Оксисоединения альдегиды и кетоны
Контрольная
работа
Оксисоединения
альдегиды и кетоны
Содержание:
Введение
.
Гомологический ряд, номенклатура
.
Способы получения оксосоединений
.
Строение оксосоединений
.
Свойства оксосоединений
.
Присоединение нуклеофилов
.
Реакции присоединения-отщепления
Литература
Введение
Оксосоединения характеризуются присутствием в
молекулах группы С = О, называемой карбонилом или карбонильной группой. Она
придает данным соединениям специфические свойства, которые разительно отличают
их от рассмотренных выше соединений других классов. Если карбонильный атом
связан с одним атомом углерода и водородом, т.е. карбонил расположен на краю
цепи, то соединение принадлежит к классу альдегидов; если карбонильная группа
связана с двумя углеродными атомами, т.е. карбонил находится внутри цепи, то
речь идет о классе кетонов. Карбонильная группа может также входить в состав
функциональной группы карбоновых кислот (карбоксила) и их производных, но эти
классы соединений будут рассмотрены в соответствующих главах.
Химические свойства альдегидов и кетонов имеют
много общего, хотя есть и различия. Как альдегиды, так и кетоны, могут иметь в
составе молекулы алифатические, непредельные и ароматические фрагменты, что
также влияет на их реакционную способность.
1. Гомологический ряд, номенклатура
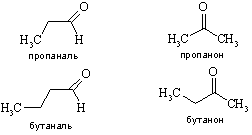
Гомологический ряд алифатических альдегидов
начинается формальдегидом (муравьиным альдегидом).

Названия альдегидов по номенклатуре
IUPAC образуют путем прибавления окончания -аль к названию соответствующего
алкана (метан + аль = формальдегид, пропан + аль = пропионовый альдегид).
Однако зачастую названия альдегидов производят от названий соответствующих
карбоновых кислот (уксусный альдегид, пропионовый альдегид, масляный альдегид).
Когда в молекуле альдегида имеется более старшая по правилам номенклатуры
группа, -СНО-группа обозначается в названии соединения приставкой «формил»,
например:

Начиная с пропаналя, наблюдается
изомерия альдегид - кетон
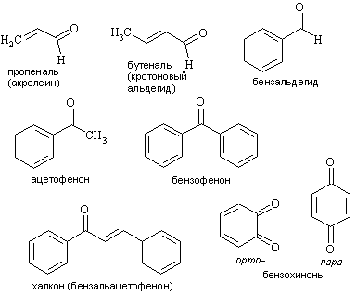
Официальные названия кетонов
образуются прибавлением окончания - он к названию алкана с указанием положения
карбонильной группы арабскими цифрами (например, пентанон-2). По другой
номенклатуре - радикальной, для составления названия кетона указывают
углеводородные радикалы и прибавляют окончание -кетон (например,
метилпропилкетон = пентанон-2, диметилкетон = пропанон-2 = ацетон).
Распространены тривиальные названия (ацетон, ацетофенон).
. Способы получения оксосоединений
Для получения альдегидов и кетонов
могут быть использованы реакции окисления-восстановления, замещения,
присоединения, отщепления,
Реакции окисления
Альдегиды могут быть получены
окислением или дегидрированием первичных спиртов, кетоны - вторичных. Альдегиды
легко окисляются дальше в карбоновые кислоты и поэтому для получения альдегидов
применяют технические приемы, позволяющие предотвратить этот процесс. Так,
образующийся альдегид можно отгонять из реакционной смеси в ходе реакции,
благодаря тому, что в подавляющем большинстве случаев температура кипения
альдегида ниже, чем у соответствующего спирта. Можно также связывать альдегид в
момент его образования в устойчивое к действию окислителей производное:
например, при проведении окисления в уксусном ангидриде альдегид превращается в
устойчивый диацетат.
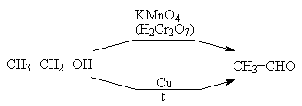
Применяют некоторые селективные
окислители, например, активированную двуокись марганца или диоксид селена,
которые не взаимодействуют с альдегидами в мягких условиях или взаимодействуют
с низкой скоростью.
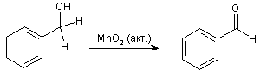
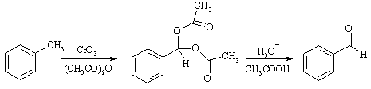
Альдегид можно получить окислением
не только спиртовой группы, но и метила, связанного с ароматическим кольцом.
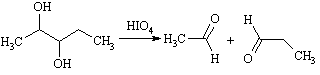
Карбонильные соединения образуются в результате
перйодатного расщепления 1,2-диолов.
Реакции восстановления
Для получения альдегидов используют
реакции восстановления карбоновых кислот и их функциональных производных.
Весьма легко претерпевают восстановление нитрилы (при действии HCOO-
NH4+), хлорангидриды и сложные эфиры (восстановитель
LiAlH4). Используется также каталитическое гидрирование.
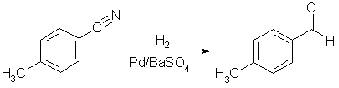
Реакции замещения
Реакции замещения для получения
альдегидов и кетонов чаще применяются в ароматическом ряду. Введение альдегидной
группы называют формилированием; в тех случаях, когда происходит замена
водорода на кетонную группу, превращение обозначают термином «ацилирование».
Для введения ацила непосредственно в
ароматическое ядро используется реакция Фриделя-Крафтса. Катализатор - кислота
Льюиса - служит для активации ацилирующего реагента, в данном случае
хлорангидрида.
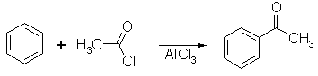
Известны различные методы
формилирования ароматических молекул, их объединяет то, что такие реакции
осуществимы лишь на активированных субстратах (фенолах и их эфирах, анилинах,
p-избыточных гетероциклах и т.д.)
Для формилирования по
Вильсмайеру-Хааку используют амиды муравьиной кислоты - N,N-диметилформамид и
N-метилформанилид, которые в виде комплексов с хлорокисью фосфора замещают
протон в ароматическом соединении формильной группой.

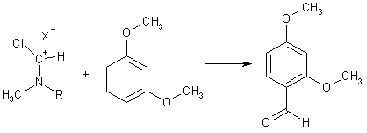
Формилирование по Реймеру-Тиману
проводят в щелочной среде действием хлороформа, который является источником
реагента - дихлоркарбена. Последний атакует электроноизбыточный анионный
субстрат.

Реакции гидратации и гидролиза
К этому типу относятся те реакции, в
ходе которых карбонильная группа формируется на основе уже имеющегося в молекуле
исходного соединения атома углерода.
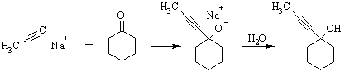
При обработке ацетилена водными
растворами кислот в присутствии солей ртути (II) образуется
ацетальдегид, другие ацетилены в этих условиях превращаются в кетоны. Эта
реакция гидратации ацетиленов носит имя Кучерова.
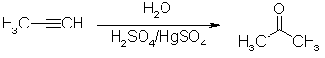
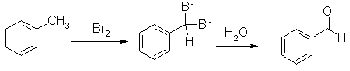
Оксосоединения могут быть получены
также гидролизом гем-дигалогеналканов. Особенно часто этот метод применяется
для синтеза бензальдегидов, т.к. соответствующие a,a-дигалогенметилбензолы
(бензилиденгалогениды) доступны благодаря возможности галогенирования боковых
цепей гомологов бензола.
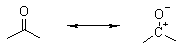
. Строение оксосоединений
Карбонил является электронным
аналогом этиленовой группы с тем отличием, что группа С=О полярна по причине
разной электроотрицательности углерода и кислорода. Спиновая плотность p-связи
смещена к атому кислорода, что наглядно иллюстрирует полярная каноническая
структура карбонильной группы.
Кроме того, по причине легкой
поляризуемости (деформируемости) p-связи в группе С=О и стремления атома
кислорода приобрести стабильную электронную конфигурацию под действием как
электрофилов, так и нуклеофилов происходит ее дополнительная поляризация.
Переход кислорода в восьмиэлектронное состояние достигается как в результате
взаимодействия с электрофилами, так и при нуклеофильной атаке по карбонильному
атому углерода.
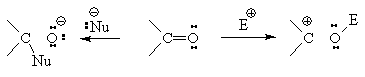
Дипольный момент альдегидов и
кетонов лежит в пределах 2,5-2,8 D. Карбонильная группа плоская, так как атом
углерода имеет гибридизацию sp2. Валентные углы несколько отличаются
от 120о. Длина связи С=О составляет около 122 пм. Атом кислорода
несет две несвязывающие пары электронов.

Метод молекулярных орбиталей
позволяет охарактеризовать электронную плотность на атомах карбонильной группы:
С 0,552; О 1,552 (эффективный заряд: С 0,448; О -0,448) и порядок p-связи,
равный 0,894. Таким образом, карбонильный атом углерода является электрофильным
реакционным центром, куда направляется атака нуклеофилов.
Дефицит электронной плотности на
карбонильном атоме углерода, обусловленный полярностью С=О-связи, является
причиной отрицательного индуктивного эффекта относительно a-углеродного атома и
наведения на нем частичного положительного заряда. В результате этого атомы
водорода при a-С-атоме способны отрываться сильными основаниями в виде
протонов. В то же время, образующийся при этом енолят-анион проявляет более
высокую стабильность по сравнению с неустойчивыми алкильными радикалами.
Причиной СН-кислотности α-углеродных
атомов альдегидов и кетонов является делокализация отрицательного заряда,
описываемая двумя резонансными стуктурами.

Если обратная реакция,
протонирование енолята, ориентируется по атому кислорода, то в результате
образуется неустойчивая форма карбонильного соединения - енол. Переход
альдегида или кетона в енольную форму и обратно, называемый кето-енольной
таутомерией, может катализироваться не только основаниями, но и кислотами.
Несмотря на то, что для обычных карбонильных соединений равновесная
концентрация енола крайне низка, его присутствие зачастую определяет
направление реакции, т.к. и енол, и тем более енолят-анион, являются сильными
С-нуклеофилами.

Таким образом, оксосоединения
способны воспринять атаку нуклеофилов по карбонильному атому углерода, сильных
оснований по a-углеродному атому. Свободные радикалы могут атаковать
a-углеродный атом алкилкетонов и алифатических альдегидов и атом водорода
альдегидной группы. Следует отметить, что альдегиды в большинстве превращений
более реакционноспособны, чем кетоны.

. Свойства оксосоединений
Окисление
Альдегиды весьма легко окисляются.
Так, бензальдегид, и другие ароматические альдегиды, особенно, содержащие
галоген в кольце, превращаются на воздухе за короткое время в бензойные
кислоты. Окисление кислородом воздуха имеет свободнорадикальный механизм.
Причина окисляемости бензальдегида (алифатические альдегиды окисляются
медленнее) заключается в том, что промежуточно образующийся бензоильный радикал
резонансно стабилизирован.
Последний далее превращается в
надкислоту, которая окисляет молекулу исходного альдегида и восстанавливается.
Оба процесса дают бензойную кислоту.
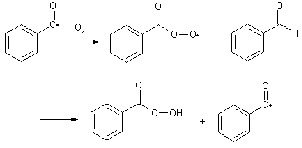

В качестве подтверждения образования
бензоильного радикала приводят реакцию хлорирования бензальдегида на свету, в
результате которой образуется бензоилхлорид.
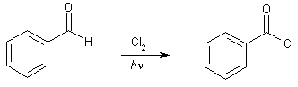
Для защиты альдегидов от окисления
при хранении добавляют стабилизаторы - антиоксиданты, т.е. те соединения,
которые способны останавливать развитие цепи свободнорадикальной реакции,
например, 2,6-ди(трет-бутил)-пара-крезол.
Альдегиды могут быть окислены даже
такими мягкими реагентами, как оксид серебра или оксид меди. В обоих случаях
реакцию проводят в щелочной среде в присутствии комплексообразователей,
способствующих растворению оксида. Используют аммиачный раствор гидроксида
серебра и щелочной раствор гидроксида меди в присутствии винной кислоты
(реактив Феллинга). В первом случае образуется металлическое серебро, которое
покрывает равномерным слоем специально подготовленную стеклянную посуду,
поэтому процесс называют реакцией серебряного зеркала.
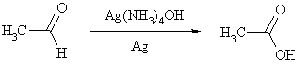
Окисление альдегидов до карбоновых
кислот проводят также действием перманганата калия и других окисляющих
реагентов.
Окисление кетонов требует жестких
условий и происходит с разрушением углеродной цепи, в результате чего
образуется смесь карбоновых кислот.
Восстановление
Восстановление альдегидов и кетонов
действием алюмогидрида лития или борогидрида натрия дает спирты. В ходе реакции
происходит нуклеофильное присоединение гидрид-иона, поставляемого комплексным
гидридом металла, к карбонильному атому углерода.
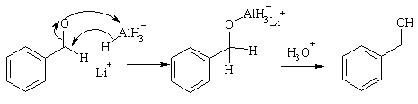
Более глубокое восстановление до
алканов может быть осуществлено двумя методами. Первый заключается в нагревании
альдегида или кетона с амальгамированным цинком в соляной кислоте (реакция
Клемменсена), второй - в щелочном расщеплении заранее полученного гидразона
карбонильного соединения (реакция Кижнера-Вольфа).

К восстановительным превращениям
оксосоединений относится реакция Меервейна-Понндорфа-Верлея. Например,
метилэтилкетон переходит в бутанол-2 при действии изопропилата алюминия.

В этой реакции происходит восстановление
кетонов и альдегидов до вторичных спиртов вторично-спиртовыми производными
алюминия путем гидридного переноса.

Ввиду того, что в реакции образуется
другой кетон, обладающий близкими свойствами, очевидно, что она обратима. Для
смещения равновесия в сторону продуктов реакции ацетон отгоняют.
Альдегиды, не содержащие атомов
водорода в a-положении, вступают в реакцию окислительно-восстановительного
диспропорционирования (реакция Канниццаро), в результате которой из двух
молекул альдегида получается молекула кислоты и молекула спирта. В первую
очередь реакция применима к ароматическим альдегидам. Синтез проводят в
спиртовом или водно-спиртовом растворе крепких щелочей.
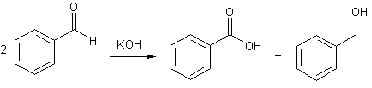
Механизм реакции включает
нуклеофильную атаку карбонильного углерода гидроксид-ионом и последующий
перенос гидрид-иона от образовавшегося анионного аддукта к другой молекуле
альдегида.

В условиях недостатка алкоголята
алюминия в реакции Мейервейна-Понндорфа-Верлея (см. выше) из альдегидов
образуются сложные эфиры, соответствующие кислоте и спирту, которые образуются
в реакции Канниццаро. Считают, что механизм данного превращения аналогичен
предыдущему и тоже связан с гидридным переносом.
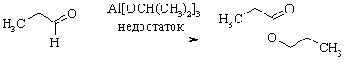
. Присоединение нуклеофилов
При действии на альдегиды и кетоны
тех нуклеофилов, в молекуле которых есть лишь один кислый атом водорода,
происходит раскрытие двойной связи и присоединение элементов реагента. В результате
присоединения нуклеофила карбонильный углерод регибридизуется в
тетраэдрический, отрицательный заряд смещается на кислород, затем к нему
присоединяется протон.
Присоединение простейшего нуклеофила
- гидрид-иона - было рассмотрено выше.
О-нуклеофилы
К карбонильной группе присоединяются
вода и спирты. Эта реакция обратима и, вследствие низкой нуклеофильности
реагентов, требует участия катализатора, чаще всего кислот, которые активируют
карбонильную группу. В результате присоединения воды образуются, неустойчивые в
большинстве случаев, гидраты альдегидов и кетонов.

Равновесная концентрация гидратной
формы для альдегидов выше, чем для кетонов, и возрастает при наличии
электроноакцепторных атомов в молекуле, особенно при a-углеродном атоме. К
соединениям, существующим в гидратированной форме, относятся трихлоруксусный
альдегид (хлоральгидрат), индантрион (нингидрин) и др.
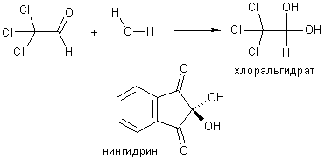
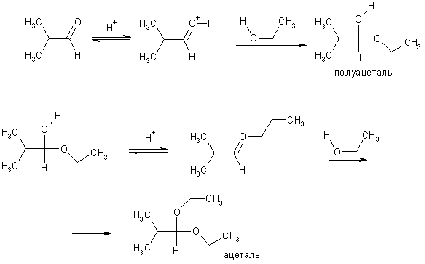
Обратимая реакция со спиртами идет в
присутствии кислот и дает полуацетали, которые в избытке спирта превращаются в
более устойчивые ацетали.
Обе стадии протекают через промежуточное
образование резонансно-стабилизированного катиона.
Ацетали гидролизуются до альдегидов в кислой
среде, но устойчивы к действию оснований. Это позволяет использовать их в
качестве защиты карбонильной группы. Ввиду того, что равновесие этой реакции
смещено влево, синтез ацеталей проводят в присутствии водоотнимающих средств,
либо используют азеотропную отгонку воды (для высококипящих карбонильных
соединений).
Другим примером присоединения
кислорода к карбонильной группе является реакция полимеризации, характерная для
низших алифатических альдегидов. Так, формальдегид существует в виде твердого
малорастворимого полимера, известного под названием параформ. Уксусный
альдегид, напротив, при хранении тримеризуется до так называемого паральдегида.

Высшие алифатические и ароматические
альдегиды существуют, главным образом, в виде мономеров. Полимеризация
альдегидов обратима и протекает под действием даже следов кислот, равно как и
обратный процесс. Для получения мономеров ацетальдегида и формальдегида
полимеры нагревают в присутствии нелетучей кислоты.
S-нуклеофилы
Для альдегидов и кетонов характерна
реакция присоединения гидросульфита натрия. Несмотря на то, что отрицательный
заряд гидросульфит-аниона сосредотоочен на атомах кислорода, реакция идет по
более нуклеофильному атому серы. При этом образуются натриевые соли
a-оксисульфокислот, или бисульфитные аддукты. Это твердые соединения, в
большинстве случаев нерастворимые в органических растворителях и кристаллизующиеся
из воды. Бисульфитные производные альдегидов и кетонов легко гидролизуются
разбавленными кислотами или основаниями до исходных оксосоединений. Благодаря
этому их используют в лабораторной практике для идентификации, а также очистки
альдегидов и кетонов.
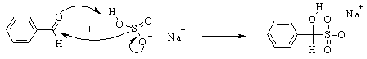
Реакция альдегидов и кетонов с
пропан-1,3-дитиолом приводит к важным в синтетическом отношении продуктам -
1,3-дитианам. Каталитическое гидрирование этих соединений приводит к замене
бывшего карбонила группой СН2.
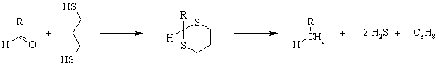
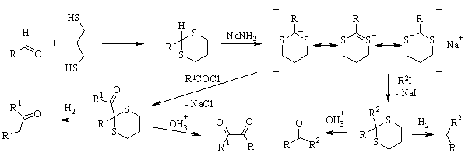
Если 1,3-дитиан синтезирован на
основе альдегида, то он обладает еще более интересными свойствами. Атом
водорода группы СН, находящейся между атомами серы, обладает повышенной
кислотностью и дитианы депротонируются под действием сильных оснований. Причина
заключается в том, что свободные d-орбитали атомов серы способны к
делокализации находящегося рядом отрицательного заряда, поэтому анион дитиана
относительно стабилен. Это явление используется в синтетических целях: анион
дитиана можно алкилировать и ацилировать, а полученный продукт затем либо
восстанавливать, либо гидролизовать. С помощью таких превращений могут быть
получены различные кетоны, a-дикетоны, алканы сложного строения и т.д.
С-нуклеофилы
Альдегиды и кетоны присоединяют
циановодород. В первом случае породукты называют циангидринами или
оксинитрилами. Реакция имеет большое значение в органическом синтезе, т.к.
гидролиз нитрильной группы позволяет синтезировать a-оксикислоты - важный класс
соединений.
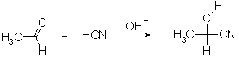
Это первая органическая реакция, для
которой был установлен механизм (Лепуорт, 1904).
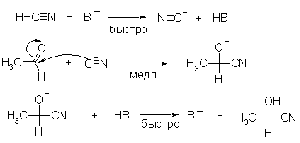
Карбонильная группа присоединяет
реактивы Гриньяра и другие металлоорганические соединения, в результате чего
образуются смешанные металлогалогенидные алкоголяты, из которых гидролизом в
кислой среде выделяют спирты. При использовании формальдегида в данной реакции
получаются первичные спирты, любого другого альдегида - вторичные, кетонов -
третичные.
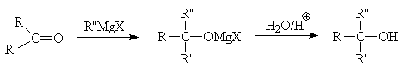
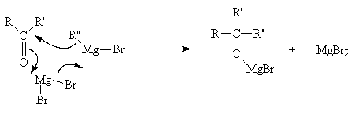
Присоединение магнийорганических
соединений идет легче в присутствии бромида магния. Это дает основания
предполагать, что превращение происходит в шестицентровом переходном комплексе.
Бромид магния катализирует процесс переноса нуклеофила к электронодефицитному
С-атому карбонильной группы.
Ацетилениды щелочных металлов,
комплексы Иоцича, также проявляют высокую активность в качестве нуклеофильных
реагентов при взаимодействии с карбонильными соединениями.
Присоединение образующегося in situ
цинкорганического соединения является ключевой стадией реакции Реформатского.

. Реакции присоединения-отщепления
N-нуклеофилы
Как было сказано выше, присоединение
нуклеофилов к карбонильной группе не всегда завершается образованием устойчивых
продуктов. Поэтому в том случае, когда исходный нуклеофил содержит при
реакционном центре два атома водорода, реакция зачастую не останавливается на
стадии присоединения и происходит отщепление молекулы воды. Например, так идет
реакция, приводящая к иминосоединениям, содержащим двойную связь с атомом азота
при бывшем карбонильном атоме углерода. Формально происходит замещение атома кислорода
азотистым фрагментом, но в ряде случаев удается выделить промежуточные аддукты
- результат присоединения, поэтому реакция не получила титул
"замещение". Реакция присоединения-отщепления наблюдается при
взаимодействии альдегидов и кетонов с такими аминосоединениями, как первичные
амины, гидроксиламин, гидразин, арилгидразины, семикарбазид, тиосемикарбазид.

В зависимости от активности
реагентов, оксосоединения и амина, реакция может идти без катализатора либо в
условиях кислого или основного катализа. Роль кислоты, как и в приведенных выше
примерах, состоит в активации карбонильной группы, что бывает необходимо, когда
применяется слабонуклеофильный (слабоосновный) амин.
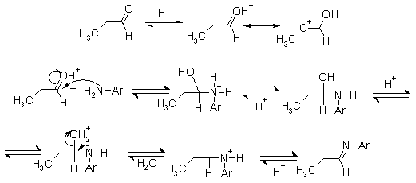
Менее распространенный катализ
основаниями заключается в отрыве протона от атома азота в аддукте, после чего
следует отщепление гидроксид-иона.
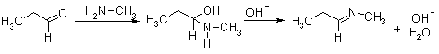
Характер продуктов реакции
оксосоединений с аминами зависит от природы реагентов. Формальдегид с аммиаком
образует неустойчивый метиленимин, который вступает во вторичные превращения,
результатом которых является образование полициклического соединения
гексаметилентетрамина (уротропина). Нужно отметить, что этот аддукт состава 6:4
образуется даже в том случае, когда используются другие соотношения реагентов.
Имины на основе алифатических
альдегидов и кетонов и алкиламинов тоже малоустойчивы; они легко гидролизуются
до исходных, поэтому реакцию проводят с отделением воды. При хранении такие
имины склонны к обратимой тримеризации. Кетоны реагируют медленнее, чем
альдегиды.
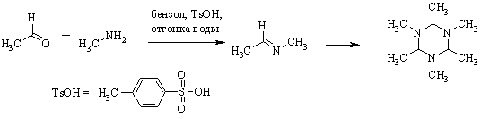
Ароматические альдегиды образуют
более устойчивые имины, особенно если амин - тоже ароматический, в данном
случае реакция идет с хорошим выходом без необходимости удаления воды. Это
связано с тем, что связь C=N стабилизирована сопряжением с ароматическим(и)
ядром(ами). Такие альдимины получили название азометины, а также по имени их
первооткрывателя и исследователя - основания Шиффа.

Наконец, диарилкетоны в обычных
условиях реагируют только с такими сильными N-нуклеофилами, как гидразины.
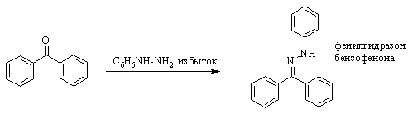
Незамещенный гидразин зачастую
реагирует с альдегидами и кетонами своими обеими аминогруппами. Возникающие в
результате такой реакции соединения называют азинами.

Реакцию только по одной аминогруппе
гидразина удается провести в условиях большого избытка последнего. Образующееся
соединение называется гидразоном альдегида (кетона).

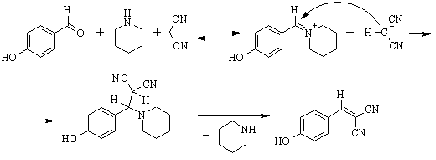
Для идентификации оксосоединений
зачастую используют 2,4-динитрофенилгидразин, который в присутствии кислот
образует трудно растворимые кристаллические гидразоны альдегидов и кетонов.
Необходимость кислотного катализа в данной реакции обусловлена низкой
нуклеофильностью 2,4-динитрофенилгидразина.

Наконец, реакция альдегидов со
вторичными аминами может привести либо к аминалям, либо к енаминам. Второй
вариант реализуется в том случае, когда альдегид имеет a-атомы водорода.

С-нуклеофилы
К реакциям присоединения-отщепления
относятся альдольно-кротоновая конденсация и родственные ей превращения.
Собственно кротоновая конденсация протекает при обработке алифатических
альдегидов крепкой щелочью. Основание превращает альдегид в резонансностабилизированный
енолят-анион вследствие депротонирования a-положения. Анион, как С-нуклеофил,
присоединяется к другой молекуле альдегида с образованием альдегидо-спирта
(альдоль), который затем отщепляет воду и превращается в кротоновый альдегид.

Таким образом, альдольная
конденсация представляет собой первую стадию альдольно-кротоновой. В
зависимости от условий, могут быть получены оба этих продукта. Конденсация идет
также в кислой среде, но в этих условиях обычно удается получить только
непредельное соединение.
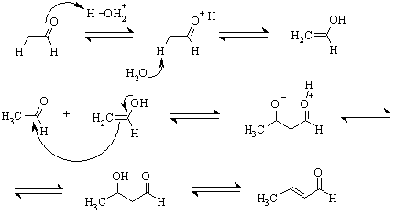
Отметим, что все стадии превращения
являются обратимыми, причем для алифатических альдегидов и кетонов равновесие
самоконденсации сильно смещено влево.
Альдольно-кротоновая конденсация
может быть перекрестной, когда используется смесь различных карбонильных
соединений. Если реагенты имеют родственную природу, например, два
алифатических кетона, имеющих a-водороды, образуется сложная смесь всех
возможных продуктов. Однако, соединения разных классов обычно конденсируются
вполне определенным образом. Так, кетон в реакции с альдегидом всегда выступает
в качестве СН-кислотной компоненты.
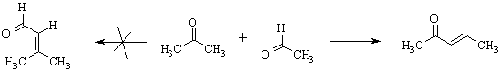
Особенно часто в перекрестную
реакцию вводят ароматические альдегиды, которые a-водородных атомов не имеют и
поэтому всегда реагируют как электрофилы.
альдегид кетон
оксосоединение нуклеофил
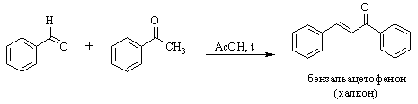
По аналогичному кротоновой
конденсации механизму идут реакции Перкина, Кневенагеля, Виттига.
Реакция Кневенагеля - взаимодействие
альдегидов в щелочной среде с соединениями, проявляющими повышенную
СН-кислотность. К ним относятся b-дикарбонильные соединения, нитроалканы,
малоновая кислота и ее производные, и т.д.
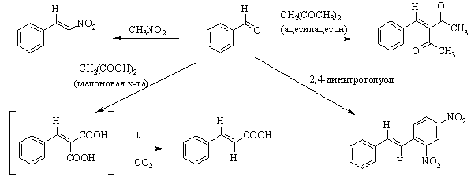
Реакция наиболее эффективно
катализируется вторичными алифатическими аминами, которые с одной стороны,
депротонируют СН-кислоту, а, с другой стороны, активируют альдегид, превращая
его в более электрофильную соль аммония.
В том случае, когда в качестве СН-кислотной
компоненты используется ангидрид алифатической кислоты, превращение называют
реакцией Перкина.

Катализатором реакции служит соль
той же алифатической кислоты, ангидрид которой используется.
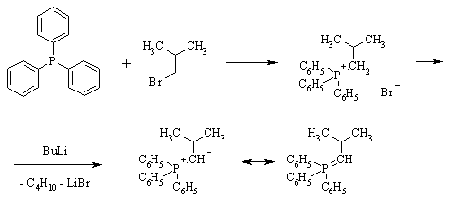
Реакция Виттига представляет собой
присоединение-отщепление к альдегиду или кетону илида фосфора. Илидами
называются соединения, имеющие связь С-гетероатом и разделенные заряды: минус
на углероде, плюс - на гетероатоме. В данном случае реагент синтезируют на
основе трифенилфосина: сначала получают четвертичную соль фосфония, затем
обрабатывают ее основанием.
Реакция сопровождается образованием
окиси трифенилфосфина и протекает, предположительно, через четырехцентровое
переходное состояние.
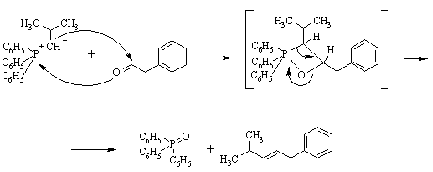
Благодаря универсальному характеру и
возможности широко варьировать структуру исходных соединений, реакция Виттига
является важным общим методом синтеза непредельных соединений.
Галогены
С галогенид-ионами карбонильные
соединения реагируют только в активированной форме, превращаясь в
гем-дигалогенопроизводные. Подходящими нуклеофилами являются галогениды
фосфора. Движущей силой реакции является активация карбонильного соединения за
счет образования связи O-P.

Реакции a-углеродного атома
Альдегиды и кетоны способны
участвовать в других реакциях замещения по a-положению. Так, они легко
галогенируются действием молекулярных галогенов.

Реакция может лишь формально рассматриваться как
замещение, на самом деле она протекает по механизму присоединения-отцепления.
Оксосоединение участвует в енольной форме, в присутствии оснований реакция
протекает легче, т.к. реагирует более активный енолят-анион.
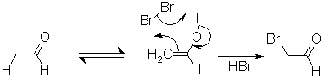
В щелочной среде при действии
избытка галогена реакция зачастую (но не всегда) не останавливается на стадии
монозамещения и идет дальше, причем обычно галогенируется все тот же атом. Это
объясняют тем, что при появлении электроотрицательного галогена СН-кислотность
возрастает. Процесс заканчивается гидролитическим разрывом С-С-связи, и
превращение называют галоформным расщеплением, т.к. образуется тригалогенметан
- галоформ.
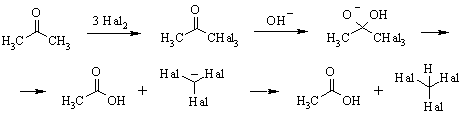
Отметим, что галогенирование
a-положения в неполярных растворителях зачастую катализируется такими
реагентами, как перекись бензоила. Это свидетельствует о том, что реакция может
протекать и по свободно-радикальному механизму. В некоторых случаях приходится
применять специфические бромирующие реагенты, например, пербромид
триметилфениламмония (PTT) реагирует с СН3-группами даже при наличии
сильно электроноизбыточных заместителей в ядре.

Повышенная подвижность a-атомов
водорода доказывается также реакцией дейтерообмена, которая идет как в кислой,
так и в щелочной среде.
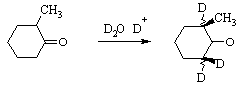
Литература
1.
Коровин Н.В. Общая химия. - М., Высш. шк. 2008
.
Фролов В.В. Химия - М., Высш. шк. 2006
.
Глинка Н.Л. Задачи и упражнения по общей химии 2006
.
Е.В. Молоток, А.Г. Назин, В.Н. Линник, С.Ф. Якубовский. Общая химия:
Учебно-методический комплекс для студентов нехимических специальностей. В 2-ух
частях. Ч-1
.
Е.В. Молоток, Л.И. Линник. Общая химия: Учебно-методический комплекс для
студентов нехимических специальностей. В 2-ух частях. Ч-2.
Дополнительная:
1.
Лучинский Г.П. Курс химии. - М., Высш. шк. 2005
.
Васильева З.Г., Грановская А.А., Таперова А.А. Лабораторные работы по общей и
неорганической химии. - Л., Химия 2006