вследствие изменения рН среды происходит
отщепление протона от имидазолиниевого катиона с образованием соответствующего
енамина. Последний, очевидно, имеет неплоскую структуру, причём влияние
2-(2-метоксикарбонилэтильного) заместителя делает выгодной конформацию, в
которой метильная группа при атоме азота направлени в противоположную сторону.
В результате направление присоединения определяется разнонаправленным влиянием
заместителей в положениях 2 и 3 гетероцикла.
СХЕМА
7

Рис.1
Строение молекулы 11А. 2-карбоксиметильный заместитель в положении 2 и
метильная группа в положении 4 имидазолидинового цикла цис-ориентированы друг
относительно друга. Соответствующие центры ассиметрии имеют конфигурации: 2R(S),4R(S).
Интересно отметить, что соединение 11А обладает рК 6.2, тогда как рК
негидролизованного сложноэфирного производного 10А составляет 3.96. Увеличение
рК при введении карбоксильной группы ранее было отмечено для рН-чувствительных
НР имидазолинового ряда. Устойчивость нового соединения к восстановлению ниже,
чем наблюдаемая для 10А (см. таблицу 1), что очевидно связано с существованием
этой аминокислоты в цвитерионной форме. Протонирование атома азота в 3-ем
положении цикла существенно повышает окислительный потенциал НР. Тем не менее,
константа скорости восстановления НР 11А аскорбатом равна 0.65 М*с-1 (ср.
константа скорости восстановления известного рН-чувствительного НР
4-амино-2,2,5,5-тетраметил-2,5-дигидро-1H-имидазол-1-оксила (ATI) равна 22.5 М*с-1, при рК 6.1 и в
1.6 раз меньшей чувствительности!) Любопытно отметить, что для НР 11А расчётная
константа скорости меняется от 0.3 M-1с-1 при низких концентрациях аскорбат-аниона до 0.7 M-1с-1 при высоких
концентрациях (рис.2).
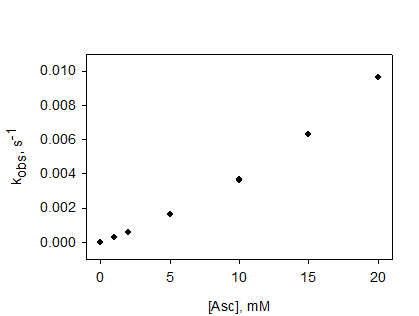
Рис.
2. Зависимость наблюдаемой скорости восстановления радикала 11А от концентрации
аскорбата (по данным Комарова Д.А., ИХКиГ СО РАН).
Поскольку
концентрация аскорбиновой кислоты обычно ниже (например, в плазме крови ~ 50 μM), ожидаемое
время жизни НР 11А в живых системах должно быть велико. Таким образом, по
совокупности параметров, приведённых в табл.1, НР 11А - один из самых удачных
на сегодняшний день спиновых зондов для исследования колебаний межклеточного
рН.
Синтезированные
затрудненные имидазолидиновые НР 2c,d, 5 представляют интерес как потенциальные
регуляторы контролируемой радикальной полимеризации стирольных и виниловых
мономеров. Полимеризация с их участием должна происходить в контролируемом
режиме при более низких температурах, нежели с имидазолиновыми производными
аналогичной структуры. Однако, наиболее перспективным для синтеза полимеров
соединением оказался всё тот же НР 11A. Этот водорастворимый радикал
оказался пригодным для проведения контролируемой полимеризации акриламида и
других водорастворимых мономеров в водной среде, давая «живые» (т.е., способные
к реиницианции) полимеры с низкой полидисперностью (см. также главу 7).
Известно,
что имидазолиниевые соли 2, содержащие метильную группу в положении 4, в нейтральной
или щелочной среде превращаются в енамины, взаимодействие которых с
хлорангидридами приводит к ацилированию по экзометиленовому атому углерода с
образованием енаминокетонов [68]. С использованием этого превращения на основе
енамина Х, образующегося при взаимодействии метилсульфата имидазолиниевого
производного (3) с гидрокарбонатом натрия (схема 5), могут быть получены
спиновые метки, присоединение которых к гидрофильным молекулам позволило бы
получить водорастворимые НР. Исчерпывающее восстановление енаминокетонной
группировки могло бы превратить эти НР в рН-чувствительные спиновые зонды.

СХЕМА
5
Действительно,
енамин Х реагирует с хлорацетилхлоридом в присутствие триэтиламина с
образованием енаминокетона 4. Однако при попытке восстановить Полученный НР
боргидридом натрия происходит восстановительное дегалогенирование (схема 6).
Отрыв атомов галогенов при восстановлении боргидридом обычно не наблюдается.
Интересно, что, подобное дегалогенирование происходит и при восстановлении
бром-енамина 5, полученного бромированием енамина Х при 0оС в смеси ЧХУ-эфир.
Таким образом, поведение соединения 5 отличается от поведения описанного в
литературе 2,2,5,5-тетраметильного аналога, который в этих условиях
превращается 4-бромметил-2,2,3,5,5-пентаметилимидазолидин. Причина этого
различия, возможно, связана с различием в стерической доступности атома
углерода С-4.
В
реакции енамина Х с оксалил хлоридом и метанолом с хорошим выходом образуется
соединение 6. Т.к. в молекуле кетоэфира 6 присутствуют функциональные группы,
которые могут восстанавливаться с разной скоростью, то в зависимости от
количества боргидрида натрия, добавляемого в реакции, и длительности его
воздействия, вероятно, могут образовываться разные продукты. Поэтому восстановление
постарались провести на максимальную глубину, прибавляя в этанольный раствор 6
сразу большой избыток боргидрида натрия. Однако через 30 мин в реакционной
смеси методом ТСХ можно было наблюдать преимущественное образование
единственного продукта и отсутствие исходного вещества. Образовавшееся
промежуточное соединение медленно превращался в другое вещество, поэтому
остатки боргидрида сразу нейтрализовали добавлением ацетона. Известно, что в
случае, если восстанавливаемое соединение содержит кето- и сложноэфирную
группы, боргидрид натрия селективно восстанавливает кетон. В таком случае,
продукт восстановления соединения 6 должен скорее всего иметь строение α-гидроксиэфира. Но в УФ спектре полученного продукта
присутствует интенсивная полоса поглощения при 305 нм, типичная для
енаминокетонов. В области, характерной для колебаний карбонильных связей, ИК
спектра соединения 6 до восстановления есть полосы при 1741, 1650, 1633 и 1539
см-1, после восстановления сигналы при 1741 и 1650 см-1 исчезают. Кроме того, в
спектре имеется полоса колебаний связи О-Н при при 3436 см-1. Основываясь на
этих особенностях и учитывая данные элементного анализа, можно предположить,
что новое соединение имеет строение 7 (см. схему 6).

СХЕМА
6
Таким
образом, нам не удалось синтезировать спиновые метки выбранным путем.
Глава 4. Синтез нитроксильных радикалов на базе
4Н-имидазол-3-оксидов
Поскольку присоединение бутиллития к α-трет-бутилнитрону ряда
2Н-имидазол-1-оксида оказалось удачным методом синтеза
пространственно-затруднённых НР имидазолинового ряда, представлялось
целесообразным применить данный подход для синтеза НР из других циклических
нитронов. Аналогичное превращение в ряду 4Н-имидазол-3-оксида по аналогии с
литературными данными позволило бы получить пространственно-затруднённые
рН-чувствительные зонды для биофизических исследований.
Для получения 4Н-имидазол-3-оксида с трет-бутильным заместителем при
атоме углерода нитронной группы 3-гидроксиамино-3-этилпентан-2-он (13а) конденсировали
с триметилуксусным альдегидом и аммиаком аналогично методике конденсации с
пропионовым альдегидом (схема 1).
2-трет-Бутил-1-гидрокси-2,2-диэтил-4-метил-2,5-дигидроимидазол (14) является
минорным продуктом, выход которого не превышает 10 %. В качестве основного
продукта было выделено соединение 15 -
6-трет-бутил-3,3-диэтил-4-метил-3,6-дигидро-2Н-1,2,5-оксадиазин, изомерное
имидазолину 14. Спектральные характеристики этого соединения близки к
характеристикам изомера 14; в качестве спектральных особенностей соединения 15
можно отметить заметный сильнопольный сдвиг сигналов узлового атома углерода
при геминальных этильных группах и атомов углерода метинового фрагмента и
иминогруппы в спектре ЯМР 13С, а также слабопольный сдвиг одного из протонов
АВ-систем метиленовых групп в спектре ЯМР 1Н (см. экспериментальную часть).
Следует отметить, что, в отличие от имидазолина 14, соединение 15 не
претерпевает изменений при действии диоксидов свинца или марганца. Строение
соединения 15 установлено на основании данных рентгеноструктурного анализа
нитрозопроизводного 16, образующегося при обработке оксадиазина 15
изопропилнитритом в присутствии триэтиламина (Рис. 1). Образования
оксадиазинового цикла реакции 1,2-гидроксиаминокетонов с карбонильными
соединениями и аммиаком никогда ранее не наблюдали.

СХЕМА
1
Соединение
15 превращается в имидазолин 14 при действии ацетата аммония. Проведение
конденсации в присутствии ацетата аммония сразу приводит к образованию
соединения 14 с высоким выходом (см.схему 1).

Рис.
1. Строение молекулы 16. Избранные длины связей (Е): O(1)-N(1)
1.392(2), N(1)-N(2) 1.299(2), N(1)-O(2) 1.233(2), C(4)-N(5) 1.269(2).
Интересно
отметить, что при конденсации 3-гидроксиамино-3-метилбутан-2-она (13b) с
пивалевым альдегидом как в присутствии аммиака, так и в присутствии ацетата
аммония, образования оксадиазина не наблюдали. Тем не менее, выход имидазолина
17 в этой реакции не превышает 14%, а основным продуктом реакции является
2,2,3,5,5,6-гексаметил-2,5-дигидропиразин (18) (схема 2). Интересно отметить,
что ИК-спектр полученного образца соединения 18 не соответствовал ИК спектру
заведомого образца. Поэтому был проведён рентгеноструктурный анализ выделенных
кристаллов, который показал, что они образованы кристаллогидратом соединения 18
(Рис.2).

СХЕМА
2
Образование
соединения 18 при конденсации 3-гидроксиамино-3-метилбутан-2-она 13b с
карбонильными соединениями в присутствии ацетата аммония или аммиака наблюдали
и ранее. Это соединение является основным продуктом реакции, если скорость
образования соответсвующего 2,5-дигидроимидазола низка по стерическим или иным
причинам.
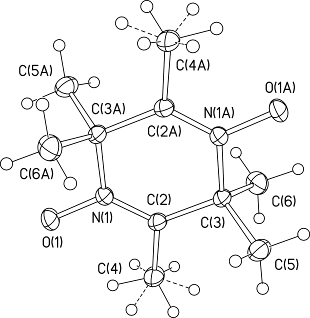
Рис.
2. Строение молекулы 18. Молекула расположена в центре симметрии, поэтому
нумерация не соответствует номенклатурной. Атомы, содержащие символ А в номере,
являются симметрично зависимыми. Избранные длины связей (Е): O(1)-N(1)
1.321(1), N(1)-C(2) 1.290(2), N(1)-C(3А) 1.497(2), C(2)-C(3) 1.506(2).
В
реакциях конденсации гидроксиаминокетона 13а, напротив, никогда не наблюдали
образования соответствующего дигидропиразина. Различие в реакционной
способности гидроксиаминокетонов 13а и 13b, очевидно,
обусловлено стерическими факторами. Пространственные затруднения, создаваемые
геминальными этильными заместителями соединения 13а и трет-бутильной группой
пивалевого альдегида делают менее выгодным переходное состояние, необходимое
для нуклеофильной атаки неподелённой пары атома азота гидроксиламиногруппы по
карбонильному атому углерода пивалевого альдегида. В результате реализуется
термодинамически менее выгодный процесс образования оксадиазина 15. В
присутствии же ацетата аммония, катализирующего как реакции конденсации, так и
обратные реакции, конечным продуктом превращения становится имидазолин 14.
Из
соединения 14 по аналогии с разработанными ранее методиками получали амидины 22
(схема 3). При окислении имидазолина 14 образуется 4Н-имидазол-3-оксид 19 -
устойчивое кристаллическое соединение, спектральные характеристики которого
близки к характеристикам полученных ранее производных этого ряда.
Для
получения оксима 20 использовали разные методики, приведённые в работе, с целью
подобрать вариант, обеспечивающий максимальный выход целевого продукта. При
этом и обработка имидазолина 14 изопропилнитритом в хлороформе в присутствии
триэтиламина, и нитрозирование соединения 19 изопропилнитритом в присутствии
изопропилата натрия в изопропиловом спирте при -10оС выходы оксима 20 составили
~ 40%. Интересно, что окисление имидазолина 14 в 4Н-имидазол-3-оксид 19
изопропилнитритом в хлороформе в присутствии триэтиламина происходит гораздо
медленнее, чем это описано для других 1-гидрокси-3-имидазолинов, и не
сопровождается бурным выделением окиси азота. При этом в спектре ЭПР
реакционной массы имеется интенсивный сигнал, представляющий собой дублет
триплетов с константами аN = 14.5 и аН = 17.8 Гс, очевидно, соответствующий НР
14R (рис. 3).
СХЕМА
3
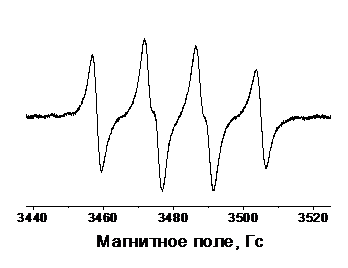
Рис.3.
Спектр ЭПР НР 14R
Известно,
что окисление гидроксиламинов в нитроны может проходить через промежуточное
образование нитроксильного радикала с атомом водорода у α-атома углерода. Последующее диспропорционирование
таких НР приводит к нитрону и исходному гидроксиламину. По-видимому,
диспропорционирование пространственно затруднённого НР 14R
идёт очень медленно, подобно описанному для 2,6-дитрет-бутилпиперидин-1-оксила
(Глава 1).
Далее
оксим 20 с хорошим выходом превращали в нитрил 21, который затем обрабатывали
различными аминами (пирролидином, N,N,N’-триэтил-1,3-диаминопропаном,
метиламином) для получения набора амидинов различного строения (схема 3).
Скорость
протекания этой реакции нуклеофильного замещения и даже ее результат сильно отличаются
для аминов разной структуры. Например, в случае небольшого метиламина процесс
замещения завершается всего за 10-20 мин в хлороформном растворе, тогда как
реакция карбонитрила 21 с N,N,N’-триэтил-1,3-диаминопропаном идет 2-е суток без
растворителя. Несколько неожиданным оказался тот факт, что нитрил 21 вообще не
реагирует с пирролидином при обычных условиях (ср. реакция 2-этил-замещенного
аналога 21 с пирролидином проходит за 12 ч с выходом 57% ). Только при
длительном нагревании до температур ~ 80-100оС без растворителя происходит
медленное разложение исходного 4Н-имидазола с образованием сложной смеси
продуктов.
Полученные
амидины 22a,b обладают спектральными характеристиками, типичными
для соединений этого класса. Интересно отметить уширение сигналов при 43.84 и
30.51 м.д., относящихся к первому и второму СН2-звеньям цепи заместителя при
азоте амидинового фрагмента, в спектре ЯМР 13С соединения 22a. Подобная форма
сигналов свидетельствует о затрудненном вращении аминогруппы в данном
соединении.
Однако,
ни обработкой полученных соединений 22 этилмагнийбромидом, ни при воздействии
бутиллития получить желаемые нитроксильные радикалы 23 не удалось. Во всех
случаях образовывалась сложная смесь неидентифицированных продуктов. Следует
отметить, что в спектрах ЭПР реакционных масс, полученных после разложения
избытка металлоорганического соединения и окисления MnO2, все же
присутствуют малоинтенсивные сигналы. Так спектр смеси продуктов реакции
амидина 22a содержит сигнал, форма которого соответствует иминонитроксильному
радикалу (ИНР) (триплет триплетов с аN ~ 3 и 7 Гс). Известно, что в
зависимости от характера заместителей металлоорганические соединения могут
присоединяться к 4Н-имидазол-3-оксидам не только в положение 2, но и в
положение 5 []. По-видимому, металлоорганический реагент присоединяется в более
доступное стерически положение 5 соединения 22a, а
образующиеся ИНР c вторичной аминогруппой или их восстановленные формы
неустойчивы. Взаимодействие амидина 22b с бутиллитием сильно осложняется малой
растворимостью исходного соединения и продукта его металлирования по
MeNH-фрагменту в растворителях (толуол, бензол, гексан), инертных по отношению
к литийорганическим агентам. По этой причине амидин вводили в реакцию в виде
суспензии в толуоле, и нагревали полученную смесь до ~ 80оС в течение 6 ч.
Такие жесткие условия проведения процесса могли привести к разложению
образующихся продуктов, но, в более мягких условиях реакция не идет вовсе.
Спектр реакционной смеси, тем ни менее, содержит триплетный сигнал, что,
очевидно, подтверждает образование нитроксильного радикала желаемого строения,
но лишь в следовых количествах.
Таким
образом, стерические препятствия, создаваемые объемными этильными заместителями
в 4-м положении и трет-бутильной группой во 2-м положении 4Н-имидазольного
цикла осложняют протекание всех стадий синтеза и негативно сказываются на
выходах. Вероятно, по той же причине реакция нуклеофильного замещения в
карбонитриле 21 столь чувствительна к природе используемого амина. Мы
попытались осуществить синтез немного иначе: сначала сформировать незамещенный
по положению 2 амидин, а затем ввести по этому положению трет-бутильный и
бутильный заместители действием металлорганических соединений (см. схему 4). В
таком варианте схема синтеза становится полностью аналогичной схеме,
применявшейся для получения НР 31 на базе 2Н-имидазол-1-оксида 16d.
Действительно,
выход нитрила 26, исходя из 3-(гидроксиамино)-3-этилпентанона-2, составляет
66%, тогда как аналогичный выход нитрила 21 не превышает 24%. Однако, отсутствие
заместителя во 2-м положении осложняет последующую реакцию нуклеофильного
замещения. Так, по литературным данным, выходы амидинов в реакциях
2-алкил(арил)-4,4-диэтил-замещенных карбонитрилов с аминами разного строения в
большинстве случаев составляют ~ 60%. Выход же амидина 27 не превышает 40%,
что, вероятно, обусловлено не только стерическими препятствиями, создаваемыми
этильными группами, но и повышенной реакционной способностью альдонитронного
фрагмента молекулы, о чем свидетельствует накопление в смеси окрашенных
побочных продуктов. Спектральные характеристики соединений 24-27 в основном
совпадают с характеристиками соответствующих 2-замещенных производных. Сигналы
альдонитронных протонов в ПМР-спектрах выходят при 4.72 м.д. для гидроксиамина
24 и в области 7.3-8.3 м.д. для 4Н-имидазол-3-оксидов 25-27. Интересно, что
положение сигнала при ~ 141 м.д. альдонитронного атома углерода в спектрах ЯМР
13С соединений 25-27 практически не меняется, хотя положение аналогичного
сигнала, выходящего в кетонитронах 19-22 на 5-25 м.д. в более слабом поле,
отличается для каждой структуры. Реакция амидина 27 с трет-бутилмагнийхлоридом
и последующее окисление соответствующего гидроксиамина происходят без
осложнений, обеспечивая неплохой выход соединения 28, аналогичного амидинам
22a,b. В отличие от 5-метиламино-производного 22b диметиламино-замещенный
4Н-имидазол 28 хорошо растворяется в бензоле, и его реакция с бутиллитием
проходит за ~ 15 ч при комнатной температуре. Процесс в этом случае также
протекает очень неоднозначно, но из образующейся после разложения и окисления
смеси продуктов хроматографией удается выделить 3 основных продукта, первый из
которых обладает характеристиками целевого НР. Так соединение 29 обладает
типичным для НР подобной структуры ИК-спектром и триплетным сигналом в спектре
ЭПР, а данные его элементного анализа удовлетворяют расчетным. Выход НР 29
составляет 8%. Это соединение действительно обладает рН-зависимым спектром ЭПР,
что подтверждается приведённой на рис. кривой зависимости конствнты СТВ на
атоме азота нитроксильной группы от рН.

СХЕМА
4
Таким
образом, стратегия введения неметаллирующегося заместителя и использования
литийорганического соединения для введения второго, не дает столь впечатляющих
результатов в синтезе затрудненных рН-чувствительных зондов на базе
4Н-имидазол-3-оксидов, как в синтезе с участием 2Н-имидазолов. Тем ни менее,
согласно этой схеме, нам удалось получить сильно затрудненный рН-чувствительный
зонд 29 с небольшим выходом.
Глава 5. Синтез нитроксильных радикалов пирролидинового ряда
НР пирролидинового ряда занимают важнейшее место среди разнообразных
спиновых меток, используемых в биофизике. Одна из причин этого - низкий
окислительный потенциал этих радикалов, благодаря чему НР этого ряда относятся
к самым устойчивым к восстановлению в биологических системах. Для синтеза таких
НР успешно используются реакции пирролин-N-оксидов с металлоорганическими соединениями. Мы попытались
получить пространственно-затруднённые НР пирролидинового ряда исходя из
описанного в литературе нитрона 24 (см. схему 1).
Нитрон 24 обрабатывали избытком трет-бутилмагнийхлорида, реакционную
массу разлагали водой и сразу окисляли кислородом воздуха в присутствии
аммиаката меди аналогично описанному в. В результате из реакционной массы был
выделен единственный продукт. В спектре ЯМР 13С полученного соединения
присутствуют сигналы двух нитронных групп (альдонитронной при 140.87 м.д. и
кетонитронной при 143.57 м.д.). Кроме того, в спектрах ЯМР имеются сигналы пяти
метильных и четырёх метиленовых групп. В спектрах ПМР сигналы протонов
метильных групп представляю собой три синглета с интенсивностью 9Н, 3Н и 3Н, но
в спектре ЯМР 13С имеется четыре сигнала атомов углерода метильных групп, что
указывает на отсутствие в молекуле трет-бутильной группы. Сигналы протонов двух
метиленовых групп - синглеты, а сигналы двух других образуют АВ-системы, что
указывает на присутствие в молекуле двух пирролиновых остатков, в одном из
которых имеется ассиметрический центр. Кроме того, в спектрах ЯМР 13С имеются
сигналы трёх узловых атомов углерода, один из которых в слабом поле (77.35
м.д.). На основании этих данных полученному соединению приписано строение
2-((3,3-диметил-1-оксидо-3,4-дигидро-2H-пиррол-5-ил)метил)-2,4,4-триметил-3,4-дигидро-2H-пиррол-1-оксида (25). Образование соединения 25 обусловлено
тем, что реакция металлирования протекает быстрее, чем присоединение
трет-бутилмагнийхлорида по атому углерода нитронной группы. В литературе
имеются соответствующие аналогии. Кроме того, подобные димерные соединения были
получены из аналогов пирролина 24 действием амида натрия в жидком аммиаке.
СХЕМА
1
В
отличие от трет-бутилмагнийхлорида, аллилмагнийбромид нормальным образом
присоединяется по нитронной группе соединения 24 в полном соответствии с
литературными данными. После окисления из реакционной массы с высоким выходом
был выделен альдонитрон 26. Полученное соединение 26 далее подвергали действию
магнийорганичеких соединений. Вопреки ожиданиям, многочасовое кипячение нитрона
26 с избытком трет-бутилмагнийхлорида не привело к каким-либо изменениям. Из
реакционной массы был выделен исходный нитрон 26 с количественным выходом. Но
при обработке нитрона 26 этилмагнийбромидом после окисления из реакционной
массы с небольшим выходом (40%) удалось выделить желаемый нитрон 27. Однако при
обработке последнего избытком этилмагнийбромида вместо ожидаемого
нитроксильного радикала из реакционной массы с высоким выходом был выделен
продукт дезоксигенирования 28.
Полученные
нами данные демонстрируют нетривиальность поставленной задачи. Неудача в
синтезе пространственно-затруднённых пирролидиновых НР из нитрона 24 заставила
нас обратиться к методу синтеза НР пирролидинового ряда, предложенному
Резниковым В.А. Ключевой стадией синтеза по этому методу является рециклизация
в кислой среде енаминокетонов имидазолидинового ряда в пирролин-N-оксиды,
которые затем обрабатывают металлорганическим соединением (схема 2).
СХЕМА
2.
Мы
попытались использовать этот подход для получения 2,2,5,5-тетраэтил-замещённого
НР пирролидинового ряда. Подходящим исходным соединением для синтеза
соответствующего енаминокетона представляется 5,5-диэтил-2,2,4-триметил-2,5-дигидроимидазол-1-ол
29, упоминавшийся в разделе 2 данной главы.
Как
правило, конденсация 1-гидрокси-3-имидазолинов со сложными эфирами приводит к
образованию енаминокетонов - производных имидазолидин-1-оксила. Соединение 29
вводили в конденсацию с избытком метилового эфира пропионовой кислоты в
присутствии диизопропиламида лития. Однако, после обработки реакционной массы
было выделено бесцветное кристаллическое соединение 30, спектральные
характеристики которого отличались от ожидаемых. Так, в ИК-спектре полученного
соединения помимо интенсивных полос при 1661 и 1552 см-1, характерных для
енаминокетонов, наблюдается интенсивная полоса при 1771 см-1, характерная для
1-ацетокси-3-имидазолинов, в спектре ПМР наблюдаются сигналы протонов четырёх
этильных групп, а в спектре ЯМР 13С - также и сигналы двух карбонильных атомов
углерода - при 199.60 м.д., характерный для енаминокетонового фрагмента и при
172.74 м.д., характерный для сложноэфирного карбонила.
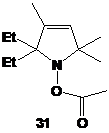
РИСУНОК
8
Интересно
что, сигналы заместителей в положениях 2 и 5 гетероцикла в спектрах ЯМР,
записанных при 300 К неэквивалентны и уширены. Такое уширение можно объяснить
затруднённой инверсией у атома азота в положении 1 гетероцикла, вызванной
введением объёмного заместителя к атому кислорода гидроксиамино-группы. Для
подтверждения этого предположения действием уксусного ангидрида на имидазолин
29 было получено модельное соединение 31 (рис. 1). В спектрах ПМР 31
действительно наблюдается зависимость формы сигналов заместителей в положениях
2 и 5 от температуры. Так, в спектре, записанном при 293 К, метильным группам
во 2-м положении соответствуют 2 уширенных синглетных сигнала с химсдвигами
1.27 и 1.36 м.д. Но уже при 301 К в спектре присутствует всего один уширенный
сигнал при 1.33 м.д. При 310 К этот сигнал заметно сужается. Уширение
соответствующих сигналов наблюдается и в спектре ЯМР 13С. Таким образом,
инверсия у атома азота в 1-м положении соединений 30 и 31 действительно
затруднена. На основании этих данных, а также данных элементного анализа
соединению 30 была приписана структура
1-(2,2-диметил-1-пропионилокси-5,5-диэтилимидазолидин-4-илиден)бутан-2-она.
Очевидно, образующиеся в условиях реакции дианион
1-(2,2-диметил-1-гидрокси-5,5-диэтилимидазолидин-4-илиден)бутан-2-она и анион
5,5-диэтил-2,2,4-триметил-2,5-дигидроимидазол-1-ола 29 претерпевают
ацилирование избытком метилпропионата. Обработка соединения 30 соляной кислотой
приводит к гидролизу сложноэфирной группы и рециклизации образующегося 1-гидрокси-производного
аналогично описанному в работе. Сопоставление спектральных характеристик
полученного пирролинон-N-оксида 32 с характеристиками ранее описанных подобных
соединений показало, что, в полном соответствии с литературными данными,
соединение 32 в растворе в хлороформе существует в виде равновесной смеси
таутомеров A и B в соотношении 7 : 3 по данным ЯМР (схема 3); спектры
ИК, УФ и ЯМР таутомерной смеси близки к приведённым для подобных соединений в
работе. Следует отметить, что аналогично известным пирролинон-N-оксидам,
соединение 32 с заметной скоростью окисляется в растворах кислородом воздуха,
что выражается в появлении красной окраски.

СХЕМА
3
Как
известно, при обработке 1-пирролин-4-он-1-оксидов реактивами Гриньяра
первоначально образуются соли енолов, причём енолизации подвергается
кето-группа, второй эквивалент реактива Гриньяра присоединяется по нитронной
группе. Действительно, прибавление пирролина 32 к раствору этилмагнийбромида
приводит к образованию осадка (очевидно, соли енола), который затем медленно
реагирует с избытком EtMgBr. Исчезновение исходного соединения (или соли енола) в
реакционной массе происходит только после 15 часов кипячения в ТГФ. После
разложения реакционной массы и окисления нам удалось выделить лишь небольшое
количество НР 33 (~ 2 %). Причина низкого выхода, очевидно, заключается в
слишком низкой скорости присоединения этилмагнийбромида к нитрону 32, что
приводит к возрастанию роли побочных процессов, например, автоконденсации аналогично
описанному выше. Понижение скорости, в свою очередь, скорее всего, обусловлено
пространственной затруднённостью нитронной группы и малой растворимостью соли
соответствующего енола в реакционной среде.
СХЕМА
4
Как
было показано в разделе 1 данной главы, использование реакции a-трет-бутилнитрона ряда 2Н-имидазола с бутиллитием вместо обработки
соответсвующего a-этилнитрона этилмагнийбромидом позволяет избежать
осложнений и резко увеличить выход нитроксильного радикала. Для получения a-трет-бутилнитрона пирролинового ряда 1-гидрокси-3-имидазолин 29
конденсировали с этиловым эфиром триметилуксусной кислоты. В этом случае не
происходит ацилирования по гидроксигруппе в положении 1 и после обработки
реакционной массы был выделен НР 34 - оранжевое кристаллическое вещество, в
ИК-спектре которого имеются характерные полосы колебаний кратных связей
енаминокетонового фрагмента при 1635 (С=O) и 1561 см-1 (C=C).
При восстановлении этого НР водородом на палладии было получено соединение 35.
В спектре ЯМР 1Н этого соединения сигналы протонов метиленовых фрагментов
этильных групп представляют собой расщеплённую АВ-систему, как и в спектре
исходного соединения 29. Кроме того, наблюдается сигнал метинового протона при
5.01 м.д. и уширенный сигнал протона NH группы при 5.35 м.д., а в
спектре ЯМР 13С - сигнал атома карбонильной группы при 205 м.д., т.о.,
соединение существует в енаминокетонной форме.
СХЕМА
5
Обработка
соединения 35 соляной кислотой приводит к его гидролизу и рециклизации при
последующем повышении рН до ~ 7 с образованием пирролинона 36, существующего в
хлороформе в виде смеси кетонитронной и енгидроксиаминокетонной таутомерных
форм (A и B), соотношение которых по данным ЯМР составляет 4 : 7.
Интересно,
что гидролиз соединения 35 при выдерживании в кислом растворе проходит
медленно. Для завершения реакции требуется не менее 82 ч часов. При обработке
рнакционной массы через 40 ч помимо ожидаемого пирролин-N-оксида было выделено
соединения I-248A с выходом 15% (см. схему 6). I-248A представляет собой желтое
кристаллическое вещество, в ИК спектре которого имеются полосы при 3320 см-1 и
3150 см-1, которые могут соответствовать колебаниям водородносвязанных N-H и
О-Н-групп, а также полосы при 1661 и 1588 см-1, по-видимому, относящиеся к
колебаниям C=NH и С=С связей в иминоенгидроксиаминной таутомерной
форме I-248A-1 (см. схему 6). В спектрах ЯМР имеется один набор сигналов.
Спектр ПМР этого соединения в хлороформе содержит уширенный сигнал двух
протонов аминогруппы при 4.63 м.д. а также узкий синглетный сигнал протона при
С=С связи цикла при 5.06 м.д.. Наконец, в спектре ЯМР 13С имеется сигнал при
91.70 м.д., соответствующий СН-группе при кратной связи, однозначно
указывающий, что соединение находится в енолизованной форме. Сигналы при 159.09
и 33.90 м.д. характерны для трет-бутилнитронной группы, сигнал же при 153.59
м.д., очевидно, соответствует атому углерода енаминного фрагмента Н2N-С=.
Таким образом, в хлороформе соединение I-248A находится в форме енаминонитрона
5-трет-бутил-2,2-диэтил-2H-пиррол-3-амин-1-оксида (I-248A-2).
Образование подобных соединений было отмечено ранее, при рециклизации
имидазолидиновых енаминокетонов с акцепторной группой при экзо-метиленовом
атоме углерода. Такие енаминокетоны удается гидролизовать только при кипячении
в смеси концентированной HCl с метанолом 1 : 3, причем соответствующий
пирролинон-1-оксид оказывается минорным продуктом рециклизации. По-видимому,
при гидролизе соединения 35 в растворе образуются β-дикетонный, так и енаминокетонный ациклические
интермедиаты, образование соединения I-248A является результатом циклизации
последнего.
Соединение
I-248A комнатной температуре на воздухе быстро превращается в смест двух новых
соединенийI-248B1 и I-248B2. Особенно быстро это превращение происходит в
растворе. Так спектр ПМР, записанный для свежеприготовленного раствора I-248A в
дейтерохлороформе, содержит только сигналы исходного соединения, но за время,
необходимое для записи спектра ЯМР 13С, в растворе уже успевает накопиться еще
одно соединение I-248B1. Соединения I-248B1 и I-248B2
обладают практически одинаковыми спектрами ПМР, в которых присутствуют только
сигналы, соответствующие протонам 2-х этильных и трет-бутильной групп. Спектры
ЯМР 13С обоих продуктов содержат по два сигнала узловых атомов углерода и по 2
слабопольных сигнала атомов углерода при кратных связях С=Х в цикле, что
свидетельствует об отсутствии протонов при С3 и С4. Сигналы при 159.63 и 33.94;
153.93 и 34.41 м.д. соответвтенно могут принадлежать трет-бутил-нитронным
группам. Положение одного из сигналов в слабом поле (при ~ 178 м.д.) одинаково
в каждом из спектров, а второй сигнал в этой области находится при 171.42 и
197.35 м.д. соответственно. В ИК спектре соединения I-248B2
присутствуют полосы колебаний водородносвязанной N-H связи при 3369 и 3207
см-1, а также несколько сигналов при 1770-1620 см-1, тогда как в спектре I-248B1 не
наблюдается полос для колебаний N-H или O-H, и присутствуют уширенные полосы
при 1769 и 1696 м.д., характерные для карбонильных соединений. На основании
этих данных, а также записанных масс-спектров (массы молекулярных ионов
соединений I-248B1 и I-248B2 соответствуют их расчетным молекулярным массам) для
соединений I-248B1 и I-248B2 предложены структуры 2-трет-бутил-5,5-диэтил-2H-пиррол-3,4-дион-1-оксида
(I-248B1) и
2-трет-бутил-5,5-диэтил-4-имино-4,5-дигидропиррол-3-он-1-оксида (I-248B2).
Образование
подобных α,β-дикетонов ранее было отмечено при окислении
соответствующих 3-хлор-замещенных пирролин-N-оксидов в водном метаноле в
присутствии основания. Реакция, очевидно, идёт через промежуточное образование
неустойчивого винилнитроксильного радикала. Винилнитроксильные радикалы
являются исключительно высокореакционноспособными и обычно сразу же вступают в
реакции рекомбинации с образованием димеров типа С-С или С-О. Различия в
поведении пирролинов 36 и I-248A, возможно, связано с различием в распределении
спиновой плотности в образующихся винилнироксильных радикалах. Кроме того,
винилог ацилнитроксильного радикала, образующийся при окислении пирролина 36,
очевидно, более реакционноспособен, чем винилог иминонитроксильного радикала,
образующийся при окислении соединения I-248A. По-видимому, взаимодействие
последнего с кислородом происходит быстрее димеризации.
Гидролизуясь,
иминокетон I-248B2 быстро превращается дикарбонильное соединение
I-248B1. Вероятно, поэтому соединения типа I-248B2
никогда ранее не выделялись. Иминокетон подобного строения описан впервые.
СХЕМА
6
Как
и другие известные 2,4-дигидропиррол-3-он-1-оксиды, соединение 36 в хлороформе
медленно окисляется кислородом воздуха, что приводит к появлению фиолетовой
окраски димера этиленового типа 37. Димер 37 образуется с высоким выходом при
окислении пирролинон-1-оксида 36 диоксидом марганца. Спектральные данные его
аналогичны данным синтезированных ранее подобных соединений. Однако многие
сигналы в спектрах ЯМР 1Н и особенно 13С оказались сильно уширены и малоинтенсивны.
Уширение сигналов ЯМР для большинства аналогичных димеров также было отмечено в
работе. Это явление обычно объясняют вкладом бирадикальной формы (схема 6).
Квинтетный спектр ЭПР, показывающий наличие в растворе бирадикальных частиц,
наблюдается для ближайшего аналога соединения 37 - димера 37а.
Рис.
2. Строение молекул соединения 37. Гетероциклы молекулы плоские в пределах
±0.032 Е, угол между плоскостями гетероциклов равен 60.0(2)°.
Рентгеноструктурный
анализ кристаллов соединения 37 (Рис.2) показал, что угол между плоскостями
пирролиновых фрагментов составляет 60°, что очевидно указывает на большой вклад
бирадикальной резонансной структуры.

СХЕМА
7
При
обработке пирролинон-1-оксида 36 бутиллитием в бензольно-гексановом растворе с
высоким выходом был получен единственный продукт - НР 38 - жёлтое
кристаллическое вещество, строение которого подтверждено данными ИК, УФ
спектров и элементного анализа.
Интересно
отметить, что присоединения этилмагнийбромида по нитронной группе соединения 36
не происходит даже при многочасовом кипячении в ТГФ. Из реакционной массы с
количественным выходом было выделено исходное соединение.
Таким
образом, стратегия введения неметаллирующегося заместителя и использования
литийорганического соединения для введения второго находит успешное применение
в синтезе НР не только имидазолинового, но и пирролидинового ряда. Полученный
по этому методу 5-трет-бутил-5-бутил-2,2-диэтилпирролидин-3-он-1-оксил 38 -не обладает
в качестве спинового зонда для биофизических исследований. Кроме того, наличие
карбонильной группы в положении 3 гетероцикла может существенно повысить
окислительный потенциал этого радикала.
В
связи с этим, мы попытались модифицировать соединение 38 различными способами,
главным образом, по кетонному фрагменту (схема 7). Неожиданным оказался тот
факт, что пиролидинон 38 не вступает в большинство типичных для
карбонильных-соединений реакций. Использование жестких условий или повышенных
температур для модификации НР 38 невозможно всилу его лабильности (см. главу
5). Вероятно, причина подобной инертности в пониженной пространственной
доступности группы карбонильного атома углерода в этом соединении. Единственное
превращение, которое нам удалось осуществить - это восстанволение карбонильной
группы в спиртовую боргидридом натрия (см. схему 7). При этом с количественным
выходом был выделен
2-бутил-2-трет-бутил-3-гидрокси-5,5-диэтилпирролидин-1-оксил (39).

СХЕМА
8 Превращения НР 38
Образцы полученного таким образом радикала 39 были переданы для изучения
его устойчивости к восстановлению в биологических средах и модельных системах.
Обсуждение биофизических экспериментов с участием НР 39 представлено в разделе
5 данной главы. Исследование свойств НР 39 показало, что этот радикал обладает
исключительно высокой устойчивостью к восстановлению, что делает
привлекательной идею использования его в качестве спинового зонда в живых
организмах (in vivo). НР 39 - липофильное соединение, очевидно, способное
преодолевать клеточную мембрану. Высокое время жизни этого НР в гомогенатах и в
изолированном сердце предполагает его устойчивость и внутри клетки.
В отличие от межклеточного пространства, в котором многие гидрофильные
(не проникающие через клеточную мембрану), нитроксильные зонды демонстрируют
приемлемые времена жизни, внутри клетки, и особенно внутри митохондрий,
восстановление НР происходит быстро, что связано с работой ферментативных
систем, обеспечивающих энергетику клетки. В то же время, работа этих и
некоторых других ферментативных систем может сопровождаться образованием
активных форм кислорода (АФК) (прежде всего - супероксидного радикала),
играющих огромную роль как в авторегуляции клетки, так и в развитии различных
патофизиологических процессов. Возникновение и развитие ряда патологий, таких
как рак, нейродегенеративные болезни, инфаркт, связывают с тем, что
естественные клеточные антиоксиданты не справляются с возрастающим количесвом
АФК. Показано, что введение некоторых соединений, связывающих АФК
(искусственных антиоксидантов), в том числе НР, может предотвратить развитие
патологических процессов.
В последнее время активно исследуется возможность направленной доставки
антиоксидантов в митохондрии с помощью липофильных катионов. При этом градиент
концентрации активного вещества создаётся за счёт мембранного потенциала
150-180 мВ (отрицательный внутри) на внутренней митохондриальной мембране.
Легко проникать сквозь липидный бислой могут липофильные катионы, заряд которых
распределен по большой площади поверхности, а градиент потенциалов позволяет
накапливать их митохондриальном в матриксе. Поглощение митохондрией липофильных
катионов возрастает в 10 раз на каждую разность мембранных потенциалов в 61.5
мВ, приводя к накоплению их 100-500 кратного избытка. Поглощение катионов
клеткой также управляется мембранным потенциалом (30-60 мВ, отрицательный
внутри). Поэтому нацеленные в митохондрии антиоксиданты могут быть получены, к
примеру, присоединением липофильного трифенилфосфониевого катиона к
антиоксидантному фрагменту. Известны успешные примеры создания таких
антиоксидантов, в том числе и на основе НР. Аналогичным образом, присоединение
трифенилфосфониевого катиона к радикалу 39 позволило бы избирательно направлять
его внутрь митохондрий клетки, что не только позволило бы исследовать его
поведение внутри клетки, но и изучить его антиоксидантную активность.
Трифенилфлсфониевые соли получают алкилированием трифенилфосфина
галоидпроизводными. Для этого были получены алкилирующие метки 40, 41 и 42
(Схема 8). НР 40 и 41 образуются при обработке НР 39 2-хлорэтилизоцианатом и
2-хлорацетилхлоридом в присутствие триэтиламина. Для получения еще одной метки
42 раствор НР 39 в ДМСО обрабатывали 1,4-дибромбутаном в присутствие гидрида
натрия (схема 8). В ИК спектрах выделенных соединений 40 и 41 присутствуют
широкие полосы колебаний сложноэфирных С=О фрагментов, при 1727 см-1 для НР 40
и при 1765, 1741 см-1 - для 41. В последнем случае наблюдается расщепление
карбонильной полосы, характерное для моно-α-галоген-замещенных сложных эфиров.
Спектр соединения 42 содержит характерный интенсивный сигнал колебаний простого
эфира при 1113 см-1. Экспериментальные данные элементного анализа НР 40, 41 и
42 соответствуют расчетным.
К сожалению, нам не удалось получить НР типа 43 с трифенилфосфониевым
фрагментом на основе НР 41. По-видимому, нагревание соединения 41 с
трифенилфосфином приводит к отщеплению хлорацетокси-фрагмента, т.к. после
завершения реакции из смеси вновь был выделен НР 39. Однако, спиновая метка 42
реагирует с трифенилфосфином желаемым образом при нагревании реакционной смеси
до 70-75оС. НР 44 был выделен с хорошим выходом в виде очень вязкого
смолообразного желтоватого вещества. Полоса колебаний R-O-R’ в ИК спектре 44 находится при 1100
см-1. С присутствием в молекуле трифенилфосфониевой группы связаны сигнал при
3059 см-1 колебаний связей H-C= фенильных колец в ИК спектре, а также полосы
поглощения при 226, 262, 268 и 275 нм в УФ спектре.
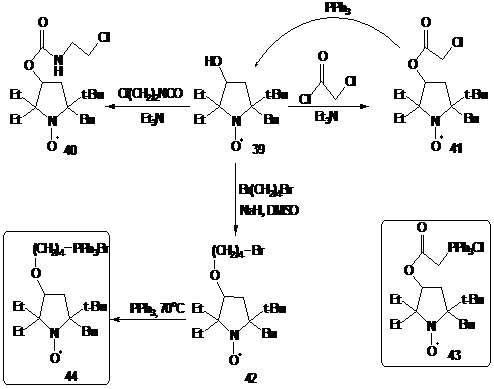
СХЕМА
9
Полученный,
таким образом, НР 44 передан в Государственный Университет Огайо (США) для
изучения поведения этого соединения внутри клетки и в митохондриях.
Глава 6. Разложение трет-бутил-бутил-замещенных НР
Благодаря высокой стерической напряжённости своей структуры НР,
содержащие объемные трет-бутильный и бутильный заместители, склонны к
нетипичным для других НР химическим превращениям. трет-Бутил-бутил-замещенный
имидазолиновый НР 31 устойчив при комнатной температуре в твёрдом состоянии, но
быстро разлагается при повышенных температурах. Необычная термическая
нестабильность НР 31 была исследована методом ЭПР. Так при нагревании
бензольного раствора НР 31 до 70оС без доступа воздуха, интенсивность сигнала
ЭПР этого радикала уменьшается в 10 раз за 30 мин. Для изучения механизма этой
реакции НР 31 нагревали в вакууме с избытком
2,2,6,6-тетраметилпиперидин-1-оксила (ТЕМРО), эффективно захватывающего
С-центрированные радикалы с образованием соответствующих алкоксиаминов (схема
р1). При 80оС реакция проходит до конца за ~ 12 ч. После удаления остатков
спиновой ловушки, из реакционной смеси были выделены
2-бутил-3-фенил-1,4-диазаспиро[4.5]дека-1,3-диен-1-оксид (р1) и
1-трет-бутокси-2,2,6,6-тетраметил-пиперидин (р2) в качестве единственных
продуктов (схема р1). Строение соединения р1 установлено на основании
спектральных данных и подтверждено встречным синтезом через присоединение
бутилмагнийхлорида к 2Н-имидазол-1-оксиду 6d и окисление. трет-Бутоксиамин р2 представляет собой летучее
бесцветное масло, спектры ЯМР 1Н и 13С которого соответствуют приведенным в
литературе данным.

СХЕМА
р1
Образование
алкоскиамина р2 доказывает, что термическое разложение НР 31 происходит по
радикальному механизму с отрывом трет-бутильного радикала. Отрыв алкильного
радикала от α-углеродного атома нитроксильной группы (реакция,
обратная спиновому захвату) никогда ранее наблюдался, хотя для некоторых
реакций спинового захвата радикалов нитронами была показана их обратимость.
Помимо
нагревания, НР 31 оказался также чувствителен к действию окислителей, и в
частности, диоксида марганца. При обработке этого радикала диоксидом марганца в
хлороформе снова был получен 2Н-имидазол-1-оксид р1. Предположительная схема
этого превращения включает обратимое окисление НР 31 в оксоаммониевый катион и
отщепление трет-бутильного катиона (схема р2).

СХЕМА
р2
НР
п1 и BBO ряда пирролидина, синтез которых был рассмотрен в
разделе 4 настоящей главы, также как и 31 содержат бутильный и трет-бутильный
заместители при α-атоме углерода нитроксильной группы. Логично было бы
предположить, что п1 и BBO также будут склонны к превращениям, наблюдавшимися
для 31. При хранении при комнатной температуре пирролидинонового НР n1
наблюдается медленное разложение радикала и окрашивание вещества в
красно-розовый цвет. Наблюдаемые явления вполне могут быть следствием
протекания реакции, обратной спиновому захвату, с последующей окислительной
димеризацией образующегося бутилпирролина. Поэтому с п1 был поставлен
эксперимент, аналогичный описанному для НР 31. Для полного разложения радикала
в этом случае потребовалось существенно больше времени - 7 дней. После
завершения процесса из реакционной смеси действительно были выделены
алкоксиамин р2 и новое соединение п2 (схема р3). Продукт п2 представляет собой
интенсивно окрашенное (темно-красное) кристаллическое вещество с характерным
металлическим блеском. Его спектральные данные аналогичны данным димера
этиленового типа 34, обсуждаемого в разделе 4, и других подобных соединений,
синтезированных ранее. На основе этих данных, а также данных элементного
анализа продукту п2 присвоена структура
4,4’-би(5-бутил-3-оксо-2,2-диэтил-3,4-дигидропирролилиден)-1,1’-диоксида (схема
р3). В отличие от трет-бутил-замещенного димера 34 молекула п2, содержащая
менее объемные бутильные группы в 5-м положении, по-видимому, имеет близкое к
плоскому строение. В результате, вклад бирадикальной форм, снижен, о чем
свидетельствует отсутствие уширения сигналов в спектрах ЯМР 1Н и 13С и
ЭПР-спектра. Очевидно при термическом разложении радикала п1 сначала образуется
соответствующий 5-бутилпирролин-N-оксид, который далее окисляется в димер п2
под действием TEMPO.

СХЕМА
р3
нитроксильный радикал разложение синтез
Разложение
при повышенных температурах по механизму, обратному спиновому захвату,
по-видимому, является общим свойством для НР описанных типов. Интересно, что
3-гидрокси-производное BBO существенно более устойчиво нежели его предшественник
п1, может храниться при комнатной температуре длительное время и выдерживает
нагревание до 70є С в течение нескольких суток. Очевидно, для отщепления
трет-бутильного радикала большое значение имеет возможность сопряжения образующейся
нитронной группы с другой кратной связью (С=N в случае p1 и
С=С енольного таутомера в случае п3).
Таким
образом, чрезмерное стерическое напряжение, создаваемое слишком объемными
заместителями при α-атомах углерода нитроксильной группы может сделать НР
слишком нестабильным, в результате чего его невозможно будет не только хранить,
но и выделить.
Глава 7. Исследование перспектив применения
полученных НР
Синтез
алкоксиаминов на базе полученных НР. Применение в контролируемой радикальной
полимеризации.
Одной из быстро развивающихся областей науки, в которой синтезированные
нами НР могут быть востребованы, является контролируемая радикальная
полимеризация. В настоящее время группами исследователей ведется поиск новых
нитроксильных радикалов, эффективных регуляторов полимеризации не только
стирола и его производных, но и алкилакрилатов и многих водорастворимых
мономеров, таких как винилацетат. Эти радикалы должны эффективно регулировать,
по крайней мере, два типа мономеров для возможности создания блок-сополимеров.
Эффективная регуляция подразумевает под собой отсутствие побочных реакций
Н-переноса при распаде и/или образовании соответствующего (макро)алкоксиамина,
а также низкое значение индекса полидисперсности продукта полимеризации.

СХЕМА
1 . I-инициатор, М - мономер, R, R' - растущие полимерные цепи, P -
полимер
Исследование контролируемой полимеризации с участием непосредственно НР
(схема1) осложняется непредсказуемостью влияния на реакцию инициатора (I) и, следовательно, невозможностью
математического описания процесса. Такой проблемы не возникает в случае, если
вместо НР и инициатора использовать алкоксиамины, производные соотвествующих НР
(схема 2).
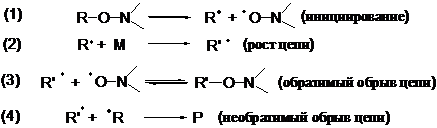
СХЕМА
2 . I-инициатор, М - мономер, R, R' - растущие полимерные цепи, P -
полимер
На сегодняшний день именно алкоксиамины чаще всего используются в
качестве инициаторов псевдоживой полимеризации, поскольку: а) они обладают
установленным соотношением (1:1) инициирующего алкильного радикала к
регулирующему нитроксильному, что позволяет легко исследовать и моделировать
кинетику процесса; б) при использовании алкоксиаминов отпадает необходимость
использования дополнительного инициирования; в) возможно получение
макроалкоксиаминов, в том числе, заранее привитых к поверхности.
В настоящее время предложено несколько эффективных подходов к синтезу
таких алкоксиаминов, причем в основном в качестве предшественника используется
соответствующий нитроксильный радикал. Одним из наиболее часто используемых
методов синтеза алкоксиаминов является простой и универсальный метод,
предложенный Матьяшжевским с соавторами (схема 1).
Согласно этому методу нами были получены алкоксиамины ряда имидазолина и
имидазолидина на основе НР 1, 2, 3, 4, 5, 6 и 7. В качестве галогенпроизводных
в синтезе применялись (1-бромэтил)бензол, трет-бутил-a-бромизобутират, этил-a-бромизобутират и 4-нитрофенил-a-бромизобутират (см. рис.1). Реакции
проводятся в дегазированных бензольных растворах, в присутствии комплексов одновалентной
меди. Скорость реакции, а также ее конверсия и выход целевых алкоксиаминов
существенным образом зависят от качества используемого медного порошка и
герметичности реакционной системы. Для достижения наилучшего результата медь
необходимо получать непосредственно перед проведением синтеза (обычно такой
медный порошок получают восстановлением сульфата меди цинковой пылью в водном
растворе).
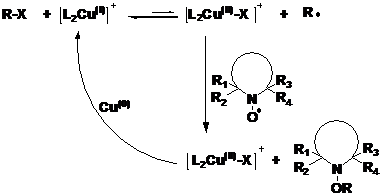
СХЕМА
3 Метод Матьяшжевского [19] для синтеза алкоксиаминов. Х - галоген (обычно Br),
R - алкильный фрагмент, L - лиганд (обычно - OTf).
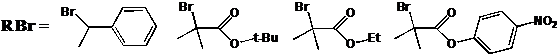
Рисунок
1 Галогенпроизводные, которые использовались в синтезе алкоксиаминов по методу
Матьяшжевского.
На основе имидазолиновых НР 6,7,8 получен набор алкоксиаминов различного
строения (схема 4). Все они представляют собой кристаллические, стеклообразные
или жидкие бесцветные вещества, не имеющие характерных полос поглощения в
УФ-спектрах. Данные спектров ЯМР 1Н и 13С этих соединений приведены в таблицах
1 и 2. Вследствие затруднённой инверсии у атома азота в положении 1, те из
соединений, в которых присутствует еще один или более ассиметрический центр,
являются неразделимой диастереомерной смесью. Так алкоксиамины 10b и 11
существуют в виде смесей 2-х диастереомеров в соотношениях 1 : 2 и 1 : 1.2
соответственно. На это указывает тот факт, что в их спектрах ЯМР имеются
удвоенные наборы сигналов соответствующих интенсивностей. Вероятно, большая
стерическая напряженность в молекулах соединений 9b,c и d, а также 12
обуславливает преимущественное существование их в виде только одного
диастереомера - в их спектрах наблюдали всего один набор сигналов.
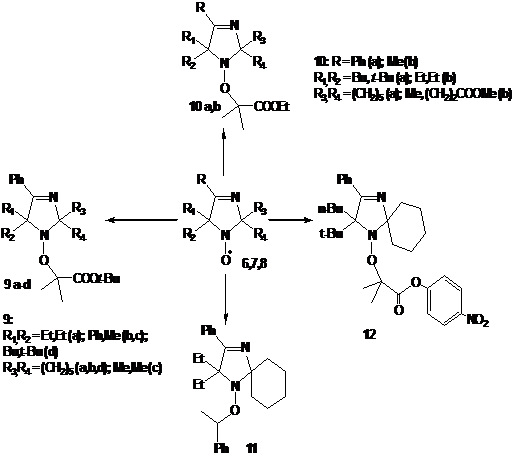
СХЕМА
4
Опыты по полимеризации различных мономеров винилового типа, проведенные с
использованием полученных имидазолиновых НР и их алкоксиаминов показали, что
они являются хорошими регуляторами синтеза гомополимеров стирола и
бутилакрилата. Особо интересные результаты были получены для алкоксиаминов НР
88. С применением этих соединений была исследована полимеризация
метилметакрилата. Добиться протекания его полимеризации в псевдоживом режиме на
сегодняшний день еще не удавалось ни с одним из известных НР. Причиной этому
считается быстрое превращение нитроксильных радикалов в гидроксиламин за счет
переноса атома водорода либо с алкильного радикала, либо внутримолекулярно при
разложении алкоксиамина, и последующий обрыв активных цепей, возникших в
результате побочных процессов гидроксиламинах. В опытах с НР 88 перенос
водорода не наблюдается вовсе, и происходит только гомолитический разрыв связи
С-О в алкоксиаминах. К сожалению, термическая нестабильность НР 88 в условиях
полимеризации (см. главу 6) (70-80оС) не позволяет добиться «живого» характера
полимеризации. Тем ни менее полимеризация метилметакрилата с НР 88 позволяет
достичь полидисперсности (PDI
< 2), меньшей, чем в случае типичных радикальных полимеризаций.
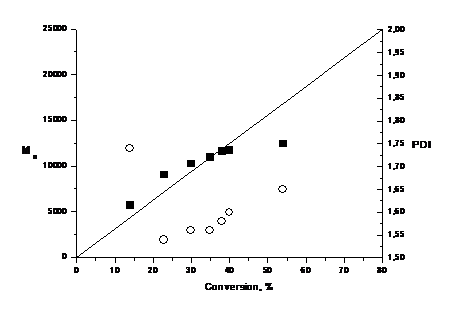
Рисунок
2 Рост среднечисленной молекулярной массы (n) и индекс полидисперстности (¡)
полимеризации метилметакрилата, инициируемой алкоксиамином 12 при 353K.
Прямая линия - теоретическая оценка молекулярной массы.
Преобразование
имидазолиновых НР в соответствующие имидазолидиновые производные сопровождается
восстановлением связи C=N, что приводит к образованию в молекуле нового
ассиметрического центра. Поэтому все, синтезированные нами, алкоксиамины ряда
имидазолидина (схема 5) также являются диастереомерными смесями. По данным ЯМР
1Н и 13С (таблицы 3 и 4 соответственно) 1-этилфенильное производное 15
существует в виде 4-х диастереомеров, а 2-метоксикарбонилэтил-замещенные
алкоксиамины 16a и b - в виде 2-х, в соотношении ~1 : 6, т.е. одной из форм
заметно больше.
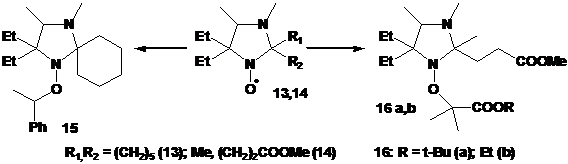
СХЕМА
5
Имидазолидиновые алкоксиамины существенно менее устойчивы по сравнению с
алкоксиаминами имидазолинового ряда, и в присутствие кислорода быстро
разлагаются с регенерацией исходного НР. Однако без доступа воздуха небольшое
количество образующегося при разложении алкоксиамина НР сдвигает равновесие
обратно и таким образом стабилизирует оставшийся алкоксиамин.
На основе алкоксиаминов 16a,b мы попытались получить водорастворимые
алкоксиамины, потенциально пригодные для инициирования и контролирования
полимеризации в водных растворах. Гидролиз проводили в водно-спиртовых
растворах щелочи, т.к. имидазолидины не устойчивы в кислых условиях. Было
обнаружено, что трет-бутиловый эфир не гидролизуется в упомянутых условиях, и
из алкоксиамина 16а нам удалось получить только продукт гидролиза метилового
эфира в составе заместителя во 2-м положении гетероцикла - аминокислоту NL1128-2. Однако алкоксиамин 16b в тех
же условиях гидролизуется по обеим сложноэфирным группам с образованием
динатриевой соли NL1149. Получены положительные предварительные результаты по
полимеризации акриламида с участием соединения NL1149 в водных растворах.

СХЕМА
6
Исследования по применимости НР 8а,b в контролируемой полимеризации
стирола проводятся также в НИИ химии Нижегородского государственного
университета Гришиным с сотр.
Биофизические исследования

Рис.0
Исследование
свойств НР 39, проведённое в ИХКиГ СО РАН, показало, что этот радикал обладает
исключительно высокой устойчивостью к восстановлению. Обработка его даже
5000-кратным мольным избытком аскорбата (концентрация радикала 39 - 0.1 мМ)
понижает интенсивность сигнала в спектре ЭПР лишь на 20 % в течение часа, затем
концентрация радикала выходит на плато и меняется очень медленно (Рис.1). Для
сравнения на рисунке 4 приведен также график зависимости концентрации радикала
от времени для незатрудненного аналога НР 39 - 3-гидрокси-2,2,5,5-тетраметилпирролидин-1-оксила
(40). Очевидно, что концентрация радикала 40 быстро снижается уже при
концентрациях аскорбиновой кислоты, на 2 порядка более низких, чем
применявшиеся для обработки НР 39. Удалось оценить примерное значение константы
скорости восстановления этого радикала аскорбатом - 0.005 M-1с-1
(т.е. почти в 40 раз меньше, чем для НР 40 (kred = 0.19 ±
0.01 M-1c-1), и примерно в 20 раз меньше, чем для
3-карбокси-2,2,5,5-тетраметилпирролидин-1-оксила (41)), константа скорости
обратной реакции (окисление соответствующего гидроксиламина аскорбатным
радикалом) - 5*104 M-1с-1 (примерно в 40 раз больше, чем для 41).
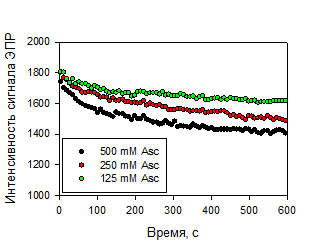
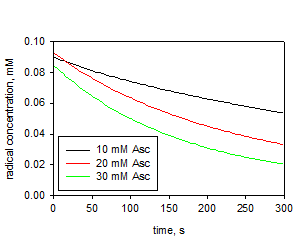
Рис.1
Кинетика восстановления аскорбатом натрия НР 39 (слева) и 40 (справа).
Эксперимент
проводился в 0.1М фосфатном буфере (рН 7.5) с 0.1мМ DTPA, в атмосфере аргона
при 23-25оС, концентрации НР 0.1мМ.
На
рисунке 2 представлены кривые падения интенсивности ЭПР-сигнала со временем под
действием аскорбиновой кислоты для циклических НР разного строения. Важно
отметить, что изображенные кривые получены для различных концентраций
аскорбата, и на основе схемы можно сделать только приблизительную оценку. Тем
ни менее, схема дает наглядное представление об относительной устойчивости
разных типов НР и из нее можно сделать вывод, что, соединение 39, очевидно,
является на сегодняшний день самым устойчивым к восстановлению нитроксильным
радикалом.
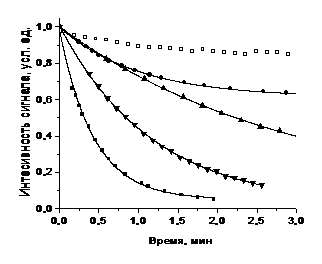
Рис.2
Кинетика восстановления аскорбатом натрия нитроксильных радикалов:
* 39
(500 мМ аскорбата натрия)
l 19a (80 мМ
аскорбата натрия)
41
(50мМ аскорбата натрия)
‚ 42
(125 мМ аскорбата натрия)
¡ 43
(4 мМ аскорбата натрия)
Образец
радикала 39 передан в Госуниверситет Огайо (США) для работ на животных и
изолированных органах. Предварительные исследования устойчивости НР 39 в
гомогенатах сердца и печени показали, что интенсивность сигнала ЭПР НР в этих
условиях понижается очень медленно даже в присутствие сукцината, активирующего
продуцирование в тканях активных восстанавительных агентов. Так, за 30 мин
радикал 39 восстанавливается менее, чем на 5%, тогда как
4-гидрокси-2,2,6,6-тетраметилпиперидин-1-оксил (44) восстанавливается полностью
за тот же промежуток времени (Рис.3), что позволяет проводить измерения
длительное время. Сердце крысы гомогенизировали на льду в присутствии 0.1 M
фосфатного буфера с 0.1 мM ДТПА, в соотношении 1 мл буфера на 1 г материала.
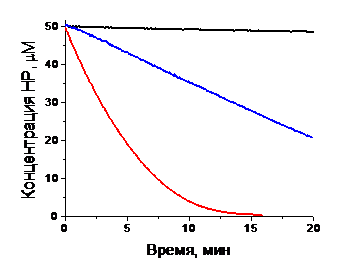
Рис.3
Кинетика восстановления НР 39 (чёрным) в гомогенате сердца крысы в присутствии
10мМ раствора сукцината натрия, для сравнения приведены кинетики восстановления
44 (красным) и 41 (синим).
Ведутся работы по изучению поведения этого НР в изолированных органах.
Для проведения экспериментов на изолированном сердце крысы были выбраны НР 39,
43 и 45. Работа выполнена на самках крыс Sprague-Dawley
массой 300±30
г. После полной анестезии фенобарбиталом (65 мг/кг i.p.) извлекали
сердца, быстро подключали катетеры и начинали обратную перфузию по методу
Лангендорфа с постоянным давлением 80 торр, используя модифицированный
бикарбонатный буфер Кребса (17 мМ глюкозы, 120 мМ NaCl, 2.5 мМ хлористого кальция, 5.9 мМ KCl, 1.2 мМ хлористого магния, и 25 мМ
бикарбоната натрия) в качестве перфузата. Все растворы для перфузии фильтровали
через 1.2 мкм фильтры Millipore и продували смесью 95% кислорода и 5% углекислого газа при 37 оС. После
перфузирования буфера в течение 30 мин. поток останавливали и вводили НР (3 мМ)
и помещали сердца в резонатор низкопольного ЭПР спектрометра (L-band), фиксируя изменение интерсивности сигнала НР при
комнатной температуре. Измерения показали, что в этих условиях интенсивность
сигналов ЭПР НР 43 и 45 быстро снижалась (до 30 и 20 % от первоначальной
интенсивности за 30 мин соответственно). В отличие от этого, падения
интенсивности сигнала НР 39 не наблюдали в течение часа (рис.4).
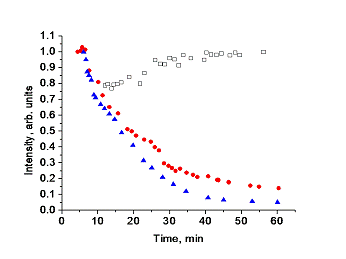
Рис.4 Кинетика восстановления НР 39 в условиях ишемии в лангендорфовском
сердце, для сравнения приведены кинетики восстановления НР 45 (красным) и 43
(синим).
Как было отмечено выше, увеличение объема заместителей в окружении
радикального центра НР 39 не только понижает скорость восстановления его
аскорбатом, но и увеличивает скорость обратной реакции. В результате при
добавлении избытка аскорбиновой кислоты к растворам некоторых НР в отсутствии
кислорода интенсивность сигнала ЭПР не падает до нуля, а выходит на плато при
некоторой «равновесной» концентрации НР (см. рис.1-4). Таким образом, влияние
заместителей не сводится к изменению кинетических параметров реакции
восстановления, но и меняет термодинамические характеристики НР. Для более
подробного изучения этого явления в МТЦ СО РАН было рассмотрено равновесие в
окислительно-восстановительной реакции между НР различного строения и
изотопно-меченым (15N)
1-гидрокси-3-карбокси-2,2,5,5-тетраметилпирролидином (СРН-15N). Для проведения эксперимента
готовили растворы исследуемых нитроксильных радикалов (0.5 мМ) и их смеси с
гидроксиламином CPH-15N (0.5 мМ) в соотношении 1:1 в
метаноле с добавлением 15 мМ КОН для исключения влияния рН и 0.1 мМ ДТПА для
удаления следов переходных металлов в изолированном боксе без доступа воздуха.
После установления равновесия (через ~ 24 ч) определяли соотношение дублетного
и триплетного сигналов в спектрах ЭПР реакционных масс (см. рис.8). Константы
равновесия рассчитывали как квадрат отношения интегральных интенсивностей
сигналов СР-15N и исследуемого НР (поскольку оба
радикала брались в эквимолярном количестве). Константа равновесия для НР 39
составляет 0.02±0.01, т.е. имеет наименьшую среди исследованных радикалов
величину. Таким образом, полученные данные показывают, что замена метильных
заместителей на более объемные приводит к ослаблению окислительных свойств НР.
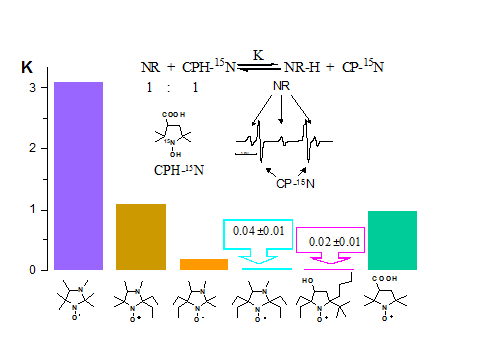
Рис.8
Измерение константы равновесия реакции обратимого восстановления некоторых НР
1-гидрокси-3-карбокси-2,2,5,5-тетраметилпирролидином (СРН-15N).
-
исследуемый нитроксильный радикал, NR-H -
соответствующий исследуемому НР гидроксиамин, CP-15N -
3-карбокси-2,2,5,5-тетраметилпирролидин-1-оксил
Глава 8. Экспериментальная часть
ИК спектры записаны на спектрометре Bruker Vector 22 FT-IR в KBr при
концентрации 1 : 150, в тонком слое, в растворах в ЧХУ при концентрации 1 :
150. УФ спектры записаны на приборе HP Agilent 8453 в EtOH (концентрация ~ 10-4 М). Спектры ЯМР 1Н записаны на
спектрометрах Bruker АС 200 (200.132 МГц), WP 200 SY (200.132 МГц), AV 300 (300.132 МГц), AM 400 (400.134 МГц), AV 600 (600.305
МГц) в 5-10 % растворах CDCl3 (δH=7.24 м.д., δС=76.69 м.д.), CD3OD (δH=
м.д., δС= м.д.), ДМСО-d6 (δH=2.50
м.д., δС=39.51 м.д.). В качестве стандарта
использовали сигнал растворителя. Спектры ЯМР 13С записаны на спектрометрах
Bruker AC 200 (50.323 МГц), AV 300 (75.467
МГц), AM 400 (100.614 МГц) при температуре 300К. Спектры ЭПР записаны на
спектрометрах Bruker ER-200D-SRC и Bruker ESP-200. Масс-спектры высокого
разрешения были записаны на спектрометре Finnigan 8200. Хромато-масс спектры записаны
на спектрометре Agilent 6890N GCD. Температуры плавления определены на
микронагревательном столике Кофлера или в капилляре. Элементный анализ
синтезированных соединений был выполнен в лаборатории микроанализа НИОХ.
Рентгеноструктурный анализ выполнен сотрудниками ЛФМИ НИОХ Ю.В. Гатиловым и
И.Ю. Багрянской на дифрактометре Bruker
P4. Контроль за ходом реакции осуществлялся с помощью тонкослойной
хроматографии (ТСХ) на пластинках Sorbfil UV-254, DC-Alufolien (элюент - хлороформ, его смеси с
гексаном, хлороформ с добавлением 2-5% этанола, диэтиловый эфир и его смеси с
гексаном, гексан, этилацетат и его смеси с гексаном, четыреххлористый углерод и
его смеси с хлороформом). Для очистки полученных веществ использовали методы
перекристаллизации, хроматографии на колонке или пластине (силикагель для
колоночной хроматографии Kieselgel 60, “Merck”, окись алюминия для хроматографии).
Изонитрозоацетон и изонитрозоацетофенон получали по литературным методикам.
Магнийорганические соединения получали по методикам, описанным в работах,
литийорганические соединения - по методикам, описанным в работе.
8.1 Синтез пространственно затрудненных
нитроксильных радикалов на базе 2Н-имидазол-1-оксидов
Н-Имидазол-1-оксиды 16а-e.
Общая методика.
H-имидазол-1-оксиды
получали по методике, приведённой в работе. Условия проведения реакции,
соотношения реагентов, методы очистки и выходы приведены в таблице 3 (с. 32).
Смесь изонитрозокетона (изонитрозоацетофенона, изонитрозоацетона,
парахлоризонитрозоацетофенона), ацетата аммония, уксусной кислоты и кетона
(диэтилкетона, циклогексанона) перемешивали при температуре, указанной в
таблице 3, контролируя ход реакции с помощью ТСХ (Sorbfil, элюент гексан - диэтиловый эфир = 2 : 1). После
исчезновения исходного соединения реакционную массу разбавляли водой в 10 раз и
экстрагировали этилацетатом или хлороформом. Экстракт сушили MgSO4, осушитель отфильтровывали,
растворитель и остатки кетона удаляли при пониженном давлении. Остаток далее
обрабатывали как указано в таблице 3.
ИК спектры 3-фенил-1,4-диаза-спиро[4.5]дека-1,3-диен-1-оксида (16a) и
3-метил-1,4-диаза-спиро[4.5]дека-1,3-диен-1-оксида (16b) соответствуют спектрам заведомых образцов.
ТАБЛИЦА
3. Синтез 2Н-имидазол-1-оксидов: условия реакции, соотношение реагентов,
способы очистки и выходы.
|
Шифр соединения
|
Условия проведения реакции
|
Мольные соотношения
реагентов
|
Способ очистки
|
Выход, %
|
|
T, oC
|
время
|
изонитрозокетон
|
кетон
|
NH4OAc
|
HOAc
|
|
|
|
16a
|
50
|
4 ч
|
1
|
9
|
6
|
8.3
|
Перекристаллизация (гексан:Et2O=2:1),
хроматография (SiO2, t-BuOMe-гексан=1:1)
|
57
|
|
16b
|
50
|
3 сут.
|
1
|
2.5
|
5.8
|
8.3
|
хроматография (Al2O3,
CH2Cl2)
|
55
|
|
16c
|
43
|
8 ч
|
1
|
2
|
2.6
|
11
|
хроматография (SiO2,
Et2O), перекристаллизация (гексан)
|
20
|
|
16d
|
47
|
8 ч
|
1
|
5
|
6
|
12
|
хроматография (SiO2,
Et2O)
|
17
|
|
16e
|
20
|
14 ч
|
1
|
7.5
|
6
|
8.5
|
Растирали с гексаном и
отфильтровывали
|
47
|
,2-Диэтил-4-фенил-2H-имидазол-1-оксид
(16c)
Розоватые кристаллы, Тпл=45-47оС. (Найдено: С, 72.05; Н, 7.30; N, 13.03. Рассчитано для C13H16N2O:
C, 72,19; H, 7,46; N, 12,95 %); νmax (KBr)/см-1 3077 (N=C-H), 2974, 2935, 2881 (CH3, CH2), 1605, 1590, 1563, 1514 (C=N, C=C); λmax (EtOH)/нм 233 (lg ε 4.27), 277 (lg ε 4.20);
δH (400 МГц; CDCl3)/м.д. 0.59 (6H, т, J 7.2
Гц, 2CH3, 2 ´ Et), 1.92, 2.08 (оба 2H, ABк JAB 14 Гц, Jк 7.2
Гц, 2 ´ CH2, 2 Et),
7.40 (3H, м, м-, п-H, Ph), 7.70 (1H, с, H-C5), 7.82 (2H, м, o-H, Ph);
δC (100 МГц; CDCl3)/м.д. 6.55 (CH3, 2 Et),
29.93 (CH2, 2 Et), 107.31 (C2),
126.49 (C5), 127.02 (o-C, Ph), 128.76 (м-C, Ph), 131.09 (и-C, Ph), 131.39 (п-C, Ph), 165.94 (C4).

,2-Диэтил-4-метил-2H-имидазол-1-оксид
(16d)
Желтое масло. (Найдено: С, 61.67; Н, 9.17; N, 17.67. Рассчитано для C8H14N2O: C, 62,31; H,
9,15; N, 18,17 %); νmax (в
тонком слое)/см-1 3078 (H-C=N), 2975, 2937, 2882 (CH3, CH2),
1596, 1512 (C=N); λmax (EtOH)/нм 283 (lg ε
3.92); δH (200 МГц; CDCl3)/м.д. 0.41 (6H, т, J 7.4,
2CH3, 2 ´ Et), 1.66, 1.84 (оба 2H, ABк JAB 20 Гц, Jк 7.4
Гц, 2 ´ CH2, 2 Et),
2.10 (3H, с, CH3-С4), 7.04 (1H, с, H-C5); δC (50 МГц; CDCl3)/м.д. 5.97 (СН3, 2 Et), 16.97 (Н3С-C4),
28.97 (СН2, 2 Et), 106.32 (С2), 128.75 (С5), 166.45
(С4).
-(4-Хлорфенил)-1,4-диаза-спиро[4.5]дека-1,3-диен-1-оксид (16e)

Бесцветные кристаллы, Тпл=140-142 оС. (Найдено: С, 63.87; Н, 5.56; Cl 13.50; N, 10.80. Рассчитано для C14H15ClN2O: C, 64.00; H, 5.75; Cl,
13.49; N, 10.66 %); νmax (KBr)/см-1
3067(N=C-H), 2935, 2859 (CH3, CH2), 1602, 1587, 1558, 1508 (C=N, С=С); λmax (EtOH)/нм 243 (lg ε 4.19), 278 (lg ε
4.19); δH (400 МГц; CDCl3)/м.д. 1.40 (3H, м, (СН2)5), 1.85 (5H, м, (СН2)5), 2.07 (2H, м, (СН2)5), 7.41, 7.79 (оба 2Н, AA’BB’, J 8.5 Гц, Ar), 7.67 (1H, c, H-C2);
δC (100 МГц; CDCl3)/м.д. 23,19, 23.05, 34,88 ((СН2)5), 104.71 (C5), 124.75 (C2), 128.26 (o-C, Ar), 129.01 (м-C, Ar), 129.78 (и-C, Ar), 137.52 (C-Сl), 163.36 (C3).
Реакция циклических нитронов (2Н-имидазол-1-оксидов,
4Н-имидазол-3-оксидов, пирролин-N-оксидов)
с реактивами Гриньяра. Общая методика.
В работе использовали растворы магнийорганических соединений,
приготовленные из 0.03 моль магниевой стружки и 0.02 моль алкилгалогенида
(арилгалогенида) в 30-40 мл абсолютного серного эфира по известным методикам.
Для проведения реакций с альдонитронами использовали ~ двукратный избыток
магнийорганического соединения, с кетонитронами - ~ четырёхкратный (если не
указано иначе).
Полученный раствор реактива Гриньяра прибавляли по каплям к раствору 0.01
моль нитрона (если не указано иначе) в минимальном количестве абсолютного
эфира, сухого ТГФ или бензола так, чтобы кипение эфира не было слишком бурным.
Реакционную смесь перемешивали или кипятили с обратным холодильником в течение
0.5-12 ч, до исчезновения исходного соединения по данным ТСХ. Затем осторожно,
по каплям прибавляли воду до образования вязкой неорганической фазы.
Органический раствор декантировали. Неорганическую массу 2-3 раза промывали
эфиром. В реакциях с пирролин-N-оксидами
вместо воды использовали насыщенный раствор хлорида аммония. Органическую фазу
отделяли на делительной воронке, неорганическую - экстрагировали 2-3 раза
эфиром. Объединённый раствор полученного соединения в органических
растворителях сушили сульфатом магния и упаривали на ротационном испарителе. В
реакциях фенилмагнийбромида с альдонитронами ряда 2Н-имидазола образующиеся при
этом 1-гидрокси-2,5-дигидроимидазолы выделяли в чистом виде, в остальных
случаях образующиеся N-гидроксипроизводные
без выделения окисляли в соответствующие нитроны или нитроксильные радикалы.
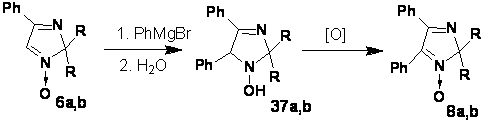

,2-Диметил-4,5-дифенил-2,5-дигидроимидазол-1-ол
(37b) Соединение получали согласно общей методике при
взаимодействии 2,2-диметил-4-фенил-2Н-имидазол-1-оксида (6b) и
фенилмагнийбромида. Продукт растерли с эфиром и отфильтровали, а затем
перекристаллизовали из метанола. Выход: 65 %. Бесцветные кристаллы,
Тпл.=186-189оС. (Найдено: С, 76.57; Н, 7.05; N, 10.58.
Рассчитано для C17H18N2O: C, 76.66; H, 6.81; N, 10.52 %); νmax (KBr)/см-1
3277 (OH), 3070, 3029 (=C-H),
2999, 2974, 2939 (CH3), 1612 (C=N); λmax (EtOH)/нм 244 (lg ε 4.10); δH
(300 МГц; CDCl3, CD3OD)/м.д. 1.32, 1.34 (оба 3H, с, 2CH3,
при C2), 3.59 (1H, ушир. с, ОН), 5.24 (1H, с,
Н-C5), 7.20 (всего 8H, м, Н Ph
при С5, м-,п-Н Ph при С4), 7.43 (2H, м, o-H, Ph
при С4); δC (75
МГц; CDCl3, CD3OD)/м.д. 19.67, 27.02 (2СН3, при C2),
77.29 (С5), 90.74 (С2), 127.47 (п-С, Ph при С5), 127.56 (м-C, Ph
при С5), 127.72 (o-C, Ph при С5), 128.09 (o-C, Ph
при С4), 128.74 (м-C, Ph при С4), 130.12 (п-С, Ph при С4),
131.80 (и-С, Ph при С4), 138.39 (и-С, Ph при С5)
167.90 (C4).
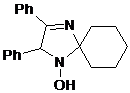
,3-Дифенил-1,4-диаза-спиро[4.5]дека-3-ен-1-ол
(37a) был получен аналогичным образом из
3-фенил-1,4-диаза-спиро[4.5]дека-1,3-диен-1-оксида (6a) и
фенилмагнийбромида. После прибавления к реакционной смеси воды, отделили
суспензию продукта в органических растворителях. Растворители отогнали при
пониженном давлении. Остаток растерли с водой и отфильтровали,
перекристаллизовали из метанола. Выход: 86 %. Мелкие бесцветные кристаллы,
Тпл.=208-211оС. (Найдено: С, 78.31; Н, 7.23; N, 8.32.
Рассчитано для C20H22N2O: C, 78.40; H, 7.24; N, 9.14 %); νmax (KBr)/см-1
3276 (OH), 3061, 3028 (=С-Н), 2961,2928, 2851 (CH3, CH2),
1620 (C=N); λmax (EtOH)/нм
245 (lg ε 4.13); δH
(300 МГц; DMSO-d6)/м.д. 1.34, 1.57, 1.74, 2.00 (всего 10Н, м, СН2, (CH2)5),
5.39 (1H, с, H-C2), 7.18-7.54 (всего 10H, м, о-, п-, м-H, 2Ph),
7.95 (1Н, с, ОН); δC (75
МГц; DMSO-d6)/м.д. 20.31, 21.95, 25.43, 30.92, 37.16 (CH2,
(CH2)5), 77.74 (C2), 93.11 (C5),
127.34 (п-C, Ph при С2), 127.78 (м-C, Ph
при С2), 128.18 (о-C, Ph при С2), 128.21 (о-C, Ph
при С3), 128.93 (м-C, Ph при С3), 130.25 (п-C, Ph
при С3), 133.13 (и-C, Ph при С3), 139.98 (и-C, Ph
при С2), 167.37 (C3).
Окисление 1-гидрокси-2,5-дигидроимидазолов. Общая методика.
К раствору или суспензии 1-гидрокси-2,5-дигидроимидазола (или остатка,
полученного после обработки реакционной массы, полученной в результате
взаимодействия 2Н-имидазол-1-оксида с металлоорганическими соединениями) в
хлороформе присыпали 5-ти кратный по массе избыток окислителя (диоксида свинца
или марганца) и перемешивали в течение 0.5-3 ч. После исчезновения пятна
исходного соединения, наблюдаемого при ТСХ, окислитель отфильтровывали и 2-3
раза промывали смесью хлороформа со спиртом (4 : 1). Растворитель отгоняли на
ротационном испарителе.

,2-Диметил-4,5-дифенил-2H-имидазол-1-оксид
(8b). Гидроксиамин 37b растворяли в
хлороформе и окисляли двуокисью свинца по описанной выше методике. Продукт
перекристаллизовали из смеси этилацетат-гексан (1 : 1). Выход: 58.5 %.
Бесцветные кристаллы Тпл.=114-116оС. (Найдено: С, 77.37; Н, 6.09; N, 10.61. Рассчитано для C17H16N2O: C,
77.25; H,
6.10; N, 10.60 %); νmax (KBr)/см-1 3054 (=C-H, Ph), 2986, 2934 (CH3), 1606, 1586, 1563, 1519 (C=N, С=С); λmax (EtOH)/нм
252 (lg ε 4.23), 314 (lg ε
3.84); δH (200 МГц; CDCl3)/м.д. 1.70 (6H, с, 2 ´ CH3 при C2), 7.36 (всего 6H, м, п-, м-H, 2Ph), 7.49 (всего 4H, м, о-H, 2Ph); δC (100 МГц; CDCl3)/м.д.
24.63 (CH3 при C2), 100.12 (C2),
126.20 (и-C, Ph при С5), 128.15 (о-C, Ph при С4), 128.31 (о-,м-C, Ph при С5), 128.95 (м-C, Ph при С4), 129.83 (п-C, Ph при С5), 130.70 (п-C, Ph при С4), 132.45 (и-C, Ph при С4), 135.25 (C5), 166.20 (C4).
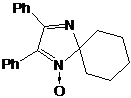
,3-Дифенил-1,4-диаза-спиро[4.5]дека-1,3-диен-1-оксид (8a) получали аналогичным образом при
окислении гидроксиамина 37a
двуокисью свинца. Продукт растерли с сухим эфиром, охладили и отфильтровали.
Выход: (83.5%). Бесцветные кристаллы, Тпл.=149-151оС. (Найдено: C, 78.99; H,
6.65; N, 9.15 %. Рассчитано для C20H20N2O: C, 78.92; H, 6.62; N, 9.20 %); νmax (KBr)/см-1 3068, 3035 (=C-H, Ph), 2948, 2852 (CH2), 1603, 1585, 1558, 1519 (C=N, C=C);
λmax (EtOH)/нм 251 (lg ε 4.24), 313 (lg ε 3.84); δH (200 МГц; CDCl3)/м.д. 1.54 (3H, м, CH2,
(СН2)5), 1.92 (5Н, м, CH2, (CH2)5), 2.25 (2H, м, CH2, (CH2)5), 7.36 (всего 6H, м, п-, м-H, 2Ph),
7.53 (всего 4H, м, о-H, 2Ph); δC (100 МГц; CDCl3)/м.д.
23.11 (CH2, (CH2)5), 24.65 (CH2, (CH2)5), 35.23 (CH2, (CH2)5),
102.79 (C5), 126.33 (и-С, Ph при С2), 128.10 (o-C, Ph при С3), 128.35
(o-C, Ph при С2), 128.44
(м-C, Ph при С2), 129.01 (м-C, Ph при С3), 129.68 (п-C, Ph при С2), 130.43 (п-C, Ph при С3), 132.89 (и-C, Ph при С3), 135.44 (С2), 166.07 (C3).
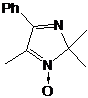
,2,5-Триметил-4-фенил-2H-имидазол-1-оксид
(9b). Соединение получали согласно общей
методике. В реакции 2,2-диметил-4-фенил-2Н-имидазол-1-оксида с
метилмагниййодидом образовывался соответствующий гидроксиамин, который без
выделения окисляли диоксидом свинца. Продукт перекристаллизовали из гексана.
Спектральные характеристики нитрона 9b соответствуют литературным данным. Выход: 95.6 %. Бесцветные кристаллы.
Тпл.=87-89 оС.
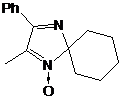
-Метил-3-фенил-1,4-диаза-спиро[4.5]дека-1,3-диен-1-оксид (9a). Соединение получали согласно общей
методике. В реакции 3 г (13.2 ммоль) 2Н-имидазола 6a с метилмагниййодидом, полученного из 0.95 г (39.6 ммоль)
магния и 1.65 мл (26.4 ммоль) метилйодида, образовывался соответствующий
гидроксиамин, который без выделения окисляли 15 г диоксида свинца. Раствор
промыли 3% водным раствором сульфита натрия для удаления йода и сушили над
сульфатом магния. Соединение 9а перекристаллизовали из гексана. Выход 3.1 г
(93%). Бесцветные кристаллы, Тпл.=75-77оС. (Рассчитано для C15H18N2O: C, 74.35;
H, 7.49; N, 11.56 %); νmax (KBr)/см-1
3056 (=C-H, Ph), 2934, 2857
(СН3, CH2), 1592, 1565, 1514 (C=N, C=C);
λmax (EtOH)/нм 229 (lg ε 4.03), 273 (lg ε 3.96).
Получение 2-Метил-2,3-дифенил-1,4-диаза-спиро[4.5]дека-3-ен-1-оксила (7a).
Метод А.
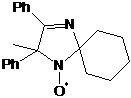
Метилнитрон 9a (2.8 г, 11.6
ммоль) обработали фенилмагнийбромидом из 2.5 г (0.1 моль) магния и 7.3 мл (69.4
ммоль) бромбензола в соответствии с общей методикой. После обработки
реакционной смеси остаток растворили в 20 мл хлороформа и перемешивали с 15.5 г
диоксида марганца 1ч. Окислитель отфильтровали, растворитель отогнали на
ротационном испарителе. Остаток перекристаллизовали из гексана. Выход
нитроксильного радикала 7a 1.37г (37 %).
Желтые кристаллы, Тпл.=161-163оС. (Найдено: С, 78.80; Н, 7.29; N, 8.76. Рассчитано для C21H23N2O•:
C, 78.96; H, 7.26; N, 8.77 %); νmax (KBr)/см-1 3067, 3031 (H-C=, Ph),
2997, 2977, 2938, 2923, 2857 (CH3, CH2), 1597, 1570 (C=N, С=С); λmax (EtOH)/нм
249 (lg ε 4.61).
Метод В

Реакцию проводили по стандартной методике. После прибавления
метилмагниййодида к раствору фенилнитрона 8a реакционную смесь кипятили с обратным холодильником при
перемешивнии еще 10 ч. После обработки реакционной смеси, остаток растворили в
хлороформе. Раствор промыли 3% водным раствором сульфита натрия для удаления
йода, высушили над сульфатом магния и окислили диоксидом марганца. Полученную
массу хроматографировали на колонке с силикагелем, элюент - хлороформ : гексан
= 1 : 1. В результате хроматографии помимо НР 7a (Выход: 40 %) был выделен
2,3-Дифенил-1,4-диаза-спиро[4.5]дека-1,3-диен (10) (Выход: 20 %). Бесцветные
кристаллы, Тпл.=102-104оС. (Найдено: C, 83,06; H, 6,97; N, 9,63 %. Рассчитано
для C20H20N2: C, 83.30; H, 6.99; N, 9.71 %); νmax (KBr)/см-1 3065 (=C-H, Ph), 2928, 2846 (CH2), 1606, 1552 (C=N, C=C);
λmax (EtOH)/нм 263 (lg ε
4.02); δH (300 МГц; CDCl3)/м.д. 1.73 (2H, м, CH2,
(СН2)5), 1.82 (3Н, м, CH2, (CH2)5), 1.96 (5H, м, CH2, (CH2)5), 7.27 (всего 6H, м, п-, м-H, 2Ph),
7.50 (всего 4H, м, о-H, 2Ph); δC (75 МГц; CDCl3)/м.д.
23.69 (CH2, (CH2)5), 25.27 (CH2, (CH2)5), 34.29 (CH2, (CH2)5),
103.65 (C5), 127.83 (o-C, Ph), 128.44 (м-C, Ph), 129.51 (п-C, Ph), 132.69 (и-C, Ph), 163.56 (C2, C3).
Метод С
К раствору метиллития, приготовленному из 0.23 г (32 ммоль) лития и 1 мл
(16.1 ммоль) метилйодида в 20 мл абсолютного эфира по известной методике,
прибавили по каплям раствор 2 г (6.6 ммоль) фенилнитрона 8a в 20 мл сухого бензола. Реакционную
смесь перемешивали под аргоном в течение 12 ч, затем осторожно разлагали водой.
Водную фазу отделили и экстрагировали эфиром 2 раза по3 мл. Объединенный
экстракт промыли 2 раза 3% раствором сульфита натрия (по 20 мл), сушили
сульфатом магния и затем окисляли 1 ч в присутствие 12 г диоксида марганца. НР
7а перекристаллизовали из гексана. Выход: 1.8 г ( 86.6 %).
Получение 2,2,5-Триметил-4,5-дифенил-2,5-дигидро-имидазол-1-оксила (7b).
Метод А

Соединение получали согласно общей методике. В реакции фенилнитрона 8b с метилмагниййодидом образовывался
соответствующий гидроксиамин, который без выделения окисляли диоксидом
марганца. Раствор продукта промывали 3% водным раствором сульфита натрия для
удаления йода и сушили над сульфатом магния. Растворитель отгоняли на
ротационном испарителе. Остаток хроматографировали на колонке с силикагелем,
элюент: хлороформ: гексан = 3 : 2. Перекристаллизовали из гексана. Выход: 60 %.
Желтые кристаллы Тпл.=128-132оС. (Найдено: С, 77.72; Н, 6.82; N, 10.12. Рассчитано для C18H19N2O•:
C, 77.39; H, 6.86; N, 10.03 %); νmax (KBr)/см-1 3065, 3032 (H-C=, Ph), 2982, 2932 (CH3), 1599, 1572 (C=N, С=С); λmax (EtOH)/нм
248 (lg ε 4.20).
Метод В
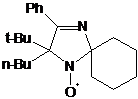
Аналогичным образом метилнитрон 9b обрабатывали фенилмагнийбромидом, в результате чего
образовывался соответствующий гидроксиамин, который без выделения окисляли
диоксидом марганца. Выделение и очистку нитроксильного радикала 7b проводили аналогично описанному в
методе А. Выход: 42%.
-трет-Бутил-2-бутил-3-фенил-1,4-диаза-спиро[4.5]дека-3-ен-1-оксил (4). К
раствору бутиллития в 40 мл сухого гексана, приготовленному из 1 г (0.143 моль)
мелко нарезанного лития и 6.8 мл (0.074 моль) бутилхлорида, прибавили по каплям
при перемешивании в атмосфере аргона раствор 3.7 г (0.013 моль)
2-трет-Бутил-3-фенил-1,4-диаза-спиро[4.5]дека-1,3-диен-1-оксида (1) в 20 мл
сухого бензола. Перемешивали 1 ч. Приливали воду до растворения неорганического
осадка (220 мл). Органическую фазу отделили, водную экстрагировали эфиром (2
раза по 15 мл). Экстракт объединили с органической фазой, барботировали
воздухом 12 ч, сушили сульфатом магния, осушитель отфильтровали, раствор
упарили. Остаток растерли с гексаном, охладили и образовавшийся осадок
отфильтровали. Выход: 3.69 (83 %). Желтые кристаллы, Тпл. 107-109оС. (Найдено:
С, 76.97; Н, 9.30; N, 8.20.
Рассчитано для C22H33N2O•: C, 77.37; H, 9.74; N, 8.20 %); νmax (KBr)/см-1 3055 (=C-H, Ph), 2969, 2937, 2862, 2852 (CH3, CH2), 1573, 1594 (C=N, С=С); λmax (EtOH)/нм 237 (lg ε 4.07).
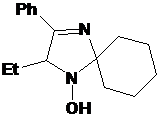
-Фенил-2-этил-1,4-диаза-спиро[4.5]дека-3-ен-1-ол (17а)
Из 2.1 г (0.0875 моль) магниевой стружки и 5 мл (0.0670 моль) этилбромида
получали этилмагнийбромид в виде раствора в 50 мл сухого серного эфира. К
раствору 3.25 г (0.0143 моль) соединения 16а в 20 мл сухого ТГФ прибавили по
каплям раствор магнийорганического соединения. Перемешивали реакционную смесь
еще 30 минут, затем прилили воду до образования вязкой неорганической фазы. При
этом выпал органический осадок продукта. Для отделения имидазолина 17а, его
растворяли при нагревании в смеси этилацетата с этанолом (5 : 1; 3 ´ 15 мл) и декантировали. Объединённую
суспензию соединения 17а в органических растворителях упарили на ротационном
испарителе. Остаток перекристаллизовали из этилацетата (30 мл). Выход: 2.1 г.
(57 %). Бесцветные кристаллы, Тпл.=184-186.5оС. (Найдено: С, 74.11; Н, 8.58; N, 10.70. Рассчитано для C16H22N2O:
C, 74,38; H, 8,58; N, 10,84 %); νmax (KBr)/см-1 3175 (OH), 2987, 2962, 2928, 2852 (CH3, CH2),
1621 (C=N); λmax (EtOH)/нм 241 (lg ε
4.14); δH (200 МГц; D3COD, CDCl3)/м.д. 0.71 (3H, т, J 7.2 Гц, CH3, Et), 1.23 (4Н, м, CH2, (CH2)5, Et),
1.42 (7Н, м, (CH2)5, Et), 1.88 (1H, м,
(CH2)5), 4.16 (1H, дд, J1 10
Гц, J2 4 Гц, H-C2), 7.12 (3H, м, п-, м-H, Ph), 7.35 (2H, м, o-H, Ph);
δC (50 МГц; D3COD, CDCl3)/м.д. 10.35 (CH3, Et), 22.67, 22.79, 25.03, 25.19, 31.11, 37.47 (CH2, Et, (CH2)5),
78.40 (C2), 95.32 (C5), 127.36 (o-C, Ph), 127.95 (м-C, Ph), 129.99 (п-C, Ph), 132.66 (и-C, Ph), 171.05 (C3).
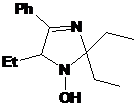
,2,5-Триэтил-4-фенил-2,5-дигидро-имидазол-1-ол (17b)
получали аналогично описанному для 17а. После прибавления воды продукт не
выпадал в осадок и, органическую фазу декантировали, неорганический осадок
промывали диэтиловым эфиром. Объединённый эфирный экстракт сушили MgSO4, осушитель отфильтровали и
растворитель отогнали на ротационном испарителе. Перекристаллизовали из
гексана. Выход: 45 %. Бесцветные кристаллы, Тпл.=137-140оС. (Найдено: С, 73.42;
Н, 9.21; N, 11.44. Рассчитано для C15H22N2O: C,
73,13;
H, 9,00; N, 11,37 %); νmax (KBr)/см-1 3167 (OH), 3032 (=C-H), 2979, 2962, 2922, 2879, 2838 (CH3, CH2), 1631 (C=N);
λmax (EtOH)/нм 237 (lg ε
4.17); δH (200 МГц; CDCl3, CD3OD)/м.д. 0.74 (3H, т, J 7.4 Гц, CH3, Et-C5), 0.87, 0.88 (оба 3H, т, J1,2
7.4 Гц, 2 ´ CH3, 2 Et-C2), 1.85 (6H, м, 3 ´ CH2, Et-C5, 2 Et-C2), 4.47 (1H, дд, J1 3.6 Гц, J2 6
Гц, H-C5), 6.79 (1H, уш.
с, ОН), 7.34 (3H, м, п-, м-H, Ph), 7.54 (2H, м, o-H, Ph);
δC (100 МГц; CDCl3)/м.д. 7.84 (СН3, Et-C5), 8.61, 8.89 (СН3,
Et-C2), 24.43, 27.25 (СН2, Et-C2), 31.75 (CH2, Et-С5), 74.90 (С5), 95.21 (С2), 127.03 (o-C, Ph), 128.15 (м-C, Ph), 130.06 (п-C, Ph), 133.31 (и-C, Ph), 171.43 (C4).
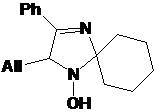
2-Аллил-3-фенил-1,4-диаза-спиро[4.5]дека-3-ен-1-ол (17c)
К раствору 4 г (0.0175 моль) соединения 16а в 25 мл сухого ТГФ прибавили
по каплям раствор аллилмагнийбромида, полученного из 7 г (0.292 моль) магниевой
стружки и 8.9 мл (0.105 моль) аллилбромида, в 120 мл абсолютного эфира.
Перемешивали 1 ч. Затем прибавляли воду до образования вязкой неорганической
фазы. Органическую фазу декантировали, сушили сульфатом магния, осушитель
отфильтровали и растворитель отогнали на ротационном испарителе. Остаток
растерли с гексаном, осадок отфильтровали. Выход: 0.71 г. (15 %) Бесцветные
кристаллы, Тпл.=137-141оС. (Найдено: С, 75.41; Н, 7.98; N, 10.38. Рассчитано для C17H22N2O:
C, 75,52; H, 8,20; N, 10,36 %); νmax (KBr)/см-1 3185 (OH), 3032 (=С-H),
2933, 2849 (CH3, CH2), 1619 (C=N);
λmax (EtOH)/нм 241 (lg ε
4.11); δH (400 МГц; CDCl3)/м.д. 1.49 (1H, м, СН2, (СН2)5), 1.69 (5H, м, СН2, (СН2)5), 1.84 (3H, м, СН2, (СН2)5), 2.09 (1H, м, СН2, (СН2)5), 2.35, 2.59 (оба 1Н, м, СН2, All), 4.64 (1Н дд J1 4.0 Гц, J2 8.8 Гц, Н-С2), 5.02, 5.06 (оба 1Н, м, =СН2, All), 5.93 (1H, м, =CH-, All), 6.21 (1H, уш. с, ОН), 7.41 (3H, м, п-, м-H, Ph), 7.73 (2H, м, о-H, Ph);
δC (100 МГц; CDCl3)/м.д. 23.38, 23.68, 25.66, 31.97, 36.88, 38.16 (CH2, (СН2)5, All), 76.62 (C2), 96.38 (C5), 116.77 (=СН2,
All), 127.95 (o-C, Ph), 128.33 (м-C, Ph), 130.25 (п-C, Ph), 133.06 (и-C, Ph), 135.23 (=СН-, All), 168.58 (C3).
Взаимодействие 4-фенил-2,2-диэтил-2H-имидазол-1-оксида (FI-1) с
трет-бутилмагний хлоридом.
Реакцию 9.5 г (43.8 ммоль) 2H-имидазол-1-оксида FI-1 с трет-бутилмагний
хлоридом провели согласно общей методике. После обработки реакционную массу
хроматографировали на колонке с силикагелем, элюент: диэтиловый эфир - гексан
(градиент от 1 : 10 до 1 : 5). В результате хроматографии было выделено 3 новых
соединения: 5-трет-бутил-1-трет-бутокси-2,2-диэтил-4-фенил-2,5-дигидроимидазол
(FI-2A), 5-трет-бутил-4-фенил-2,2-диэтил-2,5-дигидроимидазол-1-ол ( FI-2B) и 5-трет-бутил-4-фенил-2,2-диэтил-2,5-дигидро-имидазол
(FI-2C).

-трет-Бутил-1-трет-бутокси-2,2-диэтил-4-фенил-2,5-дигидроимидазол (FI-2A)
Выход: 0.38 г (3 %). Бесцветные кристаллы. Тпл= 59-62оС (гексан) (Найдено: С,
77.10; Н, 10.37; N, 8.37.
Рассчитано для C21H34N2O: C, 76.31; H, 10.37; N, 8.48 %); νmax (в тонком слое)/см-1 3060 (=C-H, Ph), 2971, 2927, 2868 (CH3, CH2),
1629, 1578 (C=N); λmax (EtOH)/нм 238 (lg ε
4.02); δH (400 МГц; CDCl3)/м.д. 1.02 (9H, с, CH3, t-Bu-C5), 1.06, 1.08
(оба 3Н, т, Jт=7.4 Гц, СН3, Et), 1.28 (9H, с, CH3, t-Bu-O), 1.40 (1H, м, CH2, Et), 1.83 (2H, м, CH2, Et), 1.99 (1H, м, CH2, Et), 4.55 (1H, с, H-C5), 7.37 (3H, м,
п-, м-H, Ph), 7.68 (2H, м, o-H, Ph);
δC (100 МГц; CDCl3)/м.д. 8.98, 10.09 (СH3, Et), 26.50, 29.59 (CH2, Et), 28.32 (CH3, t-Bu-C5), 30.91 (CH3, t-Bu-O), 34.86 (C(CH3)3, C5-t-Bu), 78.17 (C(CH3)3, O-t-Bu), 88.47 (C5), 101.13 (C2), 128.05 (o-C, Ph), 128.22 (м-C, Ph), 129.35 (п-C, Ph), 136.72 (и-C, Ph), 170.29 (C4).
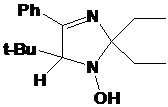
-трет-Бутил-4-фенил-2,2-диэтил-2,5-дигидроимидазол-1-ол ( FI-2B)
Перекристаллизовали из гексана. Выход: 5.4 г (45 %). Бесцветные
кристаллы, Тпл=137-138оС. (Найдено: С, 74.79; Н, 9.49; N, 10.41. Рассчитано для C17H26N2O: C, 74.41; H, 9.55; N,
10.21 %); νmax (KBr)/см-1 3241 (OH), 3080 (=C-H), 2986, 2955, 2879, 2824 (CH3, CH2), 1626 (C=N);
λmax (EtOH)/нм 229 (lg ε
4.47); δH (400 МГц; CDCl3,)/м.д. 0.83 (9Н,с, СН3, t-Bu), 0.89, 1.02 (оба 3H, т, Jт 7.3 Гц, CH3, Et), 1.70, 1.77 (2H, ABк, JAB=14.4 Гц,
Jк=7.3 Гц, CH2, Et), 1.84, 2.11 (2H, ABк, JAB=14.0 Гц, Jк=7.3 Гц, CH2, Et),
4.29 (1H, c, H-C5), 4.98 (1H, c, OH), 7.34 (3H, м, п-, м-H, Ph), 7.43 (2H, м, o-H, Ph); δC (100 МГц; CDCl3)/м.д. 8.55, 8.79 (СН3, Et-C2), 27.59, 33.34
(СН2, Et-C2), 27.72 (CH3, t-Bu), 35.50 (C(CH3)3, t-Bu), 84.81 (С5),
96.33 (С2), 127.59 (o-C, Ph), 127.88 (м-C, Ph), 129.04 (п-C, Ph), 137.31 (и-C, Ph), 171.76 (C4).
-трет-Бутил-4-фенил-2,2-диэтил-2,5-дигидро-имидазол (FI-2C)
Перекристаллизовали из гексана. Выход: 0.48 г (4%). Бесцветные кристаллы,
Тпл =46-49оС. (Найдено: С, 78.49; Н, 10.04; N, 10.06. Рассчитано для C17H26N2: C, 79.02; H, 10.14; N,
10.84 %); νmax
(KBr)/см-1 3379 (N-H), 3060, 3029 (=C-H, Ph), 2963, 2877 (CH3, CH2), 1627 (C=N);
λmax (EtOH)/нм 229 (lg ε 3.92); δH (400 МГц; CDCl3)/м.д. 0.80 (9H, c, CH3, t-Bu), 0.83, 1.04 (оба 3Н,
т, Jт=7.5 Гц, СН3, Et), 1.68, 1.84 (2H, ABк, JAB=13.7 Гц,
Jк=7.5 Гц, СН2, Et), 1.70, 1.76 (2H, ABк, JAB=13.2 Гц, Jк=7.5 Гц, СН2, Et),
4.36 (1H, c, H-C5), 7.33 (3Н, м, м-,п-Н, Ph), 7.43 (2Н, м, о-Н, Ph); δC (100 МГц; CDCl3)/м.д. 7.62, 8.82 (СН3, Et), 27.54 (CH3, t-Bu),
32.28, 33.92 (CH2, Et), 35.10 (C(CH3)3, t-Bu), 77.44 (C5), 92.83 (C2), 127.52 (o-C, Ph), 127.91 (м-C, Ph), 128.87 (п-C, Ph), 137.29 (и-C, Ph), 172.33 (C4).

-Метил-2,2,5-триэтил-2,5-дигидро-имидазол-1-ол (17а).
Гидроксиамин 17a
получали по аналогии с общей методикой для реакций циклических нитронов с
магнийорганическими соединениями. Раствор 2Н-имидазола 16a в абсолютном эфире прибавляли к
эфирному раствору этилмагнийбромида.. После разложения водой и удаления
растворителя полученную смесь продуктов растирали с гексаном, выпавший осадок
гидроксиамина 17a перекристаллизовывали из гексана. Выход 22%. Буроватые
кристаллы. νmax (KBr)/см-1 3154 (OH), 2982, 2964, 2944, 2922, 2879, 2854 (CH3, CH2), 1652 (C=N); в УФ-спектре поглощение не
наблюдается; δH (200
МГц; CDCl3)/м.д. 0.74 (3H, т, Jт= 6.4 Гц, CH3, Et-C5), 0.81, 0.85 (оба 3H, т, J1,2=
6.6 Гц, 2 ´ CH3, 2 Et-C2), 1.60 (6H, м, 3 ´ CH2, Et-C5, 2 Et-C2), 1.88 (3H, с, CH3, Me-C4), 3.76 (1H, т, Jт=
4.0 Гц, H-C5), 6.55 (1H, уш.
с, ОН); δC (50 МГц; CDCl3)/м.д. 8.22, 9.21, 9.56 (СН3, Et при C2 и C5), 17.19 (CH3, Me-C4), 23.41, 26.61, 31.02 (СН2, Et при C2 и C5), 74.76 (С5), 93.97 (С2),
171.48 (C4).
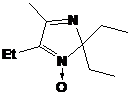
-Метил-2,2,5-триэтил-2Н-имидазол-1-оксид (18a)
Выделенный имидазолин 17a
окисляли диоксидом свинца в хлороформе, согласно общей методике окисления
имидазолинов. После завершения реакции (контролировали методом ТСХ), помимо
целевого кетонитрона 18a,
смесь содержала примеси, не отделимые хроматографически. Поэтому после удаления
избытка окислителя раствор перемешивали с активированным углем, затем промывали
10% водным раствором гидроксида натрия, затем удаляли растворитель, а остаток
сублимировали при пониженном давлении. Однако получить аналитически чистый
образец вещества не удалось. Выход: 90%. Рыже-бурое масло. δH (200 МГц; CDCl3)/м.д. 0.39 (оба 3H, т, Jт=
7.2 Гц, CH3, Et-C2), 1.02 (3H, т, Jт= 7.6 Гц, CH3, Et-C5), 1.76, 1.91 (оба 2H, ABк, JAB= 14.4 Гц, Jк= 7.2 Гц, CH2, Et-C2), 2.20 (3H, с, CH3, Me-C4), 2.43 (2H, к, Jк= 7.6 Гц, CH2, Et-C5).
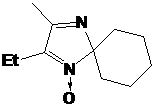
-Метил-2-этил-1,4-диаза-спиро[4.5]дека-1,3-диен-1-оксид (18b)
Этилнитрон 18b получали по аналогии с общей методикой для реакций
циклических нитронов с магнийорганическими соединениями и окисления
соответствующих гидроксиаминов диоксидом свинца. Раствор 2Н-имидазола 16b в
абсолютном эфире прибавляли к эфирному раствору этилмагнийбромида. После
окисления реакционную массу хроматографировали на колонке с силикагелем, элюент
- эфир : гексан = 2 : 1). Полученный с выходом 40%
3-метил-2-этил-1,4-диаза-спиро[4.5]дека-1,3-диен-1-оксид (18b) использовали далее без
дополнительной очистки, νmax (KBr)/см-1
2968, 2940, 2857 (CH3, CH2), 1601, 1518 (C=N); λmax (EtOH)/нм 285 (lg ε
3.85); δH (200 МГц; CDCl3)/м.д. 1.10 (3H, т, Jт=
7.4 Гц, CH3, Et-C2), 1.33 (3H, м, CH2, (CH2)5),
1.53 (5H, м, CH2, (CH2)5),
1.70 (2H, м, CH2, (CH2)5),
2.14 (3H, с, CH3-C3), 2.45 (2H, к, Jк= 7.4 Гц, CH2, Et-C2).
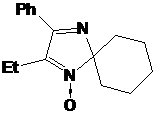
-Этил-3-фенил-1,4-диаза-спиро[4.5]дека-1,3-диен-1-оксид (18a)
К раствору 3.2 г (0.0124 моль) имидазолина 17а в 30 мл хлороформа
прибавили 16 г диоксида свинца и перемешивали в течение 2-х часов. Окислитель
отфильтровали и растворитель отогнали на ротационном испарителе. Остаток
хроматографировали на колонке с силикагелем, элюент - метил-третбутиловый эфир
: гексан=1 : 4). Выход: 1.59 г (53 %). Бурое масло. (Найдено: C, 74,68; H,
8,31; N, 10,34 %. Рассчитано для C16H20N2O: C, 74.97; H, 7.86; N, 10.93 %); νmax (в тонком слое)/см-1 3062 (=C-H, Ph), 2937, 2858 (CH3, CH2), 1564, 1515 (C=N, C=C);
λmax (EtOH)/нм 229 (lg ε 3.90), 269 (lg ε
3.73); δH (200 МГц; CDCl3)/м.д. 1.07 (3H, т, Jк 7.6
Гц, CH3, Et), 1.83 (8Н, м, CH2, (CH2)5), 2.17 (2H, м, CH2, (CH2)5), 2.69 (2H, к, Jк
7.6Гц, CH2, Et), 7.44 (3H, м,
п-, м-H, Ph), 7.67 (2H, м,
о-H, Ph); δC (50 МГц; CDCl3)/м.д. 9.59 (CH3, Et),
17.37, 23.04, 34.81 (CH2, (CH2)5), 24.62 (CH2, Et),
101.46 (C5), 127.49 (o-C, Ph), 128.67 (м-C, Ph), 130.56 (п-C, Ph), 132.72 (и-C, Ph), 140.80 (C2), 166.45 (C3).
Аналогичным образом получали 2,2,5-триэтил-4-фенил-2H-имидазол-1-оксид (18b).

Выход: 89 %. Бурое масло. (Найдено: С, 73.32; Н, 8.01; N, 11.14. Рассчитано для C15H20N2O:
C, 73,74; H, 8,25; N, 11,47 %); νmax (в тонком слое)/см-1 3062 (=C-H, Ph), 2974, 2936, 2880 (CH3, CH2),
1515, 1566 (C=N, С=С); λmax (EtOH)/нм
231 (lg ε 4.01), 273 (lg ε
3.89); δH (400 МГц; CD3OD)/м.д. 0.64 (6H, т, J 7.2 Гц, 2 ´ CH3, 2Et-C2), 1.23 (3H, т, J 7.6 Гц, CH3, Et-C5), 2.05, 2.19 (оба 2H, AВк, JAB 14
Гц, Jк 7.2 Гц, 2 ´ CH2, 2Et-C2), 2.80 (2H, к, J 7.6
Гц, CH2, Et-C5), 7.58 (3H, м, п-, м-H, Ph), 7.80 (2H, м, о-H, Ph);
δC (100 МГц; CD3OD)/м.д. 6.96 (CH3, Et-C2), 10.35 (CH3, Et-C5), 18.02 (CH2, Et-C5), 30.36 (CH2, Et-C2), 104.56 (C2), 128.22 (o-C, Ph), 129.558 (м-C, Ph), 131.71 (п-C, Ph), 132.89 (и-C, Ph), 144.38 (C5), 168.84 (C4).
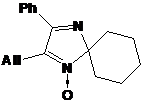
-Аллил-3-фенил-1,4-диаза-спиро[4.5]дека-1,3-диен-1-оксид (18c)
Получали аналогично описанному для 18а. Соединение 18с выделяли
хроматографией на колонке с силикагелем, элюент - диэтиловый эфир : гексан = 1
: 2. Выход: 80 %. Бесцветные кристаллы, Тпл.=54-56оС. (Найдено: С, 76.19; Н,
8.11; N, 10.54. Рассчитано для C17H20N2O:
C, 76,09; H, 7,51; N, 10,44 %); νmax (KBr)/см-1 3078, 3061 (=C-H, Ph), 2950, 2929,
2893, 2848 (CH3, CH2), 1604, 1587, 1566, 1516 (C=N, С=С); λmax (EtOH)/нм 231 (lg ε
4.06), 277 (3.96); δH (400
МГц; CDCl3)/м.д. 1.39 (3H, м, СН2 , (СН2)5), 1.87 (5H, м, СН2, (СН2)5), 2.15 (2H, м, СН2, (СН2)5), 3.44 (2H, м CH2, All),
5.00, 5.10 (оба 1Н, м, =СН2, All),
5.84 (1Н, м, =СН-, All), 7.44 (3Н м, Ph), 7.74 (2Н м, Ph);
δC (100 МГц; CDCl3)/м.д. 23.01, 24.58, 27.81, 34.92 (CH2, (СН2)5, All), 101.78 (C5), 117.44 (=СН2, All),
127.62 (o-C, Ph), 128.57 (м-C, Ph), 130.01 (п-C, Ph), 130,58 (=СН-, All), 132.45 (и-C, Ph), 136.93 (C2), 166.52 (C3).
-трет-Бутил-2,2-диэтил-4-фенил-2H-имидазол-1-оксид (FI-2D).
Соединение получили окислением 5.4 г (19.7 ммоль) гидроксиамина FI-2B
диоксидом марганца согласно общей методике.
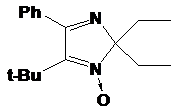
Перекристаллизовали из гексана. Выход: 5.2 г (97 %). Бесцветные
кристаллы, Тпл=95-97оС. (Найдено: С, 74.96; Н, 8.85; N, 10.35. Рассчитано для C17H24N2O: C, 74.96; H, 8.88; N,
10.28 %); νmax
(KBr)/см-1 3029 (=C-H, Ph), 2977, 2959, 2941, 2911, 2879 (CH3, CH2),
1568, 1506 (C=N, С=С); λmax (EtOH)/нм
300 (lg ε
3.85); δH (400 МГц; CDCl3)/м.д. 0.59 (6H, т, Jт=7.3
Гц, CH3, Et), 1.18 (9Н, с, CH3, t-Bu), 2.01, 2.09 (оба 2H, AВк, JAB=13.7
Гц, Jк=7.3 Гц, CH2, Et),
7.33 (2H, м, о-H, Ph), 7.40 (3H, м, п-, м-H, Ph);
δC (100 МГц; CDCl3)/м.д. 6.18 (CH3, Et),
26.69 (CH3, t-Bu), 29.88 (CH2, Et), 33.07 (C(CH3)3,
t-Bu), 103.28 (C2),
127.50 (o-C, Ph), 128.12 (м-C, Ph), 129.14 (п-C, Ph), 136.52 (и-C, Ph), 147.29 (C5), 171.31 (C4).
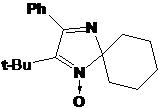
-трет-Бутил-3-фенил-1,4-диаза-спиро[4.5]дека-1,3-диен-1-оксид (18d)
Раствор третбутилмагнийхлорида в 90 мл абсолютного эфира, полученный из
2.6 г (0.108 моль) магниевой стружки и 9.5 мл (0.088 моль) третбутилхлорида,
прибавили по каплям при перемешивании к раствору 4 г (0.0175 моль) 2Н-имидазола
16а в 40 мл сухого ТГФ. Перемешивали 3 ч. Вылили реакционную смесь в холодный
раствор 40 мл ледяной уксусной кислоты в 100 мл диэтилового эфира при
энергичном перемешивании. Разбавили смесь в 2 раза водой, отделили водную фазу
и промыли её 200 мл раствора карбоната натрия с рН= 9-10. Экстракт сушили
сульфатом магния, затем прибавили 23 г (0.264 моль) диоксида марганца и
перемешивали 2 ч. Отфильтровали осадок, раствор упарили. Остаток растерли с
гексаном, образовавшийся осадок отфильтровали. Выход: 3.5 г (70 %). Бесцветные
кристаллы, Тпл.=136-138 оС (гексан). (Найдено: С, 76.01; Н, 8.54; N, 9.84. Рассчитано для C18H24N2O: C,
76,02; H, 8,51; N, 9,85 %); νmax (KBr)/см-1
3047, 3030 (=C-H, Ph), 2980, 2937,
2865, 2847 (CH3, CH2), 1560, 1505 (C=N, С=С); λmax (EtOH)/нм 297 (lg ε
3.85); δH (300 МГц; CDCl3)/м.д. 1.17 (9Н, с, 3 ´ CH3, t-Bu), 1.38 (3H, м, СН2, (СН2)5), 1.87 (5H, м, СН2, (СН2)5), 2.13 (2H, м, СН2 , (СН2)5), 7.35 (2Н, м, о-Н, Ph), 7.41 (3Н, м, м-, п-Н, Ph);
δC (75 МГц; CDCl3)/м.д. 23.09, 24.72, 35.42 (СН2, (СН2)5), 26.91 (CH3, t-Bu), 32.86 (C(CH3)3), 101.33 (C5),
127.77 (o-C, Ph), 128.25 (м-C, Ph), 129.17 (п-C, Ph), 136.71 (и-C, Ph), 144.50 (C2), 169.00 (C3).
-трет-Бутил-1-трет-бутокси-3-фенил-1,4-диаза-спиро[4.5]дека-3-ен (19)
Фильтрат, оставшийся после выделения 2Н-имидазола 18d, упарили, остаток и
хроматографировали на колонке с силикагелем, элюент - эфир : гексан = 1 : 8.
Выход: 0.1 г (1.67 %). Бесцветные кристаллы, Тпл.=88-90оС. (Найдено: С, 77.08;
Н, 10.33; N, 8.04. Рассчитано для C22H34N2O: C,
77.14; H, 10.01; N, 8.18 %); νmax (KBr)/см-1
3057 (=C-H), 2973, 2937, 2854 (CH3, CH2),
1617, 1576 (C=N, C=C);
λmax (EtOH)/нм 238 (lg ε
4.02); δH (300 МГц; CDCl3)/м.д. 1.02 (9H, с, 3 ´ CH3, t-Bu-C2), 1.29 (9H, с, 3 ´ CH3, t-Bu-O), 1.36 (1H, м, CH2, (CH2)5), 1.72 (7H, м, CH2, (CH2)5), 2.01 (2H, м, CH2, (CH2)5), 4.52 (1H, с, H-C2), 7.38 (3H, м, п-, м-H, Ph), 7.71 (2H, м, o-H, Ph); δC (75 МГц; CDCl3)/м.д. 23.31, 24.55, 28.19, 34.04, 37.25 (CH2, (CH2)5), 28.19 (CH3, t-Bu-C2), 30.92 (CH3, t-Bu-O), 34.61 (C(CH3)3, C2-t-Bu), 77.56 (C(CH3)3, O-t-Bu), 87.82 (C2),
98.20 (C5), 128.03 (o-C, Ph), 128.41 (м-C, Ph), 129.34 (п-C, Ph), 136.93 (и-C, Ph), 169.94 (C3).
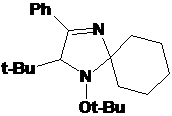
Взаимодействие 3-(4-Хлорфенил)-1,4-диаза-спиро[4.5]дека-1,3-диен-1-оксида
(16e) с
(2-(1,3-диоксолан-2-ил)этил)магнийбромидом

(2-(1,3-диоксолан-2-ил)этил)магнийбромид получали методом сопровождения
по аналогии с методикой, описанной. При этом к 1.64 г (0.068 моль) магниевой
стружки в 23 мл сухого ТГФ прибавили смесь 3.4 мл (0.03 моль) 2-(2-бромоэтил)-1,3-диоксолана,
2.6 мл (0.03 моль) дибромэтана и 4.5 мл сухого ТГФ. Нагревали 20 минут после
окончания прибавления. К раствору магнийорганического соединения прилили по
каплям раствор 4 г (0.015 моль) 2Н-имидазола 16e в 25 мл сухого ТГФ. Перемешивали, поддерживая кипение
растворителя, еще 2 суток. Вылили смесь в 100 мл насыщенного раствора хлорида
аммония, органическую фазу отделили, водную фазу, экстрагировали диэтиловым
эфиром (3 раза по 20 мл). Экстракты объединили, сушили сульфатом магния, осушитель
отфильтровали, растворитель отогнали на ротационном испарителе. Остаток
хроматографировали на колонке с силикагелем, элюент - диэтиловый эфир : гексан
= 1.5 : 1. В результате хроматографии было выделено и охарактеризовано 2 новых
соединения (2-(2-(1,3-Диоксолан-2-ил)этил)-3-(4-хлорфенил)-1,4-диаза-спиро[4.5]дека-1,3-диен-1-оксид
(18e) и
3,3'-бис-(4-Хлорфенил)-2,2’-би-(1,4-диаза-спиро[4.5]децил)-1,3,1’,3’-тетраен)
(20) ) и исходный имидазол 16е.
-(2-(1,3-Диоксолан-2-ил)этил)-3-(4-хлорфенил)-1,4-диаза-спиро[4.5]дека-1,3-диен-1-оксид
(18e)

Выход: 0.3 г (5.4 %). Желтое масло. (Найдено: С, 62.52; Н, 6.29; Cl 10.00; N, 7.35. Рассчитано для C19H23ClN2O3: C, 62.89; H, 6.39; Cl,
9.77; N, 7.72 %); νmax (в тонком слое)/см-1 3076 (=C-H), 2937, 2859 (CH3, CH2), 1600, 1585, 1558, 1514 (C=N, С=С); λmax (EtOH)/нм 297 (lg ε
4.06); δH (400 МГц; CDCl3)/м.д. 1.34 (3H, м, СН2, (СН2)5), 1.85 (5H, м, СН2, (СН2)5), 2.11 (2H, м, СН2, (СН2)5), 1.93 (2H, дт, Jд 4
Гц, Jт 8 Гц, CH2, СН-CH2),
2.82 (2H, т, Jт 8 Гц, CH2-C=N), 3.72 (4H, м, OCH2-CH2O), 4.79 (1H, т, Jт 4 Гц, CH),
7.42, 7.70 (оба 2Н, AA’BB’, J 8.5 Гц, Ar); δC (100 МГц; CDCl3)/м.д.
18.17 (CH2-C=), 23.01, 24.62, 34.64 (СН2, (СН2)5), 28.24 (СН2-СН), 64.71
(ОСН2), 101.24 (C5), 102.98 (CH), 128.86 (o-C, Ar), 129.00 (м-C, Ar), 131.34 (и-C, Ar), 138.68 (Cl-C, Ar), 136.69 (C2),
165.50 (C3).
,3'-бис-(4-Хлорфенил)-2,2’-би-(1,4-диаза-спиро[4.5]децил)-1,3,1’,3’-тетраен)
(20)
Выход: 0.08 г (1.1 %). Бесцветные кристаллы, Тпл=177-179 оС. (Найдено: С,
68.16; Н, 5.82; Cl 14,23; N, 11.26. Рассчитано для C28H28Сl2N4: C, 68,43; H,
5,74; Cl, 14,43; N, 11,40 %); νmax (KBr)/см-1 3059 (=C-H, Ar), 2936, 2854 (CH3, CH2), 1603, 1567, 1530 (C=N, С=С); λmax (EtOH)/нм 275 (lg ε
4.17); δH (400 МГц; CDCl3)/м.д. 1.82 (10H, м, СН2, (СН2)5), 7.24, 7.52 (оба 2Н, AA’BB’, J 8.5
Гц, Ar);
δC (100 МГц; CDCl3)/м.д. 23.75, 25.32, 33.82 (СН2, (СН2)5), 106.64 (C5), 128.35 (o-C, Ar), 129.88 (и-C, Ar), 130.00 (м-C, Ar), 136.61 (C-Сl), 159.24 (C2), 161.95 (C3); M/z. Найдено: 490.17123. Вычислено для C28H28Сl2N4: 490,16909.
,2-Диэтил-3-фенил-1,4-диаза-спиро[4.5]дека-3-ен-1-оксил (22a)
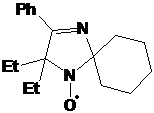
К раствору 1.6 г (0.0063 моль) имидазола 18a в 10 мл сухого серного эфира прибавили по каплям раствор
этилмагнийбромида, полученный из 1.4 г (0.0583 моль) магниевой стружки и 2.8 мл
(0.0375 моль) этилбромида в 34 мл серного эфира. Перемешивали еще 30 минут,
затем прибавили воду до образования вязкой неорганической фазы. Органическую
фазу отделили, неорганическую массу промыли диэтиловым эфиром. Объединённые
эфирные экстракты высушили сульфатом магния. К раствору прибавили 5 г (0.057
моль) диоксида марганца и перемешивали в течение 2-х часов. Осадок
отфильтровывали, растворитель отгоняли на ротационном испарителе. Остаток
хроматографировали на колонке с силикагелем, элюент - диэтиловый эфир : гексан
= 1: 4. Полученный радикал 22а перекристаллизовали из гексана (2 мл). Выход:
0.54 г (30 %). Крупные желтые кристаллы, Тпл.=97-99оС. (Найдено: С, 75.36; Н,
8.61; N, 9.68. Рассчитано для C18H25N2O•:
C, 75.75; H, 8.83; N, 9.82 %); νmax (KBr)/см-1 3060, 3024 (H-C=, Ph),
2978, 2962, 2933, 2856 (CH3, CH2), 1598, 1571 (C=N, С=С); λmax (EtOH)/нм
245 (lg ε 4.19).
Аналогичным образом получали
2,2,5,5-тетраэтил-4-фенил-2,5-дигидро-имидазол-1-оксил (22b).

Полученный радикал чистили хроматографически на колонке с силикагелем,
элюент - метилтретбутиловый эфир : гексан = 1 : 4. Выход: 50 %. Оранжевое
масло. (Найдено: С, 75.48; Н, 9.16; N, 9.98. Рассчитано для C17H25N2O•: C, 74.68; H, 9.22;
N, 10.25 %); νmax (в тонком
слое)/см-1 3059 (H-C=, Ph), 2972, 2939, 2880 (CH3, CH2),
1601, 1572 (C=N, С=С); λmax (EtOH)/нм
244 (lg ε 4.11).
Синтез 2,2-Диэтил-3-фенил-1,4-диаза-спиро[4.5]дека-3-ен-1-оксила (22a) без выделения промежуточных
продуктов
Раствор этилмагнийбромида в 20 мл абсолютного эфира, полученный из 1.42 г
(0.059 моль) магниевой стружки и 1.32 мл (0.018 моль) этилбромида, прибавили по
каплям к раствору 2 г (0.0088 моль) 16а в 20 мл сухого ТГФ. Перемешивали еще 30
минут, затем вылили смесь в охлажденный раствор, состоящий из 14 мл ледяной
уксусной кислоты и 65 мл эфира, при энергичном перемешивании, прилили 200 мл
воды. Органическую фазу отделили, водную - экстрагировали диэтиловым эфиром (3
раза по 30 мл). Объединённые экстракты промыли водным раствором карбоната
натрия с рН=9 (200 мл), высушили сульфатом магния. Перемешивали с 17 г (0.195
моль) диоксида марганца в течение 1 ч. Диоксид отфильтровали, растворитель
отогнали на ротационном испарителе. Полученную массу растворили в 30 мл
абсолютного эфира и прибавили к этому раствору по каплям при перемешивании
раствор этилмагнийбромида в 50 мл абсолютного эфира, полученный из 2.16 г
(0.090 моль) магниевой стружки и 5.8 мл (0.078 моль) этилбромида. Перемешивали
еще 1 ч, затем прибавили 200 мл насыщенного водного раствора хлорида аммония до
образования жидкой водной фазы. Органическую фазу отделили, водную
экстрагировали диэтиловым эфиром (3 раза по 50 мл). Объединённые эфирные
экстракты сушили сульфатом магния. Перемешивали с 12 г (0.138 моль) диоксида
марганца 1 ч. Окислитель отфильтровали, растворитель отогнали на ротационном
испарителе. Полученную массу хроматографировали на колонке с силикагелем,
элюент - хлороформ : гексан = 1 : 2. Выход 1 г (40 %).

-Метил-2,2,5,5-тетраэтил-2,5-дигидро-имидазол-1-оксил (19a)
Реакцию также проводили согласно общей методике. Раствор этилнитрона 18a
прибавляли к раствору этилмагнийбромида. После разложения реакционной массы,
органический слой отделяли и доокисляли избытком диоксида марганца в течение 30
мин. Для выделения НР смесь продуктов хроматографировали на колонке с
силикагелем, элюент эфир - гексан (1 : 2). НР 19 a перекристаллизовывали из
гексана. Выход: 40%.
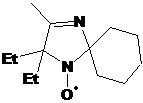
Общий выход: 8 %. Желтоватые кристаллы, Тпл=119-121оС (гексан).
Спектральные характеристики этого соединения совпадают с приведёнными в
литературе [1].
Аналогичным образом получали
2,2-диэтил-3-метил-1,4-диаза-спиро[4.5]дека-3-ен-1-оксил (19b). Выход 51 %. Суммарный выход: 20%.
Оранжевые кристаллы, Tпл=90-93oC (гексан) Спектральные характеристики
этого НР аналогичны приведённым в литературе.
Взаимодействие
2-трет-Бутил-3-фенил-1,4-диаза-спиро[4.5]дека-1,3-диен-1-оксида (18d) с этилмагнийбромидом
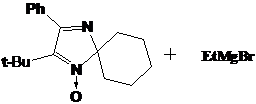
Раствор этилмагнийбромида в 20 мл абсолютного эфира, приготовленный из
0.96 г (0.040 моль) магниевой стружки и 1.4 мл (0.019 моль) этилбромида,
прибавили по каплям к раствору 0.8 г (0.028 моль) 2Н-имидазола (18d) в 15 мл сухого ТГФ. Перемешивали 2
ч при комнатной температуре и еще 4 ч при кипении растворителя. Прибавили воду
до образования вязкой неорганической фазы. Органическую фазу отделили,
неорганическую массу промыли диэтиловым эфиром (2 раза по 10 мл). Объединённые
эфирные экстракты сушили сульфатом магния, осушитель отфильтровали,
растворитель отогнали на ротационном испарителе. Остаток хроматографировали на
колонке с силикагелем, элюент - хлороформ : гексан = 2 : 1. В результате
хроматографии было выделено и охарактеризовано 2 новых соединения:
2-трет-бутил-3-фенил-1,4-диаза-спиро[4.5]дека-1,3-диен (23) и
2-трет-Бутил-3-фенил-2-этил-1,4-диаза-спиро[4.5]дека-3-ен-1-оксил (22c).
-трет-Бутил-3-фенил-2-этил-1,4-диаза-спиро[4.5]дека-3-ен-1-оксил (22c)

Выход: 0.32 г (36.5 %) Желтые кристаллы, Тпл.=77-79оС. (Найдено: С,
77.30; Н, 9.44; N, 8.92.
Рассчитано для C20H29N2O•: C, 76.63; H, 9.33; N, 8.94 %); νmax (KBr)/см-1 3058 (=C-H, Ph), 2937, 2859 (CH3, CH2), 1599, 1573 (C=N, С=С); λmax (EtOH)/нм 236 (lg ε 4.08).
-трет-Бутил-3-фенил-1,4-диаза-спиро[4.5]дека-1,3-диен (23)
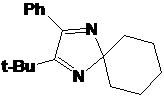
Выход: 0.43 г (57 %) Бесцветные кристаллы, Тпл.=90-93оС. (Найдено: С,
80.57; Н, 9.03; N, 10.46.
Рассчитано для C18H24N2: C, 80.55; H, 9.01; N, 10.44 %); νmax (KBr)/см-1 3061, 3028 (=C-H, Ph), 2969, 2935, 2857 (CH3, CH2), 1572, 1552 (C=N, С=С); λmax (EtOH)/нм 254 (lg ε
3.19); δH (400 МГц; CDCl3)/м.д. 1.14 (9Н, с, 3 ´ CH3, t-Bu), 1.47 (2H, м, СН2, (СН2)5), 1.65 (3H, м, СН2, (СН2)5), 1.85 (5H, м, СН2, (СН2)5), Ph: 7.33 (2Н, м, о-Н, Ph),
7.37 (3Н, м, п-, м-Н, Ph); δC (75 МГц; CDCl3)/м.д.
24,05, 25.62, 34,53 (СН2, (СН2)5), 28.76 (CH3, t-Bu), 34.83 (C(СН3)3), 101.86 (C5), 128.13 (o-, м-C, Ph), 128.59 (п-C, Ph), 136.94 (и-C, Ar), 166.60 (C2), 172.82 (C3).
-Бутил-2-трет-бутил-3-фенил-1,4-диаза-спиро[4.5]дека-3-ен-1-оксил (24)
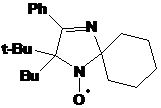
К раствору бутиллития в 40 мл сухого гексана, приготовленному из 1 г
(0.143 моль) мелко нарезанного лития и 6.8 мл (0.074 моль) бутилхлорида,
прибавили по каплям при перемешивании в атмосфере аргона раствор 3.7 г (0.013
моль) 2Н-имидазола (18d) в
20 мл сухого бензола. Перемешивали 1 ч. Приливали воду до растворения
неорганического осадка (220 мл). Органическую фазу отделили, водную
экстрагировали эфиром (2 раза по 15 мл). Экстракт объединили с органической
фазой, барбатировали воздухом 12 ч, сушили сульфатом магния, осушитель
отфильтровали, раствор упарили. Остаток растерли с гексаном, охладили и
образовавшийся осадок отфильтровали. Выход: 3.69 (83 %). Желтые кристаллы, Тпл.
=107-109оС. (Найдено: С, 76.97; Н, 9.30; N, 8.20. Рассчитано для C22H33N2O•: C, 77.37; H, 9.74; N, 8.20
%); νmax (KBr)/см-1 3055 (=C-H, Ph), 2969, 2937, 2862, 2852 (CH3, CH2), 1573, 1594 (C=N, С=С); λmax (EtOH)/нм 237 (lg ε 4.07).
Взаимодействие 5-трет-бутил-2,2-диэтил-4-фенил-2H-имидазол-1-оксида (FI-2D) с бутиллитием.
Реакцию проводили в атмосфере аргона. К смеси 13 мл 2.6 М раствора
бутиллития в толуоле и 10 мл сухого бензола прибавили по каплям раствор 1.5 г
(5.5 ммоль) 2H-имидазол-1-оксида FI-2D в 15 мл сухого бензола. Через 4 ч
реакционную массу осторожно погасили водой до формирования органического и
водного слоев, после чего продолжали энергично перемешивать еще 2 ч.
Органический слой отделили, водный - экстрагировали диэтиловым эфиром (1 раз,
20 мл). Объединенный экстракт сушили сульфатом магния, растворитель удалили при
пониженном давлении. 0.3 г полученного остатка хроматографировали на пластине с
окисью алюминия, элюент: этилацетат - гексан (1 : 8). В результате
хроматографии были выделены 2 фракции: I-291A и I-291B. Фракцию I-291A
хроматографировали дважды на пластине с окисью алюминия, элюент: этилацетат :
гексан = 1 : 100, в результате чего был выделен
5-трет-бутил-5-бутил-4-фенил-2,2-диэтил-2,5-дигидро-имидазол (I-291-A1). Из
фракции I-291B перекристаллизацией из гексана был выделен
4-трет-бутил-5-бутил-5-фенил-2,2-диэтил-2,5-дигидроимидазол-3-оксид (I-291-B1).
-трет-Бутил-5-бутил-4-фенил-2,2-диэтил-2,5-дигидро-имидазол (I-291-A1)
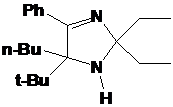
Выход: 95 мг (предположительно из ~200-300 мг реакц. смеси) (%).
Бесцветное масло. (Найдено: С, 80.96; Н, 11.12; N, 8.58. Рассчитано для C21H34N2: C, 80.20; H, 10.90; N, 8.91
%); νmax (в тонком слое)/см-1 3369 (N-H), 3060 (=C-H, Ph), 2961, 2874 (CH3, CH2), 1623 (C=N); в УФ-спектре
поглощения не наблюдается; δH (600 МГц; CDCl3)/м.д.
0.97 (3Н, т, Jт=7.3 Гц, СН3, n-Bu), 0.95, 1.02 (оба 3H, т, Jт=7.5 Гц, CH3, Et), 0.98 (9H, с,
CH3, t-Bu), 1.40, 1.44 (2H, AB6, JAB=13.0 Гц, J6=7.3 Гц, СН2-СН2-СН3, n-Bu), 1.56 (2H, м, СН2-СН2-СН3, n-Bu), 1.76 (1H, м,
CH2-C5, n-Bu), 2.40 (1H, ддд, J1=14.0 Гц, J2=11.2 Гц, J3=4.3 Гц, CH2-C5, n-Bu),
1.46, 1.75 (2H, ABк, JAB=13.0 Гц, Jк=7.5 Гц, CH2, Et), 1.74, 1.82 (2H, ABк,
JAB= 14.2 Гц, Jк= 7.5 Гц, CH2, Et), 1.91 (1H, уш. с, N-H), 7.20 (1Н, м, п-Н, Ph), 7.29 (2Н, м, м-Н, Ph), 7.34 (2Н, м, о-Н, Ph);
δC (100 МГц; CDCl3)/м.д. 8.90, 8.97 (CH3, Et), 14.10 (CH3, n-Bu),
23.22, 27.58, 32.01, 32.27, 36.45 (CH2, Et, n-Bu), 30.71 (CH3, t-Bu), 36.06
(C(CH3)3, t-Bu), 78.19 (C5), 91.46 (C2), 126.73 (п-C, Ph), 127.46 (о-C, Ph), 128.12 (м-C, Ph), 145.03 (и-C, Ph), 180.98 (C4).
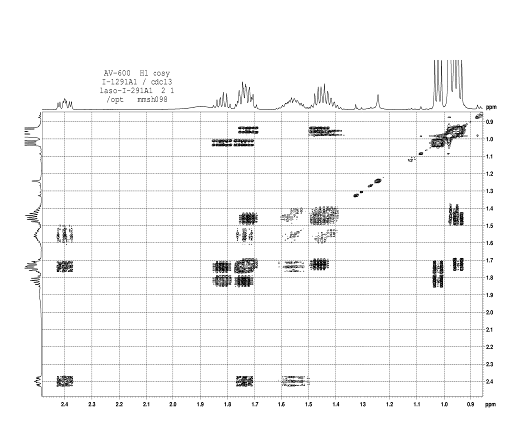
Рис. х
-трет-Бутил-5-бутил-5-фенил-2,2-диэтил-2,5-дигидроимидазол-3-оксид (I-291-B1)
Перекристаллизовали из гексана. Выход: 50 мг (%).Бесцветные кристаллы,
Тпл=79-81оС. (Найдено: С, 76.53; Н, 10.68; N, 8.48. Рассчитано для C21H34N2O: C, 76.31; H, 10.37; N, 8.48
%); νmax (KBr)/см-1 3339 (N-H), 3092, 3070 (=C-H, Ph), 2955, 2873 (CH3, CH2), 1551 (C=N);
λmax (EtOH)/нм 240 (lg ε 4.08); δH (400 МГц; CDCl3)/м.д. 0.97, 0.98 (оба 3H, т, Jт=7.5 Гц, CH3, Et),
0.99 (3Н, т, Jт=7.3 Гц, СН3, n-Bu), 1.14 (9H, с, CH3, t-Bu), 1.46 (2H, м, СН2-СН2-СН3, n-Bu), 1.64, 1.77 (оба 1H, м, СН2-СН2-СН3, n-Bu), 1.86 (1H, м, CH2-C5, n-Bu), 2.39 (1H, ддд, J1=14.3 Гц,
J2=11.2 Гц, J3=3.7 Гц, CH2-C5, n-Bu), 1.69, 1.87 (2H, ABк, JAB=13.8 Гц, Jк=7.5
Гц, CH2, Et), 1.92, 2.03 (2H, ABк, JAB=13.7 Гц, Jк=7.5 Гц, CH2, Et), 2.14 (1H,
уш. с, N-H), 7.27 (1Н, м, п-Н, Ph),
7.35 (2Н, м, м-Н, Ph), 7.38 (2Н, м,
о-Н, Ph);
δC (100 МГц; CDCl3)/м.д. 8.16, 8.20 (CH3, Et), 14.06 (CH3, n-Bu),
23.03, 27.65, 28.60, 31.12, 37.24 (CH2, Et, n-Bu), 25.68 (CH3, t-Bu), 33.76
(C(CH3)3, t-Bu), 71.06 (C5), 92.25 (C2), 127.10 (о-C, Ph), 127.63 (п-C, Ph), 128.52 (м-C, Ph), 145.08 (и-C, Ph), 151.62 (C4).
8.2 Имидазолидиновые НР
,2-Диэтил-4-метил-3-фенил-1,4-диаза-спиро[4.5]декан-1-оксил (29а)
К 0.38 г (0.0013 моль) имидазолинового радикала 22а прилили 4 мл (0.032
моль) диметилсульфата и перемешивали при нагревании (~ 60оС) в течение 3 суток.
Реакционную массу вылили на чашку Петри и оставили до высыхания. Остаток
растворили в 20 мл этанола, присыпали к раствору гидрокарбонат натрия до рН
6-7, затем присыпали несколькими порциями 0.2 г (0.0053 моль) боргидрида
натрия, перемешивали 20 мин. Спирт отогнали на ротационном испарителе, остаток
растворили в 250 мл воды и экстрагировали эфиром (3 раза по 20 мл). Экстракт
сушили карбонатом натрия, осушитель отфильтровали, растворитель отогнали на
ротационном испарителе. Остаток хроматографировали на колонке с силикагелем,
элюент - гексан. Выход: 0.07 г (17.5 %). Желтое масло. (Найдено: C, 75.43; H, 9.69; N,
9.67. Рассчитано для C19H29N2O•: C, 75.70; H, 9.70; N,
9.29 %); νmax (в
тонком слое)/см-1 3063, 3028 (=С-Н, Ph), 2935, 2852, (CH3, CH2), 2799 (N-CH3); в
УФ-спектрах поглощения не наблюдается.

-Метил-4-фенил-2,2,5,5-тетраэтил-имидазолидин-1-оксил (29b)
К раствору 0.37 г (0.0014 моль) 22b в сухом серном эфире прилили 0.38 мл (0.0040 моль)
диметилсульфата и оставили на 15 минут. Раствор профильтровали, эфир отогнали
на ротационном испарителе. Температуру бани повысили до 40оС и продолжали
перемешивание в вакууме на ротационном испарителе еще 30 минут. Образовавшуюся
массу нагревали до 50оС, контролируя ход реакции с помощью ТСХ (Sorbfil, элюент эфир-гексан 1:5). После
исчезновения исходного соединения (около суток) несколько раз промыли массу
эфиром. Т.к. растирание массы с сухим эфиром не привело к кристаллизации, её
растворили в этаноле (20 мл) охладили до 0оС и к холодному раствору при
перемешивании несколькими порциями прибавили боргидрид натрия (0.17 г, 0.0044
моль). Через 1 ч спирт отогнали на ротационном испарителе и приливали воду до
растворения остатков неорганического осадка. Нитроксильный радикал 28b экстрагировали серным эфиром (3 раза
по 10мл), экстракт сушили карбонатом натрия, осушитель отфильтровали, эфир
отогнали на ротационном испарителе. Остаток хроматографировали на колонке с
силикагелем, элюент метилтретбутиловый эфир : гексан = 1 : 8. Выход: 0.14 г (36
%). Желтое масло. (Найдено: С, 73.99; Н, 9.87; N, 9.34. Рассчитано для C18H29N2O•: C, 74.69; H, 10.10;
N, 9,68 %); νmax (в тонком
слое)/см-1 3088, 3063, 3028 (H-C=, Ph), 2969, 2937, 2880, 2856 (CH3, CH2),
2800 (CH3-N).
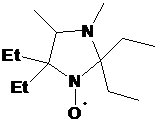
Получение 3,4-Диметил-2,2,5,5-тетраэтил-имидазолидин-1-оксилa (29d) проводили согласно методике, описанной в работе [1], выход
и спектральные характеристики полученного радикала соответствуют литературным
данным.
Аналогичным образом получали
3,4-диметил-2,2-диэтил-1,4-диаза-спиро[4.5]декан-1-оксил (29c). Выход: 90 %. Желтое масло. Cпектральные характеристики
полученного радикала соответствуют литературным данным.
-метил-3-(3-хлор-2-оксопропилиден)-2,2-диэтил-1,4-диаза-спиро[4.5]декан-1-оксил
(4)
К имидазолиниевой соли 3 (1 г, 2.87 ммоль) приливали эфир (10 мл) и
водный раствор NaHCO3 (10 мл).
При перемешивании в реакционную смесь присыпали твердый NaHCO3 небольшими порциями до прекращения
выделения газа и, пока гидрокарбонат не перестал растворяться. Органическую
фазу отделяли, водную - экстрагировали эфиром (2 раза по5 мл), затем эфирные
растворы объединяли и сушили карбонатом натрия. Растворитель удаляли при
пониженном давлении. Остаток (0.93 г, енамин 3а) растворяли в абсолютном эфире,
прибавляли к раствору триэтиламин (2 мл, 14.30 ммоль) и охлаждали смесь до 0оC
на ледяной бане. Затем приливали по каплям смесь хлорацетилхлорида (0.22 мл ,
2.80 ммоль) и 2 мл абсолютного эфира. После завершения реакции (по ТСХ, Sorbfil, хлороформ - ЧХУ 1 : 1) реакционную
смесь промывали водой и сушили карбонатом натрия. Растворитель удаляли при
пониженном давлении, остаток хроматографировали на колонке с силикагелем,
элюент хлороформ - гексан 1 : 1. Выход: 0.48 г (56% из соли 3).
νmax (KBr)/см-1
2965, 2930, 2860 (СН2, СН3), 1653, 1536 (О=С-С=С). Найдено: С 60.73, Н 8.21, Cl, 10.88, N 8.73; вычислено для C16H26ClN2O2: C, 61.23; H,
8.35; Cl, 11.30; N, 8.93.
4-метил-3-(2-гидроксипропил)-2,2-диэтил-1,4-диаза-спиро[4.5]декан-1-оксил
(4а)

К охлажденному до ~ -10оС раствору енаминокетона 4 (0.48 г, 1.60 ммоль) в
этаноле (3 мл) присыпали боргидрид натрия (0.10 г, 2.60 ммоль) несколькими
порциями. Перемешивали 30 мин, затем растворитель отгоняли при пониженном
давлении, к остатку приливали воду (5 мл) и экстрагировали эфиром (3 раза по 3
мл). Экстракт промывали соленой водой и сушили карбонатом натрия. Растворитель
отгоняли при пониженном давлении. Остаток перекристаллизовывали из гексана.
Выход: 0.37 г (86%)
Желтоватые кристаллы. Тпл= 37-42оС νmax (KBr)/см-1 3427 (уш. О-Н), 2937, 2859, 2792 (СН2, СН3).
Найдено: С 67.89, Н 11.13, N
9.98; вычислено для C16H31N2O2: C,
67.80; H, 11.02; N, 9.88. λmax (EtOH)/нм
235 (lg ε 3.29).
4-метил-3-(3-метокси-2,3-диоксопропилиден)-2,2-диэтил-1,4-диаза-спиро[4.5]декан-1-оксил
(5)
Енамин 3а получали из имидазолиниевой соли 3 аналогично описанному в
методике синтеза енаминокетона 4. Раствор енамина 3а (0.66 г, 2.78 ммоль) и
триэтиламина (3.9 мл, 26.60 ммоль) в абсолютном эфире (16 мл) прибавляли по
каплям к охлажденному на снежной бане до 0оС раствору оксалилхлорида (1.3 г,
10.3 ммоль) в абсолютном эфире (23 мл). По мере выпадения из раствора осадка
интенсивность перемешивания увеличивали. Выдерживали на снежной банееще 30 мин,
затем приливали к смеси метанол (7.7 мл) и охлаждение убирали. Перемешивали еще
1 ч. Осадок отфильтровывали и промывали эфиром, эфирный раствор сохраняли.
Затем промывали осадок этилацетатом, большую часть растворителя удаляли при
пониженном давлении, а к оставшемуся раствору приливали немного сухого эфира.
Выпавший осадок отфильтровывали, фильтрат объединяли с первым - сохраненным -
эфирным раствором, удаляли растворитель в вакууме, остаток хроматографировали
на колонке с силикагелем, элюент хлороформ - ЧХУ 1 : 1. Выход: 0.45 г (50 %).

Желтые кристаллы. Тпл = 145-150оС (этилацетат - гексан 1 : 40). νmax (KBr)/см-1 2960, 2863 (СН2, СН3), 1741, 1650, 1633, 1539
(С=С-(СО)-С=О). Найдено: С 62.34, Н 8.44, N 8.48; вычислено для C17H27N2O4: C, 63.13; H,
8.41; N, 8.66. λmax (EtOH)/нм 339 (lg ε 4.34).
-метил-3-(3-гидрокси-2-оксопропилиден)-2,2-диэтил-1,4-диаза-спиро[4.5]декан-1-оксил
(5а)

К охлажденному до ~ -10оС раствору соединения 5 (0.26 г, 0.81 ммоль) в
этаноле (20 мл) при энергичном перемешивании быстро присыпали боргидрид натрия
(0.32 г, 8.4 ммоль). Реакцию контролировали методом ТСХ (Sorbfil, хлороформ). Перемешивали 30 мин -
до исчезновения исходного соединения в реакционной смеси, затем приливали 20 мл
ацетона, чтобы вывести из реакции остаток боргидрида. Удаляли растворитель в
вакууме, остаток растворяли в воде (10 мл), экстрагировали раствор эфиром (3
раза по 5 мл), сушили экстракт сульфатом натрия. Удаляли растворитель при
пониженном давлении. Выход: 0.17 г (71%).
Желтые кристаллы. Тпл = 167-180оС (этилацетат - гексан 1 : 2). νmax (KBr)/см-1 3436 (О-Н), 2930, 2860 (СН2, СН3), 1641, 1547
(С=С-С=О). Найдено: С 64.11, Н 9.67, N 9.19; вычислено для C16H27N2O3: C, 65.06; H,
9.21; N, 9.48. λmax (EtOH)/нм 305 (lg ε 4.32).
-Гидрокси-2,4-диметил-5,5-диэтил-2-(2-карбоксиэтил)-2,5-дигидро-1Н-имидазол
(18а).

Смесь 3-гидроксиамино-3-этилпентанон-2 гидрохлорида (1 г, 5.5 ммоля),
левулиновой кислоты (2 г, 17 ммоль), ацетата аммония (2 г, 26 ммоль) и 5 мл
метанола перемешивали под аргоном 4 сут.. Смесь разбавляли насыщенным водным
раствором NaCl в 5 раз и экстрагировали хлороформом.
Объединённые экстракты промывали насыщенным водным раствором NaCl и сушили MgSO4. Хлороформ отгоняли при пониженном давлении, остаток
растирали в смеси диэтиловый эфир - гексан 1:1, выпавший осадок отфильтровывали
и перекристаллизовавали из смеси гексан - этилацетат 1:1. Выход 916 мг (69%),
бесцветные кристаллы, Тпл= 179-183 єC. ИК (KBr): n= 3187уш, 2926уш, 1706, 1654, 1462уш, 1375, 1303,
1263, 1220, 1169, 1107, 1038, 945, 888 cм-1. ПМР δH(200 MГц; CDCl3): 0.93 (6Н м, 2´СН3 Et), 1.78 и 2.02 (3H и 1Н, оба м 2´СН2 Et), 1.39 (3H, c 2-СН3), 1.57 и
2.02 (оба 1Н м, 2-СН2), 2.35 (2Н м, СН2СО), 8.90 (2Н уш, ОН и СООН). 13С ЯМР δC(100 MHz; CDCl3-СD3OD): 7.48 (СН3 Et),
9.15 (4-СН3), 21.08 (2-СН3), 24.47 (СН2СО), 28.05 и 28.69 (СН2 Et), 35.48 (2-СН2), 77.63 (С5), 90.00
(С2), 175.94 (COOH), 176.80 (C=N). Найдено, %: C, 59.42; H, 9.15; N,
11.32; вычислено для C12H22N2O3: C, 59.48; H, 9.15; N, 11.56.
,4-Диметил-5,5-диэтил-2-(2-метоксикарбонилэтил)-2,5-дигидро-1Н-имидазол-1-оксил
(19а). К 100 мл 0.1 M раствора
диазометана в трет-бутилметиловом эфире прибавляли соединение 18а (0.9 г, 3.7
ммоля). Суспензию перемешивали до образования гомогенного раствора и оставляли
на 16 ч, после чего добавляли MnO2
(2 г, 23 ммоля) и перемешивали ещё 1 ч. Окислитель отфильтровывали,
растворитель отгоняли при пониженном давлении, остаток хроматографировали на
колонке с силикагелем, элюент - смесь хлороформ - гексан 1:1. Выход 0.8 г
(85%), оранжевое масло. ИК (в тонком слое): n= 2967, 2879, 1739, 1638, 1458, 1437, 1380, 1290, 1197, 1175,
1073, 990, 879, 845, 785, 747 cм-1.
Найдено, %: C, 61.42; H, 9.08; N,
10.82; вычислено для C13H23N2O3: C, 61.15; H, 9.08; N, 10.97.
,4-диметил-2-(2-карбоксиэтил)-5,5-диэтил-2,5-дигидро-1Н-имидазол-1-оксил,
натриевая соль (8а)
Оранжевые
кристаллы. νmax (KBr)/см-1
2969, 2938, 2881 (СН2, СН3), 1639, 1609, 1569 (С=О). Найдено: С 53.40, Н 7.71, N
9.88; вычислено для C12H20N2NaO3: C, 54.74; H,
7.66; N, 10.64. λmax (EtOH)/нм
242 (lg ε 3.07).
-(2-метоксикарбонилэтил)-2,3,4-триметил-5,5-диэтил-имидазолидин-1-оксил
(10)
Метод
A
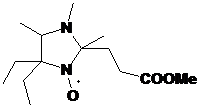
К
раствору имидазолина 8 (0.42 г, 1.65 ммоль) в 5 мл сухого диэтилового эфира
прибавляли диметилсульфат (0.23 мл, 2.48 ммоль). Диэтиловый эфир отгоняли при
пониженном давлении, остаток перемешивали при температуре 40-45 °С при пониженном давлении в течение 1 ч, затем растирали с сухим
диэтиловым эфиром. Образовавшийся осадок метилсульфата 9 промывали сухим
диэтиловым эфиром, растворяли в этаноле и к образовавшемуся раствору при
перемешивании разом прибавляли NaBH4 (0.20 г, 5.26 ммоль). За ходом реакции следили по
ТСХ (Сорбфил, эфир-гексан 1:2). После завершения реакции этанол отгоняли при
пониженном давлении, остаток разбавляли водой (10 мл) и экстрагировали
хлороформом (2 раза по 5 мл). Экстракт сушили Na2СO3,
растворитель отгоняли при пониженном давлении. Остаток хроматографировали на
колонке с силикагелем, элюент гексан- эфир 2:1. Выход ((2R(2S),4R(4S))-
2-(2-метоксикарбонилэтил)-2,3,4-триметил-5,5-диэтил-имидазолидин-1-оксила (10A)
0.3 г (67%)
Метод
B
Аналогично
описанному в методе А из имидазолина 8 получали метилсульфат 9. К раствору 9
(5.0 г, 18.5 ммоль) в этаноле (50 мл) прибавляли маленькую порцию (не более 0.1
г, 2.6 ммоль) боргидрида натрия и охлаждали до комнатной температуры, затем
прибавляли следующую порцию (также не более 0.1 г) боргидрида. В среднем
боргидрид вбрасывали в реакционную смесь с частотой раз в 300 мин. После
завершения реакции смесь обрабатывали аналогично описанному в методе А.
Хроматографией на колонке с силикагелем, элюент хлороформ, из смеси выделяли 2
соединения: ((2R(2S),4R(4S))-
2-(2-метоксикарбонилэтил)-2,3,4-триметил-5,5-диэтил-имидазолидин-1-оксил (10A)
(выход: 4.0 г, 80%) и ((2R(2S),4S(4R))-2-(2-метоксикарбонилэтил)-2,3,4-триметил-5,5-диэтил-имидазолидин-1-оксил
(10B) (выход: 0.5 г, 10%).
((2R(2S),4R(4S))-2-(2-метоксикарбонилэтил)-2,3,4-триметил-5,5-диэтил-имидазолидин-1-оксил
(10A)
Оранжевое
масло. νmax
(в тонком слое)/см-1 2977, 2882, 2798 (СН2, СН3), 1739 (С=О). Найдено: С 62.19,
Н 10.04, N 10.10; вычислено для С14Н27N2O3: C
61.96, H 10.03, N 10.32.
((2R(2S),4S(4R))-2-(2-метоксикарбонилэтил)-2,3,4-триметил-5,5-диэтил-имидазолидин-1-оксил
(10B)
Оранжевое
масло. νmax
(в тонком слое)/см-1 2976, 2941, 2881, 2849, 2797 (СН2, СН3), 1740 (С=О).
Найдено: С 61.02, Н 10.00, N 10.20; вычислено для С14Н27N2O3: C
61.96, H 10.03, N 10.32.
(2R(2S);4R(4S))-2-(2-карбоксиэтил)-2,3,4-триметил-5,5-диэтил-имидазолидин-1-оксил
(11А)
К
раствору НР 10А (885 мг, 3.3 ммоля) в 5 мл этанола прибавляли раствор NaOH
(200 мг, 5 ммоль) в 2 мл воды. Через 1 ч, после завершения реакции по ТСХ
(Сорбфил, элюент - хлороформ) через раствор пропускали ток СО2, после чего
раствор экстрагировали эфиром, сушили сульфатом натрия, растворитель удаляли
при пониженном давлении. Осадок перекристаллизовывали из гексана. Выход - 839
мг (86%).

Желтые
кристаллы. Тпл= 94-97оС. Найдено: С 60.95, Н 9.80, N 10.85;
вычислено для C13H25N2O3: C, 60.67; H, 9.79; N, 10.89. νmax (KBr)/см-1
3189 (уш. О-Н), 2977, 2945, 2879, 2804 (СН2, СН3), 1732 (С=О).
((2R(2S),4R(4S))-2-(2-карбоксиэтил)-2,3,4-триметил-5,5диэтилимидазолидин-1-оксил,
натриевая соль

К
раствору НР 10А (885 мг, 3.3 ммоля) в 5 мл этанола прибавляли раствор NaOH
(200 мг, 5 ммоль) в 2 мл воды. Через 1 ч, после завершения реакции по ТСХ
(Сорбфил, элюент - хлороформ) в раствор добавляли 500 мг NaHCO3 и
перемешивали 3 ч, затем раствор упаривали до суха при пониженном давлении, к
остатку приливали этанол, неорганический осадок отфильтровывали. Органический
раствор упаривали и растирали с сухим эфиром.
Оранжевые
кристаллы. ИК (КBr), νmax (KBr)/см-1 2976, 2939, 2799 (СН2, СН3), 1566 (уш С=О).
Найдено: С 52.49, Н 8.93, N 8.83,
золы 10.41; вычислено для С13Н24N2O3Na*Н2О: C 52.51, H 8.81, N 9.42.
8.3 НР на базе 4Н-имидазол-3-оксидов
Взаимодействие 1,2-гидроксиаминокетонов:
3-(гидроксиамино)-3-этилпентанона-2 и 3-(гидроксиамино)-3-метилбутанона-2 с
триметилуксусным альдегидом.
Метод А
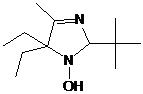
К раствору 100 ммоль гидрохлорида 1,2-гидроксиаминокетона в 10 мл
метанола прибавляли при перемешивании 2.5 мл 25% водного раствора аммиака и 100
ммоль альдегида. Образовавшуюся суспензию перемешивали 24 ч, после чего
реакционную массу экстрагировали 3 раза эфиром (по 5 мл). Объединенную
органическую фазу сушили сульфатом магния. Осушитель отфильтровывали, фильтрат
упаривали при пониженном давлении. Остаток растирали с гексаном, помещали в
холодильник (0 °С)
на 10-12 ч. Выпавший осадок отфильтровывали, перекристаллизовывали из гексана.
2-трет-Бутил-4-метил-5,5-диэтил-2,5-дигидро-имидазол-1-ол (14) Выход: 9%.
Бесцветные кристаллы, Тпл.=130-131оС. (Найдено: С, 68.03; Н, 11.84; N, 13.41. Рассчитано для C12H24N2O: C,
67.88; H, 11.39; N, 13.19 %); νmax (KBr)/см-1
3218 (OH), 2971, 2955, 2917, 2880 (CH3, СН2), 1654 (C=N); λmax (EtOH)/нм не поглощает; δH (300 МГц; CDCl3)/м.д.: 0.88, 0.89 (оба 3H, т, Jт=7.4
Гц, 2CH3, Et), 0.98 (9Н, с, 3СН3, t-Bu), 1.30, 2.13
(оба 1Н, АВк, JАВ=14.5, Jк=7.3 Гц, СН2, Et),
1.51, 1.57 (2Н, АВк, JAB=14.6,
Jк=7.4 Гц, СН2, Et), 1.91 (3Н, д, Jд=2.1 Гц, СН3 при С4), 4.43 (1Н, к, Jк=2.1 Гц, Н-С2), 4.60 (1H, ушир. с, ОН); δC (75 МГц; CDCl3)/м.д. 8.79, 9.63 (СН3, Et), 16.90 (СН3 при С4), 25.93, 30.37 (СН2, Et), 26.22 (СН3, t-Bu), 35.39 (С(СН3)3), 79.44 (С5), 100.10 (С2-Н), 174.70 (С4).
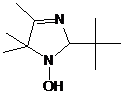
-трет-Бутил-4,5,5-триметил-2,5-дигидро-имидазол-1-ол (17). Выход: 14%.
Бесцветные кристаллы, Тпл.=150-154оС. (Найдено: С, 65.08; Н, 10.76; N, 15.34. Рассчитано для C10H20N2O: C,
65.18; H, 10.94; N, 15.20 %); νmax (KBr)/см-1
3169 (OH), 2960, 2927, 2866 (CH3), 1648 (C=N);
λmax (EtOH)/нм не поглощает; δH (300 МГц; CDCl3)/м.д. 0.86 (9Н, с, 3СН3, t-Bu), 1.05, 1.13
(оба 3H, с, 2CH3 при С5), 1.855 (3Н, д, Jд=2.1 МГц, СН3 при С4), 3.73 (1H, ушир. с, ОН), 4.13 (1Н, к, Jк=2.1 МГц, Н-С2; δC (100 МГц; CDCl3, CD3OD)/м.д. 14.41 (СН3 при С4), 16.38,
23.74 (СН3 при С5), 24.83 (СН3, t-Bu), 33.51 (С(СН3)3), 71.94 (С5), 96.55
(С2-Н), 178.64 (С4).

Для выделения 6-трет-бутил-4-метил-3,3-диэтил-3,6-дигидро-2H-1,2,5-оксадиазина (15) фильтрат
после выделения соединения окисляли 20 мин в присутствие 10 г диоксида свинца.
Отфильтровывали окислитель, отгоняли растворитель при пониженном давлении.
Остаток хроматографировыли на колонке с силикагелем, элюент: хлороформ : гексан
= 4 : 1. Выход: 55%. Бесцветные кристаллы Тпл.=41-43оС. Масс-спектр (Система
капиллярного напуска, Т = 140є С), m/z (Iотн(%)): 212 [M]+
(3.5); 181 [M-NOH]+ (33); 166 [M-NOH-CH3]+ (21); 124 [M-NOH-t-Bu]+ (100). Найдено: m/z 181.18464 [M-NOH]+. C12H23N. Вычислено: M-NOH = 181.18304. νmax (KBr)/см-1:
1662 (С=N).
δH (300 МГц; CDCl3)/м.д.: 0.80 и 0.91 (оба т, по 3H, СH3, Et, J = 7.5); 0.91 (с, 9H, t-Bu); 1.40 и 1.55 (AB к, CH2, JAB = 15.0, Jк = 7.5); 1.54 и 1.78 (AB к,
CH2, JAB = 14.5, Jк = 7.5); 1.91 (д, 3H, Me, J = 2.1); 4.43 (к, 1H, CH, J = 2.1). δC (75 МГц; CDCl3)/м.д.: 7.8 и 8.5 (СН3, Et); 21.9 (Me);
25.2 (CH3, t-Bu); 26.2 и 30.4 (CH2); 35.2 (C(Me)3); 61.8
(С(Ме)2); 96.2 (CH); 169.0 (C=N).

Для выделения 2,2,3,5,5,6-гексаметил-2,5-дигидропиразина (18) водный
раствор после экстракции 2-трет-бутил-4,5,5-триметил-2,5-дигидро-имидазол-1-ола
(17) упаривали досуха при пониженном давлении, остаток растирали со смесью
хлороформ - метанол 10 : 1, осадок отфильтровывали. Раствор сушили MgSO4 и упаривали при пониженном
давлении. Остаток перекристаллизовывали из хлороформа. Выход 53%. Спектральные
характеристики соединения 6 идентичны характеристикам заведомого образца.
Метод В
К раствору 100 ммоль гидрохлорида 1,2-гидроксиаминокетона в 10 мл
метанола прибавляли при перемешивании 3 г ацетата аммония и 100 ммоль
альдегида. Через 5-7 ч реакционную массу разбавляли водой в 2 раза и помещали
на 10-12 ч в холодильник, выпавший осадок имидазолина отфильтровывали,
промывали водой и высушивали.
Дигидропиразин 18 выделяли из водного фильтрата способом, описанным в
методе А.
При конденсации 3-(гидроксиамино)-3-этилпентанона-2 (13а) с
триметилуксусным альдегидом получен имидазолин 14 с выходом 78.5 %.
При конденсации 3-(гидроксиамино)-3-метилбутанона-2 (13b) с триметилуксусным альдегидом
получены имидазолин 17 (Выход: 7 %) и 2,5-дигидропиразин 18 (Выход: 90 %).
-трет-Бутил-4-метил-2-нитрозо-3,3-диэтил-3,6-дигидро-2H-1,2,5-оксадиазин (16)
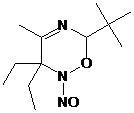
К раствору 2.73 г (12.9 ммоль) соединения 15 в 10 мл четыреххлористого
углерода прилили .5 мл (36 ммоль) триэтиламина. 6 мл (58.5 ммоль)
изопропилнитрита прибавляли порциями по 0.5-1.5 мл в течение 5 ч. Как и при
нитрозировании имидазолина 14 реакционный сосуд накрывали колпаком. Смесь
выдерживали 12 ч. Затем отгоняли растворитель при пониженном давлении, остаток
растворяли в 10 мл эфира и 3 раза промывали водой (по 5 мл) для удаления солей
триэтиламина. Органический раствор сушили сульфатом магния. Осушитель
отфильтровывали, фильтрат упаривали. Остаток перекристаллизовывали из гексана.
Выход: 2.56 г (82 %), желтоватые кристаллы, Тпл. 52-53оС. Найдено (%): С,
60.01; Н, 10.02; N, 17.45. C12H23N3O2. Вычислено (%): С, 59.72; H, 9.61; N, 17.41, νmax (KBr)/см-1: 1666 (С=N). δH (200 МГц; CDCl3)/м.д.: 0.85 и 0.86 (оба т, по 3H, СH3, Et, J = 7.5); 0.99 (с, 9H, t-Bu); 2.00 (м, 3H, СH2, Et) и
2.60 (д к, CH2, Jд = 14.7, Jк = 7.5); 2.05 (д, 3H, Me, J = 2.0); 4.63 (к,
1H, CH, J
= 2.0). δC (75 МГц; CDCl3)/м.д.: 8.0 и 8.9 (СН3, Et); 20.9 (Me);
24.7 (CH3, t-Bu); 28.0 и 32.3 (CH2); 35.7 (C(Me)3); 74.0
(С(Ме)2); 98.3 (CH); 164.5 (C=N).
-трет-Бутил-5-метил-4,4-диэтил-4H-имидазол-3-оксид (19)
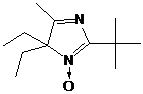
К раствору 1.92 г (9.1 ммоль) имидазолина 14 в 15 мл хлороформа,
прибавляли 5 г диоксида свинца и перемешивали до исчезновения исходного
соединения по данным ТСХ - 20 мин. Отфильтровывали окислитель, промывали его
хлороформом. Растворитель отгоняли при пониженном давлении, а затем удаляли в
струе воздуха 12 ч. Остаток перекристаллизовывали из гексана. Выход: 95 %.
Крупные оранжевые кристаллы Тпл.=72-74оС. (Найдено: С, 69.09; Н, 10.50; N, 13.10. Рассчитано для C12H22N2O: C,
68.53; H, 10.54; N, 13.32 %); νmax (KBr)/см-1
2979, 2957, 2873, 2867 (СН3, СН2), 1571, 1525 (C=N);
λmax (EtOH)/нм 216 (lg ε 3.20), 315 (lg ε
3.60); δH (300 МГц; CDCl3)/м.д. 0.45 (6H, т, Jт=7.4
МГц, 2CH3, Et), 1.36 (9Н, с, 3СН3, t-Bu), 1.62, 1.92
(оба 2H, АВк, JАВ=14.0, Jк=7.4
МГц, 2CH2, Et), 2.11 (3Н, с, СН3 при С5); δC (75 МГц; CDCl3)/м.д. 6.98 (СН3, Et), 17.17 (СН3 при С5), 26.08 (СН3, t-Bu), 28.14 (СН2, Et), 33.24 (С(СН3)3), 88.78 (С4),
157.69 (С2), 178.63 (С5).
-трет-Бутил-4,4-диэтил-5-((гидроксиимино)метил)-4H-имидазол 3-оксид (20)

К раствору 0.5 г (2.4 ммоль) соединения 14 в 4 мл хлороформа прибавляли
0.4 мл триэтиламина. Реакционный сосуд накрывали колпаком, чтобы избежать
попадания в реакционную массу азотной кислоты, образующейся на стенках сосуда в
результате окисления на воздухе выделяющегося NO. К раствору небольшими порциями прибавляли 1.1 мл (10.5
ммоля) изопропилнитрита. Через 24 часа - после исчезновения исходного
соединения, по данным ТСХ, - раствор промывали 2 раза 5% водным раствором
гидроксида натрия (по 10 мл). Водную фазу подкисляли уксусной кислотой до рН
6-7, при этом окраска раствора сменялась с рыжей на желтую. Экстрагировали 2
раза хлороформом (по 10 мл). Экстракт сушили сульфатом магния. Осушитель
отфильтровывали, растворитель отгоняли при пониженном давлении. Выпавший осадок
оксима отфильтровывали и промывали смесью гексан - этилацетат (2 : 3). Выход:
0.23 г (41 %). Рыжие кристаллы Тпл.=210-212оС. (Найдено: С, 60.02; Н, 8.45; N, 17.41. Рассчитано для C12H21N3O2:
C, 60.23; H, 8.84; N, 17.56 %); νmax (KBr)/см-1 1538, 1511 (C=N); λmax (EtOH)/нм 230 (lg ε 4.11), 279 (lg ε 3.35), 372 (lg ε
3.80); δH (300 МГц; CDCl3)/м.д. 0.51 (6H, т, Jт=7.35
МГц, 2CH3, Et), 1.45 (9Н, с, 3СН3, t-Bu), 2.15 (4H, к, Jк=7.35, 2CH2, Et), 7.98 (1Н, с, С5-СН=), 12.32 (1Н,
с, =N-OH); δC (75 МГц; CDCl3)/м.д. 7.17 (СН3, Et), 26.03 (СН3, t-Bu), 30.31 (СН2, Et), 33.70 (С(СН3)3), 89.65 (С4),
144.01 (С5-СН=), 161.41 (С2), 172.68 (С5).
-трет-Бутил-5-циано-4,4-диэтил-4H-имидазол-3-оксид (21)
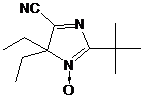
К раствору 2.71 г (11.3 ммоль) оксима 20 в 50 мл хлороформа приливали 2.3
мл триэтиламина и присыпали небольшими порциями 1.92 г (10.1 ммоль)
п-толуолсульфохлорида, избегая сильного нагревания реакционной смеси, в течение
30 мин. Ход реакции контролировали с помощью ТСХ на (sorbfil - SiO2, элюент -
хлороформ). Выдерживали реакционную массу еще 1 ч, до исчезновения исходного
соединения, затем промывали ее насыщенным водным раствором хлорида натрия,
сушили сульфатом магния. Осушитель отфильтровывали, растворитель отгоняли при
пониженном давлении. Остаток перекристаллизовывали из гексана. Выход: 1.9 г (76
%). Желтые кристаллы. Тпл.=63-65оС. (Найдено: С, 65.02; Н, 8.64; N, 18.88. Рассчитано для C12H19N3O: C,
65.13; H, 8.65; N, 18.99 %); νmax (KBr)/см-1
2966, 2937, 2880 (СН3, CH2), 2216 (СºN), 1522 (C=N);
λmax (EtOH)/нм 377 (lg ε
3.85); δH (300 МГц; CDCl3)/м.д. 0.58 (6H, т, Jт=7.4
Гц, CH3, Et), 1.38 (9Н, с, 3СН3, t-Bu), 1.91, 2.00 (4H, АВк, JАВ=14.6, Jк=
7.4 Гц, CH2, Et); δC (75 МГц; CDCl3)/м.д. 6.84 (СН3, Et), 25.79 (СН3, t-Bu), 28.75 (СН2, Et), 33.52 (С(СН3)3), 91.67 (С4),
111.70 (СºN), 146.63 (С2), 159.75 (С5).
-трет-Бутил-5-((3-(диэтиламино)пропил)(этил)амино)-4,4-диэтил-4H-имидазол-3-оксид (22)
К раствору 1.8 г (8.15 ммоль) нитрила 21 в 15 мл хлороформа прилили 3 мл
(17.09 ммоль) N,N,N’-триэтилпропан-1,3-диамина и перемешивали 12 ч. Затем
растворитель отгоняли при пониженном давлении и перемешивали еще 12 ч. Ход
реакции контролировали с помощью ТСХ (элюент - гексан : хлороформ = 1 : 3).
После исчезновения исходного соединения реакционную смесь хроматографировали на
колонке с окисью алюминия, элюент: эфир : гексан = 1 : 1. Выход: 1.8 г (63 %).
Желтоватое масло. (Найдено: С, 66.27; Н, 11.08; N, 18.14. Рассчитано для C12H19N3O´HCN: C, 66.45; H, 10.89; N, 18.45 %);
νmax (в тонком слое)/см-1 2968, 2933,
2876 (СН3, СН2), 2805 (СН2 при N),
1570 (NR2 деф.), 1540, 1505 (C=N); λmax (EtOH)/нм δH (400 МГц; CDCl3)/м.д.
0.40 (6Н, т, Jт=7.28 МГц, 2СН3, Et при С4), 0.85 (6Н, т, Jт=7.12 МГц, 2СН3, Et2N), 1.06 (3Н, т, Jт=7.00 МГц, СН3, EtN-С5),
1.29 (9Н, с, 3СН3, t-Bu), 1.62, 1.76 (оба 2Н, АВк, JAB=14.5, Jк=7.28 МГц, 2СН2, Et при С4), 2.17 (2Н, ушир. м, СН2-СН2-СН2), 2.26 (2Н, т, Jт=7.01 МГц, СН2-NEt2), 2.35 (4Н, к, Jк=7.12 МГц, 2СН2, Et2N), 3.21 (2Н, т, Jт=7.32 МГц, СН2-EtN-C5), 3.29 (2Н, к, Jк=7.00 МГц, СН2, EtN-C5); δC (75 МГц; CDCl3)/м.д. 7.55 (СН3, Et при С4), 11.63 (СН3, Et2N), 13.28 (СН3, EtN-C5), 25.45 (СН2, Et при С4), 26.22 (СН3, t-Bu), 30.51 (СН2-СН2-СН2), 33.40
(С(СН3)3), 43.47 (СН2-EtN-C5), 46.41 (СН2-NEt2), 46.81 (СН2, NEt2), 50.24 (СН2, EtN-C5), 91.69 (С4), 156.84 (С2), 170.05 (С5).
-трет-Бутил-4,4-диэтил-5-((гидроксиимино)метил)-4H-имидазол 3-оксид (20)
Метод A

К раствору 0.5 г (2.4 ммоль) соединения 14 в 4 мл хлороформа прибавляли
0.4 мл триэтиламина. Реакционный сосуд накрывали колпаком, чтобы избежать
попадания в реакционную массу азотной кислоты, образующейся на стенках сосуда в
результате окисления на воздухе выделяющегося NO. К раствору небольшими порциями прибавляли 1.1 мл (10.5
ммоля) изопропилнитрита. Через 24 часа - после исчезновения исходного
соединения, по данным ТСХ, - раствор промывали 2 раза 5% водным раствором
гидроксида натрия (по 10 мл). Водную фазу подкисляли уксусной кислотой до рН
6-7, при этом окраска раствора сменялась с рыжей на желтую. Экстрагировали 2
раза хлороформом (по 10 мл). Экстракт сушили сульфатом магния. Осушитель
отфильтровывали, растворитель отгоняли при пониженном давлении. Выпавший осадок
оксима отфильтровывали и промывали смесью гексан - этилацетат (2 : 3). Выход:
0.23 г (41 %).
Метод B
К 23.0 г охлажденного до ~ -10оС изопропилата натрия в изопропаноле,
приготовленного из 2.2 г натрия и 112 мл изопропанола по известной методике [],
приливали 1.5 мл (14.6 ммоль) изопропилнитрита и раствор 1.7 г (8.1 ммоль)
4Н-имидазол-3-оксида 19 в минимальном количестве изопропанола. Реакционную
смесь оставляли при температуре ~ -10оС на 24 ч, до исчезновения исходного соединения
в смеси (контролировали методом ТСХ на пластинках Sorbfil, элюент этилацетат - гексан 1 : 1). Затем из смеси
при пониженном давлении удаляли изопропанол. Остаток растворяли в воде (20 мл)
и экстрагировали раствор эфиром (3 раза по 5 мл), экстракт отбрасывали. Водную
фазу подкисляли уксусом до рН ~ 6 и экстрагировали хлороформом (2 раза по 5
мл). Экстракт сушили сульфатом магния, растворитель удаляли при пониженном
давлении, а остаток растирали со смесью этилацетат - гексан 1 : 2 и
отфильтровывали. Выход: 0.8 г (41%).
Рыжие кристаллы Тпл=210-212оС (этилацетат - гексан 1 : 1). (Найдено: С,
60.02; Н, 8.45; N, 17.41.
Рассчитано для C12H21N3O2: C, 60.23; H, 8.84; N, 17.56 %); νmax (KBr)/см-1 1538, 1511 (C=N);
λmax (EtOH)/нм 230 (lg ε 4.11), 279 (lg ε 3.35), 372 (lg ε
3.80); δH (300 МГц; CDCl3)/м.д. 0.51 (6H, т, Jт=7.35
МГц, 2CH3, Et), 1.45 (9Н, с, 3СН3, t-Bu), 2.15 (4H, к, Jк=7.35, 2CH2, Et), 7.98 (1Н, с, С5-СН=), 12.32 (1Н,
с, =N-OH); δC (75 МГц; CDCl3)/м.д. 7.17 (СН3, Et), 26.03 (СН3, t-Bu), 30.31 (СН2, Et), 33.70 (С(СН3)3), 89.65 (С4),
144.01 (С5-СН=), 161.41 (С2), 172.68 (С5).
Взаимодействие 2-трет-бутил-5-циано-4,4-диэтил-4H-имидазол-3-оксида (21) с пирролидином
К раствору 0.7 г (3.17 ммоль) нитрила 21 в 4 мл хлороформа приливали 2 мл
(24.00 ммоль) пирролидина и перемешивали 12 ч. Ход реакции контролировали
методом ТСХ (на пластинках Sorbfil,
элюент хлороформ). Поскольку через 12 ч в реакционной смеси наблюдали только
исходные соединения, растворитель отгоняли при пониженном давлении, а в
реакционную смесь прибавляли еще 3 мл (36.00 ммоль) пирролидина и нагревали ее
на масляной бане до 80-100оС в течение 7 дней. После исчезновения исходного
соединения реакционную смесь промывали этилацетатом. Органический раствор
экстрагировали 4-5 раз подкисленной уксусной кислотой (до рН ~ 4) водой. Водный
экстракт нейтрализовали карбонатом натрия и снова экстрагировали этилацетатом
(4 раза по 3 мл). Экстракт сушили карбонатом натрия, органический растворитель
удаляли при пониженном давлении. Остаток хроматографировали на колонке с окисью
алюминия, элюент хлороформ - гексан (градиент от 1 : 2 до 1 : 1). В результате
хроматографии удалось выделить единственный продукт -
(E)-5-(гидрокси(пирролидин-1-ил)метилен)-2-трет-бутил-4,4-диэтил-4,5-дигидроимидазол-3-оксид
(h1)
Выход: 0.05 г (5%). Желтое масло. (Найдено: С, 65.15; Н, 9.84; N, 14.88. Рассчитано для C16H29N3O2:
C, 65.05; H, 9.89; N, 14.22 %); νmax (в тонком слое)/см-1 3496 (ОН), 3346 (N-H), 2972, 2929, 2877 (СН3, СН2), 1664, 1617, 1536 (C=N, С=С); λmax (EtOH)/нм
224 (lg ε
3.67) δH (400 МГц; CDCl3)/м.д. 0.84 (6Н, т, Jт=7.5 Гц, СН3, Et),
1.40 (9Н, с, СН3, t-Bu), 1.65, 1.74 (оба 2Н, ABк, JAB= 14.2 Гц, Jк= 7.5 Гц, СН2, Et),
1.84 (4H, м, СН2-СН2-N, Pyrr), 3.48, 3.62
(оба 2Н, м, СН2-N, Pyrr); δC (75 МГц; CDCl3)/м.д. 8.84
(СН3, Et), 23.79, 26.11 (СН2CH2-N, Pyrr), 29.50 (С(СН3)3, t-Bu), 30.76 (СН3, t-Bu), 30.68 (СН2, Et),
45.86, 48.24 (СН2CH2-N, Pyrr), 78.77 (С4), 88.32 (С5), 162.95
(С2), 168.42 (=C-OH).
-трет-Бутил-5-метиламино-4,4-диэтил-4Н-имидазол-3-оксид (I-280A)
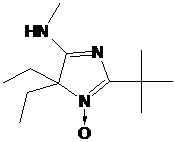
При перемешивании продували раствор 0.24 г (1.1 ммоль) нитрила 21 в 2-3
мл хлороформа газообразным метиламином, который получали, прибавляя 20% водный
раствор MeNH2 к твердому гидроксиду натрия, в течение 10-20 мин. После
завершения процесса (отсутствие исходного нитрила согласно данным ТСХ на
пластинках DC-Alufolien, элюент хлороформ) реакционную смесь
промывали насыщенным водным раствором хлорида натрия (2 раза по 3 мл), сушили
карбонатом натрия и удаляли растворитель при пониженном давлении.
Перекристаллизовывали из этилацетата. Выход: 0.23 г (94 %). Желтоватые
кристаллы. Тпл= 266-270оС (в капилляре) (Найдено: С, 63.43; Н, 10.35; N, 19.06. Рассчитано для C12H23N3O: C,
63.96; H, 10.29; N, 18.65 %); νmax (KBr)/см-1 3248, 3190, 3116 (N-H водородные связи),
2983, 2899 (СН3, СН2), 1624 (N-H деф.), 1539 (C=N);
λmax (EtOH)/нм 326 (lg ε
3.79); δH (400 МГц; CDCl3)/м.д. 0.58 (6Н, т, Jт=7.3 Гц, СН3, Et),
1.37 (9Н, с, CH3, t-Bu), 1.52, 1.91 (оба 2Н, АВк, JAB=14.0, Jк=7.3
Гц, СН2, Et), 2.93 (3Н, c, CH3-N), 4.17 (1H, уш.
с, H-N);
δC (100 МГц; CDCl3)/м.д. 6.88 (СН3, Et), 26.19 (СН3, t-Bu), 27.89 (N-CH3), 28.32 (CH2, Et),
33.68 (C(CH3)3, t-Bu), 81.04 (С4), 165.54 (С2), 173.68 (С5).
Взаимодействие 2-трет-бутил-5-метиламино-4,4-диэтил-4H-имидазол-3-оксида (I-280) с
бутиллитием
Реакцию проводили по аналогии с процессом, описанным для амидина 22a.
4Н-имидазол-3-оксид 22b прибавляли к раствору бутиллития в толуоле виде
суспензии, перемешивали при нагревании до ~ 80оС в течение 6 ч. После
разложения реакционной смеси водой, проводили окисление диоксидом марганца
согласно общей методике. Для полученной смеси продуктов записывали спектр ЭПР,
в котором присутствует малоинтенсивный триплетный сигнал (аN ~ 14 Гс).
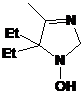
-Метил-5,5-диэтил-2,5-дигидро-имидазол-1-ол (I-295, DT-9B).
К раствору 6 г (33 ммоль) гидрохлорида 1,2-гидроксиаминокетона в 10 мл
метанола приливали 10 мл (66 ммоль) 20% водного раствора формальдегида. К
охлажденной до -15оС смеси по каплям прибавляли 9 мл 25% водного раствора
аммиака. Через 2 ч из реакционной массы при пониженном давлении удаляли
метанол, остаток промывали смесью: изопропанол - этилацетат (1 : 2) (3 раза по
20 мл). Полученный экстракт промывали насыщенным водным раствором NaCl (2 раза
по 20 мл), сушили сульфатом магния, удаляли растворитель при пониженном
давлении. Продукт использовали в дальнейшем синтезе без дополнительной очистки.
Выход неочищенного продукта: 4.9 г (95%). Для получения аналитического образца
порцию вещества хроматографировали на колонке с силикагелем, элюент хлороформ.
Бесцветное масло. (Найдено: С, 61.26; Н, 10.31; N, 17.31. Рассчитано для C8H16N2O: C, 61.50; H, 10.32; N,
17.93 %); νmax (в
тонком слое)/см-1 3207 (OH),
2922, 2879 (CH3,CH2), 1651 (C=N); λmax (EtOH)/нм не поглощает; δH (400 МГц; CDCl3)/м.д. 0.82 (6Н, т, Jт= 7.4 Гц, СН3, Et), 1.49, 1.70 (оба 2H, ABк, JAB= 14.4 Гц, Jк= 7.4 Гц, CH2, Et), 1.89 (3H, т, Jт= 1.8 Гц,
CH3-C4), 4.72 (2H, к, Jк= 1.8 Гц, N-CH2-N); δC (100 МГц; CDCl3)/м.д. 8.71 (CH3, Et), 16.86 (СН3 при С4), 26.21
(CH2, Et), 79.49 (C5), 84.80 (С2), 176.19 (С4).
-((Гидроксиимино)метил)-4,4-диэтил-4H-имидазол-3-оксид (I-296, DT-10-1, NL 1140)
Синтез оксима I-296 осуществляли по аналогии с методикой нитрозирования
имидазолина 14. Исходное соединение I-295 растворяли в минимальном объеме
гексана. Через 24 часа после прибавления изопропилнитрита, выпавший осадок
оксима I-296 отфильтровывали и промывали смесью этилацетат - гексан (1 : 1).
Выход: 87 %.
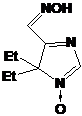
Желтые кристаллы Тпл=178разлоС (этилацетат). (Найдено: С, 52.54; Н, 7.20;
N, 22.31. Рассчитано для C8H13N3O2: C,
52.45; H, 7.15; N, 22.94 %); νmax (KBr)/см-1
1531 (C=N), 1004 (N-O); λmax (EtOH)/нм
230 (lg ε 3.92), 355 (lg ε 3.85); δH (400 МГц; ДМСО-d6)/м.д. 0.45 (6H, т, Jт=7.3 Гц, CH3, Et), 1.86, 2.04 (4H, ABк, JAB=13.6 Гц, Jк=7.3
Гц, CH2, Et), 7.94 (1Н, с, С5-СН=), 8.29 (1Н, с, =C2-H); δC (100 МГц; ДМСО-d6)/м.д. 7.41 (СН3, Et), 30.32 (СН2, Et), 88.46 (С4), 141.94 (C2), 144.41
(С5-СН=), 171.24 (С5).
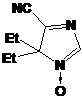
-Циано-4,4-диэтил-4H-имидазол-3-оксид
(I-297, DT-11) получали аналогично нитрилу 21. Ход реакции контролировали с
помощью ТСХ (sorbfil - SiO2, элюент хлороформ - гексан (1 : 1)). Вещество
выделяли хроматографией на колонке с силикагелем, элюент хлороформ - гексан
(градиент от 1 : 2 до 1 : 1). Выход: 80 %.
Желтые кристаллы. Тпл=43-45оС (гексан). (Найдено: С, 58.21; Н, 6.70; N, 25.61. Рассчитано для C8H11N3O: C,
58.17; H, 6.71; N, 25.44 %); νmax (KBr)/см-1
2981, 2932, 2887 (СН3, CH2), 2220 (СºN), 1521 (C=N);
λmax (EtOH)/нм 358 (lg ε
3.85); δH (300 МГц; CDCl3)/м.д. 0.62 (6H, т, Jт=
7.4 Гц, CH3, Et), 1.92, 2.02 (4H, АВк, JАВ=15, Jк=7.4, CH2, Et), 8.70 (1Н, с, Н-С2); δC (75 МГц; CDCl3)/м.д. 6.88 (СН3, Et), 28.61 (СН2, Et),
91.35 (С4), 111.38 (СºN),
141.13 (С2), 148.42 (С5).
-диметиламино-4,4-диэтил-4H-имидазол-3-оксид
(I-298)
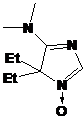
К раствору 1.62 г (9.7 ммоль) нитрила I-297 в 10 мл сухого хлороформа
приливали 3 мл (45.3 ммоль) диметиламина. Через 1 ч растворитель удаляли при
пониженном давлении, остаток разбавляли 20 мл воды и промывали эфиром (6 раз по
5 мл). Затем pH водного раствора доводили до 9-10 добавлением карбоната калия и
насыщали раствор NaCl. Полученный раствор промывали 1 раз эфиром и
экстрагировали хлороформом (4 раза по 20 мл). Растворитель удаляли при
пониженном давлении. Остаток хроматографировали на колонке с Al2O3, элюент -
хлороформ. Ход реакции контролировали с помощью ТСХ (элюент - гексан :
хлороформ = 1 : 3). Выход: 0.71 г (40 %).
Желтые кристаллы. Вещество гигроскопично. Тпл= 154-157 оС (этилацетат -
гексан (1 : 2)). (Найдено: С, 56.02; Н, 9.35; N, 21.25. Рассчитано для C9H17N3O Ч 0.5H2O: C, 56.22; H, 9.44;
N, 21.86
%); νmax (KBr)/см-1 2976, 2940, 2881 (СН3,
СН2), 1617 (NMe2 деф.), 1618, 1565 (C=N); λmax (EtOH)/нм 342 (lg ε 3.84); δH (300 МГц; CDCl3)/м.д. 0.56 (6Н, т, Jт=7.2 Гц, СН3, Et),
1.74, 2.02 (оба 2Н, АВк, JAB=
14.2 Гц, Jк= 7.2 Гц, СН2, Et), 3.01 (6H, с, CH3, NMe2), 7.39 (1H,
с, H-C2=); δC (75 МГц; CDCl3)/м.д. 7.22
(СН3, Et), 26.47 (СН2, Et), 37.41 (CH3, NMe2), 81.85 (С4),
141.65 (С2), 171.96 (С5).
-трет-Бутил-5-диметиламино-4,4-диэтил-4H-имидазол-3-оксид (I-299)
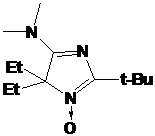
Синтез проводили в соответствии с общими методиками для реакций
циклических нитронов с реактивами Гриньяра и окисления образующихся
гидроксиаминов в кетонитроны. Исходный 4H-имидазол растворяли в минимальном
количестве сухого ТГФ. Реакция завершалась за 30 мин. Органический экстракт,
полученный после обработки реакционной массы, сушили карбонатом натрия.
Окисление выделенного гидроксиамина проводили диоксидом свинца в хлороформе в
течение 1 ч. Выход: 58 %.
Желтые кристаллы. Вещество гигроскопично. Тпл= 154-157 оС (этилацетат -
гексан (1 : 2)). (Найдено: С, 66.27; Н, 11.08; N, 18.14. Рассчитано для C9H17N3O: C, 58.99; H, 9.35; N, 22.93
%); νmax (KBr)/см-1 2976, 2940, 2881 (СН3,
СН2), 1617 (NMe2 деф.), 1618, 1565 (C=N); λmax (EtOH)/нм 342 (lg ε 3.84); δH (300 МГц; CDCl3)/м.д. 0.56 (6Н, т, Jт=7.2 Гц, СН3, Et),
1.74, 2.02 (оба 2Н, АВк, JAB=
14.2 Гц, Jк= 7.2 Гц, СН2, Et), 3.01 (6H, с, CH3, NMe2), 7.39 (1H,
с, H-C2=); δC (75 МГц; CDCl3)/м.д. 7.22
(СН3, Et), 26.47 (СН2, Et), 37.41 (CH3, NMe2), 81.85 (С4),
141.65 (С2), 171.96 (С5).
Взаимодействие 2-трет-Бутил-5-диметиламино-4,4-диэтил-4H-имидазол-3-оксида (I-299) с
бутиллитием
-трет-Бутил-2-бутил-4-диметиламино-5,5-диэтил-2,5-дигидроимидазол-1-оксил
(I-300)

8.4 Пирролидиновые НР
Окисление N-гидроксипирролидинов.
Общая методика.
К раствору 0.1 г сульфата меди в 1-2мл воды приливали 4мл водного
раствора аммиака. Полученный раствор прибавляли к раствору 8 ммоль N-гидроксипирролидина в 10-15мл метанола
и пропускали через раствор слабый ток воздуха 5-24 ч. Окончание реакции
определяли по отсутствию исходного соединения, по данным ТСХ. Далее метанол
отгоняли при пониженном давлении. Остаток растворяли в 15 мл насыщенного
водного раствора хлорида аммония и экстрагировали хлороформом 4-5 раз (по
3-5мл). Объединённый экстракт промывали 2 раза водным раствором трилона Б (0.5
г трилона Б на 100 мл воды) (по 10 мл), чтобы удалить соли меди. В объединенных
промывных водах растворяли хлористый натрий до насыщения. Экстрагировали их 2
раза хлороформом (по 5 мл). Органические экстракты объединяли и сушили
сульфатом магния. Осушитель отфильтровывали, растворитель отгоняли при
пониженном давлении.
-((3,3-Диметил-1-оксидо-3,4-дигидро-2H-пиррол-5-ил)метил)-2,4,4-триметил-3,4-дигидро-2H-пиррол-1-оксид (25)
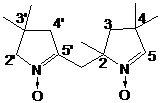
Реакцию 3,3,5-триметил-3,4-дигидро-2H-пиррол-1-оксида (24) с третбутилмагнийхлоридом проводили по
стандартной методике. Процесс проходит полностью за 24 ч. Образующееся гидрокси-производное
окисляли кислородом в присутствие медных солей и обрабатывали по описанной выше
методике. Полученное таким образом вещество хроматографировали на колонке с
силикагелем, элюент: метанол : хлороформ = 1 : 50. Продукт кристаллизовался при
стоянии в холодильнике в течение 7 дней. После чего его растирали с гексаном и
отфильтровывали. Выход 84 %. Бесцветные кристаллы. Гигроскопичное. νmax (в тонком слое)/см-1: 3064 (=С-Н),
2962, 2871 (CH3, CH2), 1581 (С=N); λmax (EtOH)/нм
235 (lg ε
4.13); δH (300 MГц; CDCl3)/м.д.:
1.02 (9H, с, 3СН3, гем. Me), 1.09 (3H, c, СН3, гем. Me), 1.39 (3Н, с, СН3, гем. Me), 1.84, 2.19 (оба 1Н, AB, JАВ=13.8 МГц, CH2
при С3), 2.46 (2H, с, СН2 при
С4’), 2.67, 2.95 (оба 1Н, AB, JАВ=13.5 МГц, CH2 экзоциклическая); 3.59 (2Н, с, СН2 при С2’), 6.52 (1Н, с,
=СН). 13С ЯМР в СDCl3 75 MГц: 26.85 (СН3 при С2), 27.69, 27.79
(Гем. СН3 при C4), 27.99 (гем. СН3 при С3’), 31.78
(С3’), 34.47 (CH2 экзоциклическая), 37.68 (С4); 44.86
(С3), 46.97 (С4’), 74.39 (C2’),
77.35 (C2), 140.87 (C5), 143.57 (C5’).
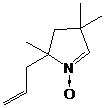
-Аллил-2,4,4-триметил-3,4-дигидро-2H-пиррол-1-оксид (26)
Соединение получали согласно общей методике. При взаимодействии
3,3,5-триметил-3,4-дигидро-2H-пиррол-1-оксида
(24) с аллилмагнийбромидом образовывался соответствующий гидроксиамин, который
без выделения окисляли кислородом в присутствие солей меди и выделяли по
приведенной выше методике. Продукт хроматографировали на колонке с силикагелем,
элюент: метанол : хлороформ = 4 : 100. Выход 84 %. Желтоватое масло. (Найдено: C, 64.54; H, 10.55; N,
7.85. Вычислено для C10H17NO´Н2О: C, 64.83; H,
10.34; N,
7.56 %); νmax (в тонком
слое)/см-1: 3075 (=С-Н, All),
2964, 2870 (CH3, CH2), 1641 (CH=CH2), 1578 (С=N); λmax (EtOH)/нм 236 (lg ε
3.96); δH (300 MГц; CDCl3)/м.д.
1.10, 1.15 (оба 3Н, с, 2СН3 при С4), 1.39 (3Н, с, СН3 при С2), 1.74, 2.04 (оба
1Н, АВ, JАВ=13.35 МГц, СН2 в цикле), 2.24,
2.53 (оба 1Н, АВд, JАВ=13.77, Jд1=8.43, Jд2=6.21 МГц,СН2, All), 5.07 (2Н, м, =СН2, All),
5.57 (1Н, м, =СН-, All), 6.55 (1Н, с,
Н-С5); dC (75 MГц; CDCl3)/м.д.
26.02 (СН3 при С2), 27.74, 28.36 (СН3 при С4), , 37.42 (С4), 42.51, 44.51 (СН2,
в цикле, All), 76.38 (С2), 119.20 (Н2С=), 132.10
(=СН-, All), 140.55 (С5).
-Аллил-2,4,4-триметил-5-этил-3,4-дигидро-2H-пиррол-1-оксид (27)

Соединение получали аналогичным образом из пирролин-N-оксида 26 и этилмагнийбромида.
Образовавшийся гидроксиамин также окисляли кислородом в присутствие солей меди
по описанной методике. Продукт выделяли хроматографией на колонке с
силикагелем, элюент: метанол : хлороформ = 4 : 100. Выход 40 %. Желтоватое
масло. (Найдено: H, 10.10; N, 6.82. Вычислено для C12H21NO: C, 73.80; H, 10.84; N, 7.17 %); νmax (в тонком слое)/см-1: 3076 (=С-Н, All), 2967, 2876 (CH3, CH2), 1641 (CH=CH2), 1587 (С=N); λmax (EtOH)/нм 235 (lg ε
3.94); δH (400 MГц; CDCl3)/м.д.
1.014 (3Н, т, Jт=7.12 МГц, СН3, Et), 1.01, 1.03 (оба 3Н, с, 2СН3 при
С4), 1.28 (3Н, с, СН3 при С2), 1.56, 1.85 (оба 1Н, АВ, JАВ=13.28 МГц, СН2 в цикле), 2.13 (2Н, м, СН2, Et), 2.03, 2.45 (оба 1Н, м, СН2, All), 4.96 (2Н, м, =СН2, All), 5.44 (1Н, м, =СН-, All); dC (75 MГц; CDCl3)/м.д. 9.39 (ушир., СН3, Et), 17.54 (ушир., СН2, Et), 26.09 (СН3 при С2), 27.57, 28.28
(ушир., СН3 при С4), 40.53 (ушир., С4), 42.93, 43.76 (СН2, в цикле, All), 74.09 (С2), 119.16 (Н2С=, All), 132.83 (=СН-, All), 153.24 (ушир., С5).

-Аллил-2,4,4-триметил-5-этил-3,4-дигидро-2H-пиррол (28)
Кетонитрон 27 обрабатывали этилмагнийбромидом по стандартной методике.
Реакционную смесь выдерживали 24 ч. После обработки реакционной массы продукт
хроматографировали на колонке с окисью алюминия, элюент: эфир : гексан = 2 : 1.
Выход 84 %. Желтоватое масло. (Найдено: C, 80.01; H, 11.69; N,
6.52. Вычислено для C12H21N: C, 80.38; H, 11.81;
N, 7.81 %); νmax (в тонком
слое)/см-1: 3075 (=С-Н, All),
2962, 2871 (CH3, CH2), 1643 (CH=CH2);
λmax (EtOH)/нм не поглощает; δH (300 MГц; CDCl3)/м.д.
1.01, 1.13 (оба 3Н, с, 2СН3 при С4), 1.16 (3Н, т, Jт=7.5 МГц, СН3, Et), 1.21 (3Н, с, СН3 при С2), 1.49, 1.76 (оба 1Н, АВ, JАВ=13.14 МГц, СН2 в цикле), 2.17 (2Н,
к, Jк=7.5 МГц, СН2, Et), 2.26 (2Н, д, Jд=7.32 МГц, СН2, All), 5.01 (2Н, м, =СН2, All), 5.72 (1Н, м, =СН-, All); dC (75 MГц; CDCl3)/м.д. 11.35 (СН3, Et), 20.84 (СН2, Et), 26.85 (СН3 при С2), 27.23, 28.56
(СН3 при С4), 47.06, 48.24 (СН2, в цикле, All), 50.65 (С4), 71.28 (С2), 116.73 (Н2С=, All), 134.96 (=СН-, All), 181.02 (С5).
-(3,3-Диметил-2-оксо-1-бутилиден)-2,2-диметил-5,5-диэтилимидазолидин-1-оксил
(31)
К раствору фениллития, приготовленному из 1 г (0.143 моль) лития и 7.6 мл
(72.6 ммоль) бромбензола в 100 мл абсолютного эфира, прибавили по каплям при
перемешивании 10 мл (74.3 ммоль) диизопропиламина. Реакцию проводили в
атмосфере аргона. Перемешивали 15 мин и прибавили по каплям при перемешивании
раствор 4.3 г (23.4 ммоль) 2,2,4-Триметил-5,5-диэтил-2,5-дигидроимидазол-1-ола
(30) в 30 мл абсолютного эфира. Через 15 мин к реакционной смеси прибавили по
каплям при перемешивании 12 мл (78.5 ммоль) этилового эфира триметилуксусной
кислоты. Далее реакционную массу нагревали до кипения при перемешивании 48 ч.
После прибавления воды органическую фазу отделили и промыли насыщенным
раствором хлорида натрия, подкисленным соляной кислотой до рН 3-4, пока раствор
после промывки не стал кислым (рН 4-5). К полученному раствору прибавили 2-х
кратный избыток по массе диоксида свинца и сульфат магния и перемешивали в
течение 20 мин, осадок отфильтровали, растворитель отогнали на ротационном
испарителе. Полученное вещество выделяли хроматографией на колонке с
силикагелем, элюент - хлороформ : гексан = 1 : 2. Выход: 2.6 г (54%). Желтые
кристаллы, Тпл.=84-86оС. (Найдено: C, 67.66; H, 10.18; N, 10.46. Вычислено для C15H27N2O2•: C, 67.38; H,
10.18; N,
10.48 %); νmax (KBr)/см-1 3274 (уш. NH), 2978, 2952, 2869 (CH3, CH2), 1635 (С=O),
1561 (C=C); λmax (EtOH)/нм 298 (lg ε 4.27).


-(1-Гидрокси-2,2-диметил-5,5-диэтил-имидазолидин-4-илиден)-3,3-диметил-бутан-2-он
(32) получали гидрированием нитроксильного радикала 31 при атмосферном давлении
в метанольном растворе на катализаторе (палладий на угле). Выход: 97 %.
Бесцветные кристаллы, Тпл.=108-111оС. (Найдено: C, 67.61; H,
10.58; N, 9.79. Вычислено для C15H28N2O2: C, 67.13; H,
10.52; N,
10.44 %); νmax (KBr)/см-1 3281 (уш. OН, NH), 2968, 2871 (CH3, CH2), 1617 (С=O), 1526 (C=C);
λmax (EtOH)/нм 304 (lg ε
4.28); δH (300 MГц; CDCl3)/м.д.
0.89 (6 H, т, J 7.3 Гц, 2 Ч CH3, Et), 1.15 (9H, с, CH3, t-Bu), 1.42 (6H, с,
2 Ч CH3, гем. CH3 ), 1.69, 1.82 (оба 2H, ABк, Jк 7.3 Гц, JАВ 14.5 Гц, 2 Ч CH2, Et), 5.01 (1H, c, CH), 5.35 (1H, уш. с, N-H), 10.12 (1H, уш. с, N-OH); dC (75 MГц; CDCl3) 8.94 (CH3, Et),
27.57 (CH3), 27.80 (CH2, Et),
27.83 (CH3, t-Bu), 41.41 (C(CH3)3), 74.03 (C5),
79.97 (C2), 83.91 (=CHCO), 165.45 (C4), 204.83 (C=O).
-трет-Бутил-2,2-диэтил-2,4-дигидро-пиррол-3-он-1-оксид (33) получали по
аналогии с известной методикой [39], образовавшийся осадок перекристаллизовали
из гексана. Выход 60 %. Бесцветные кристаллы, Тпл.=138-143оС. (Найдено: C, 67.83; H, 10.36; N,
6.32. Вычислено для C12H21NO2: C,
68.21; H, 10.02; N, 6.63 %); νmax (KBr)/см-1: 2964, 2924, 2877 (CH3, CH2),
2580 (уш. OH), 1594, 1534 (уш., С=N, C=C);
λmax (EtOH)/нм 332 (lg ε
3.97); δH (300 MГц; CDCl3)/м.д.
Форма А: 0.67 (6H, уш. м, 2 Ч CH3, Et), 1.35 (9H, c, 3 Ч CH3, t-Bu), 1.67, 1.92 (оба 2Н, уш. АВк, Jк 6.8 Гц, JAB 13.6 Гц, 2 Ч CH2, Et),
3.17 (2H, уш. с, CH2-CO); Форма В: 0.66
(6H, уш. м, J 7.6 Гц, 2 Ч CH3, Et), 1.35 (9H, c, 3 Ч CH3, t-Bu), 1.67, 1.81
(оба 2Н, уш. АВк, Jк 6.8 Гц, JAB 13.6 Гц, 2 Ч CH2, Et), 4.96 (1H, уш.
с, CH), 10.22 (1H, уш. с, N-OH; dC (75 MГц; CDCl3)/м.д. Форма А: 7.64 (CH3, t-Bu), 25.64 (CH3, Et),
28.14 (CH2, Et), 33.89 (C(CH3)3), 42.87 (C4), 85.06 (C2),
151.31 (C5), 209.84 (C=O); Форма В: 7.64
(CH3, t-Bu), 27.92 (CH3, Et), 34.38 (C(CH3)3), 79.50 (C2), 94.50 (C4),
185.96 (C5), 196.55(C=O).
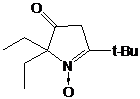
,4’-би(5-трет-бутил-3-оксо-2,2-диэтил-3,4-дигидропирролилиден)-1,1’-диоксид
(34)
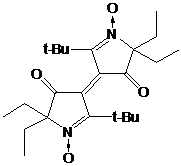
К раствору 0.15 г (0.71 ммоль) пирролина 33 в 3 мл хлороформа прибавляли
0.75 г двуокиси марганца и перемешивали 7 дней. Окислитель отфильтровывали, а
фильтрат упаривали. Остаток перекристаллизовали из этилацетата. Выход: 0.145 г
(95 %). Фиолетовые кристаллы Тпл.=157-160оС. (Найдено: С, 68.91; Н, 9.27; N, 6.71. Рассчитано для C24H38N2O4: C,
68.87; H, 9.15; N, 6.69 %); νmax (KBr)/см-1
2972, 2928, 2850 (СН3, СН2), 1701 (С=О), 1505, 1489, 1459, 1422 (C=N, С=С); λmax (EtOH)/нм
234 (lg ε 3.83), 307 (lg ε 3.93), 375 (lg ε 3.70), 579 (lg ε
4.03); δH (300 МГц; CDCl3)/м.д. 0.51 (3H, т, Jт
7.35 Гц, CH3, Et), 0.69 (3H, т, Jт 7.40 Гц, CH3, Et),
1.45 (9Н, с, 3СН3, t-Bu), 1.80, 1.95 (оба 2H, оба м, 2CH2, Et).
- трет-Бутил-5- бутил-2,2-диэтилпирролидин-3-он-1-оксил (35)
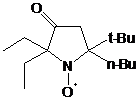
К раствору бутиллития в 30 мл сухого гексана, приготовленному из 0.7 г
(0.100 моль) мелко нарезанного лития и 3.54 мл (0.0383 моль) бутилхлорида,
прибавили по каплям при перемешивании в атмосфере аргона теплый раствор 0.5 г
(0.00237 моль) пирролина (33) в 30 мл сухого бензола. Нагревали до кипения при
энергичном перемешивании 1.5 ч. Прибавляли воду до растворения неорганического
осадка. Органическую фазу отделили, водную экстрагировали 1 раз бензолом (15
мл). Экстракт объединили с органической фазой, сушили сульфатом магния,
отфильтровали осушитель, раствор упарили. Соединение 35 выделяли хроматографией
на колонке с силикагелем, элюент - четыреххлористый углерод. Выход: 0.39 г (61
%) Желтые кристаллы, Тпл.=42-43оС. (Найдено: C, 71.90; H,
11.49; N, 4.96. Вычислено для C16H30NO2•: C, 71.59; H, 11.27; N, 5.22 %); νmax (KBr)/см-1: 2969, 2874 (CH3, CH2),
1747 (C=O); в УФ-спектре поглощения не наблюдается.
Взаимодействие 5-трет-бутил-2,2-диэтил-2,4-дигидро-пиррол-3-он-1-оксида (33)
с этилмагнийбромидом. К раствору 0.464 г (0.0022 моль) пирролина 33 в 40 мл
сухого ТГФ прибавили по каплям раствор этилмагнийбромида, полученного из 1.14 г
(0.0475 моль) магниевой стружки и 2мл (0.0268 моль) этилбромида, в 40 мл
абсолютного эфира. Раствор перемешивали при нагревании до кипения 24 ч. К
реакционной смеси прилили 30 мл воды и 3 мл уксусной кислоты до растворения
неорганического осадка, ТГФ отогнали на ротационном испарителе, остаток
разбавили 10 мл воды и экстрагировали хлороформом (3 раза по 20 мл). Экстракт
сушили сульфатом магния, отфильтровали осушитель, растворитель отогнали на
ротационном испарителе. Остаток растерли с гексаном. Осадок отфильтровали.
ИК-спектр полученного вещества соответствует спектру исходного соединения.
Выход: 0.445 г (96 %).
-трет-Бутил-5-бутил-2,2-диэтилпирролидин-3-ол-1-оксил (36).

К раствору радикала 35 (0.2 г, 0.75 ммоля) в 15 мл этанола при
перемешивании порциями прибавляли NaBH4 (40 мг, 1 ммоль) в течение 30 мин. За ходом реакции следили по ТСХ
(Сорбфил, CHCl3-гексан 1:2). После завершения реакции
этанол отгоняли при пониженном давлении, остаток разбавляли водой (10 мл) и
экстрагировали диэтиловым эфиром. Экстракт сушили МgSO4, растворитель отгоняли при пониженном давлении.
Перекристаллизовали из гексана. Выход BBO количественный. Бледно-жёлтые кристаллы. Тпл. 88-90 °С. Найдено, С 70.76; Н 11.80; N 5.09 %; вычислено для С16Н32NO2, С 71.06; Н 11.93, N 5.18 %. νmax (KBr)/см-1: 3392уш (ОН), 2963, 2936, 2877 (СН3, СН2).
,2,4-Триметил-5,5-диэтил-2,5-дигидроимидазол-1-ол (31)
г (0.055 моль) 3-гидроксиамино-3-этилпентан-2-она гидрохлорида, 14.4 г
(0.187 моль) ацетата аммония и 14.4 мл (0.360 моль) метанола продували аргоном
в течение 20 минут при перемешивании. Затем к смеси прилили 43 мл (0.593 моль)
ацетона и продували аргоном при перемешивании еще 20 минут. Быстро закрыли
колбу пробкой и выдержали 12 ч. Затем реакционную смесь разбавили водой и
охладили. Выпавший осадок отфильтровали и сушили на фильтре. Выход 6.8 г (67 %)
Бесцветные кристаллы, Т.пл.=114-118oC (гексан). (Найдено: C,
65.04; H, 10.10; N, 15.37. Вычислено для C10H20N2O: C, 65.18; H, 10.94; N, 15.20 %); νmax (KBr)/см-1 3172 (уш., ОН) , 2979, 2925, 2880 (CH3, CH2), 1661 (С=N); в
УФ-спектре поглощения не наблюдается; δH (400 MГц; CDCl3)/м.д.
0.85 (6 H, м, J 7, 2 Гц, 2 Ч CH3, Et), 1.37 (6H, с, 2 Ч CH3, (H3C)2С), 1.58, 1.70 (оба 2 H, АВк, JAB 14
Гц, Jк 7.2 Гц, 2 Ч CH2, Et), 1.86 (3 H, с, CH3C=N), 6.65 (1 H, уш. с, OH); dC (100
MГц; CDCl3)/м.д. 5.34 (CH3, Et),
12.78 (CH3-C=N), 22.52 (CH2, Et), 23.60 (( H3C)2С), 75.51 (C5), 86.49 (C2),
167.97 (C3).
-(2,2-Диметил 5,5-диэтил -1-пропионилоксиимидазолидин-4-илиден)бутан-2-он
(32)
К раствору фениллития, приготовленному из 1.6 г (0.230 моль) лития и 12
мл (0.115 моль) бромбензола в 150 мл абсолютного эфира, прибавили по каплям при
перемешивании 15.8 мл (0.115 моль) диизопропиламина. Реакцию проводили в
атмосфере аргона. Перемешивали 15 мин и прибавили по каплям при перемешивании
раствор 6.8 г (0.0369 моль) имидазолина (31) в 50 мл абсолютного эфира. Через
15 мин реакционную смесь охладили до 5-0оС и прибавили по каплям при
перемешивании 7.3 мл (0.0763 моль) метилового эфира пропионовой кислоты.
Перемешивали 30 мин, затем охладили реакционную смесь до 5-0оС и прибавили 150
мл воды. Органическую фазу отделили, водную экстрагировали 3 раза хлороформом
(по 30 мл). Объединенные экстракты сушили сульфатом магния, осушитель
отфильтровали, растворитель отогнали на ротационном испарителе. Остаток
хроматографировали на колонке с силикагелем, элюент - гексан : хлороформ = 1 :
4. Выход: 8.3 г (76 %) Бесцветные кристаллы, Тпл.=70-73оС. (Найдено: C, 64.71; H, 9.79; N,
9.47. Вычислено для C16H28N2O3: C, 64.83; H, 9.52; N, 9.45 %); νmax (KBr)/см-1: 3280 (уш., NН) , 2986, 2966, 2938, 2878 (CH3, CH2),
1771 (O-C=O), 1635 (С=O), 1553 (C=C);
λmax (EtOH)/нм 302 (lg ε
4.31); δH (200 MГц; CDCl3)/м.д.
0.89 (6 H, т, J 7.4 Гц, 2 Ч CH3,
Гем. Et), 1.03 (3Н, т, J 7.4 Гц, CH3, CH3-CH2COC), 1.12 (3H, т, J 7.4 Гц, CH3, CH3-CH2COO), 1.36 (6H, уш. c, С2-СН3), 1.51, 1.69 (оба 2Н, уш. м,
С5-СН2), 2.25 (2Н, к, J 7.4
Гц, COCCH2), 2.29 (2Н, к, J 7.4 Гц, OOCCH2), 4.80 (1Н, с, СН), 10.10 (1H, уш. с, NH); dC (50 MГц; CDCl3)/м.д.
7.92 (уш. CH3, С5-Et), 8.87 (СН3СН2СОС), 9.47 (СН3СН2СОО), 24.97, 30.15 (оба уш.
С2-CH3), 25.59 (СН2СОС), 28.24, 30.12 (оба
уш., СН2, С5-Et), 34.75 (СН2СОО), 75.44 (С5), 81.00
(С2), 86.81 (СН=), 163.69 (С4), 172.74 (COO), 199.60 (C=O).
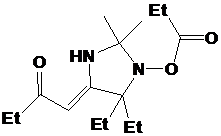
,2,5-Триэтил-2,4-дигидро-пиррол-3-он-1-оксид (33)
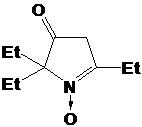
Получение и очистку соединения проводили согласно известной методике [39]
Выход: 51 %. Бесцветные кристаллы, Т.пл=87-89оС. (гексан). (Найдено: C, 65.60; H, 9.71; N,
7.51. Вычислено для C10H17NO2: C,
65.54; H, 9.35; N, 7.64 %); νmax (KBr)/см-1 2971, 2937, 2879 (CH3, CH2),
2521 (уш., ОН), 1593, 1520 (уш., C=N, C=C);
λmax (EtOH)/нм 328 (lg ε
3.93); δH (400 MГц; CDCl3)/м.д.
Форма А: 0.68 (6H, т, J 7.6 Гц, 2 Ч CH3, С2-Et),
1.14 (3H, т, J 7.6 Гц, CH3,
С5-Et), 1.69 (4Н, м, 2 Ч СН2, С2-Et), 2.70 (2Н, к, J 7.6 Гц, СН2, С5-Et), 3.12 (2Н, с, О=С-СН2); Форма В:
0.65 (6H, т, J 7.6 Гц, 2 Ч CH3,
С2-Et), 1.18 (3H, т, J 7.6
Гц, CH3, С5-Et), 1.96 (4Н, м, 2 Ч СН2, С2-Et), 2.57 (2Н, к, J 7.6 Гц, СН2, С5-Et),
4.88 (1Н, с, СН), 10.00 (1Н, уш. с, ОН); dC (100 MГц; CDCl3)/м.д. Форма А: 7.61 (CH3, С2-Et),
9.26 (CH3, С5-Et), 19.44 (CH2,
С5-Et), 27.50 (CH2, С2-Et),
42.47 (О=С-СН2), 95.76 (С2), 146.81 (C5), 208.59 (С=О); Форма В: 7.82 (CH3, С2-Et),
10.95 (CH3, С5-Et), 20.54 (CH2,
С5-Et), 28.02 (CH2, С2-Et),
78.61 (С2), 84.01 (СН), 182.06 (C5),
198.63 (С=О).
,2,5,5-Тетраэтилпирролидин-3-он-1-оксил (34)
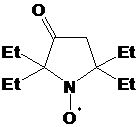
К раствору этилмагнийбромида в 100 мл сухого ТГФ, приготовленному из 3.64
г (0.152 моль) магниевой стружки и 9.8 мл (0.131 моль) этилбромида, прибавили
по каплям при перемешивании раствор 2.4 г (0.0131 моль) пирролина (33) в 10 мл
сухого ТГФ. Перемешивали при слабом кипении ТГФ 15 ч. Прибавляли воду до
образования вязкой неорганической фазы. Органическую фазу декантировали,
неорганическую массу промыли 3 раза этилацетатом (по 10 мл). Органические фазы
объединили, сушили сульфатом магния, осушитель отфильтровали, раствор упарили.
Остаток растворили в 30 мл хлороформа, присыпали 7.5 г (0.862 моль) диоксида
марганца и перемешивали в течение 1 ч., окислитель отфильтровали, раствор
упарили. Соединение 34 выделяли хроматографией на колонке с силикагелем, элюент
- метилтретбутиловый эфир : гексан = 1 : 8. Выход: 0.05 г (1.8 %) Желтое масло.
M/z, Найдено: 212.16482; вычислено для C12H22NO2: 212.16504; νmax (в тонком слое)/см-1 2971, 2929,
2882, 2856 (CH3, CH2), 1753 (C=O); в УФ-спектре поглощения не
наблюдается.
-трет-Бутил-5-бутил-3-(2-хлорэтилкарбамоилокси)-2,2-диэтилпирролидин-1-оксил
(I-252)

К раствору 0.10 г (0.37 ммоль) НР 39 в ~ 0.5 мл сухого бензола приливали
0.05 мл (0.60 ммоль) 2-хлороэтилизоцианата и каплю триэтиламина. Смесь плотно
закрывали и оставляли при комнатной температуре. Ход реакции контролировали
методом ТСХ (Sorbfil, хлороформ - гексан 1 : 1) Через сутки к смеси прибавляли
такие же количества 2-хлороэтилизоцианата и триэтиламина и выдерживали еще 1
сутки. После завершения реакции к массе приливали ~ 0.10-0.30 мл воды и
выдерживали еще сутки. Органический раствор отделяли пипеткой, неорганическую
массу промывали 1 раз бензолом (1 мл). Объединенный органический раствор сушили
сульфатом магния, растворитель удаляли при пониженном давлении. Остаток
хроматографировали на колонке с силикагелем, элюент хлороформ - гексан 1 : 1.
Выход: 0.07 мг (50 %).
Желтоватые кристаллы. Тпл=102-105оС (гексан) (Найдено C, 60.93; H, 9.64; Cl,
9.45; N, 7.35. Рассчитано для C19H36ClN2O3:
C, 60.70; H, 9.65; Cl, 9.43; N, 7.45 %); νmax (KBr) /см-1 3340 (N-H), 3059 (H-C=,
Ph), 2966, 2874 (СН3, СН2), 1727 (С=О); в УФ спектре поглощение не наблюдается.
-трет-Бутил-5-бутил-3-(2-хлорацетокси)-2,2-диэтилпирролидин-1-оксил (I-251)
К охлажденному до ~ -10оС раствору 0.11 г (0.39 ммоль) НР 39 в ~ 0.5 мл
сухого бензола, помещенному в круглодонную колбу с резиновой пробкой, вкалывали
шприцем 0.13 мл (0.93 ммоль) сухого триэтиламина и раствор 0.03 мл (0.39 ммоль)
2-хлороацетилхлорида в 0.07 мл сухого бензола. Ход реакции контролировали
методом ТСХ (Sorbfil, хлороформ - гексан 1 : 4) Через сутки в колбу вкалывали
еще столько же раствора 2-хлороацетилхлорида в бензоле. Выдерживали еще 6 ч,
после чего промывали реакционную смесь 1 раз водой (~ 1 мл), сушили сульфатом
магния и удаляли растворитель при пониженном давлении. Остаток
хроматографировали на колнке с силикагелем, элюент хлороформ - гексан 1 : 4.
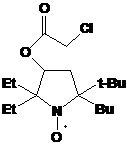
Выход: 0.09 г (67 %)
Бесцветное масло. (Найдено: C, 62.43; H, 9.86; Cl, 9.76; N, 4.05. Рассчитано для C18H33ClNO3: C, 62.32; H,
9.59; Cl, 10.22; N, 4.04 %); νmax (в тонком слое) /см-1 2962, 2875
(СН3, СН2), 1765, 1741 (С=О); в УФ спектре поглощения не наблюдается.
-(4-Бромбутокси)-5-трет-бутил-5-бутил-2,2-диэтилпирролидин-1-оксил
(I-293)
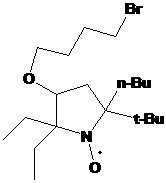
К раствору 0.3 г (1.13 ммоль) радикала 36 в 2 мл ДМСО при перемешивании
присыпали 0.2 г (8.3 ммоль) гидрида натрия. Через 10 мин к смеси прибавили 2 мл
дибромбутана и перемешивали, заткнув колбу хлоркальциевой трубкой, в течение 10
ч. Затем реакционную смесь разбавили 8 мл воды и экстрагировали ЧХУ (3 раза по
5 мл). Экстракт промыли водой (3 раза по 10 мл), сушили сульфатом магния.
Растворитель удалили при пониженном давлении. Остаток хроматографировали на
колонке с силикагелем, элюент: гексан, затем смесь: метил-трет-бутиловый эфир :
гексан = 1 : 200.
Выход: 0.37 г (81 %) Желтое масло. (Найдено: С, 60.04; Н, 9.78; Br,
20.10; N, 3.10. Рассчитано для C20H39BrNO2, C,
59.25; H, 9.70; Br, 19.71; N, 3.45 %); νmax (в тонком слое)/см-1 2961, 2932, 2874 (СН3, СН2),
1113 (R-O-R'); в УФ-спектре поглощения не наблюдается.
(4-(5-трет-Бутил-5-бутил-2,2-диэтил-3-[4-(трифенилфосфонио) бутокси]
пирролидин-1-оксил бромид (I-294B)
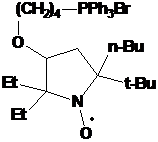
.26
г (0.65 ммоль) НР I-293 и 1 г трифенилфосфина растворили в 2 мл сухого бензола.
Раствор нагревали до 70-75 оС с обратным холодильником при перемешивании в
течение 4-х дней. Бензол удалили при пониженном давлении. Остаток
хроматографировали на колонке с силикагелем, элюент: метанол : хлороформ = 3 :
100. В результате хроматографии были выделены 2 новых соединения: I-294A и
I-294B.B
Выход:
0.21 г (%). Желтое вязкое масло. (Найдено: C, 68.35; H,
8.15; Br, 11.97; N, 2.10; P, 4.64.
Рассчитано для C38H54BrNO2P: C, 68.35; H, 8.15; Br, 11.97; N,
2.10; P, 4.64 %);
νmax (CCl4) /см-1 3059 (H-C=, Ph), 2963, 2934, 2875 (СН3,
СН2), 1100 (R-O-R'); λmax (EtOH)/нм
226 (lg ε 4.31), 262 (lg ε 3.56), 268 (lg ε 3.60), 275 (lg ε 3.52).A
Выход:
0.22 г (%). Желтое вязкое масло.
Рециклизация
енаминокетона 35
Метод
A
-трет-бутил-2,2-диэтил-2,4-дигидро-пиррол-3-он-1-оксид
(36) получали по известной методике [39]. Гидролиз енаминокетона 35 проводили
10% раствором соляной кислоты в течение 84 ч, нейтрализацию раствора
осуществляли насыщенным водным раствором карбоната натрия, осадок пирролинона
36 перекристаллизовывали из гексана. Выход 60 %.
Метод
B
К
раствору 7.6 г (28.7 ммоль) енаминокетона 35 в ~ 40 мл метанола приливали 40 мл
концентрированной соляной кислоты и выдерживали при комнатной температуре 40 ч.
Удаляли из реакционной смеси метанол при пониженном давлении, приливали к смеси
еще ~ 20 мл воды. Присыпая мелкими порциями твердый гидрокарбонат калия (всего
44 г) при перемешивании в атмосфере аргона, нейтрализовывали реакционную смесь
до рН 6-7 и перемешивали в атмосфере аргона еще 30 мин.
Водный
раствор реакционной массы экстрагировали хлороформом (3 раза по 10 мл),
экстракт промывали водой (3 раза по 20 мл), сушили сульфатом магния.
Растворитель удаляли при пониженном давлении. Остаток перекристаллизовывали из
смеси этилацетат - гексан 1 : 10. Выход пирролин-N-оксида (36) 2.9 г (47%).
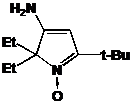
Выпавшее
на стенках колбы с нейтрализованной реакционной массой бесцветное масло смывали
хлороформом, полученный раствор промывали насыщенным водным раствором хлорида и
карбоната натрия. Хлороформный раствор сушили сульфатом магния. После удаления
растворителя при пониженном давлении остаток растерли с гексаном, выпавший
осадок 3-амино-5-трет-бутил-2,2-диэтил-2H-пиррол-1-оксида (I-248A)
перекристаллизовывали из гексана. Выход: 0.9 г (15%). Желтые кристаллы. νmax (KBr)/см-1:
3320 (N-H- водородносвязанные), 3150 (О-Н-водородносвязанные),
2972, 2877 (CH3, CH2), 1661, 1588 (С=N, C=C); δH (300 MГц; CDCl3)/м.д.
0.55 (6Н, т, Jт = 7.2Гц, СН3, Et), 1.26 (9H, c, CH3, t-Bu),
1.40, 1.92 (оба 2Н, АВк, Jк = 7.2 Гц, JAB = 13.8 Гц, CH2, Et),
4.63 (2Н, уш. с, NН2), 5.06 (1H, c, H-C4); dC (75 MГц; CDCl3)/м.д. 7.21 (CH3, Et),
26.54 (CH3, t-Bu), 28.70 (CH2, Et), 33.90 (C(CH3)3),
77.01 (C2), 91.70 (C4), 153.59 (C3),
159.09 (C5).
При
комнатной температуре соединение I-248A разлагается за несколько дней. Смесь продуктов его
распада хроматографировали на колонке с силикагелем, элюент хлороформ -гексан 1
: 4. В результате хроматографии были выделены 2 новых продукта:
2-трет-бутил-5,5-диэтил-2H-пиррол-3,4-дион-1-оксид (I-248B1) и
2-трет-бутил-5,5-диэтил-4-имино-4,5-дигидропиррол-3-он-1-оксид (I-248B2).
трет-бутил-5,5-диэтил-2H-пиррол-3,4-дион-1-оксид
(I-248B1)
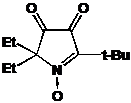
Желтые
кристаллы. Тпл = 31-31оС. (Найдено: C, 64.29; H,
8.49; N, 6.49. Вычислено для C12H19NO3: C,
63.98; H, 8.50; N, 6.22 %); Найдено: М 225, М -СН3 210; вычислено для C12H19NO3:
М 225; νmax (KBr)/см-1:
2927, 2855, 2769 (CH3, CH2), 1769, 1696 (уш. О=С-C=О); λmax (EtOH)/нм
239 (lg ε 3.83), 328 (lg ε 3.96); δH
(400 MГц; CDCl3)/м.д. 0.69 (6H, т, Jт =
7.4 Гц, CH3, Et), 1.43 (9H, c, CH3, t-Bu),
1.88, 2.00 (оба 2Н, АВк, Jк = 7.4 Гц, JAB = 14.2 Гц, CH2, Et); dC (100 MГц; CDCl3)/м.д.
7.28 (CH3, Et), 25.71 (CH3, t-Bu),
27.25 (CH2, Et), 34.41 (C(CH3)3), 81.70 (C5),
159.63 (C2), 178.56 (C3), 197.35 (C4).
-трет-бутил-5,5-диэтил-4-имино-4,5-дигидропиррол-3-он-1-оксид
(I-248B2)
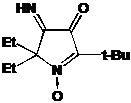
Желтое
масло. νmax
(в тонком слое)/см-1: 3369, 3207 (N-H), 2972, 2935,
2882 (CH3, CH2), 1771, 1703, 1678, 1624 (О=С, C=N); M/z,
Найдено: М 224, М -СН3 209; вычислено для C12H20N2O2: 224; δH
(400 MГц; CDCl3)/м.д. 0.63 (6H, т, Jт =
7.4 Гц, CH3, Et), 1.39 (9H, c, CH3, t-Bu),
1.93, 2.08 (оба 2Н, АВк, Jк = 7.4 Гц, JAB = 14.4 Гц, CH2, Et); dC (100 MГц; CDCl3)/м.д.
7.02 (CH3, Et), 25.71 (CH3, t-Bu),
28.71 (CH2, Et), 33.94 (C(CH3)3), 81.65 (C5),
153.93 (C2), 171.42 (C4), 178.29 (C3).
8.5 Термическое разложение
трет-бутил-бутил-замещенных нитроксильных радикалов
Термическое разложение 2-трет-бутил-2-бутил-3-фенил-1,4-диаза-спиро[4.5]дека-3-ен-1-оксила
(24) в присутствии TEMPO.
Смесь 0.31 г (0.9 ммоль) НР 35 и 1.6 г (10.3 ммоль) ТЕМРО поместили в
круглодонную колбу с краном и удалили воздух, дважды повторив замораживание
образца жидким азотом, откачивание на форвакуумном насосе и размораживание.
Дегазированную смесь нагревали до 70-80 оС при перемешивании в течение 10 ч.
Затем избыток TEMPO удалили экстракцией раствора реакционной массы в 10 мл
гексана водным раствором аскорбиновой кислоты (гексановый раствор энергично
перемешивали с ~20 мл водного раствора аскорбиновой кислоты, прибавляя новые
порции кислоты, в течение 2 ч). Далее органический слой отделили, водный -
экстрагировали этилацетатом (3 раза по 5 мл). Объединенный экстракт промыли
насыщенным водным раствором хлорида натрия и гидросульфата калия, сушили
сульфатом магния. Растворитель удалили при пониженном давлении. Остаток
хроматографировали на колонке с силикагелем, элюент: хлороформ - гексан (1 :
2). В результате хроматографии были выделены 2 продукта: 1-трет-бутокси-2,2,6,6-тетраметил-пиперидин
(t-Bu-TEMPO) и 2-Бутил-3-фенил-1,4-диазаспиро[4,5]дека-1,3-диен-1-оксид (12).
-трет-бутокси-2,2,6,6-тетраметил-пиперидина (t-Bu-TEMPO).
Выход: 0.09 г (48 %). Спектры ПМР и 13С t-Bu-TEMPO соответствуют
литературным данным.
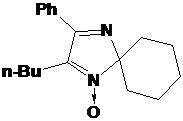
-Бутил-3-фенил-1,4-диазаспиро[4,5]дека-1,3-диен-1-оксид (12), Выход (70
%), IR, бесцветное масло, M/z. Найдено: 284.18927. Рассчитано для C18H24N2O: 289.18886; νmax (в тонком слое)/cm-1: 3061 (=C-H, Ar), 2934, 2860 (CH3, CH2), 1587, 1562, 1514 (C=N, С=С), 1481,
1447, 1387, 1290, 1226, 1168, 1068, 1016, 966, 912, 779, 699; λmax (EtOH)/нм 231 (lg ε
3.96), 271 (3.79); δH (300
МГц; CDCl3)/м.д. 0.86 (3Н, т, Jт=7.2 Гц, СН3, n-Bu), 1.34 (4Н, м, СН2-СН2-СН3, n-Bu), 1.52 (3H, м, СН2, (СН2)5), 1.90 (5H, м, СН2, (СН2)5), 2.16 (2H, м, СН2, (СН2)5), 2.74 (2Н, т, Jт=7.7 Гц, СН2-С2, n-Bu), 7.49 (3Н, м, м- и п-Н, Ph), 7.75 (2Н, м, о-Н, Ph); δC (75 МГц; CDCl3)/м.д. 13.53 (СН3), 22.45 (СН2-С2, n-Bu), 23.11 (С3’, (СН2)5), 23.61, 27.36 (СН2-СН2-СН3, n-Bu), 24.69 (С4’, (СН2)5), 34.91 (С2’, (СН2)5), 101.44 (C5), 127.53 (о-C, Ph), 128.71 (м-C, Ph), 130.55 (п-C, Ph), 132.98 (и-C, Ph), 139.66 (C2),
166.71 (C3).
Термическое разложение 5- трет-бутил-5-
бутил-2,2-диэтилпирролидин-3-он-1-оксила (35) в присутствии TEMPO.
Смесь 0.3 г (1.12 ммоль) НР 35 и 1.6 г (10.3 ммоль) ТЕМРО поместили в
круглодонную колбу с краном и удалили воздух, дважды повторив замораживание
образца жидким азотом, откачивание на форвакуумном насосе и размораживание.
Дегазированную смесь нагревали до 70-80 оС при перемешивании в течение 7 дней.
Затем реакционную смесь хроматографировали на колонке с силикагелем (элюент -
сначала гексан, затем диэтиловый эфир : гексан, градиент до соотношения 1 : 2).
В результате хроматографии были выделены 2 продукта:
1-трет-бутокси-2,2,6,6-тетраметил-пиперидин (t-Bu-TEMPO) и
4,4’-би(5-бутил-3-оксо-2,2-диэтил-3,4-дигидропирролилиден)-1,1’-диоксид (I-286A).
-трет-бутокси-2,2,6,6-тетраметил-пиперидина (t-Bu-TEMPO).
Выход: 0.09 г (38 %). Спектры ПМР и 13С t-Bu-TEMPO соответствуют
литературным данным
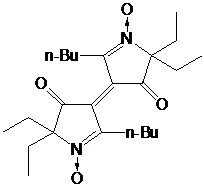
,4’-би(5-бутил-3-оксо-2,2-диэтил-3,4-дигидропирролилиден)-1,1’-диоксид (I-286A)
Перекристаллизовали из гексана. Выход:0.2 г (85 %). Темно-малиновые
кристаллы Тпл=95-100оС. (Найдено: С, 69.06; Н, 8.98; N, 6.55. Рассчитано для C24H38N2O4: C, 68.87; H, 9.15; N, 6.69
%); νmax (KBr)/см-1 2954, 2929, 2872 (СН3, СН2), 1708 (С=О), 1461,
1409, (C=N, С=С); λmax (EtOH)/нм
228 (lg ε 3.89), 284 (lg ε 3.71), 341 (lg ε 3.81), 508 (lg ε 4.19); δH (300 МГц; CDCl3)/м.д. 0.65 (3H, т, Jт 7.4
Гц, CH3, Et), 0.86 (3Н, т, Jт=7.4 Гц, СН3, n-Bu), 0.91 (3H, т, Jт 7.4
Гц, CH3, Et), 1.39 (2Н, сек, Jсек 7.4 Гц, СН2-СН2-СН3, n-Bu), 1.60 (2Н, м, СН2-СН2-СН3, n-Bu), 1.83, 1.94 (4H, АВ, JAB 14.3 Гц, Jк 7.4 Гц, 2CH2, Et), 2.95 (2Н, м, СН2-С=, n-Bu); δC (75 MГц; CDCl3)/м.д.
7.02 (СН3, Et), 13.65 (CH3, n-Bu), 22.24 (СН2-С=, n-Bu), 25.97 (СН2-СН2-СН3, n-Bu), 27.14
(СН2-СН2-СН3, n-Bu), 27.65 (СН2, Et),
101.44 (C5), 80.57 (C2), 122/45 (C4),
159.55 (C5), 199.05 (C=O).
8.6 Алкоксиамины
Синтез алкоксиаминов. Общая методика.
Алкоксиамины на основе полученных нитроксильных радикалов синтезировали
по аналогии с методикой Матьяшжевского (Matyjaszewski) [37].
Смесь НР (2.20 ммоль), алкилбромида (2.25 ммоль), порошка меди (140 мг,
2.25 ммоль), 4,4’-ди-трет-бутил-2,2’-бипиридина (24 мг, 0.09 ммоль), трифлата
меди (II) (8 мг, 0.023 ммоль) и сухого
бензола (5 мл) помещали в колбу Шленка и удаляли воздух, трижды повторяя
замораживание образца жидким азотом, откачивание на форвакуумном насосе и
размораживание. Смесь перемешивали в течение 3-24 ч в закрытой колбе при 20-75 °C. Ход реакции контролировали с
помощью ТСХ (пластинки Alufolien или Sorbfil, элюент эфир-гексан 1 : 10 или
хлороформ-ЧХУ 1:1). В случае если по данным ТСХ в реакционной смеси оставался
исходный НР, реакционную смесь снова дегазировали, как описано выше, после чего
продолжали при перемешивании нагревать колбу до указанной температуры. После
завершения процесса к реакционной смеси прибавляли 10 мл гексана, не
растворившийся осадок отфильтровывали и промывали гексаном. Гексановый раствор
упаривали при пониженном давлении. Остаток хроматографировали на колонке.
Условия проведения реакции (температура, время), а также способ выделения
алкоксиаминов и их выходы приведены в таблице Х.
Таблица Х: Синтез алкоксиаминов: условия реакции, способы очистки и
выходы
|
№
|
Условия проведения реакции
|
Выделение: колоночная
хроматография
|
Выход, %
|
|
Время, ч
|
Температура, оС
|
сорбент
|
элюент
|
|
|
30а
|
36
|
70-75
|
Al2O3
|
эфир-гексан 1 : 10
|
84
|
|
30b
|
36
|
70-75
|
Al2O3
|
эфир-гексан 1 : 10
|
37
|
|
11b
|
24
|
60
|
SiO2
|
эфир-гексан 1 : 4
|
50
|
|
11a
|
24
|
60
|
SiO2
|
эфир-гексан 1 : 4
|
48
|
|
11c
|
24
|
20-25
|
Al2O3
|
гексан
|
78
|
|
12
|
3
|
20-25
|
Al2O3
|
эфир-гексан 1 : 20
|
90
|
|
I-287A
|
10
|
50
|
SiO2
|
эфир-гексан 1 : 20
|
18
|
|
NL1147
|
3
|
40
|
Al2O3
|
хлороформ-ЧХУ 1 : 1
|
|
NL1071
|
24
|
70-75
|
Al2O3
|
эфир-гексан 1 : 10
|
50
|
|
NL1128
|
7
|
45
|
Al2O3
|
эфир-гексан 1 : 5
|
73
|
|
NL1148
|
5
|
40
|
Al2O3
|
хлороформ-ЧХУ 1:1
|
57
|
2-Метил-2-(2,2-диэтил-3-фенил-1,4-диаза-спиро[4,5]дека-3-ен-1-илокси)-пропионовой
кислоты трет-бутиловый эфир (30a)
Получали согласно общей методике из НР 22а и трет-бутилового эфира
2-бром-2-метилпропановой кислоты. Бесцветное масло. (Найдено: C, 72.51; H, 8.93; N,
6.66. Рассчитано для C26H40N2O3: C, 72.86; H, 9.41; N, 6.54); νmax (в тонком слое)/см-1 3058 (=C-H, Ph), 2935, 2858 (CH3, CH2), 1732 (С=О), 1630, 1576 (C=C, С=N);
δH (400 MГц; CDCl3)/м.д.
0.75 (3H, т, J 8 Гц, CH3, Et), 0.89 (3H, т, J 8 Гц,
CH3, Et), 1.40, 1.41 (оба 3 H, с, O-C(CH3)2CO),
1.47 (9 H, с, CH3, t-Bu), 1.69 (9Н, м, CH2, (CH2)5, Et),
1.88 (3Н, м, CH2, (CH2)5), 2.10 (1Н, м, CH2, (CH2)5), 2.27 (1Н, м, CH2, Et), 7.36 (3Н, м, м-, п-Н, Ph), 7.73 (2H, м,
о-Н, Ph); dC (75 MГц; CDCl3)/м.д. 9.54, 11.40 (CH3, Et), 25.03, 25.12 ((H3C)2C), 27.96 (C(CH3)3), 23.97, 24.56, 26.33, 27.94,
31.10, 33.65, 39.52 (СН2, (CH2)5,
Et), 80.84 (С2), 81.83 (C(CH3)3), 81.84 (С(CH3)2), 93.77 (С5), 127.81 (о-С, Ph), 128.30 (м-С, Ph), 129.50 (п-С, Ph), 135.22 (и-С, Ph), 166.82 (C3), 173.46 (C=O).
2,2-Диэтил-3-фенил-1-(1-фенилэтокси)-1,4-диаза-спиро[4.5]дека-3-ен (30b)
Получали согласно общей методике из НР 22а и (1-бромэтил)бензола. Смесь
диастереомеров (в соотношении ~ 1 : 1.2). Бесцветное масло. (Найдено: C, 80.36; H, 9.34; N,
7.12. Рассчитано для C26H34N2O: C, 79.96; H, 8.77; N, 7.17); νmax (в тонком слое)/см-1 3088, 3061, 3031 (=C-H, Ph), 2934, 2856 (CH3, CH2), 1621, 1575 (С=N, C=C); δH (400 MГц; CDCl3)/м.д.
0.50, 0.83, 0.86, 0.95 (т, J 7.4
Гц, всего 6Н, CH3, Et), 0.65, 1.40, 1.61, 1.87, 2.12, (всего 4Н, м, CH2, Et), 1.35, 1.61, 1.82, 2.04 (всего
10Н, м, СН2, (СН2)5), 1.48, 1.51 (всего 3 H, д, Jд=
6.7 Гц, O-CHCH3), 4.71, 4.75 (1Н, оба к, Jк= 6.7 Гц, H-C), 7.33 (всего 8Н, м, м-, п-Н, Ph), 7.68, 7.74 (всего 2H, м, о-Н, Ph); dC (75 MГц; CDCl3)/м.д. 9.51, 9.55, 11.28, 11.36 (CH3, Et), 21.54, 21.64 (H3C-C-O), 23.87, 23.96, 24.50, 24.68, 26.25, 26.60, 28.33 (двойной
интенсивности), 31.45, 32.56, 34.32, 34.53, 40.39 и 41.97 (CH2, (СН2)5, Et), 80.64, 80.91 (С2); 82.19 (C-H), 92.48 (С5),
127.37, 127.43 (м-С, Ph-CH), 127.62 (п-С, Ph-CH), 128.00, 128.13 (о-С, Ph-CH), 144.15,
144.21 (и-С, Ph-CH), Ph-C=N: 127.77, 127.81 (о-С, Ph-C=N), 128.30 и 128.38 (м-С, Ph-C=N), 129.59, 129.64
(п-С, Ph-C=N), 135.10 (и-С, Ph-C=N), 168.94, 169.51
(С3).
-Метил-2-(2,2,5-триметил-4,5-дифенил-2,5-дигидро-имидазол-1-илокси)-пропионовой
кислоты трет-бутиловый эфир(11b)
Получали согласно общей методике из НР 7b и трет-бутилового эфира 2-бром-2-метилпропановой кислоты.
Бесцветное масло. (Найдено: H,
8.02; N, 6.12. Рассчитано для C26H34N2O3: C, 73.90; H,
8.11; N,
6.63); νmax (в тонком слое)/см-1 3060 (=C-H, Ph), 2981, 2936 (CH3), 1732 (С=О), 1620, 1575 (C=C, С=N); δH (300 MГц; CDCl3)/м.д. 0.96, 1.05 (оба 3H, с, O-C(CH3)2CO), 1.44 (9 H, с,
3CH3, t-Bu), 1.67, 1.69
(оба 3H, с, 2CH3 при С2), 1.88 (3Н, с, CH3, при С5), 7.14, 7.24, 7.32 (каждый 2Н, м, 2Ph), 7.41 (4Н, м, 2Ph); dC (75 MГц; CDCl3)/м.д. 17.28 (СН3 при С5), 23.26,
29.64 (2СН3 при С2), 25.16, 23.54 (O-C(CH3)2CO),
27.53 (C(CH3)3), 77.04 (С5), 80.20 (C(CH3)3), 81.26 (O-C(CH3)2CO), 91.35 (С2), 126.62 (п-С, Ph при С5), 127.32 (м-С, Ph при С5), 127.50 (о-С, Ph при С5), 127.65 (о-С, Ph при С4), 127.92 (м-С, Ph при С4), 129.39 (п-С, Ph при С4), 132.35 (и-С, Ph при С4), 144.25 (и-С, Ph при С5), 169.39 (C4), 173.08 (C=O).
-Метил-2-(2-метил-2,3-дифенил-1,4-диаза-спиро[4,5]дека-3-ен-1-илокси)-пропионовой
кислоты трет-бутиловый эфир (11a)
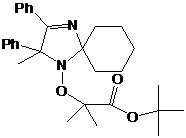
Получали согласно общей методике из НР 7a и трет-бутилового эфира 2-бром-2-метилпропановой кислоты.
Бесцветные кристаллы Тпл.=100-102оС (гексан). (Найдено: C, 75.22; H, 8.23; N,
6.10. Рассчитано для C29H38N2O3: C, 75.29; H, 8.28; N,
6.06 %);
νmax (KBr)/см-1 3062 (=C-H, Ph), 2999, 2977, 2937, 2855 (CH3, CH2), 1730 (С=О), 1613, 1575 (C=C, С=N);
δH (300 MГц; CDCl3)/м.д.
0.97, 1.03 (оба 3Н, с, O-C(CH3)2CO),
1.42, (3Н, м, CH2, (CH2)5), 1.48 (9Н, с, СН3, t-Bu), 1.87 (5Н, м, CH2, (CH2)5), 1.89 (3Н, с, СН3 при С2), 2.12 (1Н, м, CH2, (CH2)5), 2.47 (1Н, м, CH2, (CH2)5), 7.18, 7.26, 7.33 (каждый 2Н, м,
2Ph), 7.46 (4Н, м, 2Ph); dC (75 MГц; CDCl3)/м.д. 17.62 (СН3 при С2), 23.55,
25.14 (CH3, O-C(CH3)2), 27.55 (СН3, t-Bu), 23.51, 24.22, 25.70, 33.66, 38.86 (СН2, (CH2)5), 76.02 (С2), 80.19 (O-C(CH3)2), 81.31 (C(CH3)3), 93.06 (С5), 126.49 (п-С, Ph при С2), 127.42 (м-С, Ph при С2), 127.45 (о-С, Ph при С2), 127.59 (о-С, Ph при С3), 128.03 (м-С, Ph при С3), 129.19 (п-С, Ph при С3), 132.76 (и-С, Ph при С3), 144.50 (и-С, Ph при С2), 168.72 (C3), 173.22 (C=O).
-Метил-2-(2-бутил-2-трет-бутил-3-фенил-1,4-диаза-спиро[4,5]дека-3-ен-1-илокси)-пропионовой
кислоты трет-бутиловый эфир (11c)
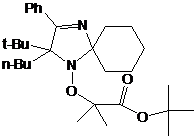
Получали согласно общей методике из НР 4 и трет-бутилового эфира
2-бром-2-метилпропановой кислоты. Бесцветные кристаллы Тпл.=103-105oC. (Найдено: C, 74.31; H,
9.72; N, 5.75. Рассчитано для C30H48N2O3: C, 74.34; H,
9.98; N,
5.78 %); νmax (KBr)/см-1: 3002 (=C-H, Ph), 2959, 2874,
2855 (CH3, CH2), 1740 (С=О); 1635, 1578 (С=N, C=C);
λmax (EtOH)/нм 230 (lg ε
3.95); δH (400 MГц; CDCl3)/м.д.:
0.76 (9H, c, t-Bu при С2); 1.00 (3H, т, Jт=7.2 МГц, СН3, n-Bu), 1.26 (3H, м, CH2, (CH2)5), 1.82 (6Н м, CH2, (CH2)5) и 2.68 (1Н м, CH2, (CH2)5), 1.42, 1.62 (оба 3H, c, OC(СН3)2), 1.46 (9H, c, СН3, Ot-Bu), 1.51 и 1.69 (2H и 4Н, оба м, (СН2)3, Bu), 7.34, 7.50 (3Н и 2Н, оба м, Ph); dC (100 MГц; CDCl3)/м.д.: 14.20 (СН3, Bu), 23.67, 27.21 (OC(СН3)2), 23.49, 24.04, 24.46, 34.81,
38.23 (CH2,(CH2)5), 26.43, 27.52, 29.64 (СН2, Bu); 27.52 (CH3, Ot-Bu), 28.62 (CH3, t-Bu при С2), 37.82 (C(CH3)3, t-Bu при С2); 80.48
(OC(CH3)3), 81.59 (OC(CH3)2), 88.96 (C2); 95.00 (C5);
127.82 (о-C, Ph), 128.55 (м-C, Ph), 128.59 (п-C, Ph), 138.90 (и-C, Ph), 170.01 (C3),
173.85 (C=O).
-Метил-2-(2-бутил-2-трет-бутил-3-фенил-1,4-диаза-спиро[4,5]децен-3-1-ил-окси)-пропионовой
кислоты 4-нитрофениловый эфир (12)

Получали согласно общей методике из НР 4 и 4-нитрофенилового эфира
2-бром-2-метилпропановой кислоты. Бесцветное стекло. (Найдено: C, 69.78; H, 7.91; N,
7.39. Рассчитано для C32H43N3O5: C, 69.92; H, 7.88; N, 7.64 %); νmax (KBr)/см-1: 2958, 2858 (CH3, CH2),
1768 (С=О); 1630, 1616, 1593 (С=N, C=C); 1531, 1348 (NO2); δH (400 MГц; CDCl3)/м.д.: 0.78 (9H, c, 3СН3, t-Bu при С2), 0.98 (3H, т, Jт=6.8 МГц, СН3, n-Bu), 1.32 (3H, м, СН2, (CH2)5),
1.87 (6Н, м, СН2, (CH2)5), 2.63 (1Н,
м, СН2, (CH2)5), 1.64, 1.86 (оба 3H, c, OC(СН3)2), 1.48,
1.77, 1.87 (каждый 2Н, м, (СН2)3, Bu), 7.36, 7.50 (3Н и 2Н, оба м, Ph), 7.31 и 8.29 (4Н, AA’BB’, J=8 Гц, OC6H4NO2); dC (100
MГц; CDCl3)/м.д: 14.16 (СН3, Bu), 23.31, 27.89 (OC(СН3)2), 23.44, 24.11, 24.48, 35.01, 38.31 (СН2, (CH2)5), 26.48, 27.56, 29.50 (СН2, Bu), 28.52 (СН3, t-Bu), 37.86 (C(СН3)3,
t-Bu) 81.23 (OC(СН3)2),
89.05 (C2), 95.29 (C5), Ph:
128.06 (о-C), 128.60 (м-C), 128.93 (п-C),
138.54 (и-C), OC6H4NO2: 122.05, 125.19 (С-Н), 145.27 (C-N), 155.47 (C-O), 172.71 (C=O), 169.97 (C3).
-Метил-2-(2-бутил-2-трет-бутил-3-фенил-1,4-диаза-спиро[4,5]дека-3-ен-1-илокси)-пропионовой
кислоты этиловый эфир (I-287A)
Получали согласно общей методике из НР 4 и этилового эфира
2-бром-2-метилпропановой кислоты. Бесцветные кристаллы Тпл = 98-99 oC (гексан). (Найдено: C, 74.07; H, 9.60; N,
6.36. Рассчитано для C28H44N2O3: C, 73.64; H, 9.71; N, 6.13 %); νmax (KBr)/см-1: 3080, 3050 (=C-H, Ph), 2959, 2925, 2855 (CH3, CH2), 1735 (С=О); 1626 (С=N), 1133 (C-O); λmax (EtOH)/нм
232 (lg ε 3.99); δH (400 MГц; CDCl3)/м.д.:
0.73 (9H, c, CH3, t-Bu); 1.03 (3H, т, Jт=7.4
Гц, СН3, n-Bu), 1.32 (3H, т, Jт=7.1 Гц, СН3, Et), 1.33 (3H, м, CH2, (CH2)5), 1.44,
1.69 (оба 3H, с, CH3, Me), 1.51 (2H, м, CH2-CH3, n-Bu), 1.71 (2H, м,
СН2-СН2-СН3, n-Bu), 1.84 (всего 8H, м, C2-CH2 в n-Bu, CH2 в (CH2)5), 2.66 (1H,
м, CH2,(CH2)5), 4.11, 4.18 (2H, ABк, JAB= 10.4 Гц, Jк= 7.1 Гц, O-CH2, OEt),
7.35 (3H, м, м-,п-Н, Ph), 7.51 (2H, м, o-Н, Ph); dC (100 MГц; CDCl3)/м.д.: 13.96 (CH3, Et), 14.23 (СН3,
Bu), 23.30, 27.70 (OC(СН3)2), 28.34 (CH3, t-Bu), 23.50,
24.18, 24.53, 34.87, 38.10 (CH2,(CH2)5), 26.52, 27.46, 29.25 (СН2, n-Bu), 37.71 (C(CH3)3, t-Bu), 60.60 (O-CH2, Et), 80.81 (OC(CH3)2), 89.03 (C2), 95.17 (C5), 127.98 (о-C, Ph), 128.65 (м-C, Ph), 128.76 (п-C, Ph), 138.82 (и-C, Ph), 170.10 (C3), 175.23 (C=O).
,4-Диметил-2-(2-(3-метокси-3-оксопропил)-5,5-диэтил-2,5-дигидроимидазол-1-илокси)-2-метилпропионовой
кислоты этиловый эфир (NL-1147)
Получали согласно общей методике из НР 19а и этилового эфира
2-бром-2-метилпропановой кислоты.
Cмесь
диастереомеров (в соотношении A : B ~ 2 : 1) Бесцветное масло. (Найдено: C, 62.19; H, 9.27; N,
7.56. Рассчитано для C19H34N2O5: C, 61.60; H, 9.25; N, 7.56); νmax (в тонком слое)/см-1 2984, 2950, 2876 (CH3, СН2), 1739, 1667 (С=О); δH (300 MГц; CDCl3)/м.д.
0.71, 0.79 (оба т, Jт= 7.4 Гц, СН3, Et, форма A), 0.65, 0.80 (оба т, Jт= 7.4
Гц, СН3, Et, форма B) всего 6Н, 1.21
(3Н, т, Jт= 7.0 Гц, СН3, OEt,
формы А и B), 1.32 (с, CH3, OC(СН3)2, форма А), 1.29 (с, CH3, OC(СН3)2, форма B)
всего 6Н, 1.31 ( c, 2-СН3, форма
В), 1.32 ( c, 2-СН3, форма А) всего 3Н, 1.21,
1.31, 1.44 (все м, СН2, Et,
СН2СН2-СО, формы А и В) всего 3Н, 1.72 (с, СН3-С2, форма В), 1.74 (с, СН3-С2,
форма А) всего 3Н, 1.73, 1.89 (все м, СН2, Et, СН2СН2-СО, формы А и В) всего 3Н, 2.16 (м, СН2СН2-СО, форма
В), 2.28 (м, СН2СН2-СО, форма А) всего 1Н, 2.48 (1Н, м, СН2СН2-СО, формы А и
В), 3.56 (с, СН3, ОМе, форма А), 3.58 (с, СН3, ОМе, форма В) всего 3Н, 4.06 (к,
Jк=7.0 Гц, CH2, ОEt,
форма В), 4.08 (к, Jк=7.0 Гц, CH2, ОEt, форма А) всего 2Н; dC (75 MГц; CDCl3)/м.д. 9.14, 10.54 (СН3, Et, форма А), 8.85, 10.64 (СН3, Et, форма В), 13.69 (СН3, ОEt, формы А и В), 16.42 (СН3, Ме-С2, форма В), 16.62 (СН3,
Ме-С2, форма А), 21.06 (СН3, Ме-С4, формы А и В), 24.28, 24.66 (СН3, C(CH3)2CO,
форма А), 24.47, 26.14 (СН3, C(CH3)2CO, форма В), 27.14 (СН2СН2СО, форма В), 27.19 (СН2СН2СО, форма
А), 29.12, 30.02 (СН2, Et, форма А), 29.37, 30.29 (СН2, Et, форма В), 31.84
(СН2СН2СО, форма В), 37.47 (СН2СН2СО, форма А), 51.12 (СН3, ОМе, форма А),
51.15 (СН3, ОМе, форма В), 60.50 (СН2, ОEt, форма В), 60.59 (СН2, ОEt, форма А), 80.88 (С5, форма А), 81.04 (С5, форма В), 81.14 (C(CH3)2CO,
форма А), 81.84 (C(CH3)2CO, форма В), 93.21 (С2, форма В), 93.27 (С2, форма А), 171.32
(С4, форма В), 171.68 (С4, форма А), 173.67 (СООEt, форма В), 173.73 (СООEt,
форма А), 174.21 (СООМе, форма А), 174.35 (СООМе, форма В).
,4-диметил-1-[(1-фенилэтил)-окси]-2,2-диэтил-1,4-диазаспиро[4,5]декан
(NL1071)
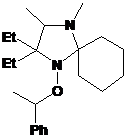
Получали согласно общей методике из НР 19b и (1-бромэтил)бензола. Смесь 4 диастереомеров, бесцветное
масло. (Найдено: C, 77.77; H, 10.13; N, 7.30. Расчитано для C22H36N2O: C, 76.69; H, 10.53; N, 8.13); νmax (в тонком слое)/см-1 3086, 3064 (Н-С=, Ph), 3031, 2930, 2876 (СН3, СН2), 2786
(СН3-N); δH (300
MГц; CDCl3)/м.д. 0.37, 0.39, 0.54, 0.78, 0.83, 1.06, 1, 10,
1.12 (всего 6H, каждый т, Jт= 7 Гц, CH3, Et), 0.92, 0.95, 1.00, 1.01 (всего 3H, каждый д, Jд= 7 Гц, 3-CH3),
1.25-2.20 (14 H, м, CH2, Et, (CH2)5), 1.51, 1.55 (всего 3H, оба д, Jд= 7 Гц, CH3, PhCHCH3), 2.27, 2.31, 2.34, 2.36 (всего 3H, каждый с, N-CH3), 2.46, 2.53,
2.88, 2.95 (всего 1H, каждый к, Jк= 7 Гц, N-CH), 4.74, 4.79,
5.24, 5.21 (всего 1H, каждый к, Jк= 7 Гц, O-CH), 7.29-7.44 (5H, м, Ph); dC (75 MГц; CDCl3)/м.д. 8.11, 8.29, 8.97, 9.36, 9.68, 9.99, 10.18,
11.04 (CH3, Et), 13.56, 13.63, 14.80, 14,93 (3-CH3), 22.11, 22.39, 22.83, 22.92 (CH3, PhCHCH3),
21.24, 22.48, 22.77, 23.77, 23.90, 24.77, 25.16, 25.28, 25.38, 25.82, 26.61,
26.88, 26.93, 27.26, 27.69, 29.19, 29.54, 30.41, 31.39, 32.48, 33.77, 35.78,
36.23 (CH2, Et, (CH2)5),
34.61, 34.69, 35.16, 35.69 (N-CH3), 58.56, 58.67, 62.66, 62.84 (N-CH), 66.63, 67.33, 69.05, 69.52 (C2), 81.77, 81.95, 82.02, 82.69 (O-CH(CH3)Ph), 81.34, 82.21 (C5), 125.96, 126.06, 126.16, 126.65, 126.73, 126.79, 126.90, 126.96,
127.15, 127.19, 127.70, 127.75, 127.84, 127.88, 127.98, 128.16, 128.51 (o-,м-,п-С, Ph), 143.96, 144.34, 144.64, 144.74 (и-C, Ph).
-(2-(3-метокси-3-оксопропил)-2,3,4-триметил-5,5-диэтилимидазолидин-1-илокси)-2-метилпропановой
кислоты трет-бутиловый эфир (NL1128)
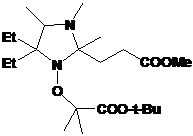
Получали согласно общей методике из НР 10 и трет-бутилового эфира
2-бром-2-метилпропановой кислоты. Смесь 2-х диастереомеров (~ 1 : 6).
Бесцветное масло. (Найдено: C,
63.74; H, 10.57; N, 6.69. Расчитано для C22H42N2O5: C, 63.74; H,
10.21; N, 6.76); νmax (в тонком слое)/см-1 2977, 2882
(СН3, СН2), 2796 (СН3-N), 1736 (C=O); δH (300 MГц; CDCl3)/м.д.
0.82, 0.95 (оба т, Jт= 7.6 Гц, СН3, Et, форма А), 0.88, 0.96 (оба т, Jт= 7.6 Гц, СН3, Et, форма В) всего 6Н, 0.91 (д, Jд= 6.4 Гц, СН3, Ме-С4, форма В), 1.00 (д, Jд= 6.4 Гц, СН3, Ме-С4, форма А) всего
3Н, 1.00 (3Н, с, СН3, Ме-С2, формы А и В), 1.33, 1.36 (оба с, CH3, OC(СН3)2, форма А), 1.27, 1.30 (оба с, CH3, OC(СН3)2,
форма В), 1.40 (с, СН3, t-Bu, форма В), 1.41 (с, СН3, t-Bu, форма А) всего 9Н, 1.13-1.84 (5Н, м, СН2, Et, СН2СН2СО, формы А и В), 1.86-2.15
(1Н, м, СН2СН2СО, формы А и В), 1.97 (с, СН3, Me-N, форма В), 1.99 (с, СН3,
Me-N, форма А) всего 3Н, 2.33-2.53 (2Н, м, СН2СН2СО, формы А и В), 2.55 (к, Jк=
6.4 Гц, Н-С4, форма В), 2.59 (к, Jк= 6.4 Гц, Н-С4, форма А) всего 1Н, 3.59 (с,
СН3, OMe, форма А), 3.60 (с, СН3, OMe, форма В); dC (75 MГц; CDCl3)/м.д.
форма А (основная): 9.08, 10.00 (СН3, Et), 13.09 (СН3, 4-Ме), 15.03 (СН3,
2-Ме), 24.32, 25.14 (OC(СН3)2),
27.60 (СН3, t-Bu), 27.29, 28.02 (СН2, Et), 31.19 (СН2СН2СО), 31.99 (СН2СН2СО),
32.38 (CH3, N-Me), 50.98 (CH3, OMe), 62.81 (C-H), 67.32 (C5), 80.58 (C(CH3)3, t-Bu), 81.57 C(CH3)2CO),
82.85 (C2), 173.43 (СООt-Bu), 175.31 (СООМе).
-(2-(3-метокси-3-оксопропил)-2,3,4-триметил-5,5-диэтилимидазолидин-1-илокси)-2-метилпропановой
кислоты этиловый эфир (NL1148)
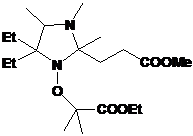
Получали согласно общей методике из НР 10 и этилового эфира
2-бром-2-метилпропановой кислоты. Смесь 2-х диастереомеров (~ 1 : 6).
Бесцветное масло. (Найдено: C,
57.68; H, 9.74; N, 6.27. Расчитано для C20H38N2O5: C, 62.15; H, 9.91; N,
7.25); νmax (в тонком слое)/см-1 2979, 2947,
2882, 2845 (СН3, СН2), 2797 (СН3-N), 1737 (C=O); δH (300 MГц; CDCl3)/м.д.
0.81, 0.93 (оба 3H, т, Jт= 7.6 Гц,
СН3, Et), 0.98 (3H, c, CH3, 2-Me), 0.99 (3H, д, Jд= 6.7 Гц, CH3, 4-Me), 1.22 (3H, т, Jт= 7.1 Гц, СН3, OEt), 1.35, 1.39 (6H, оба с, CH3, OC(СН3)2),
1.17-1.83 (6Н, м, СН2, Et,
СН2СН2СО), 1.98 (3H, с, СН3, Me-N),
2.35-2.62 (2Н, м, СН2СН2СО), 2.57 (1H, к, Jк= 6.7 Гц, Н-С4,), 3.58 (3H, с, СН3, OMe), 4.09 (2H, к, Jк= 7.1 Гц, СН2, OEt); dC (75 MГц; CDCl3)/м.д.
форма А (основная): 8.86, 9.73 (СН3, Et), 12.84 (CH3, OEt),
13.48 (СН3, 4-Ме), 14.76 (СН3, 2-Ме), 23.98, 24.69 (OC(СН3)2), 27.04, 27.66 (СН2, Et), 31.00 (СН2СН2СО), 31.66
(СН2СН2СО), 32.07 (CH3, N-Me), 50.72 (CH3, OMe), 60.28 (CH2, OEt),
62.52 (C-H), 67.20 (C5),
80.46 (C(CH3)2CO),
82.66 (C2), 173.85 (СООEt), 175.05 (СООМе).
-(3-(1-трет-бутокси-2-метил-1-оксопропан-2-илокси)-1,2,5-триметил-4,4-диэтилимидазолидин-2-ил)пропаноат
натрия (NL1128-2)
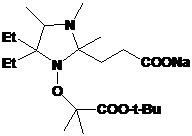
К раствору алкоксиамина NL1128
(0.50 г, 1.21 ммоль) в 6 мл этанола приливали раствор гидроксида натрия (0.25
г, 6.25 ммоль) в 2 мл воды. Через 2 ч - после исчезновения исходного соединения
по данным ТСХ (Al2O3, хлороформ - ЧХУ 1 : 1) - к
реакционной смеси присыпали гидрокарбонат натрия (~ 1 г) и удаляли этанол при
пониженном давлении. К остатку приливали изопропанол и снова упаривали смесь
при пониженном давлении для удаления остатков воды. После этого к массе
приливали изопропанол, не растворившийся неорганический остаток отфильтровывали
и промывали изопропанолом. Органический раствор упаривали при пониженном
давлении, остаток растирали с сухим эфиром и отфильтровывали полученный осадок
соли. Выход: 0.46 г (90%).
Желтоватые кристаллы, гигроскопичное. (Найдено: C, 58.84; H,
9.13; N, 6.71. Расчитано для C21H39N2NaO5:
C, 59.69; H, 9.30; N, 6.63, золы 7.34 %); νmax (в тонком слое)/см-1 2977, 2881
(СН3, СН2), 2792 (СН3-N), 1728 (C=O, в COOt-Bu), 1590 (С=О, в COONa).
-(2-(2-карбоксилатоэтил)-2,3,4-триметил-5,5-диэтилимидазолидин-1-илокси)-2-
метилпропаноат натрия (NL1149)
получали аналогично описанному для NL1128-2 при гидролизе диэфира NL1148. Выход: 95%.
Желтые кристаллы, гигроскопичное. νmax (KBr)/см-1 2973, 2943, 2880 (СН3, СН2), 2793 (СН3-N), 1561
(С=O, в COONa); δH (300 MГц; CD3OD)/м.д. 0.85, 1.03 (оба 3H, т, Jт= 7.6 Гц, СН3, Et), 1.05 (3H, д, Jд=
6.5 Гц, CH3, 4-Me), 1.05 (3H, c, CH3, 2-Me), 1.35, 1.40 (6H, оба с, CH3, OC(СН3)2),
1.30-1.48 (1Н, м, СН2СН2СО), 1.51-1.81 (4Н, м, СН2, Et), 1.98-2.16 (1H, м,
СН2СН2СО), 2.07 (3H, с, СН3, Me-N),
2.26-2.52 (2Н, м, СН2СН2СО), 2.63 (1H, к, Jк= 6.5 Гц, Н-С4,).
Выводы
1. Получен ряд пространственно-затруднённых НР имидазолинового,
имидазолидинового и пирролидинового рядов, представляющих интерес в качестве
регуляторов радикальной полимеризации виниловых мономеров и в качестве
устойчивых к восстановлению спиновых зондов для биомедицинских исследований.
2. Показано, что введение на начальном этапе синтеза объёмного
заместителя, не склонного к металлированию, к атому углерода нитронной группы
позволяет в дальнейшем использовать высокореакционноспособные литийорганические
соединения для введения второго объёмного заместителя к тому же атому.
Использование этого принципа позволило предложить стратегию синтеза
пространственно-затруднённых НР с двумя объёмными заместителями при одном α-атоме углерода.
. Установлено, что пространственно-затруднённые НР, содержащие
бутильный и трет-бутильный заместители при одном α-атоме углерода склонны претерпевать
деструкцию с отщепленеием трет-бутильной группы и образованием соотвтетсвующего
бутилнитрона. Показано, что механизм термического распада этих НР включает
гомолитическое отщепление трет-бутильного радикала, т.е., реакции, обратной
спиновому захвату.
. На примере взаимодействия 3-гидроксиамино-3-этилпентан-2-она с
пивалевым альдегидом обнаружено необычное направление реакции конденсации,
приводящее к образованию
6-трет-бутил-3,3-диэтил-4-метил-3,6-дигидро-2Н-1,2,5-оксадиазина.
Литература
1. M.A.
Foster, I.A. Grigor’ev, D.J. Lurie, V.V. Khramtsov, S. McCallum, I.
Panagiotelis, J.M. Hutchison, A. Koptioug, I. Nicholson, In vivo detection of a
pH-sensitive nitroxide in the rat stomach by low-field ESR-based techniques,
Magn. Reson. Med. - 2003. - V. 49. - P. 558-567.
. V.V.
Khramtsov, Biological imaging and spectroscopy of pH, Curr. Org. Chem. - 2005.
- V. 9. - P. 909-923.
. Kocherginsky,
N.; Swartz, H. M. Nitroxide spin labels. Reactions in biology and chemistry.
Boca Raton: CRC Press FL; 1995.
. Swartz,
H. M.; Timmins, G. S. The metabolism of nitroxides in cells and tissues. In:
Rhodes, C.J. ed., Toxicology of the human environment: the critical role of
free radicals. London: Taylor & Francis; 2000: 91-111.
. Benjamin
P. Soule, Fuminori Hyodo, Ken-ichiro Matsumoto, Nicole L. Simone, John A. Cook,
Murali C. Krishna, James B. Mitchell, The chemistry and biology of nitroxide
compounds // Free Rad. Biol. Med. - 2007. - V. 42. - P. 1632-1650.
. Розанцев,
Э.Г. Свободные иминоксильные радикалы. - М.: Химия, 1970. - 224 с.
. Hahn,
S. M.; Krishna, M. C.; DeLuca, A. M.; Coffin, D.; Mitchell, J. B. Evaluation of
the hydroxylamine Tempol-H as an in vivo radioprotector. // Free Radic. Biol.
Med. - 2000. - V. 28. - P. 953-958.
. Заремский,
М.Ю., Голубев, В.Б. Обратимое ингибирование в радикальной полимеризации // Выс.
Молек. Соед., Сер. С - 2001. - Т. 43. - № 9. - С. 1689-1728.
. Hawker,
C.J., Bosman, A.W., Hart, E. New polymer synthesis by nitroxide mediated living
radical polymerizations // Chem. Rev. - 2001. - V. 101. - N 12. - P. 3611-3660.
. G.Delaitre,
J.Nicolas, C. Lefay, et al. // Chem.Comun. 2005. - P. 614-616.
. M.Nowakowska,
A.Karewicz, M. Klos, et al. // Macromol. 2003. - V. 36. - P. 2136-2139
. Kirilyuk,
I.A., Bobko, A.A., Grigor’ev, I.A., Khramtsov, V.V. Synthesis of the tetraethyl
substituted pH-sensitive nitroxides of imidazole series with enhanced stability
towards reduction // Org. Biomol. Chem. - 2004. - N 2. - P. 1025-1030.
. Marx,
L., Chiarelli, R., Guiberteau, T., Rassat, A. A comparative study of the
reduction by ascorbate of 1,1,3,3-tetraethylisoindolin-2-yloxyl and of
1,1,3,3-tetramethylisoindolin-2-yloxyl // J. Chem. Soc., Perkin Trans. 1 -
2000. - P. 1181-1182.
. Yu.
Kinoshita, K. Yamada, T. Yamasaki, H. Sadsue, K. Sakai, & H. Utsumi,
Development of novel nitroxyl radicals for controlling reactivity with ascorbic
acid // Free Rad. Res. 2009. - V. 43. - N. 6. - P. 565-571.
. Kathryn
E. Fairfull-Smith, Farina Brackmann, and Steven E. Bottle, The Synthesis of
Novel Isoindoline Nitroxides Bearing Water-Solubilising Functionality // Eur.
J. Org. Chem. 2009. - P. 1902-1915.
. Заремский,
М.Ю., Орлова, А.П., Гарина, Е.С. и др. Псевдоживая радикальная полимеризация с
участием макромолекулярных нитроксилов на основе нитронов // Выс. Молек. Соед.,
Сер. А - 2003. - Т.45. - № 6. - С. 871-882.
. Grigor’ev,
I.A. Nitrones: Novel Strategies in Synthesis. In: Nitrile Oxides, Nitrones, and
Nitronates in Organic Synthesis. Novel Strategies in Synthesis. - 2nd Ed. / Ed.
Feuer, H. // Hoboken, New Jersey.: John Wiley & Sons, Inc. 2008. -P.
129-434.
. Имидазолиновые
нитроксильные радикалы / Володарский, Л.Б., Григорьев, И.А., Диканов, С.А. и
др. - Новосибирск: Наука, 1988. - 216 с.
. Studer,
A., Harms, K., Knoop, Ch., Muller, Ch. and Schulte, T. New Sterically Hindered
Nitroxides for the Living Free Radical Polymerization: X-ray Structure of an
R-H-Bearing Nitroxide. // Macromolecules. - 2004. - N 37. - P. 27-34.
. Nesvadba,
P., Kramer, A., Steinmann, A. and Stauffer. - 1999. - PCT Int. Appl. - WO
99/03894.
. Vertommen,
L.L., Van Den Haak, H.J. Hope, P., Lacroix, C.P., Meijer, J. and Talma, A.G.
1998. - PCT Int. Appl. - WO 98/13392.
. Braslau,
R., O’Bryan, G., Nilsen, A., Henise, J. et.at. The Synthesis and Evaluation of
New a-Hydrogen Nitroxides for ‘Living’ Free Radical Polymerization. //
SYNTHESIS. - 2005. - N 9. - P. 1496-1506.
. Розанцев,
Э.Г., Шолле, В.Д. Органическая химия свободных радикалов. _ М.: Химия, 1979. -
281 с.
. Розанцев,
Э.Г., Шолле, В.Д. О необычных продуктах окисления некоторых третичных аминов //
Изв. Акад. Наук СССР, Сер. Хим. - 1969. - № 1. - С. 149-151.
. Yoshioka,
T., Higashida, S., Murayama, K. Stable free radicals. VIII. Synthesis and
oxidation of hindered 4-oxopiperidine derivatives // Bull. Chem. Soc. Jap. -
1972. - V. 45. - N 2. - P. 636-638.
. Nesvadba,
P., Bugnon, L. and Sift, R. New 7-Membered Diazepanone Alkoxyamines for
Nitroxide-Mediated Radical Polymerization. // J. Polym. Sci. Part A: Polym.
Chem. - 2004. - V 42. - P. 3332-3341.
. Brunetti,
H., Rody, J. Neue derivate von 4-oxopiperidinen. // Ciba-Geigy. FR 2324637.
1976. DE 2621855. 1976.
. Nesvadba,
P. Beyond TEMPO. Synthesis of Cyclic Sterically Highly Hindered Nitroxides and
Alkoxyamines and their Industrial Applications. - // 4-th International
conference on nitroxide radicals: synthesis, proper-ties and implications of
nitroxides (spin-2005), Book of Abstracts, (Novosibirsk, Russia, September
20-24, 2005), P. 26
. Ma,
Z., Huang, Q. and Bobbitt, J. M. Oxoammonium Salts. 5.' A New Synthesis of
Hindered Piperidines Leading to Unsymmetrical TEMPO-Type Nitroxides. Synthesis
and Enantioselective Oxidations with Chiral Nitroxides and Chiral Oxoammonium
Salts. // J. Org. Chem. - 1993. - V 58. -N 18. - P. 4837-4843.
. Kinoshita,
Y., Yamada, H., Sadasue, T., Yamasaki, K. Sakai, K. and Utsumi, H. Stability
Evaluation for 2,6-substituted TEMPO derivatives in the Reduction Process. //
5-th International conference on nitroxide radicals. Book of abstracts. Ancona (Italy),
7-11th september 2008. SPIN 2008. - P27.
. Miura,
Y., Nakamura, N. and Taniguchi, I. Low-Temperature “Living” Radical
Polymerization of Styrene in the Presence of Nitroxides with Spiro Structures.
// Macromolecules. - 2001. - V 34. - N 3. - P. 447-455.
. Schulte,
T., Siegenthaler, K.O., Luftmann, H., Letzel, M. and Studer, A.
Nitroxide-Mediated Polymerization of N-Isopropylacrylamide: Electrospray
Ionization Mass Spectrometry, Matrix-Assisted Laser Desorption Ionization Mass
Spectrometry, and Multiple-Angle Laser Light Scattering Studies on
Nitroxide-Terminated Poly-N-isopropylacrylamides. // Macromolecules. - 2005. -
V 38. - N 16. - P. 6833-6840.
. Wetter,
Ch., Gierlich, J., Knoop, Ch.A., Mёller, Ch., Schulte, T. and Studer, A. Steric
and Electronic Effects in Cyclic Alkoxyamines-Synthesis and Applications as
Regulators for Controlled/Living Radical Polymerization. // Chem. Eur. J. -
2004. - N 10. - P. 1156 - 1166.
. Dickerman,
S.C., Lindwall, H.G. Studies in piperidone chemistry. I. A synthesis of 5-homopiperazinones
// J.Org.Chem. - 1949. - V. 14. - N 4. - P. 530-536.
. Chang,
Ch.-Ch., Siegenthaler, K.O. and Studer, A. A New Sterically Highly Hindered
7-Membered Cyclic Nitroxide for the Controlled Living Radical Polymerization.
// Helw. Chim. Acta. - 2006. - V 89. - P. 2200-2210.
. Matyjaszewski,
K., Woodworth, B.E., Zhang, X. et al. Simple and Efficient Synthesis of Various
Alkoxyamines for Stable Free Radical Polymerization // Macromolecules - 1998. -
N 31. - P. 5955-5957.
. Schulte,
T. and Studer, A. New Seven- and Eight-Membered Cyclic Alkoxyamines for the
Living Free Radical Polymerization. // Macromolecules. - 2003. - V 36. - N 9.
- P. 3078-3084.
. Michon,
J. and Rassat, A. Nitroxides. 73. Electron Spin Resonance Study of Chiral
Recognition by Cyclodextrin. // J.Am.Chem.Soc. - 1979. - P. 995.
. Morat,
C., Rassat, A. Synthesis of oxazolidines substituted by adamantine and of
stable oxazolidine nitroxide derivatives of adamantane // Tetr.Lett. - 1979. -
V. 19. - N 47. - P. 4561-4564.
. Braslau,
R., Burill, L.C., Chaplinski, V. et al. Studies in the stereoselective trapping
of prochiral carbon radicals by optically active camphoxyl nitroxides // Tetr.:
Asym. - 1997. - V. 8. - N 19. - P. 3209-3212.
. Toda,
T., Morimura, S., Murayama, K. Stable free radicals. VII. Mechanism for
cyclization reaction of .alpha.-amino nitriles with carbonyl compounds //
Bull.Chem.Soc.Jap. - 1972. - V.45. - N 2. - P. 557-61.
. Yoshioka,
T., Mori, E., Murayama, K. Stable free radicals. XIII. Synthesis and ESR
spectral properties of hindered piperazine N-oxyls // Bull.Chem.Soc.Jap. -
1972. - V. 45. - N 6. - P. 1855-1860.
. Lai,
J.T. Hindered Amines. 3,3,5,5-Tetrasubstituted-2-oxomorpholines and Derivatives
// Synth. - 1984. - P. 122-123.
. Senkus,
M. Reaction of Primary Aliphatic Amines with Formaldehyde and Nitroparaffins //
J.Am.Chem.Soc. - 1946. - N 68. - P. 10-13.
. Lai,
J.T. Hindered amines. Novel synthesis of 1,3,3,5,5-pentasubstitued
2-piperazinones // J. Org. Chem. - 1980. - V. 45. - N 4 - P. 754-755.
. Son,
P.N., Lai, J.T. Synthesis of
Hexahydro-3,3,5,5,7-pentaalkyl-2H-1,4-diazepin-2-ones from 1,3-Diamines and
Ketones // J.Org.Chem. - 1981. - N 46. - P. 323-327.
. Marx,
L., Rassat, A. Application of a spin-labeled spin-trap to the detection of
nitric oxide (NO) // Angew.Chem., Int. - 2000. - V. 39. - N 24. - P. 4494-4496.
. Ramasseul,
R., Rassat, A. Nitroxides XLIX: Steroidal nitroxides // Tetr. Lett. - 1971. - N
48. - P. 4623-4624.
. Espie,
J.C., Ramasseul, R.and Rassat, A. Nitroxides LXXXV : Nitroxydes Azetidiniques.
.// Tetr. Lett. - 1978. - N 9. - P. 795-796.
50. Graf, R.
β-Isovalerolactam-N-sulfonyl Chloride and β-Isovalerolactam. //
Organic Syntheses, Coll. - 1973. - V 5.- P.673.
51. Sбr,
C.P., Хsz, E., Jekх, J. and Hideg, K. Synthesis of Spiro[pyrrolidine-2,2’-adamantane]
Nitrones and Nitroxides. // SYNTHESIS. - 2005. - N 2. - P. 0255-0259.
. Morozov,
D.A., Kirilyuk, I.A.,Komarov, D.A., Goti, A. and Grigor’ev, I.A. Synthesis of
chiral sterically hindered nitroxide radicals via intramolecular 1,3-dipolar
cycloaddition // 5-th International conference on nitroxide radicals. Book of
abstracts. Ancona (Italy), 7-11th september 2008. SPIN 2008. - P36.
. Резников,
В.А., Володарский, Л.Б. Синтез бифункциональных производных нитроксильных
радикалов имидазолина // Хим.гетероцикл.соедин. - 1990. - № 6. - С. 772-778.
. Dцpp,
D., Dцpp, H. Nitrone. In: Houben-Weil. Methoden der Organischen Chemie. Band E
14b/Teil 2. Organische Stickstoff-Verbindungen mit einer C,N-Doppelbindung. /
Her. Von D. Klamann und H. Hagemann // Stuttgart-N.Y.: George Thieme Verlag,
1990. - P. 1372-1544.
. Hamer,
J. Nitrones // Chem.Rev. - 1964. - V. 64. - N 4. - P. 473-495.
. Мартин,
В.В. Володарский, Л.Б. Вишнивецкая, Л.А. Взаимодействие пространственно
затрудненных имидазолиноксидов - предшественников стабильных нитроксильных
радикалов с металлорганическими реагентами // Изв.Сиб.Отд.Акад.Наук - 1981. - №
4. - С. 94-103.
. Reznikov,
V.A. Interaction of Heterocyclic Nitrones With Organometallic Reagents As a
Method for the Synthesis of New Types of Nitroxides // Tetr. - 1993. - V. 49. -
N 46. - P. 10669-10692.
. Nazarski
R.B., Skowronski R. Sterically crowded five-membered heterocyclic systems. Part
3. Unexpected formation of stable flexible pyrrolodinoxyl byradicals via
nitrone aldol dimers: a spectroscopic and mechanistic study. // J.Chem.Soc.
Perkin. Tr. I. - 1989. - № 9. - P. 1603-1610.
. Braslau,
R., O’Bryan, G., Nilsen, A., Henise, J. et.at. The Synthesis and Evaluation of
New a-Hydrogen Nitroxides for ‘Living’ Free Radical Polymerization. //
SYNTHESIS. - 2005. - N 9. - P. 1496-1506.
. Studer,
A. Tin-Free Radical Cyclization Reactions Using the Persistent Radical Effect.
// Angew. Chem. - 2000. - V 39. - N 6. - P. 1108 - 1111. Supporting
Information. http://www.wiley-vch.de/contents/jc_2002/2000/z14225_s.pdf
. Reznikov,
V.A. and Volodarsky, L.B. Stable Nitroxides With Hydrogen At α-Carbon of
the Nitroxyl Group // Tetr. Lett. - 1994. - V 35. - N 14. - P. 2239 - 2240.
62. Clark,
B.A.J., Evans, T.J., Simmonds, R.G. Preparation and Some Reactions of
2,2-Diaryl-2H-imidazole 1-Oxides // J.Chem.Soc. Perkin Trans. I. - 1975. - N
18. - P. 1803-1806.
. Кирилюк,
И.А., Григорьев, И.А., Володарский, Л.Б. Синтез 2Н-имидазол-1-оксидов и
стабильных нитроксильных радикалов на их основе // Изв. Акад. АН СССР, Сер.
Хим. - 1991. - № 9. - С. 2113-2122.
. Zubenko,
D., Kirilyuk, I., Roshchupkina, G., Zhurko, I. et. al.
2,5-Dihydro-1H-imidazole-Based Nitroxides as Prospective Mediators in Living
Radical Polymerization. // Helvetica Chimica Acta. - 2006. - V 89. - P.
2341-2353.
. Potapenko,
D.I., Foster, M.A., Lurie, D.J., I.A.Kirilyuk, et. al. Real-time monitoring of
drug-induced changes in the stomach acidity of living rats using improved
pH-sensitive nitroxides and low-field EPR techniques. // J. Magn. Reson. - 2006.
- V 182. -N 1. - P. 1-11.
. Uchida,
Y., Matsuoka, N., Takahashi, H., Shimono, S., Ikuma, N. and Tamura, R.
Synthesis, Crystal Structure and Magnetic Properties of
4-(2-Methyl-1-azaspiro[4.5]deca-1-oxyl-2-yl)Phenol. // Heterocycles. - 2007. -
V 74. - P. 607-616.
. Keana,
J.F.W., Cuomo, J., Lex, L. and Seyedrezai, S.E. Azethoxyl Nitroxide
Spin-Labeled Crown Ethers and Cryptands with the N-0 0 Group Positioned near
the Cavity. // J.Org.Chem. - 1983. - N 48. - P. 2647-2654.
. A.A.Bobko,
I.A.Kirilyuk, I.A.Grigor’ev, J.L.Zweier, V.V.Khramtsov. Reversible reduction of
nitroxides to hydroxylamines: roles for ascorbate and glutathione. Free Radic.
Biol. Med., 2007, V. 42, P. 404-412.
. Григорьев,
И.А., Бакунова, С.М., Кирилюк, И.А. Взаимодействие циклических -метоксинитронов
с нуклеофильными реагентами // Изв. Акад. Наук, Сер. Хим. - 2000. - №12. - С.
2066-2071.
. Кирилюк,
И.А., Григорьев, И.А., Володарский, Л.Б. Получение 3-имидазолинов и
3-имидазолин-3-оксидов, содержащих атом водорода у углерода С-2 // Изв. СОАН
СССР. Сер. Хим. Наук - 1989. - Вып. 2. - №4. - C. 99-106.
. Пуцыкин,
Ю.Г., Володарский, Л.Б. Таутомерия кольцо - цепь 1-окси-3-имидазолин-3-оксидов
и их превращение в N-окиси имидазола и 2-изоимидазола // Изв. СО АН СССР. Сер.
Хим. Наук. - 1969. - №9. - Вып. 4. - С. 86-93.
. Володарский,
Л.Б., Лысак, А.Н., Коптюг, В.А. Химия -гидроксиаминооксимов. Х.
Конденсация анти--гидроксиаминооксимов с кетонами и получение N-окисей
2-изоимидазола // Хим.гетероцикл.соедин. - 1968. - № 2. - С. 334-338.
. Леви,
Г., Нельсон, Г. Руководство по ядерному магнитному резонансу углерода-13 для
химиков органиков // Перев. С англ. Сергеева Н.М. - Изд.: «Мир». - М.: 1975. -
296 с.
. Shun-Ichi
Murahashi,* Hitoshi Mitsui, Tatsuki Shiota, Tomoyasu Tsuda, and Shoji Watanabe
Tungstate-Catalyzed Oxidation of Secondary Amines to Nitrones. a-Substitution
of Secondary Amines via Nitrones // J. Org. Chem. - 1990. - V 55. -
P.1736-1744.
. Войнов,
М.А. Последовательность металлирование-электрофильное замещение в ряду
альдонитронов - новый подход к синтезу альфа-замещенных нитронов: Дис. … канд.
хим. наук: 02.00.03 / М.А. Войнов. Новосиб. инст. органической химии. -
Новосибирск, 2001. - 175 с.
. [
] Титце, Л., Айхер, Т. Препаративная органическая химия / Пер. с нем. К.В.
Аванесян и др. Под ред. Ю.Е. Алексеева. - М.: Мир, 1999. - 704с.
. [
] Keana J.F.W. New aspects of nitroxide chemistry. In: Spin labeling: Theory
and applications. / Berliner L.J. (Ed.). - N.Y. - London.: Academic Press.
1979. - P. 115-173.
. [
] Lee T.D., Keana J.F.W. Nitroxides derived from
3,4-dehydro-2,5-dimethyl-2H-pyrrol-1-oxide: a new series of minimum steric
pertubation lipid spin labels. // J.Org.Chem. - 1978. - V.43, №21. - P.
4226-4231.
. Имидазолиновые
нитроксильные радикалы / Володарский, Л.Б., Григорьев, И.А., Диканов, С.А. и
др. - Новосибирск: Наука, 1988. - 216 с.
. Hamer,
J. Nitrones // Chem.Rev. - 1964. - V. 64. - N 4. - P. 473-495.
. Мартин,
В.В. Володарский, Л.Б. Вишнивецкая, Л.А. Взаимодействие пространственно
затрудненных имидазолиноксидов - предшественников стабильных нитроксильных
радикалов с металлорганическими реагентами // Изв.Сиб.Отд.Акад.Наук - 1981. - №
4. - С. 94-103.
. Володарский
Л.Б, Резников В.А., Кобрин В.С..// Журн.Орган.Химии. - 1979.- Т. 15.- № 2. -С.
415-422.
. M.
A. Voinov, J. F. Polienko, T. Schanding, A. A. Bobko,V. V. Khramtsov, Y. V.
Gatilov, T. V. Rybalova, A. I. Smirnov, I. A. Grigor’ev. - Synthesis,
Structure, and X-Band (9.5 GHz) EPR Characterization
. of
the New Series of pH-Sensitive Spin Probes: N,N-Disubstituted
-Amino-2,2,5,5-tetramethyl-3-imidazoline 1-Oxyls// J. Org. Chem. 2005, 70,
9702-9711.
. Репинская-Шварцберг
. [
] I. A. Kirilyuk, T. G. Shevelev, D. A. Morozov, E. L. Khromovskih, N. G.
Skuridin, V. V. Khramtsov and I. A. Grigor'ev, Synthesis, 2003, 6, 871-878.
. [
] Kirilyuk, I.A., Bobko, A.A., Grigor’ev, I.A., Khramtsov, V.V. Synthesis of
the tetraethyl substituted pH-sensitive nitroxides of imidazole series with
enhanced stability towards reduction // Org. Biomol. Chem. - 2004. - N 2. - P.
1025-1030.
. [
] D. Zubenko, Yu. Tsentalovich, N. Lebedeva, I. Kirilyuk, G. Roshchupkina, I.
Zhurko, V. Reznikov, S. R. A. Marque, E. Bagryanskaya Laser Flash Photolysis
and CIDNP Studies of Steric Effects on Coupling Rate Constants of Imidazolidine
Nitroxide with Carbon-Centered Radicals, Methyl Isobutyrate-2-yl and tert-Butyl
Propionate-2-yl // J. Org. Chem. - 2006, - V 71. - N 16 - P. 6044-6052.
. [
] Резников В.А., Володарский Л.Б. Ацетат аммония как катализатор конденсации
пространственно-затрудненных функционально-замещенных гидроксиламинов с
кетонами // Изв. РАН сер.хим. - 1997 - N9. - C. 1654-1658.
. И.
А. Кирилюк, Д. А. Морозов, Ю. С. Табатчикова, В. С. Медведев, А. В. Лебедев, Г.
В. Романенко, Т. В. Рыбалова, И. А. Григорьев, Синтез
4Н-имидазол-5-карбальдоксим-3-оксидов и 4Н-имидазол-5-карбонитрил-3-оксидов.
Изв. АН. Сер. хим., 2008, № 7, 1487-1503.
. Boyer
J.H., Increasing the index of covalent oxygen bonding a nitrogen attached to carbon.
//Chem. Reviews, 1980,-V. 80, -No. 6, - P. 495-561.
. [
] Григорьев И. А., Кирилюк И. А., Стариченко В.Ф., Володарский Л.Б. Получение
стабильных нитроксильных радикалов с аминогруппой у -углеродного атома
радикального центра при окислительном аминировании N,N-диоксидов и N-оксидов
4Н-имидазола. // Известия АН СССР. Сер. хим. -1989. - № 3. - С. 661-667.
. [
] Keana, J.F.W., Van Nice, F.L. Influence of structure on reduction of
nitroxide MRI contrast-enhancing agents by ascorbate // Physiol. Chem. Phys.
Med. NMR - 1984. - N 16. - P. 477-480.
. [
] Keana, J.F.W. Synthesis and Chemistry of Nitroxide Spin Lables. In: Spin
Labeling in Pharmacology. / Ed. J.L. Holtzman // Orlando, Fla.: Academic Press,
1984. - P. 1-85.
95. [ ] R. Bonnet, R.F.C. Brown, V.M.
Clark, I.O. Sutherland, A. Todd Experiments towards the Synthesis of Corrins.
Patr II. The Preparation and Reactions of Δ1-Pyrroline 1-Oxides. //
J.Chem.Soc. - 1959. - P. - 2094-2102.
96. [
] Nazarski R.B., Skowronski R. Sterically crowded five-membered heterocyclic
systems. Part 3. Unexpected formation of stable flexible pyrrolodinoxyl
byradicals via nitrone aldol dimers: a spectroscopic and mechanistic study. //
J.Chem.Soc. Perkin. Tr. I. - 1989. - № 9. - P. 1603-1610.
97. [ ] R.F.C. Brown, V.M. Clark, I.O.
Sutherland, A. Todd Experiments towards the Synthesis or Corrins. Part V.
Base-catalysed Aldol-type Reactions of Δ1-Pyrroline 1-Oxides. //
J.Chem.Soc. - 1959. - P. 2109-2116.
98. [
] Bapat J.B. et al. // Aust.J.Chem. - 1972. - V. 25. - P. 2445-2450.
. [
] Резников, В.А., Володарский, Л.Б. Рециклизация енаминокетонов производных
имидазолидина в 1-пирролин-4-он-1-оксиды // Хим.гетероцикл.соедин. - 1990. - №
7. - С. 921-926.
. Резников,
В.А., Володарский, Л.Б., Рыбалова, Т.В., Гатилов, Ю.В. Стабильные винилнитроксильные
радикалы - производные пирролина. // Изв. Акад. Наук СССР Сер. Хим. - 2000. - №
1. - С. 103-111.
. Резников,
В.А., Вишнивецкая, Л.А., Володарский, Л.Б. Взаимодействие 2-замещенных
5,5-диметил-4-оксо-1-пирролин-1-оксидов с электрофильными реагентами. // Изв.
Акад. Наук СССР Сер.Хим.- 1990. - № 2. - С. 395-400.
. [
] В.А. Резников, В.В. Мартин, Л.Б. Володарский Окислительная димеризация
гетероциклических нитронов - производных пирролина и имидазолина. // Изв. АН
СССР, сер. хим. - 1990. -. N 6. - C. 1398-1404.
. M.P.
Murphy and R.A.J. Smith Targeting Antioxidants to Mitochondria by Conjugation
to Lipophilic Cations // Annu. Rev. Pharmacol. Toxicol. - 2007. - V 47. - P
629-656.
. A.T.
Hoye, J. Davoren, and P. Wipf Targeting Mitochondria // Acc. Chem. Res. - 2008.
- V 41. - N 1. - P 87-97.
105. V. E. Kagan, J. Jiang, H.
Bayır, D. A. Stoyanovsky Targeting nitroxides to mitochondria: location,
location, location, and …concentration Highlight Commentary on “Mitochondria superoxide dismutase
mimetic inhibits peroxide-induced oxidative damage and apoptosis: Role of
mitochondrial superoxide” // Free Radical Biology & Medicine - 2007. - V
43. - P 348-350.
. Dhanasekaran,
S. Kotamraju, Ch. Karunakaran, Sh.V. Kalivendi,
. S.
Thomas, J. Joseph, B. Kalyanaraman Mitochondria superoxide dismutase mimetic
inhibits peroxide-induced oxidative damage and apoptosis: Role of mitochondrial
superoxide // Free Radical Biology & Medicine. 2005. - V 39. - P 567 - 583.
. Л.
Беллами Новые Данные по ИК-Спектрам Сложных Молекул / пер. с англ. Э.Г.
Тетериной. Под ред. Ю. А. Пентина. - М: Мир, 1971. - 318 с.
. Маша
. 1H:
JOC 1994, 59, 14, 3969
. 13C:
JOC 1998, 63, 26, 9763
. Багрянская
. Mariya
V Edelevaa, Dmitry P Zubenkoa, Irina F. Zhurkob, Igor A Kirilyukb, Marque
Sylvainc, Didier Gigmesc and Elena G Bagryanskayaa H-transfer reaction in
imidazoline-, imidazolidine-, and pyrrolidine-based alkoxyamines: NMR study.
. [
] Bertin D., Gigmes D., Marque S.R.A. // Recent Res. Devel. Organic Chem. -
(2006). № 10. C. 63
. Matyjaszewski,
K., Woodworth, B.E., Zhang, X. et al. Simple and Efficient Synthesis of Various
Alkoxyamines for Stable Free Radical Polymerization // Macromolecules - 1998. -
N 31. - P. 5955-5957.
. Брюстер
Р., Грениг Т. Синтезы органических препаратов. Сб.4 / ИЛ, М. - 1953. - с.194.
. MARIYA
V. EDELEVA,1,2 IGOR A. KIRILYUK,3 DMITRY P. ZUBENKO,2 IRINA F. ZHURKO,3 SYLVAIN
R. A. MARQUE,4 DIDIER GIGMES,4 YOHANN GUILLANEUF,4 ELENA G. BAGRYANSKAYA
Kinetic Study of H-Atom Transfer in Imidazoline-, Imidazolidine-, and
Pyrrolidine-Based Alkoxyamines: Consequences for Nitroxide-Mediated
Polymerization // Journal of Polymer Science: Part A: Polymer Chemistry DOI
10.1002/pola. - 2009. - P. 6579-6595.
. Kocherginsky,
N.; Swartz, H. M. Nitroxide spin labels. Reactions in biology and chemistry.
Boca Raton: CRC Press FL; 1995.
. Swartz,
H. M.; Timmins, G. S. The metabolism of nitroxides in cells and tissues. In:
Rhodes, C.J. ed., Toxicology of the human environment: the critical role of
free radicals. London: Taylor & Francis; 2000: 91-111.
. Kuppusamy,
P.; Li, H.; Ilangovan, G.; Cardounel, A. J.; Zweier, J. L.; Yamada, K.;
Krishna, M. C.; Mitchell, J. B. Noninvasive imaging of tumor redox status and
its modification by tissue glutathione levels. Cancer Res. 62:307-312; 2002.
. Ide,
T.; Tsutsui, H.; Kinugawa, S.; Suematsu, N.; Hayashidani, S.; Ichikawa, K.;
Utsumi, H.; Machida, Y.; Egashira, K.; Takeshita, A. Direct evidence for
increased hydroxyl radicals originating from superoxide in the failing
myocardium. Circ Res. 86(2):152-7; 2000.
. [
] Володарский Л.Б., Лысак А.Н., Коптюг В.А. Химия α-гидроксиламинооксимов. Х.
конденсация анти-α-гидроксиламинооксимов с кетонами и
получение N-окисей 2-изоимидазола. // Хим. гетероцикл. соедин. 1968, - №2, - С.
334-338
. Углерод
большой - не менее процента
. Выход
взят от фонаря, т.к. полвина вещества была выделена в виде имидазолина, который
потом окисляли отдельно, а вторая - в виде 2Н-имидазола.
. A.A.Bobko,
I.A.Kirilyuk, I.A.Grigor’ev, J.L.Zweier, V.V.Khramtsov. Reversible reduction of
nitroxides to hydroxylamines: roles for ascorbate and glutathione. Free Radic.
Biol. Med., 2007, V. 42, P. 404-412.
. Zubenko,
Dmitry; Tsentalovich, Yuri; Lebedeva, Nataly; Kirilyuk, Igor; Roshchupkina,
Galina; Zhurko, Irina; Reznikov, Vladimir; Marque, Sylvain R. A.; Bagryanskaya,
Elena; Journal of Organic Chemistry; 71; 16; 2006; 6044 - 6052
. 77-81
oC
. 1.43,
1.49 (2H, AB6, JAB=14.0 Гц, J6=7.4 Гц, СН2-СН2-СН3, n-Bu),
. [
] В. А. Резников, Л. Б. Володарский // Химия гетероцикл. Соединений - 1990. -
C. 772. [Chem. Heterocycl. Compd. - 1990. - V.26. - N 6. - P. 643.]
. Нет
УФ
. Слабо
выраженная полоса
. Возможно
существует в N-OH таутомерной форме (13С сигнал 165.54 - далеко для С-нитрона,
обычно бывает 145-155 м.д.)
. Анализ
по азоту подогнан - на самом деле по азоту 19.46 - не очень хороший (его больше
почти на процент)
. С
60.26
. Нет
уверенности, что полосы отнесены верно
. Нет
уверенности, что полосы отнесены верно
. 60.64
. Нужно
попробовать записать в тонком слое
. М.
б. колбу Шленка
. 1H:
JOC 1994, 59, 14, 3969
. 13C:
JOC 1998, 63, 26, 9763
. М.
б. колбу Шленка
. 1H:
JOC 1994, 59, 14, 3969
. 13C:
JOC 1998, 63, 26, 9763