Изучение механизма побочных реакций при термолизе ряда алкоксиаминов и определение константы скорости их термолиза
Оглавление
Введение
Глава 1. Литературный обзор
.1 «Живая» контролируемая
радикальная полимеризация
.2 Основные признаки «живой»
радикальной полимеризации
.3 Метод диаграмм Фишера
.4 Побочные реакции при «живой»
радикальной полимеризации
.5 Радикальная «живая» полимеризация
гидрофильных мономеров
.6 Методы исследования
.7 Постановка задачи
Глава 2. Экспериментальная часть
.1 Объекты исследования
.2 Общий подход к определению
реакций, происходящих при термолизе алкоксиаминов.
Глава 3. Изучение механизма побочных
реакций при термолизе ряда алкоксиаминов и определение константы скорости их
термолиза
.1 Введение
.2 Результаты
.2.1 Побочные реакции при термолизе
алкоксиаминов.
.3 Полимеризация ММА, инициированная
3b
.4 Стабильность нитроксильного
радикала 3—
.5 Температурные зависимости
константы скорости термолиза алкоксиаминов kd,
определение аррениусовских параметров для 1а и 2а
.6 Обсуждение результатов
Глава 4 Исследование применимости
нитроксильных радикалов для полимеризации гидрофильных мономеров
.1 Изучение кинетики полимеризации
гидрофильных мономеров методом ЯМР
.2 Полимеризация акриламида в
присутствии 6—
Выводы и результаты
Литература
Введение
Современные технологии позволяют получать
огромное количество материалов с разнообразными свойствами, которые определяют
приложение этих материалов. Одно из важнейших мест среди них занимают
синтетические полимеры. Широко распространенно получение полимеров по
технологии радикальной полимеризации, так как она позволяет получать полимеры
высоких молекулярных масс широкого круга виниловых мономеров в мягких
реакционных условиях и широком температурном интервале. Кроме того, методом
радикальной полимеризации можно получать сополимеры, свойства которых будут
зависеть от относительного содержания того или иного мономера, входящего в
состав получающегося полимера. Основным недостатком радикальной полимеризации
является то, что практически невозможно контролировать некоторые важные
параметры получающегося полимера: молекулярную массу, полидисперсность,
функциональность концевой группы, структуру макромолекулы. С этой точки зрения
открытие в 1982 году радикальной контролируемой «живой» полимеризации явилось
настоящим прорывом, ведь этот метод позволяет получать полимеры с заданной
структурой и молекулярной массой в условиях, характерных для радикальной
полимеризации. Основной идеей радикальной «живой» полимеризации является
частичная или полная замена реакции необратимого обрыва цепи на обратимую
реакцию со стабильным радикалом, выступающим в качестве контролирующего агента
полимеризации. Одним из классов контролирующих агентов на сегодняшний день
являются нитроксильные радикалы, которые успешно применяются для полимеризации
стирола и акрилатов. Впервые нитроксильные радикалы применил Соломон в качестве
контролирующих агентов в полимеризации стирола. В настоящее время нитроксильные
радикалы находят применение для полимеризации стирола и акрилатов. Однако
проведение «живой» контролируемой полимеризации метакрилатов с использованием
нитроксильных радикалов в качестве контролирующих агентов затруднена из-за
протекания побочной реакции переноса атома водорода между нитроксильным и
третичным алкильным радикалами. Существует два механизма этой реакции -
внутримолекулярный и радикальный. Определение механизма реакции переноса атома
водорода играет важное значение в направленном синтезе универсальных медиаторов
полимеризации широкого круга мономеров. Таким образом, в настоящее время
актуальными задачами в области изучения радикальной «живой» полимеризации
являются поиск новых нитроксильных радикалов, способных эффективно
контролировать полимеризацию широкого круга мономеров, в том числе гидрофильных
мономеров, для эффективного синтеза блок-сополимеров, изучение проведения
полимеризации в эмульсии и суспензии. В настоящей работе были исследованы
побочные реакции, происходящие при гомолизе алкоксиаминов - инициаторов «живой»
контролируемой полимеризации, и определены константы скорости гомолиза методом
ЯМР (третья глава). Также показано влияние протекания побочных реакций на
полимеризацию. В четвертой главе представлены результаты по возможности
применения нитроксильного радикала имидазолидинового ряда для полимеризации
гидрофильных мономеров, инициированной персульфатом натрия.
Глава 1. Литературный обзор
.1 «Живая» контролируемая радикальная
полимеризация
Радикальная контролируемая полимеризация в
режиме «живых» цепей относиться к наиболее быстро развивающимся отраслям науки
о полимерах. Первые работы по контролируемой радикальной полимеризации
принадлежат Отсу1. Термин «живая» полимеризация возник при инзучении ионной
полимеризации Шварцем в 1959 году. Особенностью механизма ионной полимеризации
является то, что при росте цепи активные центры несут на себе заряд одного
знака, таким образом, реакция обрыва цепи за счет рекомбинации двух растущих
молекул обладает высокой энергией активации. Поэтому, особенно при невысоких
температурах обрыв цепи маловероятен. Это обстоятельство определяет уникальные
синтетические возможности ионной полимеризации: все цепи начинают расти
практически одновременно, растут в одинаковых условиях и к концу процесса
оказываются достаточно однородными по длине. Это и определяет низкую
полидисперсность получаемых полимеров. После исчерпания мономера активность
центров, ведущих полимеризацию, сохраняется долго, что позволяет возобновить
процесс полимеризации при введении новой порции мономера. При добавлении
другого мономера возможно образование блок-сополимеров с заданной длиной
блоков. Однако синтетические возможности ионной полимеризации ограничены
набором мономеров, кроме того, она чувствительна к присутствию примесей, влаги
и воздуха, что приводит к необходимости тщательной очистки исходных реагентов и
удорожанию промышленного процесса производства полимеров по этой методике.
Наиболее интересной в этом плане является радикальная полимеризация, на долю
которой приходиться большая часть промышленного производства полимеров и
которая позволяет получать полимеры широкого круга мономеров в мягких условиях.
Основным недостатком радикальной полимеризации является то, что практически
невозможно контролировать некоторые важные параметры получающегося полимера:
молекулярную массу, полидисперсность, функциональность концевой группы,
структуру макромолекулы. Поэтому всегда существовало желание улучшить процесс
радикальной полимеризации таким образом, чтобы можно было получать
высокомолекулярные полимерные материалы с контролируемой молекулярной массой,
различной архитектурой макромолекулы и низкой полидисперсностью в мягких
условиях.
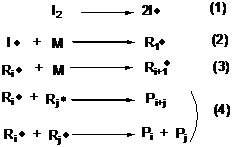
Схема 1. Механизм радикальной полимеризации
Реакция радикальной полимеризации, как любой
неразветвленный цепной процесс, состоит из трех стадий: инициирование (реакции
(1) и (2), Схема 1), рост цепи (реакция (3), Схема 1) и обрыв цепи (реакции
(4), Схема 1). При радикальной полимеризации активные центры роста цепи не
защищены от рекомбинации, которая лимитируется только взаимной диффузией
радикалов. Время роста одной полимерной цепи при обычной радикальной
полимеризации намного меньше длительности всего процесса, то есть разные
полимерные молекулы растут по-разному: меняется концентрация мономера и
инициатора, вязкость реакционной смеси. В результате меняется длина
образующихся цепей, при глубоких конверсиях значение полидисперсности может
быть много больше 5.
То есть, чтобы улучшить процесс радикальной
полимеризации, необходимо подавить реакцию обрыва цепи. Для решения этой задачи
в настоящее время предложено два различных подхода. Первый из них заключается в
ограничении подвижности растущих макрорадикалов. Второй подход заключается в
замене необратимой реакции обрыва цепи на обратимую реакцию с акцептором. То
есть макрорадикал за время полимеризации многократно превращается в
диамагнитную молекулу, но в силу обратимости этого процесса, многократно
оживает вновь. Это и есть основная идея «живой» контролируемой радикальной
полимеризации. В качестве таких акцепторов было предложено использовать
стабильные радикалы (нитроксильные, трифенилэтильные), комплексы переходных
металлов с органическими лигандами, серосодержащие соединения. К акцептору
предъявляется ряд требований: во-первых, он не должен инициировать образование
новых радикалов роста, во-вторых, ковалентная связь в аддукте рекомбинации
акцептора с растущим радикалом должна достаточно легко диссоциировать при температурах
полимеризации (100-1500С). Стабильные нитроксильные радикалы удовлетворяют всем
этим требованиям, что обусловило их применение в качестве контролирующих
агентов «живой» радикальной полимеризации. Протекание полимеризации в
контролируемом режиме требует также быстрого инициирования по сравнению со
временем всей полимеризации.
Механизм «живой» контролируемой полимеризации в
присутствии нитроксильных радикалов представлен на схеме 2. Как следует из
схемы 2, единственное отличие радикальной полимеризации в присутствии
нитроксильных радикалов от обычной радикальной полимеризации заключается в
реакциях (7.1, 7.2, 10.1, 10.2), то есть в наличии процесса обратимого
ингибирования роста полимерной цепи с образованием так называемой «спящей» цепи
Ri-Y
- высокомолекулярным алкоксиаминов. «Спящая» цепь может становиться активной с
образованием контролирующего нитроксильного радикала Y—
и растущего полимерного радикала Ri—.
В присутствии мономера последний вступает в реакцию роста цепи. За время
полимеризации происходит множество циклов рекомбинации растущего полимерного
радикала и нитроксила и термолиза алкоксиамина, таким образом, при условии
быстро инициирования каждая полимерная цепь имеет примерно одинаковую
вероятность присоединить мономер, что приводит к приблизительно одинаковой
длине образующихся полимерных молекул, а следовательно, низкополидисперсному
материалу. Инициирование реакции «живой» радикальной полимеризации может
осуществляться как с помощью инициатора I
(например азо- и пероксисоединения), так и молекулой алкоксиамина R1-Y.
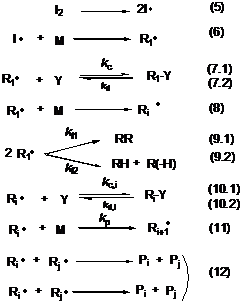
Схема 2. Механизм «живой» контролируемой
радикальной полимеризации
В работах Фишера и Фукуды была рассмотрена
кинетическая схема «живой» радикальной полимеризации и приведены условия протекания
полимеризации в контролируемом режиме. Фундаментальной основой кинетической
схемы «живой» радикальной полимеризации является эффект Фишера-Ингольда.

Схема 3.
На схеме 3 представлены реакции, происходящие в системе,
содержащей короткоживущий радикал R—
и стабильный радикал Y—.
За счет существования реакции (15) на начальных этапах радикал R—
будет расходоваться быстрее, за счет чего возникнет избыток Y—.
После этого основным продуктом реакции будет R-Y,
а вероятность протекания реакции (15) будет снижаться. Преимущественное
образование продукта R-Y
и составляет суть эффекта Фишера-Ингольда. Для возникновения в системе эффекта
Фишера-Ингольда необходимо определенное соотношение констант скорости реакций
(13) диссоциации R-Y
kd и (14)
рекомбинации R—
и Y—
kc (10-11 М<kd/kc<10-9М)8.
Экспериментально эффект Фишера-Ингольда наблюдался в работе при термическом
разложении алкоксиамина
2-фенил-2-(2’,2’,6’,6’-тетраметилпиперидин-1’-оксил)пропан. При полимеризации
снижается вероятность реакции (12), хотя она не исчезает полностью. При этом
доля накопленных продуктов реакции двух алкильных радикалов обычно не превышает
нескольких процентов за время полимеризации.
.2 Основные признаки «живой» радикальной
полимеризации
.2.1 Характеристики получаемого полимера
Важными характеристиками полимера
являются средняя молекулярная масса и полидисперсность (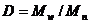 , где
, где 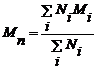 -
среднечисленная молекулярная масса полимера,
-
среднечисленная молекулярная масса полимера,  - средневзвешенная молекулярная
масса полимера) Полидисперсность характеризует распределение по длинам
полимерных цепей в образце - молекулярно-массовое распределение. В обычной
радикальной полимеризации значение полидисперсности как правило больше 2.
Высокая полидисперсность полимера является результатом высокой вероятности
реакции обрыва цепи по второму порядку. Для «живой» радикальной полимеризации
типичные значения полидисперсности находятся между 1,1 и 1,5.
- средневзвешенная молекулярная
масса полимера) Полидисперсность характеризует распределение по длинам
полимерных цепей в образце - молекулярно-массовое распределение. В обычной
радикальной полимеризации значение полидисперсности как правило больше 2.
Высокая полидисперсность полимера является результатом высокой вероятности
реакции обрыва цепи по второму порядку. Для «живой» радикальной полимеризации
типичные значения полидисперсности находятся между 1,1 и 1,5.
Особенностью полимеров, получаемых
методом «живой» полимеризации (радикальной или ионной) является сохранение
возможности роста цепи при добавлении новой порции мономера. Таким образом,
можно получать различные сополимеры, включая статистические, градиентные и блок-сополимеры.
Нужно отметить, что по окончанию полимеризации макромолекулы содержат концевую
лабильную группу -Y, что позволяет получать полимеры,
имеющие различные функциональные группы на конце цепи. За счет изменения
структуры инициатора можно влиять на трехмерную структуру материала. На рисунке
1 представлены некоторые структуры, которые можно получить при «живой»
радикальной полимеризации.
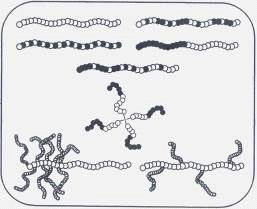
Рисунок 1. Синтетические возможности
«живой» контролируемой полимеризации.
.2.2 Признаки протекания
полимеризации в контролируемом режиме
Основным признаком протекания
полимеризации в контролируемом режиме является линейная зависимость
молекулярной массы получаемого полимера от конверсии мономера. Отклонение от
линейной зависимости свидетельствует о протекании побочных реакций переноса
цепи на мономер или о медленном инициировании. Протекание полимеризации в
контролируемом режиме позволяет предсказывать молекулярную массу образующегося
полимера.
«Живым» полимер называется тогда,
когда большая часть цепей содержит нитроксильную группу на конце. Это позволяет
реинициировать полимеризацию.
.3 Метод диаграмм Фишера
Как уже обсуждалось выше, для
успешного применения того или иного алкоксиамина и нитроксильного радикала в
качестве инициатора и медиатора «живой» контролируемой полимеризации
необходимо, чтобы константы скорости гомолиза/рекомбинации алкоксиамина
находились в определенных пределах. В работе 7 найдено аналитическое решение
кинетики «живой» полимеризации и приведены аналитические выражения, позволяющие
предсказать время полимеризации t90% (16), индекс полидисперсности
получаемого полимера PDI (17) и долю «мертвых» цепей φ (18), то есть
продуктов реакции необратимого обрыва цепи, зная константы скорости гомолиза
алкоксиамина kd, рекомбинации
нитроксильного и алкильного радикалов kc,
рекомбинации двух алкильных радикалов kt и
присоединения мономера к растущей цепи kp.
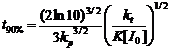 (16)
(16)
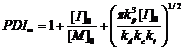 (17)
(17)
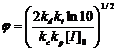 (18),
(18),
где К=kd/kc, [I]0 -
начальная концентрация инициатора, [М]0 - начальная концентрация мономера.
Таким образом, можно определить
характеристики полимеризации в присутствии данного алкоксиамина, используя в
уравнения (16)-(18). Для этого в координатах kd от kc строятся
графики уравнений (16)-(18) с желаемыми параметрами полимеризации и отмечается
положение точки, соответствующее значению констант данного алкоксиамина. Она
попадает в одну из зон А-X (Рисунок 2). Зона А соответствует
протеканию полимеризации в контролируемом режиме с заданным числом «живых»
цепей, но в течении длительного времени. Зона В соответствует протеканию
полимеризации с заданным количеством «живых» цепей, но со слабым контролем.
Время полимеризации также большое. Зона С - «живая» полимеризация со слабым
контролем, но с коротким временем полимеризации. В зону D войдут
алкоксиамины, обеспечивающие оптимальные параметры скорости, контроля и
количества «живых» цепей. Зона Х - «неживая» и неконтролируемая полимеризация.
Подобная визуализация характеристик полимеризации в зависимости от констант
гомолиза/рекомбинации алкоксиаминов называется диаграммой Фишера. При
построении диаграммы Фишера константы скорости kd и kc полагают
независимыми от длины растущей полимерной цепи, что в общем случае не
соответствует действительности. В случае полимеризации метилметакрилата чтобы
учесть изменения констант скорости при росте полимерной цепи kc уменьшают в
10 раз, а kd увеличивают
в 15.

Рисунок 2. Диаграмма Фишера.
.4 Побочные реакции при «живой»
радикальной полимеризации
По механизму «живой» радикальной
полимеризации удается получать полимеры различных виниловых мономеров -
производных стирола и акрилата. Однако пока не удается провести полимеризацию
метилметакрилатов (ММА) в контролируемом режиме с образованием «живого»
полимера. Полимеризация останавливается при малой конверсии мономера, имеет
«неживой» характер, а полученный полимер имеет высокую полидисперсность.
Причиной такого поведения полимеризации является протекание побочной реакции
отрыва атома водорода нитроксильным радикалом от алкильного. Г.С.Ананченко с
соавторами была показано отсутствие реакции переноса атома водорода при
полимеризации ММА в присутствии нитроксильного радикала N-(2-диметилпропил)-N-(1-диэтилфосфоно-2,2-диметилпропил)-N-оксил (SG1). Однако в
этом случае высокое значение константы равновесия К= kd/kc приводит к
увеличению концентрации активных цепей, а следовательно, отсутствию контроля
полимеризации. Была показана возможность проведения полимеризации ММА в
присутстиви ≈4% стирола. В настоящее время наилучший результат был
получен при полимеризации ММА с нитроксильными радикалом
2,2-дифенил-3-фенилиминоиндол-1-оксил (DPAIO) С
использованием этого радикала удалось получить полиметилметакрилат с
полидисперсностью 1,3 - 1,4 при конверсии 60%. Однако было показано, что
образующийся полимер не является «живым».
Таким образом, актуальной задачей на
сегодняшний день является изучение протекания побочной реакции переноса атома
водорода для алкоксиаминов различной структуры. Выявления зависимости
протекания реакции переноса атома водорода от структуры нитроксильного радикала
позволило бы подобрать подходящий контролирующий агент для полимеризации ММА в
контролируемом режиме.
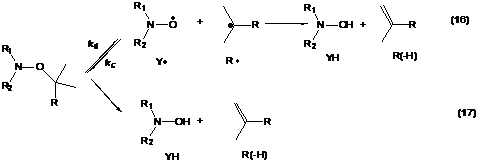
Схема 4. Механизмы протекания
реакции переноса атома водорода по радикальному (16) и внутримолекулярному (17)
пути.
Механизм реакции переноса атома
водорода при инициировании полимеризации ММА алкоксиамином представлен на схеме
4. Существует два пути: первый заключается во взаимодействии нитроксильного Y— и
алкильного R— радикалов с
образованием гидроксиламина YH и алкена R(-H) -
радикальный перенос атома водорода (реакция (16)), второй - в распаде исходного
алкоксиамина с образованием алкена R(-H) и
гидроксиламина YH, минуя радикальную стадию, -
внутримолекулярный перенос атома водорода (реакция (17)).
Наличие в системе побочной реакции,
приводящей к образованию «мертвых» цепей, играет существенную роль в процессе
полимеризации и изменяет ее характер: уменьшается количество нитроксильного
радикала Y— и радикалов
роста Ri—, и в
конечном итоге полимеризация останавливается.
Изучение реакции переноса атома
водорода и других побочных реакций, протекающих при полимеризации в присутствии
нитроксильных радикалов, является на сегодняшний день важной задачей в этой
области. В конечном итоге это позволит проводить полимеризацию ММА в
присутствии новых нитроксильных радикалов в контролируемом режиме.
.5 Радикальная «живая» полимеризация
гидрофильных мономеров
Один из классов полимеров,
обладающих ценными свойствами и находящих широкое применение в различных
отраслях экономики, являются полимеры гидрофильных мономеров. Полимеры на основе
таких мономеров как акриламид, акрилат натрия, 4-стиролсульфонат натрия,
4-гидроксиметилметакрилат, винилпирролидон имеют огромную практическую
значимость, находят широкое применение, в частности, в медицине:
полистиролсульфонат натрия применяется в качестве лекарственного средства при
гиперкалиемии, поли-(4-гидроксиметилметакрилат) - как заменитель нервной ткани
при ее повреждении. Эти полимеры широко применяются: в нефтедобывающей
промышленности для более полного извлечения нефти из скважин; в горнодобывающей
промышленности для обогащения и регенерации полезных ископаемых, для очистки
промышленных вод; как добавки при производстве бумаги и т.д. К числу
неоспоримых достоинств относится также то, что их производство не вызывает
загрязнения окружающей среды, так как оно не связано с использованием
токсичных, огне- и взрывоопасных растворителей.
Таким образом, крайне важно
получение полимеров гидрофильных мономеров высокого качества. Одним из методов
достижения этого может стать контролируемая «живая» полимеризация в водной
среде.
В литературе описано, что
полимеризация водорастворимых мономеров, контролируемая нитроксильными
радикалами, осуществлялась в среде мономера и в водно-органической эмульсии. В
качестве медиаторов использовались TEMPO и его
водорастворимые аналоги (4-гидрокси и 4-амино производных). Так23 при
полимеризации ССNa в присутствии 4-гидрокси-TEMPO, а затем
частичной замене сульфо-группы на порфириновое кольцо был получен полимер,
способный поглощать свет в видимой области. В водных растворах он образует
наноразмерные сферы, которые можно использовать в качестве нанореакторов. В
работе24 описана полимеризация акриловой кислоты, а в работе описана полимеризация
2-гидроксилакрилата в присутствии SG1.
Известно много случаев использования
макроинициаторов на основе водорастворимых полимеров для проведения
эмульсионной и миниэмульсионной полимеризации стирола и акрилатов. В работе для
получения сополимеров использовался макроинициатор на основе акриловой кислоты,
полученный методом радикальной «живой» полимеризации в присутствии SG1. Таким
способом авторы синтезировали амфифильный блок-сополимер. Еще один пример
подобного синтеза можно найти в работе.25 Авторы использовали макроинициатор на
основе углеродной нанотрубки с привитым TEMPO для
модификации нанотрубок поливинилпиридином и полистиролсульфонатом.
В работе было показано, что
нитроксильные радикалы изоиндолинового и пиперидинового ряда являются
эффективными медиаторами полимеризации стиролсульфоната натрия и акрилата
натрия в водной среде. Их использование при полимеризации позволило получить
соответствующие полимеры и блок-сополимер с узким молекулярно-массовым
распределением (PDI<1.5).
.6 Методы исследования
.6.1 Измерение константы скорости
гомолиза kd и изучение
кинетики термолиза алкоксиаминов методом 1Н ЯМР спектроскопии
Большую роль на характер
полимеризации оказывает величина константы равновесия реакции распада
алкоксиамина, которая определяется константой скорости реакций (7.1) и (7.2). И
если значение константы скорости рекомбинации нитроксильного радикала Y— и
алкильного радикала R— порядка диффузионной, то константа
скорости гомолиза алкоксиамина Y- R сильно
зависит от температуры, структуры алкоксиамина. Поэтому важно точное
определение константы скорости kd.
Определение константы скорости
гомолиза kd можно
проводить, наблюдая за накоплением продуктов рекомбинации алкильных радикалов.
Однако при этом должны быть точно измерены константы скорости рекомбинации
нитроксильного и алкильного радикалов и двух алкильных радикалов. Для более
точного и простого определения константы скорости гомолиза алкоксиаминов
необходимо подавить реакцию рекомбинации алкильного и нитроксильного радикалов
(7.1). Добиться этого можно путем добавления ловушки алкильных радикалов или
восстановителя нитроксильных радикалов. Наиболее часто используемым подходом
является детектирование накопления нитроксильного радикала методом ЭПР. При
этом в качестве ловушки алкильных радикалов используется кислород,
гальвиноксил, гидрохинон и др. Также применяется хроматографические методы -
прямой анализ продуктов гомолиза алкоксиамина в присутствии различных ловушек
алкильных радикалов или восстановителей нитроксильных радикалов и косвенный -
по анализу изменения полидисперсности полимера при термолизе алкоксиамина в
системе, содержащей мономер. Также применяется метод 1Н и 31Р ЯМР для анализа
кинетики расходования алкоксиамина при термолизе в присутствии различных
ловушек алкильных радикалов и восстановителей нитроксильных радикалов.
Использование метода ЯМР очень
удобно тем, что позволяет измерять константу скорости гомолиза kd, наблюдая
за скоростью исчезновения алкоксиамина. При этом на проведение измерений не
влияет стабильность образующегося нитроксильного радикала. В работе была
применена 31Р ЯМР спектроскопия для определения константы скорости гомолиза
фосфорсодержащих алкоксиаминов. При этом использовались различные ловушки
алкильных радикалов - нитроксильные радикалы, отличные от нитроксильного
радикала в составе алкоксиамина, фенилгидразин, тиофенол, феноксазин. В работе
Джеорджса представлено определение константы скорости гомолиза kd методом 1Н
ЯМР спектроскопии с использованием кислорода в качестве ловушки алкильных
радикалов.
.6.2 Анализ продуктов термолиза и
определение побочных реакций при термолизе алкоксиаминов
Применение 1Н ЯМР спектроскопии для
наблюдения кинетики термолиза алкоксиаминов позволяет решить еще одну задачу -
по анализу продуктов реакции и кинетики их накопления определить, какие реакции
происходят при термолизе алкоксиамина в присутствии и отсутствии радикальной
ловушки. Этот подход, в частности, позволяет разделить вклады
внитримолекулярного и радикального пути реакции переноса атома водорода.
Идентификация продуктов термолиза осуществляется по 1Н, 13С ЯМР спектру или
методом хроматографического разделения реакционной смеси с последующим
определением продуктов методом УФ-спектрометрии или масс-спектрометрии.
1.6.3 Изучение кинетики
полимеризации in situ методом 1Н
ЯМР спектроскопии
Наблюдение полимеризации в режиме
реального времени методом 1Н ЯМР спектроскопии позволяет получать детальную
информацию о кинетике полимеризации и механизмах реакций, происходящих во время
полимеризации. Эти эксперименты просты в исполнении, что делает их незаменимыми
при изучении кинетики полимеризации без изменения состава системы, анализе
продуктов, определении конверсии мономера, механизме реакций, происходящих при
полимеризации.
радикальный
полимеризация живой термолиз
1.6.4 Определение молекулярной массы
и полидисперсности полимера
Определение молекулярно-массовых
характеристик полимеров осуществляется на основе их фракционирования с
последующим определением молекулярной массы каждой фракции и расчетом
молекулярно-массового распределения и полидисперсности.
В настоящее время для определения
молекулярно-массовых характеристик полимера используется метод гель -
проникающей хроматографии (ГПХ), как в лабораторных исследованиях, так и на
производстве. В основе метода гель - проникающей хроматографии лежит разделение
молекул полимера по размерам за счет диффузии макромолекул в поры сорбента из
подвижной фазы, который представляет собой полимерные частицы с порами
различного диаметра. Внутрь пор сорбента могут с определенной вероятностью
проникать различные макромолекулы. Эта вероятность зависит от размеров
макромолекул, а скорость проникновения определяется диффузионной, подвижностью
макромолекул. Для малых макромолекул вероятность диффузии в поры выше, чем для
больших. Очень большие молекулы не проникают внутрь пор сорбента, а очень малые
попадают туда с вероятностью, близкой к единице. То есть разделение полимерных
молекул происходит не по массе, а по размеру. Этот метод позволяет легко
получать молекулярно-массовое распределение полимера простым пересчетом
хроматограммы с учетом калибровочной кривой, полученной для узкополидисперсных
образцов полимера с известными молекулярно-массовыми характеристиками.
Детектирование в гель - проникающей хроматографии осуществляется различными путями.
Самыми распространенными методами являются: детектирование УФ или оптического
поглощения элюата по сравнению с чистым элюентом; определение
рефрактометрического индекса (рефрактометрическое детектирование), определение
вязкости (вискозиметрическое детектирование).
.6.5 Определение «живого» характера
полимеризации
Отличием радикальной «живой» полимеризации
является возможность использования макроалкоксиамина в качестве инициатора, то
есть полимерная цепь способна к дальнейшему росту после полного расходования
мономера. Именно это свойство определяет уникальные синтетические возможности
радикальной «живой» полимеризации в области блок-сополимеров. Образующиеся
блок-сополимеры, таким образом, не загрязнены мономерными звеньями другого
типа.
Количество полимерных цепей, способных к
инициированию (фракция «живых» цепей) является важным параметром полимеризации.
В идеальном случае (нет бимолекулярной рекомбинации алкильных радикалов)
количество «живых» цепей равно общему количеству полимерных цепей. Однако, как
уже обсуждалось выше, при проведении полимеризации в присутствии нитроксильных
радикалов снижается вероятность рекомбинации двух алкильных радикалов, однако
исключить ее полностью нельзя. Таким образом, в реальных системах количество
«живых» цепей никогда не достигает общего количества полимерных цепей.
К определению количества «живых» цепей
существует 2 подхода. Первый из них заключается в реинициировании полимеризации
добавлением новой порции обычно другого мономера и анализе полидисперсности и
молекулярной массы получающегося блок-сополимера. На основании сравнения
расчетной молекулярной массы образующегося блок-сополимера и экспериментальной
делается вывод о количестве «живых» полимерных цепей.
Второй подход заключается в оценке количества
нитроксильного радикала, высвобождающегося при термолизе высокомолекулярного
алкоксиамина в присутствии кислорода, методом количественного ЭПР. Этот подход
эквивалентен определению константы скорости диссоциации высокомолекулярного
алкоксиамина.
Оба подхода используются в равной степени, выбор
того или иного метода зависит от возможности провести тот или иной эксперимент.
.7 Постановка задачи
Из приведенного обзора следует, что актуальной
задачей является поиск новых контролирующих агентов для полимеризации ММА по «живому»
радикальному механизму. Важнейшим требованием к этим контролирующим агентам
является отсутствие побочных реакций при полимеризации. Таким образом, изучение
термолиза алкоксиаминов позволяет дать быстрый ответ о потенциальной
возможности применения того или иного инициатора в полимеризации.
В настоящей работе решались следующие задачи:
. Протестировать метод радикальной ловушки для
установления механизма переноса атома водорода и других побочных реакций и
измерения констант скорости диссоциации kd
методом 1Н ЯМР спектроскопии на молекуле алкоксиамина
4-нитрофенил-2-(2,2,6,6-тетраметилпиперидин-1-илокси)-2-метилпропионат (1а);
применить этот метод для определения механизма термолиза ряда алкоксиаминов -
перспективных инициаторов радикальной «живой» полимеризации;
. Измерить константы скорости термолиза kd
ряда алкоксиаминов методом 1Н ЯМР спектроскопии в присутствии радикальной
ловушки.
. Исследовать возможность применения
нитроксильных радикалов, содержащих гидрофильные группы для проведения
полимеризации акриламида, акрилата натрия и стиролсульфоната натрия в водной
среде.
Глава 2. Экспериментальная часть
.1 Объекты исследования
В третьей главе работы исследовались реакции,
протекающие при термолизе алкоксиаминов (Рисунок 3.1), содержащих различные
нитроксильные фрагменты в своем составе. Нитроксильный радикал 1—
(ТЕМПО - 2,2,6,6-тетраметилпиперидин-1-оксил), входящий в состав алкоксиамина
1а (4-нитрофенил 2-(2,2,6,6-тетраметилпиперидин-1-оксил)-2-метилпропионат)
успешно применяется для полимеризации стирола и акрилатов. Нитроксильный
радикал 2— (DPAIO
- 2,2-дифенил-3-фенилиминоиндол-1-оксил), входящий в состав алкоксиамина 2а
(4-нитрофенил 2-(2,2-дифенил-3-фенилиминоиндол-1-оксил)-2-меитлпропионат), дает
на сегодняшний день наилучшие результаты при полимеризации ММА. Однако
полимеризация в присутствии этого нитроксильного радикала все же не протекает в
режиме «живых» цепей. Алкоксиамины 3а (4-нитрофенил
2-(5-бутил-5-третбутил-2-циклогексил-4-фенил-имидазол-1-оксил)-2-метилпропионат),
3b (трет-бутил-2-(5-бутил-5-третбутил-2-циклогексил-4-фенил-имидазол-1-оксил)-2-метилпропионат),
4b
(трет-бутил-2-(2,2-дибутил-5,5-диметил-пирролидин-1-окси)-2-метилпропионат), 5b
(трет-бутил-2-(5,5-диэтил-2-циклогексил-4-фенил-имидазол-1-оксил)-2-метилпропионат)
являются представителями классов алкоксиаминов на основе имидазолиновых и
пирролидиновых нитроксильных радикалов. В работе поиск эффективного медиатора
полимеризации ММА проводился среди данного класса веществ.
В четвертой главе работы изучена возможность
применения нитроксильных радикалов для проведения полимеризации водорастворимых
мономеров в контролируемом режиме. Изучена полимеризация 4-стиролсульфоната
натрия (СС Na), акрилата
натрия (A Na) и
акриламида (Рисунок 3.4), инициированная персульфатом натрия, в присутствии
нитроксильного радикала
4-(4,4-диэтил-1,2,5-триметилимидазолидин-2-оксил)-бутират натрия (6—).
Определены характеристики получаемого полимера.
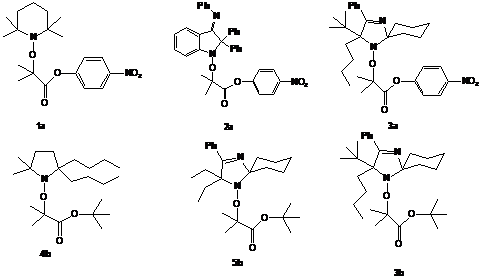
Рисунок 3.1. Исследуемые алкоксиамины.

Рисунок 3.2. Нитроксильные радикалы.

Рисунок 3.3. Продукты термолиза алкоксиаминов.

Рисунок 3.4. Гидрофильные мономеры.

Рисунок 3.5. Структура нитроксильного радикала 6—.
2.2 Общий подход к определению реакций,
происходящих при термолизе алкоксиаминов
Эксперимент 1

Эксперимент 2
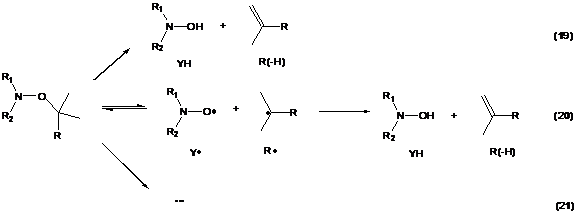
Схема 5. Схема реакций, происходящих при
термолизе алкоксиамина в присутствии (Эксперимент 1) и в отсутствии
(Эксперимент 2) тиофенола.
Подход, примененный в данной работе, аналогичен
подходу Джорджса и колл.30 Для определения побочных реакций при термолизе
проводилось 2 эксперимента для каждого алкоксиамина. Первый эксперимент
заключается в термолизе алкоксиамина в присутствии донора атомов водорода с
последующим анализом продуктов реакции. В качестве донора атомов водорода был
выбран тиофенол (PhSH).
Тиофенол имеет высокие константы скорости реакции с углеродцентрированными, kPhSH((CH3)3C•)
= 1.47 108 М-1с-1 и нитроксильными радикалами kPhSH(R1R2NO•)
» 100 М-1с-1, кроме того, сигнал протона SH-группы
в 1Н ЯМР спектре (3,1 м.д.) не перекрывается с сигналами алкильных и винильных
протонов алкоксиаминов и продуктов термолиза. Ожидается, что основной реакцией
при термолизе алкоксиаминов является разрыв связи С-О с образованием
нитроксильного Y—
и углеродцентрированного радикалов R—.
В присутствии радикальной ловушки - донора атомов водорода - нитроксильный и
алкильный радикал будут образовывать соответствующий гидроскиламин YH
и алкан RH - реакция
(18), Схема 5. Однако в случае протекания реакции переноса атома водорода по
внутримолекулярному механизму, на которую на влияет присутствие ловушки, также
будет происходить образование соответствующего алкена R(-H)
- реакция (19), Схема 5. Если реакция переноса атома водорода не протекает,
накопления алкена в реакционной смеси не происходит. Таким образом, эксперимент
1 позволяет сделать выводы о протекании внутримолекулярного переноса атома
водорода, в случае отсутствия этой реакции, определить константу скорости kd.
Второй эксперимент представляет собой термолиз
алкоксиамина в отсутствии радикальной ловушки с последующим анализом продуктов.
В случае отсутствия реакции переноса атома водорода и других побочных реакций в
системе возникает эффект Фишера-Ингольда, и уменьшения концентрации
алкоксиамина практически не происходит в течение долгого времени. Если
протекает реакция переноса атома водорода, то накапливаются гидроксиламин YH
и алкен RH (реакции
(19) и (20), Схема 5). Сопоставляя данные экспериментов 1 и 2, можно сделать
вывод о механизме протекания реакции переноса атома водорода -
внутримолекулярный или радикальный. Помимо реакции переноса атома водорода при
термолизе алкоксиаминов могут происходит другие побочные реакции, например
распад нитроксильного радикала или разрыв связи N-O
в алкоксиамине. Обычно эти реакции протекают с меньшей скоростью, чем обратимый
разрыв связи С-О в алкоксиамине. Анализируя продукты термолиза алкоксиамина в
отсутствии радикальной ловушки с применением различных методов, можно
определить эти реакции.
Алкоксиамины 1а, 2а и 6с были синтезированы по
стандартным методикам в лаборатории проф. Тордо (Марсель, Франция).
Алкоксиамины 3а, 3b, 4b,
5b были предоставлены
нам д.х.н. И.А.Кирилюком (Лаборатория азотистых соединений НИОХ СО РАН).
Запись кинетики разложения алкоксиаминов
проводилась для 0,02 М растворов алкоксиаминов в бензоле-Д6 с 10-20 кратным
избытком тиофенола в случае эксперимента в присутствии радикальной ловушки
методом времяразрешенной 1Н ЯМР спектроскопии на спектрометре DRX-
Avance-200 Bruker
с термоприставкой BVT2000.
Из образцов тщательно удалялся растворенный кислород методом заморозки -
откачки - разморозки, затем образцы запаивались в стандартных ЯМР ампулах.
Образцы помещались в нагретый датчик ЯМР спектрометра, после чего осуществлялась
запись массива 1Н ЯМР спектров через определенные интервалы времени.
Эксперименты проводились при температуре 700С. Длительность эксперимента
варьировалась в зависимости от молекулы алкоксиамина от 1 часа до 60 часов.
Перед началом и после окончания эксперимента производилась запись 1Н ЯМР
спектров образца при комнатной температуре для анализа продуктов реакции.
Идентификация продуктов реакции проводилась исходя из сигналов протонов 1Н ЯМР
спектров. Для идентификации продуктов термолиза алкоксиаминов 2с и 6с была
проведена методом жидкостной хроматографии с УФ- и масс спектроскопическим
детектированием.
Для получения температурной зависимости kd
эксперименты по термолизу алкоксиаминов проводили в смеси
трифтор-трихлор-замешенных бензолов (C6F3Cl3)
при различных температурах. Кинетические кривые получали автоматическим
интегрированием сигналов 1Н ЯМР протонов исходного алкоксиамина. Затем строили
зависимость логарифма сигнала, нормированного на начальную интенсивность, от
времени. Параметры энергии активации и предэкспоненциального множителя получали
линейной аппроксимацией аппроксимацией полученной зависимости.
Глава 3. Изучение механизма побочных реакций при
термолизе ряда алкоксиаминов и определение константы скорости их термолиза
.1 Введение
В данной главе представлены результаты
исследования термолиза алкоксиаминов в присутствии и отсутствии радикальной
ловушки тиофенол и анализа продуктов теромолиза. Определены механизмы реакции
переноса атома водорода для алкоксиаминов 1а, 3а, 4b,
5b. Показано отсутствие
реакции переноса атома водорода для 2а, 3b
и 6с. При проведении экспериментов в присутствии радикальной ловушки тиофенола
определены константы скорости гомолиза алкоксиаминов kd.
Для алкоксиаминов1а, 2а и 6с, находящих широкое применение в качестве медиаторов
полимеризации, были найдены температурные зависимости kd
и измерены энергия активации и предэкспоненциальный множитель для реакции
термолиза.
.2 Результаты
.2.1 Побочные реакции при термолизе
алкоксиаминов
.2.1.1 Термолиз 1а
Описанный в п. 2.2. подход был применен для
исследования реакций, протекающих при термолизе алкоксиамина 1а. На рисунке 3.1
представлены спектры до и после термолиза алкоксиамина 1а в присутствии избытка
тиофенола при 351 К.
Продолжительность эксперимента составила 1 час,
при этом наблюдалось полное разложение алкоксиамина 1а. В результате термолиза
алкоксиамина 1а в присутствии тиофенола образовались алкан аН и гидроксиламина
1Н. При этом в спектре ЯМР реакционной смеси после термолиза не наблюдаются
сигналы винильных протонов (стрелки на рисунке 3.1 указывают на отсутствие
сигналов винильных протонов), что говорит об отсутствии алкена а(-Н). Таким
образом, основным процессом при термолизе алкоксиамина 1а в присутствии
радикальной ловушки является радикальный разрыв связи С-О с последующим
взаимодействием нитроксильного и алкильного радикалов с тиофенолом. Реакция
переноса атома водорода по внутримолекулярному пути не протекает.
Термолиз алкоксиамина 1а в отсутствии
радикальной ловушки при 351 К протекал значительно медленнее: время проведения
эксперимента составило 15 часов, конверсия алкоксиамина 1а - 75%. Основные
продукты термолиза 1а в отсутствии тиофенола - алкен а(-Н) и гидроксиламин 1Н в
стехиометрическом соотношении по отношению к прореагировавшему алкоксиамину 1а,
что свидетельствует о протекании реакции переноса атома водорода. Суммируя
выводы экспериментов по термолизу алкоксиамина 1а в присутствии и отсутствии
радикальной ловушки, можно сделать вывод, что для алкоксиамина 1а побочная
реакция переноса атома водорода протекает по радикальному механизму. Схема
реакций, происходящих при термолизе алкоксиамина 1а в отсутствии радикальной
ловушки, представлена на схеме 6.


.1 3.2
Рисунок 3. 1Н ЯМР спектры, зарегистрированные до
и после термолиза раствора алкоксиамина 1а при 351 К в присутствии (3.1) и в
отсутствии (3.2) тиофенола. Стрелками отмечено положение сигналов алкена а(-Н)
и гидроксиламина 1Н.
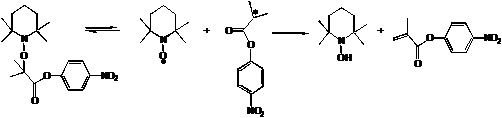
Схема 6. Термолиз алкоксиамина 1а. Реакция
переноса атома водорода по радикальному пути.
.2.1.2 Термолиз 2а
Термолиз алкоксиамина 2а в присутствии тиофенола
при 351 К протекает намного медленнее, чем 1а. Основным продуктом термолиза 2а
в присутствии радикальной ловушки является алкан аН. О накоплении
гидроксиламина 2Н можно судить по уширению сигнала протона SH
тиофенола за счет обмена. В 1Н ЯМР спектре системы после термолиза отсутствуют
сигналы протонов соединения а(-Н). Таким образом, реакция переноса атома водорода
по внутримолекулярному механизму не протекает при термолизе алкоксиамина 2а.


.1 4.2
Рисунок 4. 1Н ЯМР спектры, зарегистрированные до
и после термолиза раствора алкоксиамина 2а при 351 К в присутствии (4.1) и в
отсутствии (4.2) тиофенола. * отмечено появление сигналов продуктов реакции.
1,1,2,2-тетрахлорэтан использован в качестве стандарта интегрирования в эксперименте
с радикальной ловушкой
Для идентификации продуктов термолиза был
проведен хроматографический анализ с УФ и масс-спектрографическим
детектированием продуктов реакции. Анализ выполнен в Лаборатории экологических
исследований и хроматографического анализа НИОХ СО РАН. По результатам
хроматографического анализа в реакционной смеси было обнаружено 14
индивидуальных веществ (Рисунок 5). 2 из них были идентифицированы как
нитроксильный радикал 2— и исходный
алкоксиамин 2а путем сопоставления времен удерживания и УФ-спектров
индивидуальных образцов 2— и 2а. Стоит
отметить, что в образце исходного алкоксиамина в качестве примеси
присутствовало соединение 15 (Таблица 1). Присутствие амина 2amine
в реакционной смеси было установлено сопоставлением УФ-спектров индивидуального
образца 2amine.
Идентификация остальных соединений, присутствующих в реакционной смеси,
проводилась путем сопоставления данных масс-спектроскопического анализа и
УФ-спектроскопии. Структура возможных продуктов реакции предполагалась из схемы
термолиза алкоксиамина 2а, включающей в себя разрыв связи N-O
наряду с разрывом связи С-О. Данные ВЭЖХ/МС суммированы в Таблице 1.
Таким образом, данные анализа продуктов методом
ВЭЖХ/МС подтвердили гипотезу, согласно которой при термолизе алкоксиамина 2а
наряду с разрывом связи С-О протекает медленная реакция необратимого разрыва
связи N-O.
Образующиеся N- и
О-центрированные радикалы взаимодействуют с нитроксильным и алкильным
радикалами, давая продукты 4,5,8,10,11. Реакции, происходящие при термолизе 2а,
представлены на схеме 7.
Рисунок 5. Хроматограмма реакционной смеси после
термолиза алкоксиамина 2а в отсутствии тиофенола при 351 К.

Таблица 1. Данные масс-спектроскопического
анализа продуктов термолиза алкоксиамина 2а.
|
Номер
пика (Рис. 5)
|
Молекулярная
масса (по данным ВЭЖХ/МС анализа)
|
Соединение
|
|
4
|
434
|
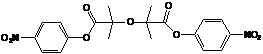
|
|
5
|
416
|

|
|
8
|
360
|
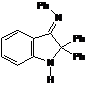
|
|
9
|
375
|
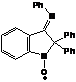
|
|
10
|
583
|

|
|
11
|
576
|

|
|
12
|
582
|
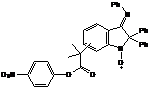
|
|
13
|
583
|
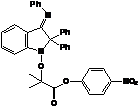
|
|
15
|
790
|

|
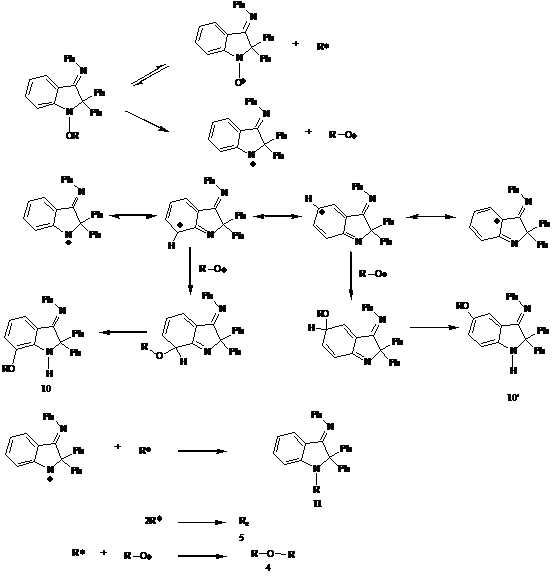
Схема 7. Термолиз алкоксиамина 2а.
.2.1.3 Термолиз 3а и 3b
Термолиз алкоксиамина 3а протекает аналогично
1а. В проведении термолиза 3а присутствии радикальной ловушки при 343 К
происходит количественное образование алкана аН и гидроксиламина 3Н, накопление
алкена a(-Н) не происходит.
Таким образом, при термолизе алкоксиамина 3а происходит разрыв связи С-О с
образованием алкильного и нитроксильного радикалов, которые затем вступают в
реакцию с радикальной ловушкой тиофенол. Реакции переноса атома водорода по
внутримолекулярному пути не происходит. Кинетика изменения концентрации
алкоксиамина 3а описывается моноэкспонентой, что позволяет определить константу
скорости kd = (1,5±0,2)
10-3 с-1 для алкоксиамина 3а при 343К.
Аналогично протекает термолиз алкоксиамина 3b:
происходит количественное образование bН
и гидроксиламина 3Н. Таким образом, основным процессом является разрыв связи
С-О с образованием алкильного и нитроксильно радикалов. Накопление алкена b(-Н)
не происходит, что однозначно свидетельствует об пренебрежимо малом вкладе
реакции переноса атома водорода по внутримолекулярному механизму. Кинетика
изменения концентрации алкоксиамина 3b
является моноэкспоненциальной. Определенная константа скорости диссоциации
алкоксиамина при 343 К составляет kd
= (6,0±0,3) 10-4 с-1.


.1 6.2
Рисунок 6. 1Н ЯМР спектры, зарегистрированные до
и после термолиза раствора алкоксиамина 3а при 343 К в присутствии (6.1) и в
отсутствии (6.2) тиофенола. Стрелками отмечено положение сигналов алкена а(-Н)
и гидроксиламина 3Н.
При термолизе 3а в отсутствии тиофенола
происходит накопление алкена a(-Н)
и гидроксиламина 3Н, что говорит о протекании реакции переноса атома водорода
по радикальному механизму.


.1 7.2
Рисунок 7. 1Н ЯМР спектры, зарегистрированные до
и после термолиза раствора алкоксиамина 3b
при 343 К в присутствии (7.1) и в отсутствии (7.2) тиофенола. Стрелками
отмечено положение сигналов алкена b(-Н)
и гидроксиламина 3Н.
Схема термолиза алкоксиамина 3а представлена на
схеме 8.

Схема 8. Термолиз алкоксиамина 3а.
Термолиз алкоксиамина 3b
в отсутствии радикальной ловушки протекает в 10 раз более медленно по
стравнению с 3а при той же температуре. Изменение концентрации алкоксиамина 3b
при термолизе при 343 К в течение 3 дней составило 15%. При этом накопление
алкена b(-Н) и
гидроксиламина 3Н не произошло.
Таким образом, основным процессом, происходящим
при термолизе алкоксиамина 3b
является обратимый разрыв связи С-О с последующей рекомбинацией образовавшихся
нитроксильного и алкильного радикалов с образованием исходного алкоксиамина.
Алкоксиамины 3а и 3b
имеют одинаковый нитроксильный фрагмент в своем составе. Наличие реакции
переноса атома водорода по радикакльному для алкоксиамина 3а, говорит о том,
что реакция переноса атома водорода должна происходить и при термолизе 3b.
Отсутствие накопления алкена b(-Н)
и гидроксиламина 3Н говорит о том, что протекание реакции переноса атома
водорода пренебрежимо мало. Основным процессом является рекомбинация
нитроксильного и алкильного радикалов с образованием исходного алкоксиамина.
Потенциально алкоксиамин 3b
может быть использован в качестве инициатора полимеризации ММА. Далее (раздел
3.3) приведены результаты полимеризации ММА, инициированной алкоксиамином 3b.
3.2.1.4 Термолиз 4b


.1 8.2
Рисунок 8. 1Н ЯМР спектры, зарегистрированные до
и после термолиза раствора алкоксиамина 4b
при 343 К в присутствии (8.1) и в отсутствии (8.2) тиофенола. Стрелками
отмечено положение сигналов алкена b(-Н)
и гидроксиламина 4Н.
Основными продуктами термолиза алкоксиамина 4b
в присутствии радикальной ловушки при 343К являются алкан bН
и гидроксиламин 4Н (рисунок 7.1). Однако в спектре ЯМР, зарегистрированном
после термолиза 4b имеются
сигналы винильных протонов алкена b(-Н).
Количество алкена в смеси после термолиза, оцененное по интегралу сигналов
винильных протонов bН,
составляет около 3% от количества разложившегося алкоксиамина 4b.
Таким образом, при термолизе алкоксиамина 4b
в присутствии радикальной ловушки наряду с образованием нитроксильного и
алкильного радикалов и их последующей реакцией с тиофенолом происходит процесс
внутримолекулярного переноса атома водорода с образованием алкена b(-Н)
и гидроксиламина.
Определенная константа скорости диссоциации
алкоксиамина 4b при 343 К
составляет kd = (2,2±0,1)
10-4 с-1.
При термолизе 4b
в отсутствии тиофенола образуется гидроксиламин и алкен, то есть происходит
реакция переноса атома водорода.
Таким образом, для алкоксиамина 4b
при его термолизе реакция переноса атома водорода протекает как по радикальному
механизму, так и по внутримолекулярному. Внутримолекулярная реакция переноса
атома водорода составляет 3% от реакции разложения алкоксиамина на нитроксильный
и алкильный радикалы. (Схема 10)
Схема 10. Реакции при термолизе 4b.
.2.1.5 Термолиз 5b
Основными продуктами термолиза алкоксиамина 5b
в присутствии тиофенола при 343К является алкан bН
и гидроксиламн 5Н. Таким образом, при термолизе 5b
основным процессом является разрыв связи С-О и последующая реакция алкильного и
нитроксильного радикалов с тиофенолом. Кинетика изменения концентриции 5b
моноэкспоненциальна, аппроксимация ее позволяет получить константу скорости
диссоциации алкоксиамина 5b
kd = (6,3±0,2) 10-5
с-1 при 343К.


Рисунок 9. 1Н ЯМР спектры, зарегистрированные до
и после термолиза раствора алкоксиамина 5b
при 343 К в присутствии (9.1) и в отсутствии (9.2) тиофенола. Стрелками
отмечено положение сигналов алкена b(-Н)
и гидроксиламина 5Н.
При термолизе 5b
в отсутствии тиофенола количественно образуются алкен b(-Н)
и гидроксиламин 5Н, что свидетельствует о протекании реакции переноса атома
водорода по радикальному механизму, как и при термолизе 1а и 3а. Схема
термолиза 5b показана на схеме
11.

Схема 11. Термолиз алкоксиамина 5b
.3 Полимеризация ММА, инициированная 3b
Отсутствие реакции переноса атома водорода при
термолизе алкоксиамина 3b
позволяет говорить о потенциальной применимости этого алкоксиамина для
полимеризации ММА. Чтобы предсказать протекание полимеризации ММА,
инициированной 3b был
использован метод диаграмм Фишера. Константа скорости kd
для алкоксиамина 3b при 343 К
была измерена в эксперименте по термолизу 3b
в присутствии тиофенола. Значение kd
при более высокой температуре было получено пересчетом константы с
предэкспоненциальным фактором А = 2 1014 с-1 , характерным для реакций распада
алкоксиаминов на нитроксильный и алкильный радикалы. Значение kс
для нитроксильного радикала, аналогичного по структуре 3—
измерена в работе. Значения констант kd
и kс было
скорректировано с учетом увеличения длинны цепи полимерного радикала
согласно14. При построении диаграммы Фишера использовались значения констант
скорости kp и kt,
приведенные в 14. Согласно диаграмме Фишера были выбраны следующие условия для
полимеризации ММА в присутствии 3b:
температура 363 К, концентрация инициатора 3b
- 3 10-2М.
.3.1 Методика полимеризации
Раствор алкоксиамина в мономере поместили в
круглодонную двугорлую колбу, снабженную обратным холодильником. Раствор
продували аргоном в течение 30 минут, после чего колбу помещали в нагретую
масляную баню. Реакция проводилась при постоянном перемешивании в атмосфере
аргона. Температура поддерживалась постоянной с точностью ±10С. Через
определенные промежутки времени отбирались пробы реакционной смеси, которые
затем быстро охлаждались во льду. Для отделения полимера от мономера первый
осаждался из пробы полимерной смеси в холодном метаноле (10-кратный избыток по
сравнению с объемом пробы), отфильтровывался и высущивался. Для определения
среднечисленной и средневзвешенной молекулярной массы навеска осажденного и высушенного
полимера растворялась в тетрагидрофуране и анализировалась методом ГПХ на
жидкостном хроматографе Agilent
LC 1200 с
термостатированной колонкой PLGel
Mixed-C,
300х7,5 мм, 5 мкм. Тетрагидрофуран использовался в качестве элюэнта, скорость
потока 1 мл/мин. Калибровка была произведена по узкодисперсным стандартам
полистирола (Polymerlabs).
Конверсия определялась методом ЯМР спектроскопии для раствора 5 мкл пробы
полимерной смеси в 700 мкл бензола-Д6.

Рисунок 10. Диаграмма Фишера для полимеризации
метилметакрилата в присутствии 3b
при 363 К. Концентрация инициатора 3 10-2 М. kp
= 1600 М-1с-1, kt
= 3,4 107 М-1с-1
.3.2 Результаты и их обсуждение
Во время полимеризации было отобрано 2 пробы
через 15 и 30 минут после начала реакции. После этого реакционная смесь стала
густой, что сделало невозможным дальнейший отбор проб. На рисунке 12
представлен график зависимости среднечисленной молекулярной массы и изменение
полидисперсности полученного полиметилметакрилата от конверсии.
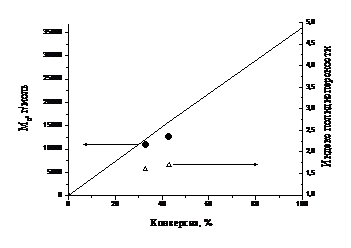
Рисунок 11. Зависимость среднечисленной
молекулярной массы (l) и индекса полидисперсности (r)
от конверсии мономера для полимеризации ММА, инициированной 3b.
Сплошная линия на рисунке 12 соответствует
теоретической молекулярной массе полимера, рассчитанной по формуле (19)
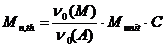 (19)
(19)
где  - начальное количество вещества
мономера,
- начальное количество вещества
мономера, 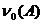 - начальное
количество вещества алкоксиамина,
- начальное
количество вещества алкоксиамина, 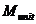 - молекулярная масса мономерного
звена, С - конверсия мономера. Из рисунка 11 следует, что при конверсии больше
40% молекулярная масса полимера начинает заметно отклоняться от рассчитанной
теоретически. При этом полидисперсность получаемого полимера остается высокой
(1,6 при конверсии 33% и 1,7 при конверсии 43%). То есть полимеризация
метилметакрилата, инициированная 3b, не
протекает в контролируемом режиме. Таким образом, неустойчивость нитроксильного
радикала 3— при
повышенной температуре имеет значительное влияние на ход полимеризации. Однако
устойчивость нитроксильного фрагмента в составе алкоксиамина, в том числе и
высокомолекулярного, позволяет получать менее полидисперсный
полиметилметакрилат (полидисперсность < 2), чем в процессах обычной
радикальной полимеризации.
- молекулярная масса мономерного
звена, С - конверсия мономера. Из рисунка 11 следует, что при конверсии больше
40% молекулярная масса полимера начинает заметно отклоняться от рассчитанной
теоретически. При этом полидисперсность получаемого полимера остается высокой
(1,6 при конверсии 33% и 1,7 при конверсии 43%). То есть полимеризация
метилметакрилата, инициированная 3b, не
протекает в контролируемом режиме. Таким образом, неустойчивость нитроксильного
радикала 3— при
повышенной температуре имеет значительное влияние на ход полимеризации. Однако
устойчивость нитроксильного фрагмента в составе алкоксиамина, в том числе и
высокомолекулярного, позволяет получать менее полидисперсный
полиметилметакрилат (полидисперсность < 2), чем в процессах обычной
радикальной полимеризации.
.4 Стабильность нитроксильного
радикала 3—
Стабильность нитроксильного радикала
3— была
проверена методом ЭПР. Раствор 3— в бензоле нагревался в масляной
бане при температуре 700С. Предварительно из образца был тщательно удален
кислород методом заморозки - откачки - разморозки, после чего образец был
запаян. Через определенные интервалы времени образец доставался из масляной
бани, остужался, затем записывался ЭПР спектр.
За 30 минут нагревания сигнал ЭПР
раствора 3— уменьшился
в 10 раз, при этом появления других сигналов ЭПР не произошло, что говорит о
разложении 3— с
образованием нерадикальных продуктов (Рисунок 12). Таким образом, нитроксильный
радикал 3— является
нестабильным при нагревании.

Рисунок 12. Изменение сигнала ЭПР
при нагревании раствора нитроксильного радикала 3— в отсутствии кислорода ( -
начальный сигнал; --- сигнал после 30-минутного нагревания при 700С).
3.5 Определение констант скорости
гомолиза алкоксиаминов kd
На рисунке 13 представлены
зависимости концентрации алкоксиаминов 3а, 3b, 4b, 5b от времени
в логарифмической шкале при 343 К. Кинетику разложения алкоксиаминов можно
аппроксимировать моноэкспонентой с параметром, соответствующим константе
скорости гомолиза при данной температуре согласно уравнению (20). Значение
констант скорости kd приведено в таблице 2.
 (20)
(20)
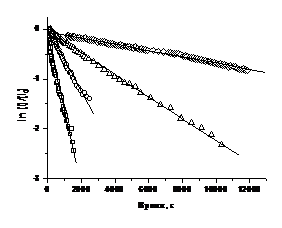
Рисунок 13. Зависимости концентрации
алкоксиаминов 3а (o), 3b (š), 4b (r) и 5b (¯) от времени
при 343 К в логарифмической шкале при термолизе алкоксиаминов в присутствии PhSH.
Таблица 2. Константы скорости kd для реакции
гомолиза алкоксиаминов 3а, 3b, 4b и 5b в
присутствии тиофенола при температуре 343 К.
|
Алкоксиамин
|
kd (10-3, с-1)
|
|
3а
|
1,5±0,2
|
|
3b
|
0,6±0,03
|
|
4b
|
0,22±0,01
|
|
5b
|
0,063±0,002
|
На рисунке 14 представлены в логарифмической
шкале зависимости концентрации алкоксиаминов 1а и 2а от времени при различной
температуре. Аппроксимация полученных экспериментальных зависимостей по
уравнению (20) позволяет получить значение константы скорости гомолиза kd
при различной температуре. Значения констант скорости гомолиза kd
алкоксиаминов при различных температурах представлено в таблице 2. Следует
отметить, что значение константы скорости гомолиза алкоксиамина 1а хорошо
согласуется с измеренным методом ЭПР (kd
(343 K) = 6.8 10-4 s-1
and Ea
= 115.5 kJ
mol-1.
Исходя из температурной зависимости констант
скорости, были найдены энергия активации и предэкспоненциальный множитель для
реакций термолиза алкоксиаминов по уравнению (21).
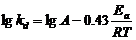 (21)
(21)
(а) (б)
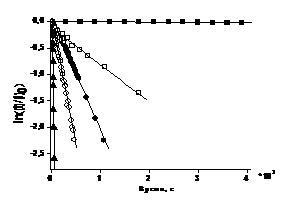
Рисунок 14. Зависимости концентрации
алкоксиаминов 1а (а), 2а (б), от времени при 323 К (¢), 351 К (o), 361 К (), 373 К (š), 398 К (▲)
в логарифмической шкале при термолизе алкоксиаминов в присутствии PhSH.
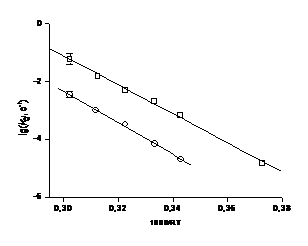
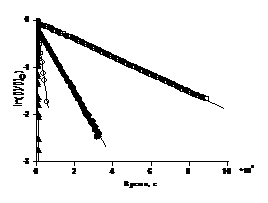
Рисунок 15. Аррениусовская
зависимость для реакции термолиза алкоксиаминов 1a (o) и 2a (š).
Таблица 3. Константы скорости
гомолиза kd при
различной температуре и аррениусовские параметры для реакции гомолиза
алкоксиаминов 1а и 2а.
|
T (K)
|
kd (10-3 с-1)
|
|
1a
|
2a
|
|
327
|
0.015±0.005
|
-
|
|
351
|
0.65 ± 0.5
|
0.02 ± 0.008
|
|
361
|
2.0 ± 0.6
|
0.07 ± 0.008
|
|
373
|
5.0 ± 0.8
|
0.32 ± 0.08
|
|
386
|
15.0 ± 0.4
|
1.0 ± 0.05
|
|
398
|
60.0 ± 4.0
|
3.5 ± 0.5
|
|
A (с-1)
|
(8.7 ±0.8 ) 1013
|
|
Ea (КДж/моль)
|
115.5 ± 0.5
|
125.0 ± 0.5
|
.6 Обсуждение результатов
.6.1 Реакция переноса атома водорода при
термолизе алкоксиаминов
В общем случае, скорость исчезновения
алкоксиаминов при термолизе в присутствии радикальной ловушки больше, чем в
отсутствии тиофенола. В присутствии тиофенола основными продуктами термолиза
алкоксиаминов являются соответствующий алкан и гидроксиламин. Эти продукты
образуются за счет взаимодействия нитроксильного и алкильного радикалов,
возникших при гомолизе связи С-О алкоксиамина, с тиофенолом. Соответствующий
алкен может образовываться только как продукт реакции внутримолекулярного
переноса атома водорода. В случае термолиза алкоксиамина в отсутствии ловушки
продуктами реакции являются нитроксильный радикал, продукты
рекомбинации/диспропорционирования алкильных радикалов (реакции (9.1) и (9.2)
Схемы 2), а в случае протекания реакции переноса атома водорода - алкен и
гидроксиламин. При отсутствии реакции переноса атома водорода при термолизе
алкоксиамина должен наблюдаться эффект Фишера-Ингольда, таким образом должно
наблюдаться незначительное уменьшение концентрации алкоксиамина в течение
длительного времени. Изменение концентрации алкоксиамина в условиях
возникновения эффекта Фишера-Ингольда описывается уравнением (22):
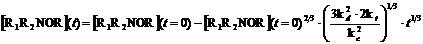 (22)
(22)
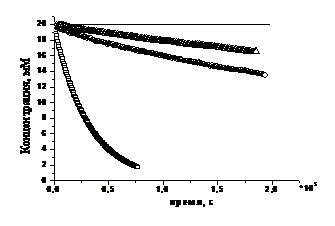
Рисунок 16. Экспериментально
зарегистрированное изменение концентрации алкоксиаминов 1а (o), 2а (š), 3b (r) при
термолизе в отсутствии радикальной ловушки и изменение концентрации
алкоксиамина в условиях эффекта Фишера-Ингольда (-) согласно уравнению (22). В
уравнении (22) использованы следующие параметры: kd = 9 10-4
с-1 (константа термолиза для алкоксиамина 1а), kc = 6 108 л∙моль-1∙с-1,
kt ≈ 109
л∙моль-1∙с-1
На Рисунке 16 представлены
зависимости изменения концентрации алкоксиаминов 1а, 2а и 3b при
термолизе в отсутствии радикальной ловушки по сравнению с изменением
концентрации алкоксиамина в условиях существования эффекта Фишера-Ингольда. Как
следует из рисунка, уменьшение концентрации алкоксиаминов 2а и 3b происходит
намного быстрее, чем следовало ожидать, то есть в системе не возникает эффекта
Фишера-Ингольда. То есть, полимеризация, инициированная этими алкоксиаминами,
не будет в полной мере носить «живой» контролируемый характер.
.6.2 Влияние побочных реакций на
протекание полимеризации в контролируемом режиме. Разрыв связи N-O при
термолизе 2а и термическая нестабильность нитроксильного радикала 3—
Как уже отмечалось ранее,
алкоксиамин 2а был использован авторами18 для инициирования полимеризации ММА.
Использование этого алкоксиамина в качестве инициатора позволило провести
полимеризацию в контролируемом режиме. Однако образовавшийся полимер обладал
малым процентом «живых» цепей. Причиной этого может служить существование
побочной реакции необратимого разрыва связи N-O. Кроме
того, образование в системе N- и O-центрированных
радикалов приводит к инициированию роста новых цепей. Таким образом, если
протекание побочной реакции разрыва связи N-O пренебрежимо
мало в течение времени полимеризации, то полимеризация будет протекать в
контролируемом режиме. Однако на больших временах реакции будет сказываться
влияние побочной реакции, что приведет к увеличению полидисперсности
получаемого полимера.
Все вышесказанное справедливо и в
случае термической нестабильности нитроксильного радикала 3—. Было
показано, что алкоксиамины, содержащие нитроксильный фрагмент 3, не
претерпевают побочных реакций. Однако свободный нитроксильный радикал 3— необратимо
разлагается достаточно быстро. Тот факт, что растущие полимерные цепи проводят
большую часть времени полимеризации в составе высокомолекулярного алкоксиамина,
позволяет провести полимеризацию ММА, контролируемую нитроксильным радикалом 3—, и получить
полимеры с достаточно низкой полидисперсностью. Однако эта полимеризация не
носит «живой» характер.
Таким образом, кроме соответствия
констант скорости гомолиза/рекомбинации алкоксиамина, для успешного протекания
полимеризации в присутствии того или иного нитроксильного радикала необходимо
также отсутствие побочных реакций. Чтобы влиянием побочных реакций можно было
пренебречь, они должны протекать много медленнее процесса полимеризации.
Глава 4. Исследование применимости
нитроксильного радикала 4-(4,4-диэтил-1,2,5-триметилимидазолидин-2-оксил)-бутират
натрия для полимеризации гидрофильных мономеров
.1 Введение
Полимеры гидрофильных мономеров
находят на сегодняшний день широкое применение во многих отраслях экономики.
Распространение технологии радикальной «живой» контролируемой полимеризации на
область водорастворимых мономеров является актуальной задачей, так как полимеры
гидрофильных мономеров с контролируемой молекулярной массой нельзя напрямую
получить с помощью ионной полимеризации. Функциональные группы, отвечающие за
растворимость в воде, необходимо химически защищать перед полимеризацией, что
приводит к усложнению процесса получения полимера. В этой главе представлены
первые результаты по применению нитроксильного радикала
4-(4,4-диэтил-1,2,5-триметилимидазолидин-2-оксил)-бутират натрия для
полимеризации 4-стиролсульфоната натрия, акриламида и акрилата натрия, изучены
кинетики полимеризации этих мономеров в присутствии и отсутствии нитроксильного
радикала методом ЯМР. Изучено изменение молекулярной массы и полидисперсности
при полимеризации акриламида в присутствии нитроксильного радикала.
.2 Изучение кинетики полимеризации
гидрофильных мономеров методом ЯМР
.2.1 Методика проведения
полимеризации
Для определения эффективности
контроля полимеризации водорастворимых мономеров нитроксльным радикалом
4-(4,4-диэтил-1,2,5-триметилимидазолидин-2-оксил)-бутират натрия 6— (Структура
представлена на рисунке 17) были проведены эксперименты по полимеризации в
водной среде в присутствии и отсутствии контролирующего агента. Нитроксильный
радикал 6— был
предоставлен нам д.х.н. И. А. Кирилюком (Лаборатория азотистых соединений НИОХ
СО РАН).
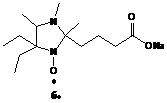

Рисунок 17. Нитроксильный радикал
4-(4,4-диэтил-1,2,5-триметилимидазолин-2-оксил)-бутират натрия и гидрофильные
мономеры 4-стиролсульфонат натрия (СС Na), акрилат
натрия (А Na), акриламид
(АА).
Эксперименты по полимеризации
проводились для 1,5 М раствора мономера в воде. В качестве инициатора
полимеризации выступал персульфат натрия, добавленный в количестве 1% (мол) от
исходного количества мономера. Нитроксильный радикал добавлялся в количестве 2%
(мол). Реакционная смесь продувалась аргоном в течение 15 минут для удаления
кислорода из раствора. Эксперимент проводился в датчике ЯМР спектрометра DRX- Avance-200. Запись
спектров проводилась с интервалом 10 минут в случае полимеризации в присутствии
нитроксильного радикала с интервалом в 1 минуту при полимеризации без
нитроксильного радикала. Кинетические кривые получали автоматическим
интегрированием соответствующих сигналов ЯМР.
.2.2 Результаты и обсуждение
.2.2.1 Кинетика полимеризации
4-стиролсульфоната натрия
На рисунке 18 представлено сравнение
ЯМР спектров, зарегистрированных при полимеризации СС Na натрия в
присутствии (Рисунок 18 а) и в отсутствии (Рисунок 18 б) нитроксильного
радикала 6— при 800С, а
на рисунке 19 представлены кинетики расходования мономера и накопления полимера
для обоих случаев. Кинетические кривые были построены по значениям интегральной
интенсивности сигналов ЯМР, обозначенных 1 и 2 для мономера, 3 для полимера
(Рисунок 19).
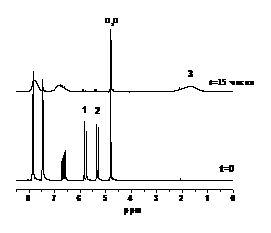
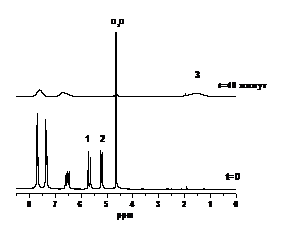
(а) (б)
Рисунок 18. 1Н спектры ЯМР,
зарегистрированные при полимеризации СС Na в
присутствии (а) и отсутствии (б) нитроксильного радикала 6—. Цифрами 1
и 2 отмечены сигналы ЯМР протонов СН2-группы мономера, по которым была измерена
кинетика расходования мономера (Рисунок 19 а), 3 - сигнал ЯМР протонов полимера
(Рисунок 19 б).
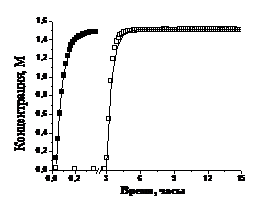
В случае полимеризации без
нитроксильного радикала кинетика расходования мономера описываются
моноэкспонентой с характерным временем t1 = (3,4±
0,1) ž102 с.
При полимеризации СС Na в
присутствии нитроксильного радикала 6— кинетическая кривая расходования
мономера не описывается моноэкспонентой. Характерное время спада кинетики
расходования мономера при полимеризации в присутствии нитроксильного радикала 6— составляет
(1,55 ± 0,05) ž103 с.
Характерное время накопления полимера также равно (1,5 ± 0,05) ž103 с.
Накопление полимера имеет
индукционный период. Наличие индукционного периода характерно для проведения
«живой» контролируемой полимеризации в смеси инициатор - нитроксильный радикал
- мономер28 и связано с накоплением алкоксиамина.

(а) (б)
Рисунок 19. Кинетика расходования
мономера (а) и накопления полимера (б) при полимеризации СС Na в
отсутствии (¢) и
присутствии (o)
нитроксильного радикала 6—.
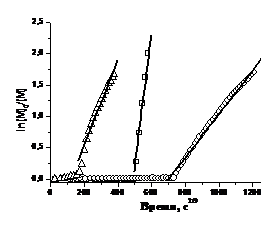
Рисунок 20. Кинетика полимеризации
СС Na (o), АА (š), А Na (r) в
присутствии нитроксильного радикала 6— в координатах Фишера.
Кинетику «живой» контролируемой
радикальной полимеризации принято анализировать в так называемых координатах
Фишера. Согласно решению кинетики для реакций «живой» контролируемой
радикальной полимеризации10, концентрация мономера в течение полимеризации
зависит от времени согласно уравнению (23):
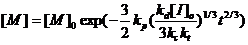 (23)
(23)
где  - концентрация мономера в текущий
момент времени и в начальный момент времени соответственно. Таким образом,
зависимость
- концентрация мономера в текущий
момент времени и в начальный момент времени соответственно. Таким образом,
зависимость 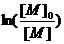 от времени
в степени 2/3 представляет собой прямую для контролируемой «живой»
полимеризации.
от времени
в степени 2/3 представляет собой прямую для контролируемой «живой»
полимеризации.
Зависимости логарифма приведенной
концентрации мономера от времени в степени 2/3 представлены на Рисунке 20.
Линейность кинетики полимеризации СС Na в
координатах Фишера сохраняется до конверсии 90%. При более глубоких конверсиях
мономера увеличивается ошибка определения концентрации мономера, что приводит к
отклонению кинетики от линейной зависимости. Таким образом, основную часть
времени полимеризация СС Na в присутствии нитроксильного
радикала 6— протекает в
контролируемом режиме.
.2.2.2 Кинетика полимеризации
акриламида
Эксперименты по полимеризации
акриламида проводились при 700С. На рисунке 21 представлено сравнение ЯМР
спектров, зарегистрированных при полимеризации АА отсутствии (а) и присутствии
(б) нитроксильного радикала 6—,
а на рисунке 22 представлены кинетики расходования мономера и накопления
полимера для обоих случаев. Кинетические кривые были построены по значениям
интегральной интенсивности сигналов ЯМР, обозначенных 1 для мономера, 2 и 3 для
полимера на рисунке 21.


(а) (б)
Рисунок 21 1Н спектры ЯМР,
зарегистрированные при полимеризации АА в присутствии (а) и отсутствии (б)
нитроксильного радикала 6—.
В случае полимеризации АА без
нитроксильного радикала кинетика расходования мономера удовлетворительно
описывается моноэкспонентой с характерным временем 200 секунд, кинетика
накопления полимера также описывается моноэкспонентой с таким же характерным
временем. Конверсия мономера при полимеризации АА без нитроксильного радикала
составила 90%.
При проведении эксперимента в
присутствии нитроксильного радикала 6— кинетика полимеризации сильно
удлиняется. Характерное время расходования мономера в присутствии
нитроксильного радикала составляет (1,0±0,05)ž104 с, накопления полимера -
(1,0±0,05) ž104 с. Из
заметного снижения скорости полимеризации можно сделать вывод, что
нитроксильный радикал 6— обратимо
присоединяется к радикалам роста при полимеризации АА.
На рисунке 20 представлена кинетика
полимеризации АА в координатах Фишера. Линейность зависимости приведенной
концентрации от времени в степени 2/3 сохраняется до глубокой конверсии
мономера (85%). Линейность кинетики полимеризации АА в координатах Фишера
говорит о реализации контролируемого режима в случае полимеризации АА в
присутствии нитроксильного радикала 6—.


(а) (б)
Рисунок 22. Кинетика расходования
мономера (а) и накопления полимера (б) при полимеризации АА в отсутствии (¢) и
присутствии (o)
нитроксильного радикала 6—.
.2.2.3 Кинетика полимеризации
акрилата натрия
Полимеризация акрилата натрия (A Na)
проводилась при 800С. На рисунке 23 представлены спектры ЯМР,
зарегистрированные до и после полимеризации, в присутствии (рисунок 23 а) и
отсутствии (рисунок 23 б) нитроксильного радикала 6—, а на
рисунке 24 представлены кинетики расходования мономера и накопления полимера
для обоих случаев. Кинетики изменения концентрации мономера регистрировались по
интегральной интенсивности сигнала, обозначенного 1, винильной группы мономера,
и сигнала, обозначенного 2 для полимера на рисунке 23. Как следует из рисунка
24, кинетика расходования мономера при полимеризации А Na без
нитроксильного радикала моноэкспоненциальная с характерным временем (1,5±0,1) ž102 с,
кинетика накопления полимера также описывается моноэкспонентой с таким же
характерным временем. Конверсия мономера при полимеризации АNa без
нитроксильного радикала составила 99%
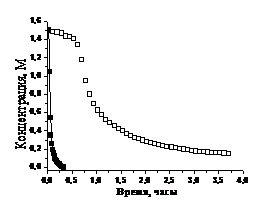
Кинетика расходования мономера и
накопления полимера при полимеризации А Na с
нитроксильным радикалом 6— имеет
характерное время спада (3,0±0,03) ž104 с. Конверсия достигает 90%. Как видим, в
случае полимеризации А Na в присутствии нитроксильного
радикала 6— кинетика
полимеризации сильно удлиняется, что свидетельствует об обратимом присоединении
нитроксильного радикала к растущей полимерной цепи.
На рисунке 20 представлена кинетика
полимеризации А Na в присутствии нитроксильного
радикала 6— в
координатах Фишера. Кинетика линейна до глубокой конверсии мономера, что
говорит о контролируемом режиме полимеризации. Начальный участок кинетики
расходования мономера соответствует по времени индукционному периоду, в течение
которого происходит образование алкоксиамина.
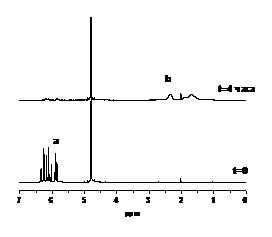

(а) (б)
Рисунок 23 1Н спектры ЯМР,
зарегистрированные при полимеризации А Na в
присутствии (а) и отсутствии (б) нитроксильного радикала 6—.

(а) (б)
Рисунок 24. Кинетика расходования
мономера (а) и накопления полимера (б) при полимеризации А Na в
отсутствии (¢) и
присутствии (o)
нитроксильного радикала 6—.

Рисунок 25. Кинетика образования
полимера при полимеризации А Na (o) в присутствии
нитроксильного радикала 6— и
образования сополимера (поли- А Na) -блок- (поли-СС Na) (š) при
реинициировании полимеризации при добавлении СС Na к реакционной
смеси.
Живой характер полимеризации А Na в
присутствии нитроксильного радикала 6— был показан методом реинициирования
полимеризации и приготовления блоксополимера (поли- А Na) -блок-
(поли-СС Na). После
того, как конверсия мономера при полимеризации А Na достигла
95%, к реакционной смеси было добавлено 0,7 М СС Na.
Реакционная смесь продувалась агроном в течение 15 минут для удаления
кислорода. Затем было продолжено наблюдение за кинетикой полимеризации методом
ЯМР. Полимеризация проводилась при 800С. Наблюдалось расходование мономера и
появление в спектре ЯМР сигналов, характерных для поли-СС Na. Кинетика
образования поли-СС Na аналогична кинетике образования
полимера при полимеризации СС Na, инициированной персульфатом
натрия в присутствии 6— (Рисунок
25). Таким образом, возможно использовать образовавшийся поли-А Na в качестве
макроинициатора для синтеза блок-сополимеров. Это подтверждает «живой» характер
образующегося полимера.
.3 Полимеризация акриламида в
присутствии 6—
.3.1 Методика полимеризации
Методика полимеризации гидрофильных
мономеров в целом аналогична описанной в п.3.3.1. Для проведения экспериментов
по полимеризации гидрофильных мономеров водный раствор 1,5 М мономера,
содержащий 6 мМ инициатора персульфата натрия и 10 мМ нитроксильного радикала 6— при рН = 7,
продувался аргоном в течение 15 минут для удаления кислорода. Реакция
проводилась при постоянной температуре. Через определенные промежутки времени
отбирались пробы реакционной смеси. Для определения конверсии мономера проводилась
запись 1Н ЯМР спектра раствора 50 мкл реакционной смеси в D2O. Анализ
полидисперсности полученных полимеров проводился методом ГПХ на жидкостном
хроматографе Agilent LC 1200 с
термостатированной колонкой PLaquagel-OH Mixed, 300х7,5
мм, 8 мм. Вода использовалась в качестве элюэнта, скорость потока 1 мл/мин.
Калибровка была произведена по узкодисперсным стандартам полиэтиленоксида (Polymerlabs).
4.3.2
Результаты
и обсуждение
Рисунок 26 Типичная хроматограмма
пробы полимерной смеси при полимеризации АА в присутствии 6— (-) и
отсутствии нитроксильного радикала (---).
Были проведены эксперименты по
полимеризации АА в присутствии и отсутствии нитроксильного радикала 6— при
температуре 800С.
На рисунке 26 (сплошная линия)
представлена типичная хроматограмма, полученная при анализе пробы объемом 50
мкл полимерной смеси, растворенной в 0,5 мл деионизованной воды. Как следует из
рисунка, образующийся полимер имеет сложное молекулярно-массовое распределение.
Были отдельно проанализированы характеристики более и менее высокомолекулярной
фракции, изучена их эволюция во времени (Рисунок 27). Стоит отметить, что обе
фракции имеют достаточно низкую полидисперсность. С увеличением конверсии
полидисперсность не превышает значение 1,5. По сравнению с полимеризацией АА,
проведенной в отсутствии нитроксильного радикала 6—,
наблюдается существенное уменьшение полидисперсности образующегося полимера
(Рисунок 25, пунктирная линия). Полидисперсность поли-АА, полученного в
отсутствии нитроксильного радикала составляет порядка 14. Как видно из Рисунка
27, при конверсии мономера 0,6 - 0,9 можно говорить о линейном росте
молекулярной массы образующегося полимера с конверсией. Отклонение от линейной
зависимости последней точки обусловлено, по-видимому, агрегацией частиц
полимера в растворе за счет увеличения его вязкости. Линейный рост молекулярной
массы образующегося полимера с конверсией говорит о реализации контролируемого
режима полимеризации. Однако для точного подтверждения контролируемого
характера полимеризации требуется более детальное изучение кинетики
полимеризации.

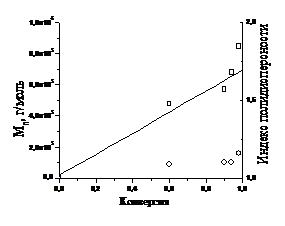
(1) (2)
Рисунок 27. Изменение
среднечисленной молекулярной массы (¢,o)
и индекса полидисперсности (,š) при
полимеризации АА в присутствии нитроксильного радикала 6— для
фракции, обладающей большей (1) и меньшей (2) молекулярной массой.
Таким образом, применение
нитроксильного радикала 6— в качестве
контролирующего агента полимеризации АА позволило получить полимер, обладающий
лучшими характеристиками, чем поли-АА полученный при полимеризации в отсутствии
нитроксильного радикала. Однако требуется дальнейшая оптимизация условий
полимеризации для получения мономодального молекулярно-массового распределения
образующегося полимера.
Выводы и результаты
1. Методом 1Н ЯМР спектроскопии в сочетании
с методом радикальной ловушки изучен термолиз ряда алкоксиаминов. На примере
алкоксиамина 4-нитрофенил
2-(2,2,6,6-тетраметилпиперидин-1-оксил)-2-метилпропионат показана применимость
метода радикальной ловушки для определения механизма реакции переноса атома
водорода при термолизе алкоксиаминов. Проведен анализ продуктов термолиза
алкоксиаминов на основе нитроксильных радикалов пирролидинового и
имидазолидинового ряда, а также нитроксильного радикала
2,2-дифенил-3-фенилиминоиндол-1-оксил, и метакрилатного алкильного фрагмента,
установлены побочные реакции, протекающие при термолизе, определены механизмы
реакции переноса атома водорода при термолизе алкоксиаминов.
2. Измерены кинетические параметры для
реакции термолиза для 6 алкоксиаминов;
. Проведена полимеризация
метилметакрилата, инициированная алкоксиамином
трет-бутил-2-(5-бутил-5-третбутил-2-циклогексил-4-фенил-имидазол-1-оксил)-2-метилпропионат,
определена полидисперсность полученного полимера, показано, что полимеризация
ММА, инициированная этим алкоксиамином, не протекает в контролируемом режиме за
счет термической нестабильности нитроксильного радикала, однако, происходит
снижение индекса полидисперсности образующегося полимера.
. Показано, что полимеризация
гидрофильных мономеров (4-стиролсульфонат натрия, акрил амид, акрилат натрия) в
присутствии нитроксильного радикала
4-(4,4-диэтил-1,2,5-триметилимидазолидин-2-оксил)-бутират натрия протекает в
контролируемом режиме до конверсии 85%. Изучено изменение молекулярной массы и
полидисперсности в ходе полимеризации акрил амида в присутствии нитроксильного
радикала, показано, что происходит существенное сужение полидисперсности
образующегося поли-акрил амида по сравнению с полимером, полученым при
полимеризации в отсутствии нитроксильного радикала. На примере полимеризации
акрилата натрия показано образование блок-сополимера (поли-акрилат
натрия)-блок-(поли- стиролсульфонат натрия), что говорит о «живом» характере
полимеризации.
Литература
5. Otsu,
T., Yoshida, M., Tazaki, T., Macromol. Rapid Commun., 1982, 3, 133.
6. US
Patent 4,581,429; Solomon, D. H.; Rizzardo, E; Cacioli, P. Chem. Abstr. 1985,
102, 221335q
. Szwarc
M. ‘Living’ polymers. Nature, 1956, 176, p 1168-9
. Georges,
M. K.; Veregin, R. P. N.; Kazmaier, P. M.; Hamer, G. K. Macromolecules 1993,
26, 2987-2988
. Bledzki,
A., Macromol. Chem., 1983, 184, 745
. Matyjaszewski,
K., Macromolecules, 1998, 31, 4710
. Reghunadhan,
N., Clouet, N., Ghaumont, G., J. Polym. Sci. Polym. Chem., 1989, 27, 1795
. Souaille,
M., Fischer, H., Macromolecules 2000, 33, 7378-7394
. Goto,
A., Fukuda, T., Prog. Polym. Sci., 2004, 29, 329-385
. Hanns
Fischer, Macromolecules 1997, 30, 5666-5672
. Kothe,
T., Marque, S., Martschke, R., Popov, M., Fischer, H., J. Chem. Soc., Perkin
Trans. 2, 1998, 1553-1559
. Fisher,
H., Journal of Polymer Science: Part A: Polymer Chemistry, 1999, 37, 1885-1901
. Fisher,
H., Criteria for livingness and control in Nitroxide-Mediated and Related
Radical polymerizations, ACS Symposium Series, 2003, 854, 10-23
. Beuermann,
S., Buback, M., Prog. Polym. Sci., 2002, 27, 191-254
. Benoit,
D., Chaplinski, V., Braslau, R., Hawker, C., J. Am. Chem. Soc. 1999, 121,
3904-3920
. Burguiere,
C., Dourges. M.-A., Charleux, B., Vairon, J.-P., Macromolecules 1999, 32,
3883-3890
. G.
S. Ananchenko, M. Souaille, H. Fischer, C. Le Mercier, P. Tordo, J. Polym.
Sci., Part A: Polym. Chem. 2002, 40, 3264.
. Charleux,
B., Nicolas, J., Guerret, O., Macromolecules, 2005, 38, 5485-5492
. Guillaneuf,
Y., Gigmes, D., Marque, R. A. S., Astolfi, P., Greci, L., Tordo, P., Bertin,
D., Macromolecules 2007, 40, 3108-3114
. Dalton,
P., Flynn, L. Shoichet, M., Biomaterials, 2002, 23, (18), 3843-3851
. Куренков
В.Ф., Соросовский образовательный журнал №5, 1997, с. 48-53
. Мелик-Нубаров
Н.С. «Взаимодействие водорастворимых полимеров с липидными мембранами»
автореферат, М. 2007
27. Nowakowska,
M., Karewicz, A., Kłos, M., Zapotoczny, S., Macromoleculs, 2003, 36, 4134-4139;
28. Couvreur,
L., Lefay, C., Belleney, J., Charleux, B., Guerret, O., Magnet, S.,
Macromolecules, 2003, 36, 8260-8267
. Zhao,
X., Lin, W., Song, N., Chen, X., Fan, X., Zhou, Q., J. Mater. Chem.,
2006,16,4619-4625
. Bian,
K., Cunningham, M., Macromolecules, 2005, 38, 695-701
. Delaittre,
G., Nicolas, J., Lefay, C., Save, M.,Charleux, B., Chem Commun, 2005, 614-616
. Huang,
W., Charleux, B., Chiarelli, R., Marx, L., Rassat, A., Vairon, J.-P., Macromol.
Chem. Phys. 2002, 203, 1715-1723
. Bon,
S. A. F., Chambard, G., German, A. L., Macromolecules 1999, 32, 8269-8276
. Fukuda,
T.; Goto, A. Macromol. Rapid Commun. 1997, 18, 683-688.
. Li,
L., Hamer, G., Georges, M., Macromolecules 2006, 39, 9201-9207
. Bertin,
D.; Gigmes, D.; Marque, S.; Tordo, P. e-Polym. 2003, 002,1-9.
. Беленький
Б. Г., Виленчик Л. 3. Хроматография полимеров. - M.: Химия, 1978.
. Guillaneuf,
Y.;Gigmes, D.; Marque, S. R. A.; Bertin, D.; Tordo, P. Macromol. Chem.Phys.
2006, 207, 1278-1288.
. Chauvin,
F., Dufils, P., Gigmes, D., Guillaneuf, Y., A. Marque, S.R.A., Tordo, P.,
Bertin, D., Macromolecules 2006, 39, 5238-5250
. Dmitrii
Zubenko, Igor Kirilyuk, Galina Roshchupkina, Irina Zhurko, Vladimir Reznikov,
Sylvain R. A. Marque, and Elena Bagryanskaya, Helvetica Chimica Acta, 2006, v.
89. p.2341
. Matyjaszewski,
K., Woodworth, B., Zhang, X., Gaynor, S., Metzner, Z., Macromolecules 1998, 31,
5955-5957
. Marque,
S.R.A.,Le Mercier, C., Tordo, P., Fischer, H., Macromolecules 2000, 33,
4403-4410
. Зубенко
Д.П. - Изучение ключевых реакций в псевдоживой полимеризации для ряда
имидазолидиновых и имидазолиновых нитроксильных радикалов - Диссертация на
соискание степени кандидата химических наук, Новосибирск, 2008, стр. 63
. Там
же, стр. 72