Использование процесса ультрозвукового диспергирования в технологии получения оксидов урана
ОБОЗНАЧЕНИЯ И СОКРАЩЕНИЯ
ОЯТ АЭС - отработавшее ядерное топливо атомных
электростанций,
ВАО -высокоактивные отходы,
ТБФ трибутилфосфат.
Процессы денитрации с образованием твердых
веществ:- ammonium diuranat, процесс с образованием диураната аммония,-
ammonium uranium carbonate, процесс с образованием уранилтрикарбоната аммония,-
modified direct denitration - модифицированная прямая денитрация,-
nitrate-oxide, процесс с образованием шестиводного уранил нитрата,- microwave
decomposition - разложение в микроволновом поле,- coprecipitation-calcination -
соосаждение с последующим термическим разложением,- direct press spheroidized
feed - непосредственное прессование сфероидального исходного материала.
СОДЕРЖАНИЕ
ВВЕДЕНИЕ
1.
АНАЛИТИЧЕСКИЙ ОБЗОР
1.1
Получение оксидов урана
1.2
Получение смешанных оксидов урана и плутония
1.3
Общая оценка известных процессов денитрации
1.4
Более детальный анализ процессов прямой денитрации
1.5
Исследование процессов диспергирования растворов
1.5.1
Использование пневматического распыления растворов
1.5.2
Использование ультразвукового распыления
2.
ЦЕЛИ И ЗАДАЧИ РАБОТЫ
3.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
3.1
Методика приготовления растворов и проведения лабораторных экспериментов
3.2
Объекты и методы исследования
4.
РЕЗУЛЬТАТЫ ЭКСПЕРИМЕНТОВ И ИХ ОБСУЖДЕНИЕ
4.1
Проверка эффективности ультразвукового распыления водных растворов
4.2
Исследование включения актинидов (на примере Th) в оксиды урана при денитрации
4.3.
Проведение прессования оксидов урана, полученных денитрацией на прямоточном аппарате-печи
с определением параметров таблеток
ЗАКЛЮЧЕНИЕ
СПИСОК
ИСПОЛЬЗОВАННЫХ ИСТОЧНИКОВ
Приложение
А
Технико-экономическая оценка результатов исследования
Приложение
Б
Стандартизация
Приложение
В
Охрана труда и окружающей среды
ВВЕДЕНИЕ
Образующиеся в результате переработки
облученного ядерного топлива растворы нитратов уранила могут быть использованы
для изготовления ядерного топлива или направлены на хранение. В любом случае
они должны быть переведены в оксиды, из которых либо будут формироваться таблетки
ядерного топлива либо твердые матрицы для длительного хранения. Эта задача
может быть решена двумя способами: 1) осаждение металлов с последующей
фильтрацией и термическим разложением осадков, в результате чего образуется
значительные объемы маточных растворов; и 2) использование процесса прямой
термической денитрации растворов урана и плутония с образованием оксидов этих
металлов и паров азотной кислоты, которая может быть сконденсирована и
возвращена в технологический процесс. По способу нагрева исходного раствора
различают термическую (или термохимическую), плазмохимическую и СВЧ-денитрацию.
В зависимости от дальнейшего обращения с получаемыми при денитрации оксидами
(изготовление топлива или утилизация отходов) предъявляются различные требования
к качеству образующихся продуктов. А это, в свою очередь, определяет условия
процесса денитрации.
Подача раствора урана в аппарат для денитрации
может осуществляться различными способами. При порционном режиме денитрации
может быть использовано распыление раствора нитратов в форсунке с узким соплом.
Интересные и, по-видимому, перспективные возможности открываются при применении
ультразвукового распыления жидкостей и растворов. Этот метод известен с 50-х
годов прошлого столетия и завоевал устойчивые позиции во многих технологических
процессах в самых разнообразных областях промышленности, несмотря на то, что
преимущественным способом остается распыление сжатым воздухом.
Ультразвуковое распыление реализуется при
контакте жидкости с поверхностью, вибрирующей с частотой более 20 кГц. При этом
очень мелкие капли образуются без использования газообразного или жидкого
распылителя под высоким давлением. Одним из наиболее существенных преимуществ
ультразвукового распыления считают его большую экономичность по сравнению с
другими способами. Кроме того, ультразвуковое распыление позволяет регулировать
размер капелек и их распределение по размерам путем изменения частоты
ультразвука при распылении.
Проведенный анализ зарубежной литературы по
различным методам денитрации в радиохимических производствах показал, что на
современных предприятиях по переработке облученного ядерного топлива
технологические узлы денитрации растворов нитратов тяжелых металлов постепенно
вытесняют узлы осаждения и фильтрации, невзирая на сложность процессов и
технологического оборудования. Одной из наиболее серьезных проблем в процессах
высокотемпературной денитрации является подача раствора в зону нагрева. Решению
этой проблемы и посвящена данная работа.
1. АНАЛИТИЧЕСКИЙ ОБЗОР
.1 Получение оксидов урана
Оксиды урана - диоксид (UO2), закись-окись
(U3O8) и триоксид (UO3) -представляют важнейшие в технологическом отношении
соединения урана. Диоксид применяется для изготовления тепловыделяющих
элементов (ТВЭЛов) ядерных реакторов. Высокая термическая стойкость диоксида
урана, устойчивость к действию ионизирующего излучения и относительная
инертность являются главными факторами, обусловливающими его применение в
качестве ядерного топлива.
Диоксид урана совместно с закисью-окисью может
использоваться в качестве исходного продукта в производстве металлического
урана. Шихту в этом случае готовят из оксидов урана, металлического кальция и
флюсующих добавок (например, хлорида кальция). После восстановления
порошкообразный уран отмывают от примесей, прессуют и обжигают при высоких
температурах до компактного металла или переплавляют.
Закись-окись и триоксид урана служат
промежуточными продуктами в производстве диоксида, тетрафторида, гексафторида и
металлического урана. Кроме того, вследствие высокой стабильности при обычных
условиях закись-окись и триоксид урана можно использовать для длительного
хранения.
Получение высших оксидов урана из солей уранила
В большинстве случаев технологические операции
по получению оксидов урана проводят после аффинажа урана. Так как наиболее
распространенным методом очистки служит экстракционный процесс с применением
трибутилфосфата, в конечном итоге получают растворы уранилнитрата, очищенные от
примесей других элементов.
Для получения оксидов урана из азотнокислых
растворов можно использовать осаждение урана в виде перекиси уранила, оксалата
уранила, уранилтрикарбоната аммония или ураната аммония с последующим их
прокаливанием до оксидов урана. Во всех этих случаях соединения урана
прокаливают на воздухе; в результате получают высшие оксиды урана: триоксид
(при температуре прокаливания 400-450 0C) и закись-окись (при более высокой
температуре). Полученные в этих процессах высшие оксиды урана можно
восстановить водородом до диоксида урана. Кроме того, если достигнута
достаточная очистка урана от примесей на аффинаже, то возможно получить высшие
оксиды урана прямым термическим разложением гексагидрата уранилнитрата.
Свойства двуокиси урана существенно зависят от
методов ее получения, а такие свойства, как структура, отношение O/U, удельная
поверхность и активность порошков при спекании, являются решающими при
использовании UO2 для изготовления твэлов для ядерных реакторов.
Получение оксидов урана методами осаждения
Уранилоксалат
При обработке растворов уранилнитрата щавелевой
кислотой образуется осадок уранилоксалата, гидратированный тремя молекулами
воды:
UO2 (NO3)2+H2C2O4 + 3H2O →
UO2C2O4·3H2O+ 2HNO3
Термическое разложение этой соли в атмосфере
воздуха при температуре выше 500 0C приводит к образованию закиси-окиси урана.
Диоксид урана может быть получен при прокаливании без доступа воздуха.
Разложение уранилоксалата в этом случае описывается уравнениями:
C2O4 → UO3 + CO + CO2,+ СО → UO2 +
CO2.
Из приведенных реакций следует, что
восстановитель получается только в стехиометрических количествах. При
проведении процесса разложения тригидрата уранилоксалата в атмосфере аргона в
интервале 400-900 0C состав газовой фазы изменяется следующим образом: CO2 от
52,0 до 57,2 %; СО от 10,5 до 4,8 % (оставшееся количество составляют аргон и влага).
Эти данные свидетельствуют о том, что скорость разложения уранилоксалата выше
скорости восстановления триоксида оксидом углерода. Следовательно, получить
диоксид урана из уранилоксалата в статических условиях невозможно. Для
получения диоксида урана необходим непрерывный процесс, причем в аппарате
должно быть две температурные зоны: зона разложения и зона восстановления.
Продолжительность процесса тем меньше, чем выше температура в зоне
восстановления.
При разложении уранилоксалата без доступа
воздуха наблюдаются следующие термические эффекты:
а)при температуре до 270 0C удаляется
кристаллизационная влага (сначала две молекулы при 120 0C, а затем третья);
б)в интервале 310-340 0C уран восстанавливается
до диоксида (в продуктах обнаруживается оксид урана состава UO2,25);
в)при 500-550 0C в продуктах разложения
присутствуют диоксид урана и оксид состава UO2,25;
г)в интервале 620-900 0C в твердом продукте
обнаруживается лишь диоксид урана, образовавшийся из фазы состава UO2,25
вследствие восстановления.
Образующийся в результате термического
разложения уранилоксалата диоксид обладает высокой реакционной способностью;
при обычных условиях он пирофорен, поэтому необходимы специальные меры по
стабилизации. Это достигается совмещением процессов прокаливания, восстановления
и гидрофторирования. Непосредственно после прокаливания диоксид урана
обрабатывают фтористым водородом; при этом образуется тетрафторид урана.
Аммонийуранилтрикарбонат
Кристаллы аммонийуранилтрикарбоната выделяют из
растворов, содержащих избыток карбоната или бикарбоната аммония.
Кристаллизационной воды они не содержат. Термическое разложение
аммонийуранилтрикарбоната на воздухе при температуре выше 500 0C сопровождается
образованием закиси-окиси урана
3(NH4)4[UO2(CO3)3] → U3O8 +
10NH3 + 9CO2 + N2 + 7H2O + 2H2.
Как видно из уравнения реакции, в ходе процесса
выделяется значительное количество аммиака, углекислоты, воды, водорода и
азота.
Кристаллы аммонийуранилтрикарбоната при
нагревании без доступа воздуха начинают разлагаться при 170 0C. Полное
разложение соли до триоксида урана происходит в интервале 240-350 0C. При этом
образуется аморфный триоксид урана
(NH4)4[UO2(CO3)3] → UO3 + 3CO2 + 4NH3 +
2H2O.
Повышение температуры приводит сначала к
перекристаллизации триоксида урана, а затем к образованию закиси-окиси урана.
Начиная с 620 0C появляется диоксид урана. Восстановление до диоксида
происходит за счет водорода, выделяющегося при разложении аммиака. При
разложении соли без доступа воздуха образуется четыре моля аммиака на один моль
триоксида урана. Получающийся избыток водорода приводит к образованию водяного
газа.
Разложение аммонийуранилтрикарбоната без доступа
воздуха, приводящее к получению диоксида, выражается следующей реакцией:
(NH4)4[UO2(CO3)3] → UO2 + 2NH3
+ 3CO2 + 2H2 + N2 + 3H2O.
Состав газовой фазы зависит от температуры
процесса.
Получение оксидов урана методом денитрации
уранилнитрата
Получение оксидов прямой термической денитрацией
гексагидрата уранилнитрата - достаточно тонкий технологический процесс. Метод
термической денитрации растворов уранилнитрата при переработке ядерного топлива
находит широкое применение на заводах в Великобритании []. Обычно процесс
денитрации проводят в две стадии: упаривание раствора уранилнитрата в выпарных аппаратах
с получением гексагидрата уранилнитрата и последующая термическая обработка
упаренного до 70-100 % продукта до трехоксида урана.
На операцию концентрирования поступают растворы
уранилнитрата с содержанием от 90 до 280 г урана на 1 л. При этом содержание
свободной азотной кислоты может изменяться от нуля до 0,05 моль/л. Для
упаривания растворов используют вертикальные трубчатые выпарные аппараты,
обогреваемые внутренними паровыми змеевиками. Работу каждого аппарата постоянно
контролируют по температуре кипения раствора для получения такой концентрации
уранилнитрата, которая необходима для операции денитрации. Эта температура
колеблется от 120 до 143 0C. При 120 0C кипит раствор, в котором содержание
урана примерно 1175 г/л, при 143 0C раствор с концентрацией урана около 1500
г/л.
Температура начала кристаллизации менее
концентрированного раствора около 60 0C, более концентрированного - около 116
0C. Поэтому всю систему транспортирования таких растворов во избежание
затвердевания уранилнитрата в трубопроводах обогревают паром. Все трубопроводы
монтируют наклонными, чтобы при необходимости были обеспечены слив раствора и
промывка их сильной струей воды.
Перед подачей на денитрацию такие
концентрированные растворы уранилнитрата хранят в обогреваемых емкостях.
Аппараты концентрирования изготавливают из нержавеющей стали.
Отходящие пары и газы конденсируют; если в
конденсате обнаруживается некоторое количество урана, растворы возвращают на
упаривание. При достаточно высоком содержании азотной кислоты конденсат
возвращают в процесс; конденсат, не содержащий урана и азотной кислоты,
используют в качестве раствора для проведения процесса реэкстракции урана из
трибутилфосфата.
В настоящее время разрабатывается еще более
эффективный аппарат - денитратор кипящего слоя. В этом варианте аппаратурного
оформления процесса обеспечивается лучшая теплопередача [, ]. Упаренный раствор
уранилнитрата (800 г/дм3) нагревают в сборнике до 70 0C и подают
насосом-дозатором одновременно со сжатым воздухом в форсунки, расположенные в
корпусе аппарата. Раствор впрыскивается форсунками непосредственно в
псевдоожиженный слой нагретой гранулированной трехокиси урана. Ожижающим
агентом служит горячий воздух, нагреваемый в калорифере. В реакторе образуется
парогазовая смесь, содержащая мелкодисперсную трехокись урана. Эта смесь из
реактора проходит циклон, в котором происходит осаждение трехокиси урана,
которая из бункера циклона питателем возвращается обратно в псевдоожиженный
слой. Вывод трехокиси урана из реактора осуществляют с помощью питателя с
уровня псевдоожиженного слоя в приемник.
Процесс денитрации проводят при температуре
300-500 0C, при скорости ожижающего агента (воздуха) 0,46 м/с при числе
псевдоожижения, равном 6. Расход тепла на 1 т U составляет 26,2 · l05 кДж. При
денитрации в аппаратах псевдоожиженного слоя не возникает местных перегревов,
процесс протекает с высокой скоростью денитрации и хорошо регулируется.
Физические характеристики получаемых гранулированных частиц UO3 (сферические
размеры, насыпную плотность и др.) можно регулировать в определенных пределах
технологическими параметрами, т. е. скоростью ожижающего агента, температурой
псевдоожиженного слоя, условиями распыления раствора уранилнитрата (скоростью
истечения, удельным расходом распыливающего агента), а также температурой и
концентрацией поступающего на денитрацию уранилнитрата.
Кроме перечисленных достоинств этого метода
следует отметить возможность получения продукта высокой чистоты, незначительную
коррозию аппаратуры и сравнительно невысокую стоимость этого метода денитрации
[].
Остаточное содержание нитрат-иона и воды в
триоксиде урана зависит от температуры кипящего слоя; так как длительность
пребывания материала в аппарате очень невелика и измеряется минутами,
содержание этих компонентов несколько выше, чем в триоксиде урана, полученной в
желобчатом денитраторе при той же температуре.
Гранулометрический состав триоксида
урана определяется температурой кипящего слоя и концентрацией уранилнитрата в
питающем растворе. В начале работы установки частицы триоксида урана
укрупняются либо цементацией их каплями питающего раствора в агрегаты, либо
укрупнением отдельных частиц при разложении на их поверхности уранилнитрата. В
дальнейшем гранулометрический состав триоксида урана стабилизируется; при этом
скорости укрупнения и измельчения частиц становятся одинаковыми [3].
Максимальный размер частиц
наблюдается при наиболее концентрированном питании, минимальный - при
сравнительно разбавленном. Уменьшение температуры кипящего слоя приводит к
некоторому измельчению частиц триоксида, что объясняется проникновением воды в
поры агрегатов и последующим их разрывом; при более высокой температуре
разложение протекает лишь на поверхности твердого вещества. Вследствие этого
обстоятельства (разложения уранилнитрата на поверхности или в порах триоксида
урана) аппаратурное оформление процесса в кипящем слое выгодно отличается от
любого другого аппаратурного решения значительно меньшей коррозией аппарата в
ходе процесса и, следовательно, меньшим загрязнением готового продукта.
Денитрацию уранилнитрата
осуществляют также в пламенных реакторах. В этом случае раствор уранилнитрата
перерабатывается в пламени, образующемся при сжигании в воздухе или кислороде
газообразных углеводородов парафинового ряда, например метана, пропана, бутана
и т. п. []
В пламенном реакторе можно получать
закись-окись или даже двуокись урана в зависимости от того, что подается в
реактор в избытке - восстановитель или окислитель. К преимуществам пламенных
реакторов относятся простота конструкции, возможность полной автоматизации
процесса, относительно высокая коррозионная стойкость аппарата и экономичность
технологического процесса. Недостатком является взрывоопасность, а также
возможность образования на стенках аппарата гарнисажа - слоя твёрдых продуктов.
.2 Получение смешанных оксидов урана
и плутония
Разработки плутоний-содержащего
топлива для реакторов на быстрых нейтронах (РБН) начались в 50-е годы и
проводятся до настоящего времени.
В 70-е гг. была разработана
технология изготовления смешанного уран-плутониевого топлива, основанная на
процессе механического смешивания порошкообразных UO2 и PuO2, а в 1980г. начала
работать установка "Пакет" на комбинате "Маяк". На этой
установке было изготовлено 10 сборок топлива для реактора БН-350. Содержание
плутония в топливе достигало 21-27,5%.
К недостаткам процесса с
использованием механического смешивания и совместного размола порошков можно
отнести наличие пылеобразующих операций, необходимость тщательного измельчения
и диспергирования частиц PuO2 в матрице диоксида урана, чтобы при спекании
образовался твердый раствор (UPu)O2.
Чтобы исключить присутствие пыли,
был разработан золь-гель процесс, и в 1985г. введена в эксплуатацию установка
"Жемчуг", действующая на основе этого процесса. На установке в
1987-1988 гг. были изготовлены 24 сборки для реактора БН-350. Однако попытки
провести испытания золь-гель процесса в большем масштабе выявили недостатки
этого процесса, основным из которых была нестабильность физико-химических
характеристик и структуры образующегося топливного материала. Непостоянство
характеристик золь-гель порошка вызывало трудности с регулированием операций
прессования и спекания; в процессе золь-гель наблюдался большой процент брака.
С учетом этого было решено, что золь-гель процесс не пригоден для внедрения на
установках промышленного масштаба.
Стремление разработать экологически
приемлемый технологический процесс (c минимальным количеством пыли) привело к
созданию процесса с использованием соосаждения гидроксидов урана и плутония
гидроксидом аммония и грануляции осадка в присутствии поверхностно-активных
веществ. Этот процесс был использован при создании установки "Гранат"
(гранулированное атомное топливо), которая была построена в течение одного года
и в 1988 г. сдана в эксплуатацию.
Производительность этой установки в
период с декабря 1988 г. по февраль 1989 г. составила 50 кг топлива из
смешанных оксидов, а к 1993 г. было наработано 700 кг смешанного оксида.
Плотность изготовленного топлива составила 10,5±0,09 г/см3; выход достигал
95-96 %. На установке "Гранат" были изготовлены экспериментальные
сборки реактора БН-600. Облучение 15 сборок в течение 6 месяцев с последующим
изучением характеристик облученного топлива позволило уточнить условия спекания
и время изотермического прокаливания топлива.
Процесс с использованием
аммоний-карбонатного соосаждения в лабораторных условиях позволяет получить
порошки с хорошими керамическими и физико-химическими характеристиками. Однако
этот процесс еще недостаточно изучен и не испытан в большом масштабе.
Образующиеся порошки имеют более высокий уровень пыления, чем порошки,
получаемые с помощью золь-гель процесса [].
Однако наиболее пылящими являются
порошкообразные оксиды, обрабатываемые в процессах механического смешивания и
совместного размола [].
Процесс плазмохимической конверсии,
в основе которого лежит высокотемпературная денитрация нитратов урана и
плутония, обладает рядом привлекательных особенностей. Одной из важных
положительных особенностей этого процесса является высокая скорость
производства порошкообразного материала, а также отсутствие операции
фильтрации, промывки, сушки и других операций с тепловой обработкой.
Для реализации плазмохимической
конверсии была разработана установка "Зенит", на которой было
получено 34 кг порошка оксидов урана и плутония. Из этого порошкообразного
материала на установке "Пакет" были изготовлены таблетки и твэлы.
Полученное топливо использовали в реакторе БН-600.
Наряду с достоинствами
плазмохимический процесс имеет и серьезные недостатки, одним из которых
является высокая степень дисперсности порошка. Такие порошки обладают
способностью к адгезии и когезии и не являются текучими. Отсюда следует
необходимость предварительного прессования и гранулирования материала,
используемого при изготовлении топливных таблеток. Высокая степень пыления
порошков требует организации эффективной системы газоочистки, что обусловливает
большое количество отходов в виде нерегенерируемых фильтров.
На основании сказанного выше можно
сделать вывод о том, что при современном уровне развития технологии плазмохимический
процесс не может быть рекомендован для промышленного использования.
Пироэлектрохимический процесс был
разработан в НИИАР (Димитровград) для получения смеси, содержащей 30 % PuO2.
Последующие исследования показали, что можно изготовить топливную композицию,
содержащую до 75 % PuO2 при сохранении требуемого качества гранулированного
материала.
Для осуществления
пироэлектрохимического процесса была создана автоматизированная дистанционно
управляемая установка. На этой установке в Димитровграде было изготовлено более
420 сборок топлива для реактора БОР-60 и 24 сборки для реактора БН-600. Для
этого было получено примерно 3 т гранулированного оксидного топлива, в том
числе 2 т топлива с PuO2[6].
.3 Общая оценка известных процессов
денитрации
Денитрацией можно назвать любой
процесс, при котором интересующие нас элементы отделяются от соединений азота
(в частности, - от нитрат-ионов). Рассматривая процессы перехода от
азотнокислых растворов урана и плутония к оксидам, имеет смысл различать два
основных типа реакций: первый - это прямая (непосредственная) денитрация, т.е.
превращение нитратов в оксиды без получения каких-либо промежуточных твердых
соединений, второй же тип включает две стадии - кристаллизацию промежуточного
соединения и его термическое разложение до оксида.
Первый путь представляется заведомо
более предпочтительным. Как отмечается в работе [], прямую денитрацию в первую
очередь и стали использовать, когда потребовалось в промышленном масштабе
производить диоксид урана для изготовления топлива энергетических ядерных
реакторов. Однако из порошков, получаемых таким способом, таблетки с
плотностью, превышающей 90% от теоретической, удавалось получать, только
используя большие давления при прессовании и температуры порядка 1750-1800 0С
при спекании таблеток.
В дальнейшем были разработаны и
многие другие варианты процессов денитрации. Для их сравнительной оценки
целесообразно использовать какие-то объективные критерии. Полезный и достаточно
простой набор критериев предложил Хаас [], для сравнения различных процессов
получения оксидов урана, пригодных для изготовления топливных таблеток. Для
технико-экономической оценки технологии учитывалось число операций, необходимых
для перехода от раствора уранилнитрата средней концентрации к порошку оксида
требуемого качества, и сложность этих операций. Критерии качества порошков к
середине 80-х уже в основном определились.
Было установлено, что площадь
поверхности порошка UO2 в интервале 6±4 м2/г наиболее благоприятна для
изготовления топливных таблеток. Порошки с меньшей поверхностью спекаются и
уплотняются хуже, тогда как порошки с большей поверхностью дают таблетки с
пониженной плотностью при прессовании, а также с излишней усадкой и с
образованием трещин линзообразной формы при спекании. Качество порошка с
большой площадью поверхности может быть улучшено его прокаливанием перед
прессованием таблеток. Эта операция уменьшает поверхность порошка и обычно
увеличивает текучесть. Растирание порошка UO2 увеличивает поверхность.
Предпочтительно получать порошки с
узким распределением частиц по размерам и со средним размером частиц 4±3 мкм.
Излишнее количество частиц с диаметром меньше 1 мкм имеет следствием пониженную
плотность спрессованных таблеток и проблемы с усадкой. Для уменьшения размеров
UO2-агломератов полезно растирать порошки.
Отдельную проблему представляют
количества и природа различных примесей в порошках. Допустимое содержание
примесей строго регламентируется. Как правило, чистота исходного раствора
нитрата уранила достаточна для получения чистых порошков UO2, и примеси обычно
вносятся в ходе технологических процессов изготовления и обработки порошка.
Существенным источником примесей является процедура измельчения и растирания
UO2-порошка.
Количественная оценка сложности
технологических операций превращения уранилнитрата в UO2 не может быть строго
объективной, но в работе [9] достаточно обоснованно принято, что их сложность
возрастает при необходимости:
обрабатывать пастообразные или
влажные материалы и перемещать твердые вещества между технологическими
операциями,
обрабатывать все потоки отходов до
состояния, пригодного для рецикла, сброса или долговременного хранения,
механически обрабатывать прокаленные
оксиды, например, проводить измельчение, смешивание или просеивание порошка,
предотвращать при аппаратурном
оформлении ядерную критичность; необходимость предотвращения критичности
ограничивает размеры технологического оборудования и делает более практичными
непрерывные технологические процессы с использованием оборудования уменьшенных
размеров.
Обобщенная технологическая схема
процесса изготовления порошка UO2 приведена на рис. 1. Она включает 13
возможных операций, часть из которых при рассмотрении конкретной технологии
может быть исключена. В публикации [9] рассмотрены восемь различных процессов денитрации,
типичные характеристики которых приведены в табл. 1.
В табл. 1 приведены лишь те процессы
денитрации, которые позволяют из чистых растворов нитрата уранила получить UO2,
пригодный для дальнейшего изготовления топливных таблеток. Метод термической
денитрации простым нагреванием раствора уранилнитрата в табл. 1 не упомянут,
т.к. получающийся при этом оксид урана, как уже отмечалось, имеет
неудовлетворительные характеристики для изготовления топливных таблеток.
Ниже приведены также уравнения
реакций, на которых основаны рассматриваемые технологические процессы.
Список химических реакций,
используемых в процессе денитрации:
. Восстановление до UO2 при 500-800
0C (все методы):
UO3 + Н2 → UO2 + Н2О.
. Стабилизация при температуре
примерно 100-400 0C (все методы):
+ хСО2 → UO2+х + хСО, х =
~0,15.
. ADU, осаждение и разложение:
2UO2(NO3)2 +
6NH4ОН → (NH4)2U2O7 + 4NH4NO3 + 3Н2О,
(NH4)2U2O7 → 2UO3 + 2NH3 +
Н2О.
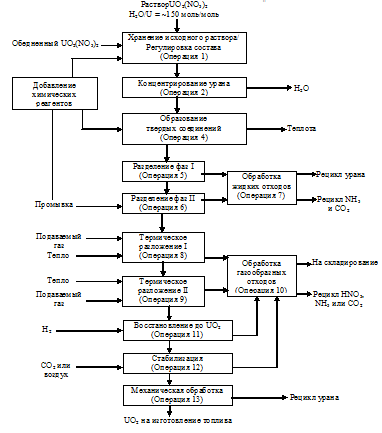
Рисунок 1 - Обобщенная
технологическая схема процесса денитрации [9]
Таблица 1 - Характеристики различных
процессов денитрации [9]
|
Процесс
|
Операция
2. Отношение Н2О/U после концентрирования, моль/моль
|
Операция
3 Добавляемые химические реагенты
|
Операция
4 Образующиеся твердые вещества для последующего термического разложения
(операции 8)
|
Операции
5 и 6 Разделительное оборудование
|
Операция
7 Состав жидких отходов
|
Операции
8 и 9 Оборудование для термического разложения
|
Операция
13 Механические процедуры
|
|
ADU
|
25
|
NH3
|
(NH4)2U2O7
|
Центрифуга
|
NH4OH,
NH4NO3
|
Шнековая
печь
|
Рубка,
размол, гранулирование
|
|
AUC
|
30
|
NH3,
CO2
|
(NH4)4UO2(CO3)3
|
Фильтр
|
(NH4)2CO3, (NH4)OH, NH4NO3
|
Ожиженный
слой
|
Нет
|
|
MDD
|
8
|
NH4NO3
|
NH4UO2(NO3)3
|
Нет
|
Нет
|
Вращающающаяся
трубчатая печь
|
Гранулирование
|
|
NITROX
|
6
|
Нет
|
UO2(NO3)2·6H2O
|
Нет
|
Нет
|
Вакуумный
сушитель
|
Растирание
|
|
MWD
|
40
|
Нет
|
UO2(NO3)2·2H2O
|
Нет
|
Нет
|
Микроволновой
сушитель
|
Размол,
перемалывание в мельнице
|
|
COPRECAL
|
30
|
NH4OH
|
(NH4)2U2O7
|
Нет
|
Нет
|
Ожиженный
слой
|
Растирание,
просеивание
|
|
DIPRES
|
20
|
Мочевина,
HMTA, NH4OH
|
UO2(OH)2·xH2O
|
Неподвижный
или движущийся слой
|
NH4NO3,
NH4OH, мочевина, HMTA
|
Бандажный
сушитель
|
Нет
|
|
Другие
осадительные процессы
|
100
|
H2O2
или другие
|
UO4·2H2O
или другие
|
Фильтр
|
HNO3,
H2O2, другие
|
Печь
периодического действия
|
Растирание
|
. AUC, осаждение и термическое разложение:
(NO3)2 + 3Н2О + 6NH3 + 3СО2 →
(2NH4)4UO2(СО3)3 + 2NH4NO3,
(NH4)4UO2(СО3)2 → UO3 + 4NH3 + 3СО2 +
2Н2О.
. MDD, образование двойной соли и разложение:
UO2(NO3)2 + NH4NO3 NH4UO2(NO3)3,
NH4UO2(NO3)3 → UO3 + N2O + 2Н2О + хNO2 +
(2-х)NO + (1.5-x)О2, х = ~0,2.
. NITROX, вероятная реакция:
(NO3)2·Н2О → UO3 + 2HNO3.
. MWD, как в простой денитрации
(NO3)2·6Н2О → UO3 + 6Н2О + хNO2 + (2-х)NO
+ (1.5-x)О2, х = ~0,14.
. COPRECAL, осаждение и разложение избытка
реагентов:
UO2(NO3)2 + 7NH4OH + Н2О → (UO3)2·NH3·5Н2О
+ 6NH4NO3,
NH4NO3 → 2N2 + 4Н2О + О2,
NH4(OH) + 3О2 → 2N2 + 10Н2О.
. DIPRES, реакции с ураном:
+ + 2ОН- → UO2(OH)2 → UO3·Н2О →
UO3 + Н2О.
. Другие осадительные методы. Реакции могут быть
самыми различными, например реакция осаждения перекисью водорода:
(NO3)2 + Н2О2 + 3Н2О → UO4·2Н2О +
2HNO3,·2Н2О → UO3 + 2Н2О + 1/2О2.
.4 Более детальный анализ процессов прямой
денитрации
процесс.
Модифицированный метод прямой
денитрации был разработан в Ок-Риджской национальной лаборатории в США [] с
целью использования надежного и несложного оборудования для изготовления
UO2-продукта с хорошими керамическими свойствами. MDD-процесс основан на
разложении двойной твердой соли NH4UO2(NO3)3 []. Двойная соль образуется до
начала денитрации и расплава UO2(NO3)2 в процессе не образуется. Так как
твердая двойная соль образуется при термической денитрации в кальцинаторе по
мере испарения воды, нет необходимости отделять, или обрабатывать твердые
соединения. Продукты разложения имеют тот же химический состав, что и продукты
простой термической денитрации раствора UO2(NO3)2 плюс некоторое количество N2O
и Н2О из NH4NO3 [11, ]. Площадь поверхности, размер частиц и поведение
MDD-продукта при изготовлении таблеток весьма близки к этим параметрам хорошего
ADU-продукта []. Вращающаяся печь, используемая в MDD-методе денитрации,
удовлетворяет таким же требованиям к теплопереносу и защите от критичности, что
и печь, применяемая в ранее освоенном ADU-процессе, однако операции осаждения и
промывки в MDD-процессе отсутствуют. Поэтому для реализации MDD-процесса
достаточно семи из тринадцати технологических операций, показанных на рис. 1,
хотя могут понадобиться и операции измельчения и гранулирования продукта перед
прессованием таблеток. Казалось бы, этот процесс заслуживал дальнейшего
изучения и промышленного применения, однако прошло более 20 лет после первых
публикаций, но какие-либо новые сведения о нем обнаружить не удалось. процесс.
Этот метод разработан фирмой
Comurhex во Франции и предполагает использование термической обработки
UO2(NO3)2·6Н2О в две стадии. На первой стадии проходит дегидратация под
вакуумом при температурах ниже 62 0C, температуры плавления уранилнитрата. На
второй стадии дегидратированная соль разлагается в вакууме в атмосфере водяных
паров до UO3. [, ] Разложение в этом случае проходит без образования расплава,
поэтому полученные оксиды должны иметь благоприятные технологические свойства.
Показано, что NITROX-процесс может быть использован и для получения порошков
UO2 - PuO2 с последующим изготовлением смешанного топлива [], в этом случае
добавляется еще операция восстановления, а в целом NITROX-процесс применительно
к нитратам урана и плутония включает следующие операции.
. Смешивание растворов нитратов
урана и плутония до заданного соотношения (если такой раствор не получен естественным
образом в ходе переработки ОЯТ).
. Концентрирование раствора нитратов
до примерно 1200 г/л тяжелых металлов.
. Дегидратация полученной смеси
нитратов при пониженном давлении и при начальной температуре 50 0С, которая
постепенно увеличивается до 270 0С. При этом в материале остается 1,2 масс. %
воды и 75 масс. % начального содержания нитрата.
. Прокаливание при температуре выше
600 0С до полного превращения нитратов в оксиды.
. Восстановление оксидов смесью
аргона и водорода с переводом урана и плутония в четырехвалентное состояние.
. Стабилизация смешанных диоксидов
контролируемым окислением.
В целом NITROX является
многоступенчатым и включает довольно сложные термические процедуры, в том числе
при пониженном давлении. Несмотря на некоторые успехи, достигнутые при
разработке технологий типа “NITROX”, по-видимому, исследования в этом
направлении были прекращены.процесс.
Этот процесс был разработан и
продемонстрирован в Японии Корпорацией по разработке энергетических реакторов и
ядерного топлива []. Раствор нитратов урана и плутония в этой технологии
обрабатывается СВЧ-излучением с частотой 2450 МГц, что приводит вначале к
удалению воды и азотной кислоты, а затем при температуре выше 300 0С образуется
смесь UO3 и PuO2. Получаемый промежуточный продукт представляет собой
губкообразный кек, который передают в печь для окончательного прокаливания и
восстановления. Смешанные диоксиды подвергается размолу в шаровой мельнице с
целью получения порошка с размерами частиц, не превышающими 1-2 мкм.
Прессованием и спеканием из полученного порошка успешно изготавливались
топливные таблетки.
При разработке микроволновой
денитрации постоянно совершенствовалось используемое оборудование. В ранних
вариантах аппаратурного оформления применяли двухступенчатую систему термической
обработки исходного раствора []. На первой ступени раствор нагревался во
вращающемся цилиндрическом аппарате с внешним или внутренним нагревательным
элементом и лишь на второй ступени полученный материал обрабатывался
микроволновым излучением, что приводило к повышению температуры и денитрации.
Денитрации проходила при достижении температуры 350-400 0С. В дальнейшем
предварительный подогрев отменили, а в публикации [] было предложено полученный
на начальном этапе денитрации UO3 продолжать нагревать в микроволновом поле до
температуры 500 0С, когда образуется U3O8. Этот оксид не содержал остатков NOх
и имел удельную площадь поверхности 3 м2/г.
Особый интерес представляют
разработки оборудования для денитрации непрерывного действия. В сравнительно
недавно (1996 г.) опубликованном патенте [] участок обработки нитратного
раствора разделен на три зоны - зона концентрирования, где происходит
выпаривание и концентрирование раствора, зона денитрации, где осуществляется
разложение нитратов и образование оксидов и, наконец, зона сушки продукта.
Перемещение продукта от зоны к зоне осуществляется с помощью шнека. Облучение
обрабатываемого материала СВЧ радиоизлучением осуществляется от генератора
через пластину, имеющую окно из проницаемого для микроволнового излучения
материала. В процессе обработки пластина с окном перемещается, позволяя
облучать различные технологические зоны.
В принципе процесс микроволнового
разложения нитратов во многом сходен с NITROX-процессом, однако использование
микроволнового нагрева накладывает определенные ограничения на материалы и
конфигурацию оборудования. Пока нет определенных данных о том, какая
максимальная производительность может быть достигнута на промышленных
установках такого типа. Отсутствуют также оценки экономичности технологии
MWD.процесс.
Этот процесс был разработан в
Ядерном центре компании Дженерал Электрик [] и по формальным признакам попал в
разряд экономичных. В этой технологии, как и в ADU-процессе, проводится
осаждение урана (обычно с плутонием) действием аммиака, но полученная суспензия
подвергается термической обработке без отделения от раствора. Согласно
источнику [18], осаждение проводят смешиванием раствора нитрата уранила (400
г/л) и аммиака в аппарате для осаждения непрерывного действия. Отношение
NH3/NO3 при осаждении должно быть близким к 1,67 моль/моль. Полученная взвесь
подвергается термической обработке при 550 0C в слое инконелевых шариков
диаметром 700 мкм, который служит средой для теплопереноса. Разлагаются также
нитрат аммония и избыток NH3, UO3 непрерывно выводится из псевдоожиженного слоя
и собирается на металлических фильтрах, которые периодически очищаются. В
качестве ожижающего газа используется азот, UO3 восстанавливается до UO2 смесью
N2 + Н2. Двуокись урана стабилизируется углекислым газом. Затем продукт
просеивается для отделения слишком крупных частиц оксида и инконелевых шариков,
попавших из ожиженного слоя. Цилиндрический слой диаметром 15 см и высотой 50
см имеет проектную производительность примерно 0,1 т урана в день. При
использовании COPRECAL-процесса для изготовления смешанных оксидов с
содержанием плутония 20-30 % предполагалось иметь производительность 0,5 кг
оксидов в час [].
Оксиды, получаемые этим методом,
хуже спекались по сравнению с продуктом технологии ADU даже после интенсивной
механической обработки. Работа псевдоожиженного слоя с инконелевыми шариками
была нестабильна даже в тщательно сконструированной установке. Поэтому
дальнейшего развития это направление не получило.
Среди рассмотренных процессов прямой
денитрации по объему проведенных исследований и количеству полученной продукции
явно лидирует японская технология с применением СВЧ-излучения. Однако остается
неясным, связан ли этот успех именно с указанным способом нагрева или
существенную роль сыграла проведенная в Японии очень тщательная разработка
оборудования. Возможно некоторые процессы типа “Nitrox” при столь же масштабной
многолетней работе могли бы успешно конкурировать с технологией MWD. Но такая
работа не проводилась просто потому, что разработка процессов прямой денитрации
на некоторое время была признана не столь актуальной.
Среди российских работ по денитрации
известны работы, выполненные на СХК (г. Северск) и в Радиевом институте (СПб),
с использованием нагрева с помощью ВЧ-плазмы []. Для генерации плазмы в работах
последних лет чаще всего используют электродуговые и высокочастотные
плазмотроны, хотя иногда упоминаются установки с применением сверхвысоких
частот и других более экзотических способов генерации плазмы. Преимущество
дуговых плазмотронов - несколько более высокий коэффициент полезного действия
по сравнению с высокочастотными и более простая электротехническая схема.
Ресурс работы электродов таких плазмотронов считается удовлетворительным: так,
по имеющимся данным, удельная эрозия катода составляет 2·10-9 кг/Кл, анода -
5·10-11 кг/Кл, ресурс плазмотрона обычно оценивается в 200 часов и может быть
доведен до 500-700 часов дополнительными приемами. Плазменный шнур при дуговом
разряде имеет малый диаметр; для преодоления этого недостатка нередко один плазмохимический
реактор используют в сочетании с тремя симметрично расположенными дуговыми
плазмотронами.
Ресурс плазмотрона с высокочастотной
генерацией плазмы считается практически неограниченным [], и плазменный факел в
этом случае имеет достаточно большой диаметр. Поэтому выбор плазмотронов такого
типа для установок института им. В. Г. Хлопина можно считать достаточно
обоснованным.
Несмотря на многочисленные сообщения
о применении плазмохимических процессов, трудно найти точные сведения о
масштабах производства порошков керамических соединений. Вероятно, в основном
это объясняется неустойчивым состоянием экономики на территории бывшего
Советского Союза. Например, на СХК предполагалось производить 250 т в год
оксидов различных нерадиоактивных элементов, но фактическая производительность
установок сейчас определяется не техническими возможностями, а платежеспособным
спросом на продукцию. Пока нет необходимых установок для изготовления таблеток,
ТВЭЛов и ТВС реакторов ВВЭР-1000, порошки оксидов урана и плутония также
требуются лишь в небольших количествах. На СХК на установках с высокочастотной
индукционной генерацией плазмы до сих пор было изготовлено 20 кг диоксида
плутония, 40 кг смешанных оксидов плутония и урана и около 1000 кг оксидов
урана. На одной из плазмохимических установок Радиевого института им. В. Г.
Хлопина в г. Гатчине (с генератором мощностью 60 кВт) получено около 8 кг
оксидов урана и столько же оксидов циркония и некоторых других металлов в
качестве компонентов кристаллических матриц для захоронения радиоактивный
отходов.
.5 Исследование процессов
диспергирования растворов
Одной из ключевых операций процесса
денитрации растворов уранилнитрата, определяющей качество образующихся
порошков, является подача исходного раствора в печь. Поэтому во всех процессах
денитрации большое внимание уделяется выбору способа и конкретных конструкций
узлов распыления растворов, которые должны обеспечить необходимую степень
диспергирования.
Под распылением обычно понимают
перевод жидкостей или их смесей в газовую фазу в виде мелких капелек. Для
этого, как правило, используют метод пропускания жидкости, движущейся с высокой
скоростью, через малое отверстие.
.5.1 Использование пневматического
распыления растворов
Наибольшее распространение в
различных областях техники нашли до настоящего времени пневматические
распылители. Принцип действия пневматических распылителей [] состоит в
следующем. У места выхода сжатого газа из отверстия находится под углом трубка
для подачи раствора; в кольцевых распылителях трубка, подающая раствор,
окружена другой трубкой и газ выходит из кольцевого зазора. Вследствие
расширения газа у места его выхода создается разрежение, и рабочий раствор
засасывается в трубку. Выходящий с большой скоростью газ увлекает, благодаря
трению, струи раствора, вытягивая их в тонкие нити, которые быстро распадаются,
образуя сферические капли. Чем больше скорость истечения газа, тем тоньше
получаются нити и тем большая степень дисперсности аэрозоля.
Среди пневматических распылителей
наибольшее распространение получили распылители концентрического типа. Обычно
распылители этого типа используются со свободной подачей раствора, но в
последнее время все шире применяется принудительная подача распыляемого
раствора с помощью различного вида насосов или других каких-либо способов.
Используются распылители как неразборной конструкции (цельносварные из кварца
или стекла) так и разборные. Достоинством этих распылителей является
относительная простота конструкции, а также возможность работы без
принудительной подачи раствора.
К недостаткам его можно отнести
следующую принципиальную особенность. Для получения капель малого диаметра
необходимо увеличивать скорость распыляющего газа, для чего приходится
увеличивать давления в газовой системе и уменьшать кольцевой зазор между
трубками подачи газа и раствора. При уменьшении кольцевого зазора размер капель
уменьшается, но появляется опасность засорения распылителя, особенно при работе
с концентрированными растворами. На Западе сейчас широкое распространение
получили распылители, производимые фирмой Мейнхарда, стандартизированные и
имеющие воспроизводимые свойства (расход раствора 1-2 или 3 л/мин, диаметр
газового отверстия от 15 до 35 мкм). Допустимое содержание солей в растворах
20-40 г/л. При больших концентрациях начинается отложение солей в кольцевом
зазоре между центральной и внешней трубками. Особенно легко засоряют
распылители легкогидролизуемые соли.
Можно увеличить зазор между трубками
и соответственно диаметр трубки, подающей раствор. Но тогда придется работать
при больших расходах распыляющего газа, если мы хотим получать малый диаметр
капель аэрозоля.
Эффективность распылителей невелика
(1,5-3,5 %), но может быть увеличена до 60-80%, если вплотную к соплу
распылителя поместить шарик импактора и увеличить расход газа. Угловые
пневматические распылители основаны на том же принципе, что и концентрические.
В большинстве случаев раствор в них подается по вертикальному, а газ - по
горизонтальному каналам. На средний диаметр капель влияет соотношение диаметров
капилляров, расстояние между их кончиками и давление газа.
В 70-х годах были рассмотрены
регулируемые распылители, но в настоящее время предпочитают использовать
распылители с жестко закрепленными капиллярами, так как у них лучше
долговременная стабильность, при этом оба капилляра часто изготавливаются в
виде единого блока.
Распылитель Бабингтона
В распылителях Бабингтона распыление
происходит при прохождении струи газа через тонкую пленку жидкости. Из
коммерческих зарубежных распылителей этого типа наибольшее распространение
получил распылитель GMK, применяемый в эмиссионной спектрометрии. Распылитель
работает с перистальтическим насосом или другим способом принудительной подачи
жидкости.
Распылитель обычно используется со
специальной камерой распыления, в которой аэрозоль изменяет направление
движения более чем 2 раза, за счет чего удаляются почти все тяжелые и
неоднородные капли. Количество вводимого аэрозоля по сравнению с пневматическим
распылителем при одинаковом расходе раствора (1,8 мл/мин) увеличивается в·1,5-2
раза, примерно в 1,5 раза повышается эффективность распыления. Примерно на 1
порядок уменьшается средний диаметр капель аэрозоля. Распылители этого типа
чрезвычайно устойчивы к засорению и позволяют распылять концентрированные
растворы солей. Опыт работы с этими распылителями во ВНИИ ТВЧ показал, что в
течение примерно 5 лет он не засорился ни разу, в то время как пневматический
концентрический распылитель требовал регулярной промывки (несколько раз в
неделю).
Недостатком этих распылителей при
использовании является наличие камеры распыления и необходимость удаления или
организации возврата на распыление отсеченных камерой крупных капель.
Сетчатый распылитель
С высокосолевыми растворами и
взвесями позволяет работать сетчатый распылитель Типдебранда (производитель
США). В этом распылителе мелкая сетка из инертного материала (например, из
платины) расположена вблизи сопла сверхзвуковой струи газа. Раствор подается на
сетку по трубке сравнительно большого сечения и жидкость, стекая по ней,
попадает под струю газа, которая срывает раствор с сетки, образуя тонкий
аэрозоль. На расстоянии 2 мм от первой сетки устанавливают вторую для
демпфирования пульсаций перистальтического насоса (если подача раствора
осуществляется с его помощью) и разбивания крупных капель. Распылитель
используется с камерой распыления конической формы.
Стабильность работы распылителя
этого типа на порядок выше, чем в обычных пневматических распылителей.
Эффективность распыления 4,0 %.
Недостатком его в случае применения
в плазмохимии также является необходимость удаления или возврата конденсата.
Распылители с пористой пластиной
В этом типе устройств распыление
происходит на пористой пластинке диаметром 10-30 мм, с одной стороны которой
подается газ, с другой - раствор. Пластинки спекаются из стекла или оксидов
металлов (алюминия, циркония и т.п.) и имеют размер пор 4-8 мкм. Эти
распылители образуют очень тонкий аэрозоль (средний размер капель около 0,1
мкм) с узким распределением. Эффективность распыления 10-94 %, скорость подачи
раствора 5-50 мкл/мин.
Недостатком распылителя является
большая память (в случае необходимости смены реагента - продолжительность
промывки-3-4минут).
Распылитель также требует решения
задачи удаления или возврата конденсата.
.5.2 Использование ультразвукового
распыления
Ряд исследователей для получения
аэрозоля используют ультразвуковые (УЗ) распылители. Этот процесс может
обеспечить образование очень мелких капелек без образования большого перепада
давления между жидкостью и газом и без использования распыляющего воздушного потока.
Наиболее существенным достоинством ультразвукового распыления по сравнению с
обычными методами считают существенную экономию энергопотребления []. Методы
ультразвукового распыления позволяют получать аэрозоли с узким распределением
капель жидкости по размерам и имеющие очень низкую кинетическую энергию. При
использовании ультразвукового распыления, меняя частоту ультразвука, можно
регулировать размеры капелек. Этого же можно добиться, изменяя скорость
флуктуирующего потока жидкости. В отличие от пневматических распылителей
распыление в ультразвуковых распылителях происходит за счет акустических
колебаний и газовый поток нужен только для переноса полученного аэрозоля.
Эффективность генерации аэрозоля в этих распылителях в 10-20 раз больше, чем у
пневматических распылителей. Эти распылители образуют тонкий аэрозоль со
средним размером капель до 1-5 мкм. При этом размер аэрозоля зависит от частоты
ультразвука. В статье Эше [] приведено описание некоторых закономерностей
процесса образования аэрозолей в ультразвуковом фонтанчике, получающемся в
результате действия сфокусированного ультразвука на поверхность раздела
жидкость-газ. Прежде всего, такой аэрозоль является тонко- и практически
монодисперсным. Так, например, при использовании ультразвука с частотой 2,5 МГц
35 % всех капелек образующегося аэрозоля имели размеры от 1,7 до 2,5 мкм,
размеры 85 % всех капелек заключены в пределах от 1,0 до 4,8 мкм.
Повышение частоты ультразвука
приводит к существенному уменьшению размера капелек аэрозоля. Поэтому для получения
мелкодисперсного аэрозоля целесообразно применять частоты более 1 МГц. Степень
дисперсности аэрозоля не зависит от мощности ультразвука.
В работе [] приводятся данные
экспериментальных исследований с помощью скоростной макро- и микрокиносъемки
процесса туманообразования в ультразвуковом фонтанчике. Фонтанчик получатся при
помощи плоского излучателя из керамики титаната бария, имеющего частоту
собственных колебаний 2 МГц. Ультразвук фокусировался плексиглазовой линзой.
Показано, что образующаяся под влиянием ультразвука струя жидкости состоит из
отдельных бусинок диаметром 0,95 мм. Область туманообразования расположена в
нижней части струи, причем верхняя граница области находится на расстоянии 4-5
мм от конца вспучивания. Наиболее интенсивно туманообразование происходит в
средней части указанной области. С увеличением мощности излучения ультразвука
возрастает высота фонтанчика, однако в пределах изменения высоты его от 5 до 12
см размеры и местоположение области туманообразования на струе практически не
меняются. Съемки показали также, что процесс туманообразования не является
непрерывным. Туман выбрасывается редкими кратковременными (<400 мксек)
взрывами, промежутки между которыми значительно больше, чем время, занимаемое
туманообразованием. Вместе с туманом, а иногда и без него, из струи фонтана
выбрасываются капли воды, размеры которых значительно больше размеров капелек
тумана. Для предотвращения попадания крупных капель в тракт подачи аэрозоля
используют, как и в пневматических распылителях камеры распыления, а также
устанавливают отбойные заслонки.
В [] предложен способ распыления
менисковой пленки жидкости, смачивающей наклонную излучающую поверхность
поршневого УЗ преобразователя, частично погруженного в распыляемую жидкость. В
качестве преобразователя использовался магнитостриктор, работающий на основной
частоте собственных колебаний 25 кГц. В этих условиях при подводимой мощности
650 Вт длина столба водяного тумана доходила до 100 см, причем в аэрозоль в I ч
переходило примерно 150 л воды с 1 дм излучающей поверхности преобразователя,
то есть столько же. При этом получающийся аэрозоль был довольно грубым:
капельки имели размер от 100 до 250 мкм. С увеличением мощности величина капель
возрастала.
Анализ приведенных данных
показывает, что этот способ распыления (распыление тонких слоев жидкости)
позволяет достичь очень высокой производительности. Однако использование частот
порядка 20 кГц недопустимо, так как получаемый аэрозоль является крупными и
полидисперсным.
Применяемые ультразвуковые распылители
встречаются двух видов:
получение ультразвукового фонтанчика
на поверхности жидкость-газ
распыление тонких пленок, омывающих
поверхность магнитострикционного преобразователя.
В работе [] подробно описана
конструкция распылителя первого типа и также приведена схема ультразвукового
генератора. Пьезоэлектрический туманообразователь, приводимый в действие
генератором высокочастотных электрических колебаний, превращает рабочий раствор
в туман. Раствор помещают в туманообразователь в стеклянных кюветах, дном которых
служит тонкая пленка тефлона. Пространство между кюветой и пьезоэлементом
(источником ультразвуковых колебаний) заполнено водой. Ультразвуковые волны
фокусируются вблизи свободной поверхности раствора, в результате чего
происходит непрерывное фонтанирование раствора и вызванное этим образование
тумана. Стеклянный конический отсекатель препятствует попаданию крупных капель
раствора в тракт. Туман подается воздушной или газовой струей при небольшом
избыточном давлении. Параметры генератора: мощность питания около 200 Вт,
рабочая частота 2,5 МГц. Размер капель тумана уменьшается с повышением частоты
ультразвука. При выбранной частоте капли имели диаметр от 1 до 3 мкм. Повышение
рабочей мощности генератора усиливало туманообразование. Однако использованию
генератора в режиме его максимальной мощности препятствует непропорционально
быстрое возрастание количества воздушных пузырьков, прилипающих ко дну кюветы,
которые экранируют и рассеивают ультразвук. Повышение температуры раствора
также приводит к усиленному туманообразование.
В работе [] между УЗ распылителем и
плазменной горелкой установлено устройство для десольватации аэрозоля.
Назначение этого устройства - удалить из аэрозоля растворитель. Устройство для
десольватации включает в себя трубчатую печь в виде кварцевой трубки длиной 200
мм и диаметром 25 мм. Температура печи около 400 0C. Из печи аэрозоль попадает
в конденсатор (охладитель), охлаждаемым проточной водой. Слив конденсата
осуществляется в нижней части конденсатора. В боковой отвод конденсатора
потоком газа осушенный аэрозоль транспортируется в плазмотрон. 80% твердого
составляющего в распыленном растворе переносится в плазму. Эмиссионный анализ
конденсата раствора железа (50 мг/мл) показал, что остальные 20% задерживаются
в конденсате.
Следует заметить, что десольвататоры
усложняют систему ввода вещества в плазмотрон и применение их по нашему мнению
оправдано лишь в случае работы с плазмотронами малой мощности при вводе
вещества непосредственно в струю плазмы. Если аэрозоль вводится в ПХР, сочлененный
с плазмотроном большой мощности, по-видимому, применение десольвататора
нецелесообразно.
В литературе описывается еще
несколько разновидностей УЗ распылителей первого типа, но, к сожалению, для
малых количеств раствора.
Дальнейшее развитие конструкций УЗ
распылителей шло по пути создания моделей, работающих по 2 схеме - то есть
методом образования тонкой пленки на поверхности УЗ преобразователя,
защищенного стеклянной пластиной. Основополагающей в этой области можно считать
работу []. В ней разработан УЗ распылитель горизонтальной конструкции, в
котором распыляемый образец (жидкость) подается перистальтическим насосом через
фторопластовую трубку на поверхность УЗ преобразователя. Распылитель работает
при частоте 1,43 МГц. Выходная мощность 40 Вт. Преобразователь охлаждается
водой. Скорость подачи образца 2,4 мл/мин. Распылитель расположен внутри камеры
распыления и сочленен с системой десольватации. Показано, что эффективность
данного распылителя в 10 раз выше, чем пневматического распылителя, работающего
при тех же условиях, и составляет ~ 11 %.
Кроме того известно, что
ультразвуковое диспергирование широко применяется в мире в процессах получения
порошков наноразмеров. Так, например, в работе [] наночастицы диоксида церия
были приготовлены из водного раствора, содержащего полимерное исходное вещество
пиролизом аэрозоля, полученного ультразвуковым распылением, при различных
условиях приготовления, таких как высокая скорость газа-носителя и,
соответственно, короткое время пребывания аэрозоля в зоне нагрева. В результате
были получены наночастицы диоксида церия при 1200 0C без применения помола.
Средний размер первичных частиц увеличивался с нескольких десятков нм до
субмикронного размера при повышении температуры от 800 до 1300 0С.
В работе [] нанокомпозитные порошки
гaммa-Fe2O3/MgO, которые могут быть использованы для биомедицинских, магнитных
и каталитических применений были приготовлены методом пиролиза аэрозоля,
полученного с использованием ультразвукового диспергатора. Исходный раствор был
приготовлен из нитратов железа и марганца, растворенных в чистой воде.
Полученный раствор с помощью ультразвукового диспергатора превращался в
аэрозоль, который газом-носителем (воздух) переносился в предварительно
нагретую камеру (500-800 0С). В камере капли аэрозоля превращались в
наночастицы гамма- Fe2O3/MgO. Все операции проводились при давлении 1 атм. Было
показано, что полученный порошок гамма- Fe2O3/MgO прекрасно кристаллизуется при
температуре 800 0С. Размер частиц порошков нанокомпозита Fe2O3/MgO приготовленных
при 800 0С был около 10 нм.
В работе [] были получены
наночастицы чистого феррита бария BaFe12O19 термообработкой при 700 0C аэрозоля
исходного раствора, содержащего цитратные комплексы бария и железа.
Нейтрализованный водный раствор, содержащий цитратные хелаты Ba+ и Fe+ был
превращен в аэрозоль с помощью ультразвукового диспергатора. Аэрозоль был
подвергнут термическому разложению в потоке воздуха при температуре 250 0C.
Полученные таким образом твердые частицы были прокалены при различной температуре,
после чего были определены химические и физические характеристики полученных
образцов. Было показано, что кристаллические ферриты бария образуются при такой
низкой температуре как 650 0C, но для получения порошка чистого феррита бария
требуется температура прокаливания выше 680 0C.
В работе [] был использован метод
пиролиза аэрозоля в пламени (FSP) для синтеза наночастиц оксида олова из
исходного раствора - этилгексаноата олова в этаноле. Полученные частицы были в
высокой степени кристалличны и имели исходный размер около 17 нм.
В работе [] описаны свойства
наночастиц никеля, приготовленных пиролизом аэрозоля раствора гексагидрата
нитрата никеля, полученного при низком давлении (LPSP). Было исследовано
влияние давления, скорости тока газа-носителя, концентрации исходного раствора
и типа восстановителя на размер, морфологию и кристалличность наночастиц
никеля. Было показано, что давление и скорость тока газа-носителя играют
решающую роль при восстановлении оксида никеля до металла в LPSP-процессе.
Относительно высокая концентрация исходного раствора способствует образованию
наночастиц никеля. Сорастворители, такие как муравьиная кислота и этанол, также
водород, вводимый с потоком газа носителя, трактуются в данной работе как
восстановители в процессе получения наночастиц металлического никеля.
В работе [] были синтезированы
наночастицы оксида титана с номинальным размером около 10 нм прямо из трех
органических исходных веществ: тетраизопропоксида (TTP), водорастворимых
источников титана ТС-300 и TC-400 методом пиролиза аэрозоля, полученного
ультразвуковым диспергированием при низком давлении (LPSP). Было исследовано
влияние температуры, растворителя, концентрации и типа исходного вещества на
получение наночастиц оксида титана. Было показано, что более высокая
температура и концентрация исходного вещества способствуют образованию
наночастиц оксида титана. Добавка этанола, как сорастворителя, способствует
формированию наночастиц.
Таким образом, использование
ультразвуковых распылителей для получения аэрозолей жидкостей имеет ряд
преимуществ по сравнению с традиционными пневматическими распылителями, так как
позволяет получить аэрозоли с размером частиц 1-4 мкм (при использовании
частоты УЗ генератора порядка 1-2 МГц). Для получения монодисперсного аэрозоля
большинство описанных в литературе УЗ распылителей (так же как пневматические
распылители) работают совместно с камерой распыления, предназначенной для
отсекания крупных капель аэрозоля и последующего слива. Наличие камеры
распыления усложняет конструкцию и снижает количество подаваемого аэрозоля.
Этот вопрос подлежит рассмотрению применительно к конкретным процессам.
Большинство приведенных в обзоре УЗ распылителей работают при низких скоростях
подачи жидкости в распылитель. Вопросы возможности увеличения производительности
распылителя при сохранении монодисперсности аэрозоля с малым диаметром капель
требуют дополнительной проработки. Из рассмотренных УЗ распылителей большую
производительность имеют распылители второго типа, то есть те распылители, в
которых распыляемая жидкость омывает в виде тонкого слоя излучающую поверхность
ультразвукового преобразователя, при этом для получения мелкодисперсного
аэрозоля необходимо работать при частоте генератора 1-2 МГц.
2. ЦЕЛИ И ЗАДАЧИ РАБОТЫ
Цель работы - разработка метода
получения оксидов урана в виде конечных форм, пригодных к прессованию и
последующему длительному и безопасному хранению до повторного использования в
тепловых или быстрых реакторах.
Задачи:
Отработка методик определения урана
в растворах переменного состава.
Определение условий ультразвукового
диспергирования растворов уранилнитрата на лабораторной установке.
Проверка эффективности
ультразвукового распыления растворов.
Проведение экспериментов по
термохимической денитрации реэкстракта урана в прямоточно-трубчатой электропечи
с получением оксидов.
Изучение возможности включения
оксидов актинидов в оксиды урана при денитрации смешанных нитратных растворов
этих элементов.
диспергирование
денитрация реэкстракт уран
3. ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Экспериментальная часть выполнялась
в лаборатории № 96 ГУП НПО “Радиевого института им. В.Г. Хлопина”.
.1 Методика приготовления растворов
и проведения лабораторных экспериментов
В работе использовалась азотная
кислота (Реахим, ГОСТ 4461-77) марки "ХЧ"; аммиак водный (ЗАО
"НПО ЭКРОС", ГОСТ 3760-79) марки "ЧДА", СТ Трилон Б 0,1 н
(ЗАО "ВЕКТОН", ТУ 2642-001-07500602-97), изопар (ECNO 292-44-6),
калий хлористый (Реахим, ТУ 6-09-3678-74) марки "ОСЧ", аммоний
углекислый (ЗАО "НПО ЭКРОС", ГОСТ 3770-75) марки "ЧДА",
алюминий азотнокислый (Реахим, ГОСТ 3757-65) марки "ЧДА"; соляная
кислота (ЗАО "ВЕКТОН", ГОСТ 3118-77) марки «ХЧ», ТБФ (Реахим, ТУ
6-09-08-1219-7) марки «ХЧ», уранил азотнокислый (СПб ФГУП «Изотоп») марки
«ЧДА», торий азотнокислый (СПб ФГУП «Изотоп») марки «ЧДА». Декан очищался путем
последовательных промывок концентрированной серной кислотой, раствором 2,5 г/л
KMnO4 в 10% серной кислоте, водой, 0,1 моль/л раствором KMnO4 в 10% гидроксиде
натрия, водой, 10 г/л щавелевой кислотой и снова водой.
Лабораторные опыты проводили при
температуре 20 ± 2 °С. Отбор проб на анализ
производился стеклянными пипетками ГОСТ 20292-74 (2 класс точности).
Определение урана и тория в
органической и водной фазах проводили по следующим методикам:
Определение урана.
Концентрация урана в экспериментах
определялась двумя методами:
Весовым методом, для определения
больших концентраций урана.
В данной методике используются
следующие реактивы:(конц.)OH
Отбирается проба объёмом 1 мл и
вносится в стакан на 100 мл. Анализируемый раствор подкисляют азотной кислотой
и кипятят до полного удаления CO2. Затем добавляют в небольшом избытке NH3 1:4,
не содержащий карбонатов, до появления отчетливого запаха аммиака. Раствор
осторожно нагревают до полной коагуляции осадка, добавляя в случае необходимости
ещё небольшое количество аммиака. Раствор выдерживается при температуре 70-80
0C. Дают осадку диураната аммония U2O7(NH4)2 осесть и фильтруют на бумажном
фильтре «белая лента». После полной отмывки осадка урана воронка с фильтром
переносится в сушильный шкаф и высушивается при температуре 90-100 0C. Затем
фильтр с осадком помещают в тигель, предварительно взвешенный. Тигель с осадком
прокалывают в муфельной печи при температуре 800-900 0C. Потом тигель
охлаждается в эксикаторе, а затем взвешивается. Уран в осадке находится в виде
U3O8 [].
Спектрофотометрическим методом, для
определения урана в рафинатах.
В данной методике используются
следующие реактивы:(конц.)
Арсеназо III
Метод основан на цветной реакции
уранил - иона с Арсеназо III. Стандартный раствор готовится с концентрацией
урана 200 мкг/мл. Путём разбавления этого раствора получаются рабочие растворы
с концентрацией урана 40, 80,120,180 мкг/мл. Измеряется оптическая плотность на
спектрофотометре. И по полученным значениям строят калибровочную кривую зависимости
оптическая плотность от концентрации урана в растворе показанную на рис. 2 [].
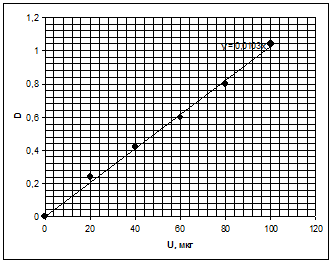
Рисунок 2 - Зависимость оптической
плотности от концентрации урана в растворе
Определение урана в тории.
В данной методике используются
следующие реактивы:
8 г
KNO3 или
Al(NO3)3
Титр трилона Б - 0,1 н
ТБФ в разбавителе 30 %
(NH4)2CO3 - 5 % раствор
Аликвоту раствора, содержащую не
более 1000 мг урана, переводят в азотнокислый раствор до рН=2-3 по
потенциометру. К приготовленному таким способом раствору добавляют 8 г
высаливателя (KNO3 или Al(NO3)3) марки «ХЧ» и ТБФ (30 %) в керосине из расчёта
1 мл на 250 мг урана, перемешивают с помощью магнитной мешалки 5 минут. Затем
органическую фазу отделяют от водной и реэкстрактируют уран из органической
фазы (ТБФ) в 5 % раствор (NH4)2CO3 (1-2 промывки водой и 2-3 раствором
карбоната аммония) [].
После реэкстракции урана, определяем
его концентрацию весовым способом.
Определение тория.
Концентрация тория в экспериментах
определялась двумя методами:
Последовательное
комплексонометрическое определение тория.
В данной методике используются
следующие реактивы:
Буферный раствор с рН=1,6 (0,2 н HCl
(131,5 мл) + 0,2 н KCl (250 мл) довести до 1 л дистиллированной водой)
Индикатор ксиленоловый оранжевый
Индикатор метиленовый голубой
Титр трилона Б - 0,1 н
Аликвоту раствора с содержанием
металла больше 50 мг помещают в коническую колбу на 250 мл, добавляют 100 мл
воды и 10 мл буферного раствора с рН=1,6; также добавляют ксиленовый оранжевый
и метиленовый голубой. Затем титруют 0,05 М раствором трилона Б от фиолетового
до изумрудно-зелёного цвета [].
Определение тория в уране.
В данной методике используются
следующие реактивы:
8 г
KNO3 или
Al(NO3)3
Индикатор ксиленоловый оранжевый
Титр трилона Б - 0,1 н
ТБФ очищенный
Аликвоту раствора, содержащую не
менее 1 мг тория, но не более 1000 мг урана, переводят в азотнокислый раствор
до рН=2-3 по потенциометру. К приготовленному таким способом раствору добавляют
индикатор ксиленовый оранжевый. Затем добавляют 8 г высаливателя (KNO3 или
Al(NO3)3) марки «ХЧ» и очищенного обычным способом ТБФ из расчёта 1 мл на 250
мг урана, перемешивают с помощью магнитной мешалки 5 минут и титруют раствором
комплексона III из микробюретки до перехода малиновой окраски в желто-зелёную [41].
.2 Объекты и методы исследования
Для проведения испытаний процесса
получения оксидов урана термохимической денитрацией азотнокислых растворов
уранилнитрата, получаемых при экстракционной переработке ОЯТ, создан
лабораторный стенд, состоящий из следующих основных узлов (рис. 3):
ультразвукового распылителя раствора (1), трубчатой электропечи (4),
позволяющей получать температуру в рабочей зоне до 700 0С, сепаратора
разделения твердой и парогазовой фазы (7), а также конденсатора паров азотной
кислоты (10).
Процесс получения оксидов урана из
растворов уранилнитрата состоял из следующих операций:
ультразвуковое диспергирование
раствора уранилнитрата с превращением его в аэрозоль с размером частиц менее 10
мкм;
транспортирование полученного
аэрозоля в трубчатую печь, что осуществлялось с помощью газа-носителя, в
качестве которого использовался воздух, поток газа-носителя на стенде
создавался прокачкой воздуха с помощью водоструйного или форвакуумного насоса.
Контроль скорости газового потока осуществлялся по высоте водяного столба в
U-образном манометре.
термообработка аэрозоля раствора
уранилнитрата в трубчатой печи при температуре 500 - 7000С, температура в
электропечи измерялась при помощи термопары.
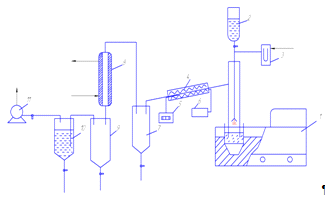
Рисунок 3 - Аппаратурная схема
лабораторного стенда денитрации азотнокислого раствора уранилнитрата
- ультразвуковой распылитель; 2 -
питательная емкость; 3 - U-образный манометр; 4 - трубчатая электропечь; 5 -
цифровой прибор определения температуры; 6 - источник питания; 7 - сепаратор
конечного продукта; 8 - конденсатор паров азотной кислоты; 9 - приемная емкость
азотной кислоты; 10 - щелочная ловушка; 11 - форвакуумный насос.
Получение аэрозоля раствора
уранилнитрата осуществлялось с помощью ультразвукового диспергатора, схема
которого представлена на рис. 4
Распыляемый раствор уранилнитрата
помещался в кювету (2), в дно которой была вмонтирована проницаемая для
ультразвука мембрана.
Ультразвуковой излучатель,
изготовленный из титаната бария, был помещен в камеру, заполненную
дистиллированной водой, которая обеспечивала охлаждение излучателя и передачу
УЗ в кювету с распыляемым раствором.
Образовавшийся аэрозоль отделялся от
крупных капель раствора с помощью конического стеклянного отсекателя (5) и с
потоком газа-носителя подавался в цилиндрическую электропечь (поз. 4 на рис. 3)
для термической денитрации.

Рисунок 4 - Общий вид
ультразвукового распылителя
- ультразвуковой излучатель; 2 -
стеклянная кювета для раствора; 3 - полиэтиленовая мембрана; 4 - охлаждающая
вода; 5 - стеклянный конический отсекатель; 6 - распыляемый раствор.
В предварительных экспериментах для
распыления водных растворов использовался ультразвуковой диспергатор, который
имел следующие основные параметры:
Частота ультразвука - 2,6 МГц;
Потребляемая мощность - 25 Вт.
Размер 90 % частиц полученного
аэрозоля не более 10 мкм;
Температура распыляемой жидкости 18
- 40 0С.
Проведенные предварительные опыты
показали, что такой диспергатор не обеспечивает достаточной производительности
по распыляемому раствору, поэтому в дальнейших экспериментах был использован
более мощный УЗ распылитель с производительностью до 400 мл/час по воде.
Проверка распылительного устройства
на воде подтвердила его достаточно высокую эффективность.
4. РЕЗУЛЬТАТЫ ЭКСПЕРИМЕНТОВ И ИХ
ОБСУЖДЕНИЕ
.1 Проверка эффективности
ультразвукового распыления водных растворов
С целью выбора условий, обеспечивающих
достаточно высокую производительность ультразвукового распыления реэкстракта
урана, было изучено влияние условий распыления на эффективность этого процесса.
Эксперименты проводились на воде и растворах уранилнитрата. Во всех опытах
использовался УЗ распылитель, схема которого была представлена выше.
Распыляемая жидкость помещалась в стеклянную кювету, дно которой было выполнено
из полимерной пленки, проницаемой для ультразвука. Результаты этих
экспериментов представлены в таблицах 2 и 3.
|
№
опыта
|
Время
работы устр-ва, мин.
|
Скорость
газового потока, л/мин.
|
Электрическая
мощность УЗ- генератора, Вт
|
Объем
воды во внутренней чашке, мл
|
Распылено
воды, % от исх.
|
|
|
|
|
исх.
|
кон.
|
|
|
1
|
60
|
1,8-2,0
|
10
|
25
|
21,5
|
14
|
|
2
|
60
|
1,8-2,0
|
20
|
20
|
8,5
|
57,5
|
|
3
|
60
|
1,9
|
20
|
25
|
15,5
|
36,0
|
|
4
|
60
|
0,9-1,0
|
20
|
25
|
21
|
16,0
|
|
5
|
60
|
0,9-1,0
|
30
|
25
|
19
|
32,0
|
|
6
|
60
|
1,8-2,0
|
30
|
25
|
15
|
40,0
|
|
7
|
60
|
0,9-1,0
|
40
|
25
|
17,5
|
30,0
|
|
8
|
60
|
0,9-1,0
|
20
|
25
|
21
|
16,0
|
|
9
|
60
|
0,9-1,0
|
40
|
25
|
20,5
|
18,0
|
Таблица 3 - Результаты проверки
аэрозольного распыления раствора уранилнитрата при электрической мощности УЗ-генератора
20 Вт (Концентрация урана в распыляемых растворах 50-220 г/л)
|
№
опыта
|
Время
работы уст-ва, мин.
|
Скорость
газового потока, л/мин.
|
Кол-во
распы-ленного р-ра, мл
|
Концентрация
UO2(NO3)2 в распыляемом растворе, г U/л
|
|
|
|
|
исх.
|
кон.
|
|
1
|
150
|
0,5-1,0
|
9,7
|
154
|
164
|
|
2
|
330
|
0,5-1,0
|
16,5
|
129
|
181
|
|
3
|
330
|
0,5-1,0
|
7,8
|
53
|
67
|
|
4
|
120
|
1,8-
2,0
|
15,0
|
53
|
61
|
|
5
|
135
|
1,8-2,0
|
14,2
|
219
|
326
|
|
6
|
300
|
0,9-1,0
|
4,0
|
96
|
103
|
Данные по влиянию режима работы УЗ диспергатора
на эффективность распыления воды, представленные в табл.2, не позволяют судить
однозначно о влиянии мощности ультразвука и скорости газового потока на
эффективность диспергирования воды. Из результатов проведенных экспериментов
можно сделать следующие выводы:
. Введение в систему УЗ распыления кюветы для
распыляемого раствора, отделяющего его от источника ультразвука с помощью
пленки, проницаемой для ультразвука, приводит к резкому снижению эффективности
распыления воды. Так, если для прямого распыления воды на данном устройстве при
максимальной мощности УЗ количество распыленной воды составляет 400 мл в час,
то в этих же условиях при использовании кюветы для распыляемого раствора,
скорость распыления воды снижается в 50 - 90 раз, что связано с
перераспределением потока УЗ энергии по сечению устройства и его потерей при
прохождении через пленочную мембрану в дне кюветы для распыляемого раствора.
. Увеличение скорости потока газа - носителя
приводит, при одинаковой мощности УЗ, к заметному увеличению скорости
распыления воды. При этом будет снижаться время нахождения аэрозоля в зоне
нагрева трубчатой электропечи.
Сравнение данных, представленных в табл.2 и 3
показывает, что при одинаковой мощности УЗ и одинаковом потоке газа носителя
скорость распыления растворов уранилнитрата примерно в 1,5 раза ниже, чем
скорость распыления воды.
Распределение урана по компонентам установки в
опытах, результаты которых представлены в табл. 3, приведены в табл. 4.
Таблица 4 - Распределение урана по
компонентам узла ультразвукового диспергирования раствора уранилнитрата
|
№
опыта
|
Количество
урана
|
|
в
ловушке
|
в
чашке
|
в
распылительной камере
|
|
г.
|
%
к исх.
|
г.
|
%
к исх.
|
г.
|
%
к исх.
|
|
1
|
0,64
|
12,8
|
4,07
|
81,4
|
0,02
|
0,4
|
|
2
|
0,13
|
9,3
|
1,24
|
88,6
|
0,006
|
0,4
|
|
3
|
0,69
|
66,3
|
0,28
|
26,9
|
<0,001
|
<0,1
|
|
4
|
2,29
|
52,3
|
1,89
|
43,1
|
<0,005
|
<0,1
|
.2 Исследование включения актинидов (на примере
Th) в оксиды урана при денитрации
Для получения оксидов урана или смеси оксидов
урана и тория использовался метод термической денитрации аэрозолей растворов
нитратов этих элементов, полученных методом УЗ распыления, описанным в разделе
3.3.1.
Эффективность процесса термической денитрации
аэрозолей растворов нитратов урана и тория зависит от размеров капель жидкой
фазы этих аэрозолей, от температуры в печи и времени пребывания аэрозоля в зоне
нагрева. Размер капель жидкой фазы аэрозолей, подвергавшихся термообработке в
прямоточной печи, в проведенных экспериментах был фиксирован, поскольку он
определяется частотой УЗ, используемого для получения аэрозоля, которая была
постоянна и физико-химическими характеристиками распыляемых растворов (плотностью
и поверхностным натяжением), которые в рамках составов использованных в
экспериментах растворов менялись незначительно. Основными параметрами, которые
могли меняться в ходе экспериментов по термической денитрации, являлись
температура в зоне нагрева аэрозоля, и время пребывания в ней аэрозоля.
Для определения распределения температуры в зоне
нагрева аэрозоля была проведена серия замеров температуры по длине реакционной
зоны печи, которая представляла собой кварцевую трубку диаметром 8 или 19 мм, в
статических условиях и при прокачке через нее воздуха и аэрозоля воды.
Полученные результаты представлены в таблицах 5 и 6.
Таблица 5 - Распределение
температуры по длине реакционной зоны электропечи (внутренний диаметр
электропечи 35 мм, длина зоны нагрева -350 мм, внутренний диаметр реакционной
зоны (кварцевой трубки) - 8 мм, поток газа-носителя - 1 л/мин, скорость
распыления воды - 7-8 мл/час)
|
Положение
точки замера температуры по длине реакционной зоны.
|
Температура,
оС
|
|
без
продувки
|
с
продувкой воздухом
|
с
воздушно- аэрозольной продувкой
|
|
На
расстоянии 4 см до начала зоны нагрева.
|
100
|
95
|
86
|
|
На
расстоянии 1 см от начала зоны нагрева.
|
390
|
345
|
320
|
|
На
расстоянии 4 см от начала зоны нагрева.
|
485
|
475
|
480
|
|
На
расстоянии 8 см от начала зоны нагрева.
|
575
|
575
|
575
|
|
На
расстоянии 15 см от начала зоны нагрева (середина электропечи)
|
585
|
585
|
585
|
|
На
расстоянии 29 см от начала зоны нагрева.
|
460
|
470
|
470
|
Таблица 6 - Распределение
температуры по длине реакционной зоны при использовании двухсекционной печки
(общая длина зоны нагрева - 53 см)
|
Положение
термопары в контрольной точке нагрева
|
Температура,
в оС (с продувкой зоны нагрева воздухом)
|
|
На
расстоянии 4 см до начала зоны нагрева 1ой электропечи
|
250
|
|
В
начале зоны нагрева 1ой электропечи
|
400
|
|
В
середине зоны нагрева 1ой электропечи
|
530
|
|
В
середине зоны нагрева 2ой электропечи
|
530
|
|
В
конце зоны нагрева 2ой электропечи
|
375
|
|
На
расстоянии 4 см после зоны нагрева 2ой электропечи
|
250
|
Как видно из данных, представленных в табл. 5,
существенного изменения температуры по длине реакционной зоны не наблюдается.
Приведенные данные свидетельствуют, что в рабочей зоне нагрева, составляющей
около 30 см по длине трубки, температура поддерживалась в пределах 485-585 0С.
Аналогичные данные были получены для реакционной зоны кварцевой трубки с
внутренним диаметром 19 мм. В дальнейшем длина зоны нагрева была увеличена до
53 см за счет присоединения к основной печке дополнительной секции. Данные по
распределению температуры по длине реакционной зоны при использовании такой
печки приведены в табл. 6.
С учетом полученных результатов была проведена
серия опытов на стенде по термической обработке аэрозолей раствора
уранилнитрата и смешанного раствора уранилнитрата и нитрата тория. Условия
проведения этих экспериментов представлены в табл. 7. Результаты, полученные в
ходе этих экспериментов, приведены в табл. 8.
Таблица 7 - Режим проведения
экспериментов по термообработке аэрозолей раствора уранилнитрата и смешанного
раствора уранилнитрата и нитрата тория
|
№
п/п
|
Продолжи-тельность
эксперим., час.
|
Поток
воздуха, л/мин.
|
Температура
в реакторе, оС
|
Диаметр
реактора, мм
|
Длина
зоны нагрева, см.
|
Состав
исходного раствора.
|
|
1
|
3
|
0,9-1,0
|
500
|
8
|
29
|
UO2(NO3)2
Cu - 305 г/л
|
|
2
|
3
|
0,9-1,0
|
500
|
19
|
29
|
UO2(NO3)2
360 г/л
|
|
3
|
5
|
0,9-1,0
|
500
|
19
|
29
|
UO2(NO3)2
Cu - 300 г/л
|
|
4
|
9,5
|
0,9-1,0
|
550
|
19
|
53
|
UO2(NO3)2
Cu - 165 г/л
|
|
5
|
10
|
0,9-1,0
|
550
|
19
|
53
|
UO2(NO3)2 Cu - 213 г/л
Th(NO3)4 CTh
- 26,3г/л
|
|
6
|
6
|
0,9-1,0
|
550
|
19
|
53
|
UO2(NO3)2 Cu - 133 г/л
Th(NO3)4 CTh
- 26,3г/л
|
В табл. 8 представлены данные по распределению
урана и суммы урана и тория по аппаратам узла денитрации.
Таблица 8 - Распределение урана и
суммы урана и тория по аппаратам узла денитрации
|
№
п/п
|
Количество
металлов в аэрозоле, г.
|
Распределение
металлов по аппаратам узла денитрации
|
|
|
Сепаратор
|
Ловушка
|
Стенки
реактора
|
|
|
г
|
%
от исх.
|
г
|
%
от исх.
|
г
|
%
от исх.
|
|
1
|
0,9
|
0,043
|
4,8
|
0,027
|
3,0
|
0,068
|
7,9
|
|
2
|
0,3
|
0,017
|
5,7
|
0,008
|
2,7
|
0,007
|
2,3
|
|
3
|
0,2
|
0,001
|
5,0
|
0,014
|
7,0
|
0,022
|
11,0
|
|
4
|
1,5
|
0,22
|
14,7
|
0,18
|
15,0
|
0,86
|
57,3
|
|
5
|
3,1
(в том числе 0,4 г тория)
|
0,17
(в том числе 0,02г Th)
|
5,5
|
0,40
(в том числе 0,05г Th)
|
12,9
|
2,15
(в том числе 0,25г Th)
|
69,4
|
|
6
|
0,75(в
том числе 0,13 г Th)
|
<
0,06
|
<
8
|
<
0,06
|
<
8
|
0,50
(в том числе 0,08г Th)
|
66,7
|
Данные представленные в табл. 8 показывают, что
продукты термической денитрации растворов уранилнитрата распределяются по
аппаратам узла термической денитрации, при этом до 57 % оксидов оказываются на
поверхности кварцевого реактора, причем с увеличением продолжительности
эксперимента растет и доля оксидов, находящаяся на поверхности кварцевого
реактора. В случае денитрации смешанного раствора уранилнитрата и нитрата тория
распределение продуктов денитрации по аппаратам узла денитрации носит
аналогичный характер.
В опыте 4 было проведена оценка распределения
оксидов урана, осаждающихся на внутренней поверхности кварцевого реактора по
зонам его нагрева при термической денитрации аэрозоля раствора. Полученные
данные представлены в табл. 9.
Таблица 9 - Распределение оксидов
урана по зонам нагрева кварцевого реактора (опыт 4 в табл. 8.)
|
Зона
нагрева кварцевого реактора
|
Распределение
оксидов урана по зонам нагрева реактора, % от общего количества оксида
|
|
UO3
|
U3O8
|
|
Первая
зона (до входа в печь)
|
41,2
|
-
|
|
Вторая
зона (зона нагрева 1-ой печи)
|
47,9
|
<
0,1
|
|
Третья
зона (между 1ой и 2ой печками)
|
0,74
|
<
0,2
|
|
Четвертая
зона (зона нагрева 2ой печи)
|
9,5
|
98,7
|
|
Пятая
зона (после выхода из 2ой печки)
|
1,4
|
<
0,1
|
Следует отметить, что 60 % распыленного
уранилнитрата осела на стенке кварцевого реактора перед зоной нагрева 1-ой
печи, где температура составляет 250 0С. В этой же зоне обнаружено и
значительное количество триоксида урана. Небольшие количества уранилнитрата
обнаружены по всей длине кварцевого реактора. Причиной осаждения уранилнитрата
из аэрозоля на входе реактора может быть потеря стабильности аэрозоля раствора
уранилнитрата при удалении из капель аэрозоля воды, для чего вполне достаточна
температура 250 0С, существующая в этой части кварцевого реактора (кстати,
косвенным подтверждением этого предположения может служить снижение
эффективности распыления раствора при высокой концентрации уранилнитрата,
обнаруженное в экспериментах на стенде). Осаждения уранилнитрата на входе в
зону нагрева вероятно можно избежать, значительно уменьшив диаметр трубки,
подводящей поток аэрозоля к кварцевому реактору на входе в зону нагрева 1-ой
печи, что повысит линейную скорость аэрозоля и, возможно, предотвратит
осаждение уранилнитрата. Кроме того, может оказаться полезным охлаждение
трубки, проводящей аэрозоль в реактор.
Основным (по величине выхода) оксидом урана, образующимся
при термообработке аэрозоля уранилнитрата, является трехокись урана (37,5 %).
Основная часть трехокиси находится в первых двух зонах нагрева (перед входом в
1-ую печь и в зоне ее нагрева)
Закись-окись урана (15,8 %), образующаяся при
денитрации аэрозоля раствора уранилнитрата практически полностью локализуется в
зоне нагрева второй печи.
.3 Проведение прессования оксидов урана,
полученных денитрацией на прямоточном аппарате-печи с определением параметров
таблеток
В ходе экспериментов по таблетированию
полученных порошков оксидов урана было проведено прямое холодное прессование
исходного порошка оксида урана без добавки связующего, с целью уменьшения
объема хранимых отходов.

Рисунок 5 - Внешний вид таблеток оксидов урана,
полученных посредством холодного прессования
Результаты проведенных экспериментов подтвердили
возможность таблетирования оксидов урана, полученных прямой денитрацией
аэрозоля уранилнитрата в прямой электропечи.
ЗАКЛЮЧЕНИЕ
Результаты проведенных экспериментов показали
принципиальную возможность проведения термохимической денитрации растворов
уранилнитрата в прямоточном аппарате-печи при подаче раствора в печь в виде
аэрозоля, полученного методом ультразвукового распыления раствора
уранилнитрата.
Использование ультразвука для диспергирования
растворов уранилнитрата и других актинидов позволяет получать аэрозоли
растворов этих элементов с размерами капель, находящимися в весьма узких
пределах. При этом размер капель аэрозоля, а после термообработки и размер
полученных твердых частиц прямо зависит от частоты ультразвука и
физико-химических свойств распыляемого раствора (плотность, вязкость,
поверхностное натяжение). Изменение этих параметров позволит получать частицы
требуемого размера.
Прямая термическая денитрация аэрозолей
азотнокислых растворов урана или смешенных растворов нитрата уранила с
нитратами других актинидов (что было показано на примере смешанного раствора
уранилнитрата и нитрата тория) позволяет получить микродисперсный порошок
оксидов этих элементов. Полученные оксиды могут быть использованы для
длительного хранения.
Существенным преимуществом процессов получения
твердых соединений элементов термообработкой аэрозолей их растворов является
то, что, поскольку превращение капель аэрозоля в твердые частицы происходит в
газовом потоке, то при соответствующем выборе условий процесса, можно исключить
накопление получаемых продуктов на стенках реакционной камеры.
СПИСОК ИСПОЛЬЗОВАННЫХ ИСТОЧНИКОВ
1
Давыдов В. И., Гамрекели М. Н., Добрыгин П. Г. Термические процессы и аппараты
для получения окислов редких и радиоактивных металлов. - М.: Атомиздат, 1977, -
207 с.
Аппарат
псевдоожижженого слоя для термической переработки замасленной окалины трубных
производств/ Г. Д. Янкин, В. А. Чемезов, В. А. Кудусов и др.// Труды
свердловского научно-исследовательского института химического машиностроения.-
1997.- Т. 68,вып. 4.- С. 103-105.
Чемезов
В. А., Каримов Р. С., Шевелин Б. П. Конструкторские проработки оборудования для
ядерного топливного цикла. Обзор// Труды свердловского
научно-исследовательского института химического машиностроения.- 2005.- Т. 76,
вып. 12.- С. 321-342.
Судариков
Б. Н., Раков Э. Г. Процессы и аппараты урановых производств. - М.:
Машиностроение, 1969. - 38 с.
Радиохимическая
переработка ядерного топлива АЭС/ В. И. Землянухин, Е. И. Ильенко, А. Н.
Кондратьев и др. - М.: Энергоатомиздат, 1983. - 232 с.
Копырин
А. А., Карелин А. И., Карелин В. А. Технология производства и радиохимической
переработки ядерного топлива: Учеб. Пособие для вузов. - М.: ЗАО «Издательство
Атомэнергоиздат», 2006. 576 с.: ил.
Жиганов
А. Н., Гузеев В. В., Андреев Г. Г. Технология диоксида урана для керамического
ядерного топлива. - Томск: Изд-во STT, 2002.
Копельман
Б. Материалы для ядерных реакторов. М.: Гос. изд. лит. в обл. ат. науки и
техники. 1962. С.117.
Haas
P.A. A Comparison of Processes for the Conversion of Uranyl Nitrate into
Ceramic-Grade UO2 / Nuclear Technology, 1988, v. 81, No. 3, p. 393-406.
Haas
P.A., Stines W.B. Method for Improved Decomposition of Metal Nitrate Solution /
US Patent 4,409,157 (Oct. 11, 1983).
Notz
K.J., Haas P.A. Properties and Thermal Decomposition of the Double Salts of
Uranyl Nitrate - Ammonium Nitrate / ORNL/TM-7820, Sep. 1981.
Haas
P.A. et al. Development of Thermal Denitration to Prepare Uranium Oxide and
Mixed Oxide for Nuclear Fuel Fabrication / ORNL-5735, Sep. 1981.
Davis
N.C., Griffin C.W. Pellet Fabrication Development Using Thermally Denitrated
UO2Powder / PNL-4305, Apr. 1982.
Bachelard
R., Lakodey P. Procėdė d’obtention de
UO3 de grand surface spėcifique a partir de
nitrate d’uranyle hydratė / Patent 2 526 006
(France). 4.11.1983.
Bachelard
R., Joubert J. Procėdė de prėparation d’oxydes mėtallique
pulverulent a partir de solution aqueses ou de mėlanges solides de
nitrates mėtalliques. / Patent 2 555 566
(France). 31.05.1985.
Bachelard
R., Germanaz P. Procėdė d’obtention d’oxide mixte frittės
solublles dans l’acide nitrique à partir de solutions de nitrates. /
Patent 2 596 384 (France). 2.10.1987.
17
Koizumi M. et al. Development of a Process for the Co-Conversion of Pu-U
Nitrate Mixed Solutions to Mixed-Oxide Powder Using a Microwave Heating Method
/ Nucl. Technol., 1983, v. 61, p. 55.
Sato
H., Morisue T. Microwave Heating Denitration Apparatus / Report Rockwell
International Corp., Golden, CO(USA), Rocky Flats Plant, Jan. 1983. Цит.
по
INIS Atomindex 14-754294.
Bao-Weimin,
Chang-Baoxiang, Guo-Zehong The Research for Applying Microwave Denitration on
the Conversion of High-Enriched Uranium / Atomic Energy Science and Technology,
1995, v. 29, May, p. 268-274/ Цит.
по
INIS Atomindex 27-004350.
Takahashi
Y. Continuous Denitration Apparatus / Patent US 5589140 31.12.1996.
Breisch
R.C. et al. Technical Data Summary for a COPRECAL Conversion in a Light Water
Reactor Fuel Reprocessing - Refabrication Plant Complex / NEDC-21746, July
1979.
Partridge
J.A., Lerch R E., Bosuego G. P. Dissolution of Coprecal Mixte-Oxide Fuel./
Trans/ Am/. Nucl. Soc.1981. V. 38. N. 6. P.205-206.
D.
N. Bykhovski. Basic Technological Solutions for Plutonium Management at the
RT-2 Plant. Nato ASI. Series 1: Disarmament Technologies - Vol. 2. P. 25.
Сурис
А.
Л.
Плазмохимические
процессы
и
аппараты.
- М.:
«Химия»,
1984. - 19 с.
25
Полуэктов Н.С. Методы анализа по фотометрии пламени. М.:
изд.
«Химия»,
1967.
Gaikwas
S. G., Reddy C., Pandit A. B. Ultrasonic Atomization: A Novel Technique for
Surface coatings/ Surface Coatings International Part B; Coating Transactions,
International. - 2005. - V. 88, B 3.- P. 189-196.
Esche.
Erzeugung von hochwertigen aerosolle mittels ultraschall - Communiations du
congres international sur les traitements per les ultrasons.// Marsielle.
- 1955, p. 179-183.
Розенберг
Л.Д., Экнадиосянц О.К.// Акустический журнал - 1960, т. 6, вып. 3, с. 370-375.
29
Crawford A.E.. Aoust J.// Soc. America - 1955, 27, № 1, p.
176.
Теркен
У.Б., Иванцов Л.М., Костин Б.И. Заводская лаборатория - 1962, т. 28, № 12, с.
1451-1454.
31
Diekenson G.W., Fassel V.A.// Anal. Chem. - 1969 - V. 41, № 8. - P. 1021-1024.
Olson
K.W., Haas W.J., Fassel V.A.//Anal. Chem. - 1977 - V. 49, № 4. - P. 632-637.
Nano-sized
ceria particles prepared by spray pyrolysis using polymeric precursor solution/
H. S. Kang, Y. C. Kang, H. Y. Koo et al.// Materials Science and Engineering B.
- 2006. - V. 127. - P. 99-104.
Synthesis
and characterization of isolated iron oxide nanoparcticle dispersed in MgO
matrix/ Y.-H. Choa, J.-K. Yang, W.-J. Yang, K.-H. Auh// Journal of Magnetism
and Magnetic Materials. - 2003. - V. 266. - P. 20-27.
Yu
H.-F., Lin H.-Y. Preparation and thermal behavior of aerosol-derived BaFe12O19
nanoparticles// Journal of Magnetism and Magnetic Materials. - 2004. - V. 283.
- P. 190-198.
Flame
spray synthesis of tin dioxide nanoparticles for gas sensing/ T. Sahm, N.
Barsan, S. E. Platsinis et al.// Sensors and Actuators B. - 2004. - V. 98. - P.
148-153.
Nickel
and nickel oxide nanoparticles prepared from nickel nitrate hexahydrate by a
low pressure spray pyrolysis/ W.-N. Wang, Y. Itoh, I. W. Lenggoro, K. Okuyama//
Materials Science and Engineering B. - 2004. - V. 111. - P. 69-76.
One-step
synthesis of titanium oxide nanoparticles by spray pyrolysis of organic
precursors/ W.-N. Wang, I. W. Lenggoro, Y. Terashi et al.// Materials
Science and Engineering B. - 2005. - V. 123. - P. 194-202.
Уран.
Методы его определения/ В. К. Марков, А. В. Виноградов, С. В. Елинсан и др. -
М.: Атомиздат, 1960. - 263 с.
Марченко
З. Фотометрическое определение элементов. - М.: Изд. «Мир», 1971. - 511 с.
Рябинин
А. И., Афанасьев Ю. А., Комплексонометрическое определение тория в уране в
присутствии ТБФ// Журнал аналитической химии.- 1966.-Т. XXI.- вып. 3.
Чернихов
Ю. А., Трамм Р. С., Певзнер К. С., Последовательное комплексонометрическое
определение тория и суммы редких земель// Зав. лаб.- 1960.-Т. XXVI.- № 8.
ГОСТ
12.0.001-74 ССБТ. Опасные и вредные производственные факторы. Классификация. -
В кн. : и др. - М.: Издательство стандартов, 1981.- 10 - 13с.
ГОСТ
12.1.019-79 ССБТ. Электробезопасность. - М.: Издательство стандартов, 1985. -
44с.
Нормы
радиационной безопасности (НРБ-99) и Основные санитарные правила обеспечения
радиационной безопасности (ОСПОРБ-99): Гигиенические нормативы. - М.: Центр
санитарно-эпидемиологического нормирования, гигиенической сертификации и
экспертизы. Министерство Здравоохранения РФ, 1999. 116 с.
Вредные
вещества в промышленности/ Под ред. Лазарева Н.В.В 3-х т. - М.: Химия, 1977.-
677с.
Пожаровзрывобезопасность
веществ и материалов и средства их тушения// Справ., изд. В 2-х книгах// Под
ред. Баратова А.Н., Котильченко А.Я. - М.: Химия, 1997.
НПБ
105-03. Определение категорий помещений и зданий по взрывопожарной и пожарной
опасности. - М.: Гл. Упр. Гос. противопожарной службы. МВД РФ, 1996. - 322с.
ГОСТ
12.1.004 - 76 ССБТ. Пожарная безопасность. Общие требования. - М.: Издательство
стандартов, 1992. - 78с.
ГОСТ
12.4.021-75 ССБТ. Системы вентиляции. Общие требования. М.: Издательство
стандартов, 1975. - 5 - 8с.
СНиП
23-05-95. Естественное и искусственное освещение. Нормы проектирования.-
М.-Стройиздат, 1995. - 48с.
Л.П.
Захаров. Техника безопасности в химической лаборатории: справ.
-Л.:Химия,1991.-336с.
Правила
устройства электроустановок (ПУЭ). - СПб.: Издательство ДЕАН, 2001. 928с.
Приложение А
Технико-экономическая оценка результатов
исследования
Важным показателем проводимых
исследований является договорная цена на научно- исследовательскую
работу (НИР) Цн:
 (А. 1)
(А. 1)
где Зн - затраты на
выполнение исследования, предусмотренного планом дипломной работы, руб.;
Р - уровень рентабельности
исследования, %;
К - коэффициент, учитывающий
поощрительную надбавку за качество разработки.
Для определения договорной цены
рассчитаны текущие затраты на выполнение НИР.
А.1 Затраты на сырье, материалы и
реактивы
Затраты на сырье, материалы и
реактивы (Зм), израсходованные на проведение исследования, определены расчетом
по формуле (А.2), результаты которого сведены в таблице А.1.
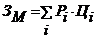 (А. 2)
(А. 2)
где Рi - расход i-го вида
материальных ресурсов, нат. ед.;
Цi -планово-заготовительная
цена i-го вида материальных ресурсов, руб.
Таблица А.1 - Расчет затрат на
сырье, материалы, реактивы
|
ГОСТ,
техническая характеристика реактивов и веществ
|
Единица
измерения
|
Израсходовано
материала
|
Цена
по прейску-ранту, руб.
|
Сумма,
руб.
|
|
Азотная
кислота
|
ХЧ,
ГОСТ 4461-77
|
кг
|
1,30
|
44,0
|
57,2
|
|
Гидроксид
аммония
|
ЧДА,
ГОСТ 3760-79
|
дм3
|
1,30
|
30,0
|
39,0
|
|
ТБФ
|
ХЧ,
ТУ 6-09-08-1219-7
|
дм3
|
0,4
|
580,00
|
232
|
|
Соляная
кислота
|
ХЧ,
3118-77
|
кг
|
0,4
|
35,0
|
14,0
|
|
Хлорид
калия
|
ОСЧ,
ТУ 6-09-3678-74
|
кг
|
0,3
|
93,0
|
27,9
|
|
Нитрат
алюминия
|
ЧДА,
ГОСТ 3757-65
|
кг
|
0,1
|
64,0
|
6,4
|
|
Карбонат
аммония
|
ЧДА,
ГОСТ 3770-75
|
кг
|
0,1
|
90,0
|
9,0
|
|
Трилон
Б
|
ТУ
2642-001-007500602-97
|
упак.
10 амп.
|
0,1
|
260,0
|
26,0
|
|
Нитрат
уранила
|
ЧДА
|
кг
|
0,15
|
40000,00
|
6000,0
|
|
Нитрат
тория
|
ЧДА
|
кг
|
0,1
|
8500,00
|
850,0
|
|
Изопар
|
ECNO
292-44-6
|
дм3
|
1,3
|
660,00
|
858,0
|
|
Бумага
фильтровальная
|
-
|
упак.
|
2
|
75,00
|
150
|
|
Итого
|
8269,5
|
А.2 Энергетические затраты
В состав энергетических затрат включены затраты
на электроэнергию (За), горячую, холодную и дистиллированную воду.
Затраты на электроэнергию определены следующим
образом:
ЗЭ = М · КИ · Т · Ц(А. 3)
где М - паспортная мощность
электроприборов, использованных при проведении исследования, кВт;
Ки - коэффициент
использования мощности электрооборудования;
Т - время эксплуатации
электрооборудования за весь период выполнения исследования, ч;
Ц - цена 1 кВтЧЧч
электроэнергии, руб. (Ц = 1,5 руб./кВт·ч)
Результаты расчета представлены в
таблице А.2. Затраты на электроэнергию составили:
За = 1515,0 · 1,5 = 2272,4 руб.
Таблица А.2 - Расчет затрат на
электроэнергию
|
Прибор
|
Паспортная
мощность, кВт
|
Коэффициент
использования мощности
|
Время
работы, ч
|
Расход
энергии, кВт ч
|
|
Дистиллятор
|
3,0
|
0,9
|
100
|
270
|
|
ФЭК
|
0,2
|
0,9
|
80
|
14,4
|
|
Вытяжной
шкаф
|
1,5
|
0,7
|
400
|
420
|
|
ПЭВМ
|
0,5
|
0,9
|
100
|
45
|
|
Электроплитка
|
1,0
|
0,9
|
400
|
360
|
|
Муфельная
печь
|
1,6
|
0,9
|
50
|
72
|
|
Сушильный
шкаф
|
2,0
|
0,8
|
200
|
320
|
|
Мешалка
|
0,02
|
0,8
|
3
|
0,048
|
|
Весы
аналитические
|
0,008
|
0,9
|
10
|
0,0072
|
|
ЛУ
Денитрации
|
0,1
|
0,9
|
150
|
13,5
|
|
Итого
|
1515,0
|
А.3 Затраты на воду
Затраты на воду (Зв) определены, исходя из ее
ориентировочного расхода и цены для условий института (26 руб/м3 с учетом
канализации). Источником горячей и дистиллированной воды в лаборатории является
дистиллятор, поэтому расход холодной воды представляет собой сумму расходов на
лабораторные нужды, на охлаждение дистиллятора и на получение дистиллированной
воды.
Для данной работы суммарный расход
составил 20,6 м3, а затраты:
Зв = 20,6 · 26 = 535,6 руб.
А.4 Затраты на заработную плату
Затраты на основную заработную плату исполнителя
(Зи) исследования определены умножением размера месячной заработной платы на
число месяцев, отводимых на выполнение дипломной работы:
Зи = 5000 · 6 = 30000 руб.
Основная заработная плата руководителей работы
(Зрук) и консультантов (Зконс) определена, исходя из суммарной нормы затрат их
рабочего времени на одну дипломную работу (33 и 2 ч соответственно) и
среднечасовой заработной платы 300 руб/ч и 200 руб/ч соответственно:
Зрук = 33 · 300 =9900 руб.
Заработная плата консультанта по дисциплине
«Охрана окружающей среды» (Зконс) составляет:
З8 =200 · 2=400 руб.
Заработная плата консультанта по экономике (З9)
составит:
Зэкон =200 · 2=400 руб.
Дополнительная заработная плата (Здоп)
определена в размере 20 % к сумме затрат по статье «Основная заработная плата»:
Здоп = (30000+9900+400+400) ·0,2 = 8140 руб.
Отчисления на социальное страхование (Зсс)
принимаются в размере 26 % от суммы основной и дополнительной заработной платы:
Зсс = (30000+9900+400+400+ 8140) · 0,26 =
12698,4 руб.
А.5 Затраты на стеклянные приборы и посуду
Затраты на стеклянные приборы и посуду
определены в зависимости от вида и емкости стеклянных изделий, использованных
для выполнения исследовательской работы и цен на них по данным лаборатории.
Текущие затраты определены, как 50% от суммарной стоимости этих изделий,
приведенной в таблице А.3.
Таблица А.3 - Затраты на стеклянные приборы и
посуду
|
Наименование
|
Объем,
см3
|
Кол-во,
шт.
|
Цена,
руб.
|
Сумма,
руб.
|
|
Пипетка
|
1
|
4
|
34,00
|
136,00
|
|
10
|
2
|
46,00
|
92,00
|
|
Колба
мерная
|
25
|
3
|
35,00
|
105,00
|
|
1000
|
2
|
355,00
|
710,00
|
|
Цилиндр
мерный
|
10
|
1
|
73,00
|
73,00
|
|
25
|
1
|
93,00
|
93,00
|
|
100
|
1
|
143,00
|
143,00
|
|
250
|
1
|
162,00
|
162,00
|
|
Воронка
лабораторная
|
75
|
1
|
37,00
|
37,00
|
|
100
|
2
|
68,00
|
136,00
|
|
150
|
2
|
244,00
|
488,00
|
|
Стакан
|
100
|
2
|
24,00
|
48,00
|
|
250
|
3
|
37,00
|
111,00
|
|
Колба
коническая
|
250
|
8
|
39,00
|
312,00
|
|
Тигель
фарфоровый
|
18
|
5
|
21,00
|
105,00
|
|
Воронка
делительная
|
100
|
1
|
65,00
|
65,00
|
|
250
|
1
|
470,00
|
470,00
|
|
Итого
|
3286,00
|
Так как после завершения исследования посуда
была сдана, то в состав текущих затрат включается только сумма амортизационных
отчислений, норма амортизационных отчислений 40 %.
А.6 Сумма амортизационных отчислений
Сумма амортизационных отчислений на реновацию А
определена, исходя из стоимости по данным лаборатории использованных для
выполнения исследовательской работы оборудования и приборов Ф, годовых норм их
амортизации на реновацию Нар и времени их использования (мес.) для данного
исследования Т по формуле (А.4):
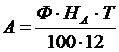 (А. 4)
(А. 4)
Результаты расчета представлены в
таблице А.4.
Таблица А.4. - Расчет
амортизационных отчислений на реновацию
|
Прибор
|
Стоимость,
руб.
|
Годовые
нормы амортизации на реновацию, %
|
Время
работы, мес.
|
Сумма
амортизационных отчислений, руб.
|
|
Дистиллятор
|
11520,00
|
8
|
4
|
307,2
|
|
ФЭК
|
50778,00
|
12
|
4
|
2031,12
|
|
Шкаф
вытяжной
|
35310,00
|
9
|
1059,3
|
|
ЭВМ
|
15000,00
|
17
|
3
|
637,5
|
|
Электроплитка
|
520,00
|
12
|
4
|
20,8
|
|
Муфельная
печь
|
34000,00
|
12
|
4
|
1360,0
|
|
Сушильный
шкаф
|
16270,00
|
9
|
4
|
488,1
|
|
Мешалка
|
1460,00
|
9
|
2
|
21,9
|
|
Весы
аналитические
|
32150,00
|
12
|
4
|
1286,0
|
|
ЛУ
Денитрации
|
25954,00
|
12
|
4
|
1038,16
|
|
Итого
|
8250,1
|
Для определения общей суммы текущих затрат
составлена смета, представляющая собой свод всех текущих затрат Зн на
выполнение исследовательской работы за весь период ее выполнения (таблица А.5).
Таблица А.5 - Полная смета затрат на проведение
научно-исследовательской работы
|
Наименование
статей затрат
|
Сумма,
руб.
|
Удельный
вес отдельных статей затрат, %
|
|
Сырье,
материалы, реактивы
|
8269,5
|
0,10
|
|
Энергетические
затраты (вода + электроэнергия)
|
2808,0
|
0,034
|
|
Основная
зарплата
|
40700
|
0,495
|
|
Дополнительная
зарплата
|
8140
|
0,10
|
|
Отчисления
на соцстрахование
|
12698,4
|
0,155
|
|
Амортизация
стеклянной посуды
|
1314,4
|
0,016
|
|
Амортизация
оборудования
|
8250,1
|
0,10
|
|
Итого:
|
82180,4
|
100
|
Уровень рентабельности исследования принимается
равным 25%. Величина научно-технического эффекта (Энт) определена по формуле
(А.5).
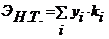 (А.5)
(А.5)
где yi - величина частного
показателя, балл; - весовой коэффициент i-того частного показателя
степень новизны, научно-технический уровень, глубина воздействия на процесс.
В результате работы подтверждены
некоторые известные положения и получена новая информация, y1 = 20; достигнуто
решение поставленных задач на основе анализа связей между полученными данными,
y2 = 20; в целом исследование подтверждает известные, но недостаточно
проверенные положения, y3 = 10. Работа носит теоретический характер, поэтому
значения весовых коэффициентов частных показателей равны 0,50; 0,25 и 0,25.
Величина научно-технического эффекта
составляет:
Энт = 0,50 · 20 + 0,25 · 20 + 0,25 ·
10 = 17,5 балла
На основании данной величины научно -
технического эффекта коэффициент К, учитывающий поощрительную надбавку за
качество разработки принимается равным 1,1.
А.7 Договорная цена на НИР
Договорная цена на НИР Цн равна:
Цн = 82180,4 · (1 + 25/100) · 1,1 =
112998,05 руб.
Данная дипломная работа является
частью научно - исследовательских работ, проводимых в лаборатории №92 Радиевого
института имени Хлопина. Исследования по данной теме имеют важное значение для
совершенствования технологии получения оксидов урана в виде конечных форм.
Экономия от внедрения полученных результатов в промышленное производство может
значительно превзойти стоимость данной НИР.
ПРИЛОЖЕНИЕ Б
Стандартизация
При выполнении настоящей дипломной
работы учитывались требования СТП ЛТИ 03-85
КС УКДВ. Порядок введения и контроля за соблюдением стандартов в институте.
Состав работы и содержание ее
основных частей соответствует СТП ЛТИ 2.605.017-85 КС УКДВ. Виды учебных занятий. Дипломный
проект, работа и работа-проект. Общие требования ГОСТ 15.101-80 СРПП. Порядок проведения
научно-исследовательских работ и ГОСТ 7.32-2001
СИБИД. Отчет о научно-исследовательской работе. Общие требования и правила
оформления.
Разделы работы составлены согласно
ГОСТ 7.1-2003 СИБИД.
Библиографическое описание документа. Общие требования и правила составления,
ГОСТ 7.12-93 СИБИД.
Сокращение русских слов и словосочетаний в библиографическом описании, ГОСТ
8.417-2002. Единицы физических величин.
При выполнении экспериментальной
части дипломной работы соблюдались санитарные нормы и правила согласно:
ГОСТ 12.0.003-74 ССБТ. Опасные и вредные
производственные факторы. Классификация.
ГОСТ 12.1.005-88 ССБТ. Воздух рабочей зоны.
Общие санитарно-гигиенические требования.
СНиП 23-05-95 Естественное и
искусственное освещение производственных помещений.
ГОСТ 12.4.021-75 ССБТ. Системы
вентиляционные. Общие требования.
Использованные в работе сырье и
материалы соответствовали приведенным ниже в таблице Б. 1 ГОСТам:
Таблица Б.1
|
Азотная
кислота
|
ГОСТ
4461-77
|
|
ТБФ
|
ТУ
6-09-08-1219-7
|
|
Соляная
кислота
|
ГОСТ
3118-77
|
|
Аммиак
водный
|
ГОСТ
3760-79
|
|
Трилон
Б
|
ТУ
2642-001-07500602-97
|
|
Калий
хлористый
|
ТУ
6-09-36978-74
|
|
Алюминий
азотнокислый
|
ГОСТ
3757-65
|
|
Аммоний
углекислый
|
ГОСТ
3770-75
|
|
Изопар
|
ECNO
292-44-6
|
При постановке экспериментов и обработке их
результатов использовалось:
СТП ЛТИ 2.075.008-81 КС УКДВ.
Методы обработки результатов наблюдений. Прямые измерения с многократными
наблюдениями.
ПРИЛОЖЕНИЕ В
Охрана труда и окружающей среды
Дипломная работа проводилась в лаборатории
"Радиевого института им Хлопина" Проведение экспериментов было
связано с работой с источниками электрического тока и химическими веществами. В
работе применялось различное лабораторное оборудование, в том числе
электрические приборы с питанием от сети переменного тока, с напряжением 220 В
и промышленной частотой 50 Гц.
В.1 Опасные и вредные производственные факторы
Опасные и вредные производственные факторы в
лаборатории можно разделить по своей природе действия на группы:[]
физические;
химические;
биологические;
психофизиологические.
К физическим факторам можно отнести:
Электромагнитное излучение.
Интенсивная и продолжительная работа с использованием ЭВМ может стать
источником тяжелых профессиональных заболеваний, так заболевания, обусловленные
травмой повторяющихся нагрузок, представляют собой постепенно накапливающиеся
недомогания, возникающие при длительной работе с устройством ввода-вывода. Труд
людей, постоянно работающих с ПЭВМ, характеризуется отсутствием воздействия
высоких уровней распространенных на производстве неблагоприятных факторов (пыль,
шум, плохая освещенность и др.), но на них влияет сложный комплекс условий,
которые при среднем и низком уровне за счет комплексного воздействия, все же,
вызывают появление неблагоприятных последствий. Следует учитывать также
воздействие факторов, не получивших еще сегодня общественного признания, в то
время как научные исследования показывают возрастание их роли в психических
формах труда, к числу таких факторов относят высокий уровень психического
напряжения, монотонность труда, отсутствие физических нагрузок,
обездвиженность, некорректная форма освещения, специфическое воздействие на
орган зрения колеблющегося свечения экрана [43].
Электрический ток. Действие
электрического тока на человека носит многообразный характер. Проходя через
организм человека, электрический ток вызывает термическое, электролитическое, а
также биологическое воздействие. Это многообразие действий электрического тока
может привести к двум видам поражения: электрическим травмам и электрическим
ударам. Путь прохождения тока через тело человека играет существенную роль в
исходе поражения, так как ток может пройти через жизненно важные органы:
сердце, легкие, головной мозг и др. Влияние пути тока на исход поражения
определяется также сопротивлением кожи на различных участках тела. Постоянный
ток примерно в 4-5 раз безопаснее переменного. Это вытекает из сопоставления
пороговых ощутимых, а также не отпускающих токов для постоянного и переменного
токов. Это положение справедливо лишь для напряжения до 250 - 300 В. ПЭВМ
соответствует этому напряжению. Для обеспечения электробезопасности применяют,
в данном случае, изоляцию токоведущих частей [].
Ионизирующее излучение. Работы с
применением источников ионизирующего излучения являются особо опасными
вследствие их комплексного воздействия на организм, которое носит
аккумулятивный характер. При воздействии ионизирующего излучения на организм
человека в тканях могут происходить сложные физические и биологические
процессы. В результате ионизации живой ткани происходит разрыв молекулярных
связей и изменение химической структуры различных соединений. Так малые
кратковременные дозы способны вызывать обратимые изменения в составе крови, в
то время как большие кратковременные дозы являются причиной развития
белокровия, рака костного мозга и гибели клеток и организма в целом. Облучение
организма малыми дозами в течение многих лет приводит к преждевременному
старению организма, развитию болезней органов зрения, пищеварения,
сердечнососудистых заболеваний. Причем согласно современным представлениям
выход неблагоприятных эффектов в диапазоне «малых доз» мало зависит от величины
дозы. Это означает, что эффект определяется прежде всего суммарной накопленной
дозой вне зависимости от того, получена ли она в течение одного дня или в
течение 50 лет []. Характеристики используемых в работе радионуклидов приведены
в таблице В.1.
Таблица В.1 - Характеристика
радионуклидов, применяемых в работе
|
Радионуклид
|
Т1/2,
лет
|
Тип
распада
|
Группа
Радионуклидной опасности
|
Минимально-значимая
активность, Бк
|
|
232Th
|
1,4
· 1010
|
β-, α
|
Г
|
103
|
|
238U
|
4,5
· 109
|
β-, α
|
Г
|
104
|
К химическим факторам относятся
воздействия на человеческий организм химических веществ и их соединений [43].
Работа с едкими, токсичными и
различными химическими веществами связана с опасностью получения организмом
таких травм, как:
расстройство функций нервной системы
(углеводороды, спирты жирного ряда, сероводород и др.);
поражение верхних и глубоких
дыхательных путей (хлор, аммиак, диоксид серы, туманы кислот и др.);
поражение кожных покровов
(неорганические кислоты, щелочи и др.);
нарушение структур ферментов
(синильная кислота, мышьяк и др.);
изменения в реактивной способности
организма (соединения никеля и др.).
Часть проводимых работ носила
характер химических, то есть с применением разнообразных химических веществ.
Используемые в работе химические вещества, представляющие собой опасность,
приведены в таблице В.2 [].
Таблица В.2 - Характеристика физико-химических и
токсических свойств веществ, используемых в работе
|
Вещества
|
Физико-химические
свойства
|
Пожаровзрывоопасные
свойства
|
Токсические
свойства
|
|
Плотность,
кг/м3
|
Агрегатное
состояние
|
Температура
кипения,0С
|
Температура
плавления,0С
|
Температура,0С
|
Пределы
распространения пламени
|
Характеристика
действия на организм человека
|
Класс
опасности[]
|
ПДК,
мг/м3[47]
|
|
|
|
|
|
вспышки
|
самовоспламенения
|
Температурные,0С
|
Концентрационные,
об %
|
|
|
|
|
|
|
|
|
|
|
нижний
|
верхний
|
нижний
|
верхний
|
|
|
|
|
Азотная
кислота
|
1502
|
Жидкость
|
83,4
|
-42
|
-
|
-
|
-
|
-
|
-
|
-
|
Раздражение
верхних дыхательных путей, ожоги кожных покровов
|
3
|
2,0
|
|
ТБФ
|
970
|
Жидкость
|
289
|
-80
|
144
|
175
|
131
|
192
|
19,7
|
-
|
Нарушение
деятельности сердечно сосудистой системы
|
2
|
0,5
|
|
Гидроксид
аммония
|
900
|
жидкость
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
Химический
ожог, слезоточивость
|
4
|
0,8
|
|
Хлорид
калия
|
1989
|
твердое
|
1406
|
768
|
-
|
-
|
-
|
-
|
-
|
-
|
Способен
вызывать аллергические заболевания в производственных условиях
|
3
|
5
|
|
Соляная
кислота
|
1690
|
жидкость
|
1109
|
-102
|
-
|
-
|
-
|
-
|
-
|
-
|
Раздражение
и ожоги слизистых оболочек дыхательных путей, помутнение роговицы, при
попадание на кожу вызывает химические ожоги
|
2
|
5
|
|
Изопар
|
729,9
|
жидкость
|
174,1
|
-28
|
86
|
535
|
6
|
37
|
1,27
|
7,0
|
Раздражение
верхних дыхательных путей, ожоги кожных покровов
|
4
|
50
|
|
Карбонат
аммония
|
1729
|
твердое
|
-
|
разл.
58
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
|
Нитрат
алюминия
|
1890
|
твердое
|
-
|
разл.
66
|
-
|
-
|
-
|
-
|
-
|
-
|
Поражение
лёгких, лимфоцитоз
|
3
|
2
|
|
Трилон
Б
|
1872
|
твёрдое
|
-
|
130
|
-
|
-
|
-
|
-
|
-
|
-
|
Оказывает
общетоксическое (поражение почек) мутагенное действие
|
2
|
0,5
|
В лаборатории, где выполнялась дипломная работа,
были приняты следующие меры безопасности:
все кислоты и основания хранились,
соответственно, в стеклянных и полиэтиленовых бутылях и банках с плотно
притертыми крышками в вытяжном шкафу;
все емкости с реактивами имели четкие надписи;
работа в лаборатории проводилась в спецодежде
(халат).
Биологические опасные и вредные производственные
факторы включают в себя патогенные микроорганизмы (вирусы, бактерии, грибы,
бактерии и др.) и продукты их жизнедеятельности, а также макроорганизмы
(растения и животные).
Психофизиологические опасные и вредные
производственные факторы по характеру действия подразделяются на физические
(статические и динамические) и нервно-психические нагрузки (умственное
перенапряжение, монотонность труда), эмоциональные перегрузки. К
психофизиологическим факторам можно отнести работу на ПЭВМ, которая в свою
очередь может сопровождаться умственными перенапряжениями, постепенно
накапливающимися недомоганиями, заболеваниями суставов.
В.2 Обеспечение пожарной безопасности
Перед началом работы были сданы экзамены по
технике безопасности, по пожарной безопасности, получен допуск по работе в
лаборатории химического анализа. Расчет пожарной нагрузки осуществляется по
формуле (В.3):
=∑Gi ∕S,(В.3)
где G - теплота сгорания МДж; - площадь 20 м2.
Теплота сгорания различных веществ
рассчитывается по формуле (В.4): =qi×mi ,(В.4)
где qi - удельная теплота сгорания МДж/кг; -
масса вещества кг.
Теплота сгорания дерева, бумаги, пластика,
соответственно:
д=10×2500=25000 МДж
Gб=18×4=72 МДжп=0,033600×20=0,672
МДж
Суммарная теплота сгорания ∑Gi = 25073 МДж
Пожарная нагрузка q=25073/20=1254 МДж×м-2
Значения пожарных нагрузок приведены в таблице
В.3.
Таблица В.3 - Значения пожарных нагрузок
|
Категория
|
Пожарная
нагрузка, МДж×м-2
|
|
В1
|
>2200
|
|
В2
|
1401-2200
|
|
В3
|
181-1400
|
|
В4
|
1-180
|
Помещения лаборатории относятся по степени
пожарной опасности к категории В3, к классу взрывоопасности В-1б[].
На случай пожара лаборатория оснащены средствами
пожаротушения: пожарный гидрант ГК-5, асбестовое одеяло, ящик с песком,
огнетушители У-2 [].
В.3 Вентиляция
В зависимости от особенностей производственного
процесса должны присутствовать встроенные устройства для удаления выделяющихся
в процессе производства любых вредных взрыво- и пожароопасных веществ
непосредственно от мест их образования или скопления.
Обеспечение нормальных метеорологических условий
и чистоты воздуха на рабочем месте в значительной степени зависит от правильно
организованной вентиляции [].
В помещениях лаборатории установлены вытяжные
шкафы 2Ш-11Ж (скорость вытяжки не менее 1,5 м/с).
Кратность воздухообмена рассчитывается по
формуле (В.1):
К=Vнв/Vп, (В.1)
где Vнв - необходимый объем воздуха для
воздухообмена м3/ч; п - объем помещения 70 м3.
Необходимый объем воздуха для воздухообмена
определяется по формуле (В.2):
нв= Vф×S, (В.2)
где Vф - скорость фильтрации м/ч;
S - площадь м2 [45].
нв= Vф×S=1.5×0.2×3600=1080 м3/ч
К=1080/70=15.4 ч-1
В.4 Освещенность
В помещении лаборатории применяется
смешанное: естественное и искусственное освещение. Рабочее освещение следует
предусматривать для всех помещений зданий, а также участков открытых
пространств, предназначенных для работ, прохода людей и движения транспорта.
Для освещения помещений лаборатории используются газоразрядные лампы дневного
света. Разряд зрительных работ IVв-IVг. Нормы освещенности - 200 - 250 лк [].
В.5 Аптечка
В лаборатории на случай оказания
первой доврачебной помощи имеется аптечка, которая находится в рабочем
помещении лаборатории. В аптечке находятся следующие материалы []:
стерильный бинт,
вата,
нашатырный спирт,
% раствор йода,
раствор борной кислоты,
раствор питьевой соды,
раствор перманганата калия.
В.6.1 Обеспечение
электробезопасности
Все применяемые приборы были
заземлены, сопротивление заземления не превышало 4 Ом. Перед началом работы
постоянно проводился контроль качества изоляции, все проводки были выполнены
проводом, рассчитанным на мощность электроприборов. Контактные клеммы
располагались недоступно для неосторожного касания. Класс помещения по
опасности поражения человека электрическим током - без повышенной опасности [].
В.6.2 Обеспечение радиационной безопасности
Радиационная безопасность персонала
от воздействия ионизирующих излучений и радиоактивных загрязнений
обеспечивается техническими и организационными мероприятиями. К техническим
мероприятиям относятся оснащение системами безопасности, предупреждающими
аварии и ограничивающими их радиационные последствия, высококачественной
проектной документацией, строительно-монтажными работами в соответствии с
проектной документацией, техническим контролем состояния оборудования и его
работоспособности и т.д.
К организационным мероприятиям
относятся:
эксплуатация, в соответствии с
действующей нормативно-технической документацией, технологическими инструкциями
по эксплуатации;
соблюдение персоналом норм и правил,
а также конкретных требований по радиационной безопасности, содержащихся в
технологических инструкциях;
методическое руководство
мероприятиями по обеспечению радиационной безопасности, осуществляемое отделом
охраны труда и техники безопасности;
контроль за соблюдением норм, правил
и требований по радиационной безопасности.
Перед началом работы были сданы
экзамены по правилам охраны труда, радиационной безопасности, пожарной
безопасности, получен допуск по работе в радиохимической лаборатории.
Эксперименты проводились в
радиохимической лаборатории, по отношению к работам с радиоактивными веществами
лаборатория относится к III класс помещений. Вход в помещения зоны строгого
режима осуществлялся через санитарный пропускник, При нахождении в зоне
строгого режима обязательно наличие индивидуального дозиметра, предназначенного
для измерения индивидуальной эквивалентной дозы внешнего облучения. На выходе
из санпропускника осуществлялся контроль загрязнений радионуклидами средств
индивидуальной защиты и тела с помощью стационарных или переносных приборов.
Суммарная активность используемых
радиоактивных источников, содержащих радионуклиды групп Г на рабочем месте, не
превышала 108 Бк (суммарная активность приведена к группе А).
Работа с активностью проводилась в
вытяжном шкафу 2Ш-НЖ (скорость вытяжки не менее 1,5 м/с), в котором установлена
биологическая защита из свинцовых блоков. Также применялись средства
индивидуальной защиты: халат и резиновые перчатки. По окончании работ с
источниками ионизирующего излучения постоянно проводился радиометрический
контроль с помощью прибора УИМ. Во всех случаях уровни загрязнения кожных
покровов, спецбелья по бета-активным радионуклидам не превышали контрольных и
допустимых уровней (соответственно 100 и 200 част/см2 мин.), а для основной
спецодежды - соответственно 1500 и 2000 част/см2 мин., что не превышает
требований НРБ-99 по допустимым уровням радиоактивного загрязнения [45].
В. 7 Анализ технологических операций с точки
зрения опасностей и вредностей при их проведении
Анализ производственных операций приведен в
таблице В.4.
Таблица В.4 - Анализ технологических операций с
точки зрения опасностей и вредности при их проведении
|
Наименование
технологической операции
|
Оборудование
и вещества для проведения операции
|
Возможные
опасности (вредное влияние при проведении данной технологической операции)
|
Причины
проявления данной опасности (вредного влияния)
|
Меры
по предотвращению опасности при проведении данной операции
|
|
Работа
с ЭВМ
|
Машина
ЭВМ
|
Поражение
электрическим током
|
Нарушение
изоляции или отсутствие заземления
|
Заземление
приборов, целостность изоляции
|
|
|
Утомляемость
глаз
|
Старое
или неисправное оборудование
|
Использование
защитного экрана, а также сокращение времени работы на ЭВМ: 4 часа работы -15
минут перерыва
|
|
Приготовление
растворов из едких и токсичных веществ
|
Вытяжной
шкаф, пипетка с грушей
|
Химический
ожог
|
Попадание
на кожу без СИЗ
|
Работа
в СИЗ
|
|
|
Раздражение
слизистых, отек легких
|
Вдыхание
паров
|
Использование
вытяжного шкафа
|
|
Проведение
измерений
|
Весы,
ФЭК
|
Поражение
электрическим током
|
Нарушение
изоляции или отсутствие заземления
|
Заземление
приборов, целостность изоляции
|
|
Нагрев
растворов
|
Электрическая
плитка
|
Поражение
электрическим током
|
Нарушение
изоляции или отсутствие заземления
|
Заземление
приборов, целостность изоляции
|
|
|
Термический
ожог
|
Соприкосновение
кожи с горячим паром
|
Работа
с выключенной остывшей печью, аккуратное проведение работ
|
|
Прокаливание
осадков
|
Муфельная
печь
|
Поражение
электрическим током
|
Нарушение
изоляции или отсутствие заземления
|
Заземление
приборов, целостность изоляции
|
|
|
Термический
ожог
|
Соприкосновение
кожи с горячими частями печи
|
Работа
с выключенной остывшей печью, аккуратное проведение работ
|
|
Приготовление
растворов для экстракции
|
Мерная
посуда, делительная воронка, автоматическая пипетка
|
Химический
ожог
|
Попадание
на кожу
|
Работать
в халате, резиновых перчатках
|
|
|
Порезы
|
Бой
посуды
|
Осторожно
пользоваться посудой
|
|
|
Отравление,
раздражение верхних дыхательных путей и слизистых оболочек глаз, носа и рта
|
Попадание
внутрь организма химических соединений
|
Работать
под тягой
|
В.8 Оказание первой помощи
)При работе с радиоактивными веществами.
Дезактивация пролива радиоактивной жидкости.
Собрать жидкость сорбирующим материалом (ветошь или фильтровальная бумага) и
поместить в емкость для сбора отходов. Произвести дезактивацию загрязненной
поверхности дезактивирующим раствором. Обмыв произвести сверху вниз и от чистых
к грязным участкам. При дезактивации вести радиометрический контроль смывов и
участка загрязнения.
Дезактивирующие средства, применяемые для
дезактивации кожных покровов, оборудования и рабочих поверхностей:
% раствор ЭДТА, 2% раствор порошка «Лотос»
(Th232, U238);
Для дезактивации рабочих поверхностей и
оборудования концентрация растворов может быть увеличена в 2-3 раза и добавлено
0,5 % раствора гексаметафосфата натрия.
Дезактивация участка зараженного радиоактивным
порошком. Собрать порошок тампонами, смоченными минеральным маслом.
Окончательно дезактивацию необходимо проводить спиртом, раствором стирального
порошка в воде. Провести радиометрический контроль. При остающемся стойком
радиоактивном загрязнении возможно удаление части загрязненной поверхности.
В связи со спецификой работы наибольшую
опасность для организма представляет загрязнение кожного покрова радиоактивными
веществами и поражение электрическим током.
К очистке кожного покрова в случае попадания на
него радиоактивных веществ приступают немедленно и действуют следующим образом:
при малых загрязнениях руки или другие части
тела моют теплой водой с мылом и применением волосяной щетки;
при больших загрязнениях обрабатывают
загрязненные части тела тампоном, смоченным дезактивирующим раствором
марганцовокислого калия, а затем промывают 5%-ным раствором азотнокислого
натрия, прополаскивают водой;
при загрязнении раны нужно промыть ее под струей
воды, вызвав при этом кровотечение, а затем промыть 1-2% раствор борной
кислоты.
) При поражении электрическим током
Прежде всего, необходимо снять
напряжение с электрооборудования, вызвавшего поражение, освободить
пострадавшего от токоведущих частей и, если имеются дыхание и пульс, положить
пострадавшего в удобное положение и обеспечить покой до прихода врача. Если
пульс и дыхание отсутствуют, пострадавшему необходимо искусственное дыхание
методом «изо рта в рот» [52].
В лаборатории на случай оказания
первой медицинской помощи имеется аптечка, которая находится в рабочем
помещении лаборатории.
) При химических ожогах
Химические ожоги возникают при
местном воздействии химически активных веществ на кожу, слизистую оболочку
дыхательных путей и глаз. Степень ожога зависит от химической активности и
токсичности веществ, его концентрации, температуры, продолжительности действия,
а также чувствительностью пострадавшего. Первая помощь при химических ожогах и
отравлениях сводится к следующему: при ожогах кислотами и щелочами пораженный
участок кожи - промывают струёй холодной воды в течение 15 мин. При ожогах
щелочами - из 2 % раствора уксусной кислоты, лимонной или виннокаменной кислот.
В.9 Мероприятия по охране окружающей
среды
К мероприятиям по охране окружающей
среды, проводимым в ходе данной работы, относятся следующие:
очистка воздуха, удаляемого из
шкафов;
слив концентрированных кислот и
щелочей в специальные емкости с последующей нейтрализацией и утилизацией;
помещение твердых отходов в
специально предназначенные емкости с последующей переработкой.
Характеристика производственных
отходов и методов их утилизации приведена в таблице В.5 [45].
Таблица В.5- Характеристика
производственных отходов
|
Наименование
отходов
|
Агрегатное
состояние
|
Наименование
вредных примесей
|
Примечание
|
|
Водные
растворы после анализа
|
Жидкость
|
Растворы UO2(NO3)2,
Th(NO3)4, кислоты
|
Собираются
в специальные емкости для последующей переработки в соответствии со
спецдокументацией
|
|
Вода
после мытья посуды, рук
|
Жидкость
|
-
|
В
канализацию
|
|
Органическая
фаза после проведения опыта
|
Жидкость
|
ТБФ
|
Сливаются
в специальные емкости, после нейтрализации выливаются в канализационную сеть
|
|
Твердые
отходы после проведения опыта
|
Твердое
тело
|
Осадки
U3O8
|
Собираются
в специальные емкости для последующей переработки в соответствии со
спецдокументацией
|
|
Твердые
отходы
|
Битое
стекло, бумага
|
-
|
В
контейнер для мусора
|