Особенности использования метода высокоэффективной жидкостной хроматографии в фармацевтике
Содержание
Введение
1. Определение компонентов препарата "БИЦИЛЛИН-3" методом
ВЭЖХ
2. ВЭЖХ в анализе препаратов, содержащих пропифеназон
3. Стандартизация препарата "Аданол"
Список использованной литературы
Введение
Хроматографический анализ является критерием однородности
вещества: если каким-либо хроматографическим способом анализируемое вещество не
разделилось, то его считают однородным (без примесей).
Принципиальным отличием хроматографических методов от других
физико-химических методов анализа является возможность разделения близких по
свойствам веществ. После разделения компоненты анализируемой смеси можно
идентифицировать (установить природу) и количественно определять (массу,
концентрацию) любыми химическими, физическими и физико-химическими методами.
Хроматография широко применяется в лабораториях и в
промышленности для качественного и количественного анализа многокомпонентных
систем, контроля производства, особенно в связи с автоматизацией многих
процессов, а также для препаративного (в том числе промышленного) выделения
индивидуальных веществ (например, благородных металлов), разделения редких и
рассеянных элементов.
В соответствии с агрегатным состоянием элюента различают
газовую (ГХ, GC) и жидкостную хроматографию (ВЭЖХ, HPLC).
Высокоэффективная жидкостная хроматография (ВЭЖХ, HPLC)
используется для анализа, разделения и очистки синтетических полимеров,
лекарственных препаратов, детергентов, белков, гормонов и др. биологически
важных соединений. Использование высокочувствительных детекторов позволяет
работать с очень малыми количествами веществ (10-11-10-9
г), что исключительно важно в биологических исследованиях.
Метод ВЭЖХ осуществляется на различных жидкостных
хроматографах. Современные жидкостные хроматографы предназначены для разделения
сложных смесей веществ на отдельные компоненты и проведения качественного и
количественного анализа компонентов разделяемой смеси.
высокоэффективная жидкостная хроматография пропифеназон
В связи с введением в практику фармацевтического производства
России GMP. повышается значимость использования современных унифицированных
методов анализа, как на предприятиях-производителях:, так и в системе
государственного контроля качества лекарственных средств. Базовым методом
анализа качества субстанций и готовых лекарственных средств в странах с
развитой фармацевтической промышленностью (США, Англия. Япония, страны ЕС)
является высокоэффективная жидкостная хроматография (ВЭЖХ). Данный метод по
своим характеристикам соответствует требованиям количественного анализа около 80-90%
препаратов.
К технике выполнения любых
хроматографических определений предъявляются некоторые общие требования. Прежде
всего, необходимо отметить те из них, которые вызывают у начинающих
специалистов больше всего вопросов.
. Кондиционирование помещения. В
помещении, где устанавливается жидкостной хроматограф, не должно быть резких
колебаний температуры.
Изменение температуры может привести к
изменению удерживания, эффективности, и даже селективности разделения.
В летнюю жару в некондиционированных помещениях
сильно затрудняется работа с нормально-фазовыми легкокипящими подвижными
фазами. В течение дня происходит их постепенное испарение, которое приводит к
изменению состава элюента.
При пониженных температурах появляются
проблемы в работе с элюентами, обогащенными водой и/или содержащими спирты.
Вязкость таких элюентов резко возрастает при понижении температуры, что
приводит к повышению давления в системе.
Влияние небольших колебаний температуры на
разделение можно устранить, термостатируя хроматографическую колонку, или всю
жидкостную систему (что возможно не для всех хроматографов).
. Качество электропитания. Большинство
современных хроматографов оснащено системами стабилизации питания, однако,
качество электропитания на месте также должно быть высоким. При недостаточно
хорошем электропитании любой запуск серии определений в автоматическом режиме
может окончиться неудачей из-за сбоя.
. Чистота растворителей. Для приготовления
подвижных фаз следует применять особо чистые растворители.
В общем случае, требования, предъявляемые
к чистоте подвижной фазы, зависят от способа детектирования, метода элюирования
(изократического или градиентного), чувствительности детектора к целевому
аналиту и его концентрации.
При применении УФ детектирования
требования к чистоте растворителей повышаются при переходе к коротковолновому
диапазону, менее 230-240 нм. Для изократического элюирования при УФ
детектировании на длинах волн, больших 220-240 нм, можно применять растворители
марки "ос. ч." и воду-дистиллят. Все реагенты, добавляемые в
подвижную фазу, также должны быть достаточно чистыми; кристаллические реагенты
перед применением бывает полезно перекристаллизовать.
Для градиентного элюирования необходимо
применять растворители марки "для жидкостной хроматографии"
("for liquid chromatography") и воду-бидистиллят. Особые требования в
методе градиентного элюирования (в обращенно-фазовой хроматографии)
предъявляются к чистоте водного буфера и воды в частности. Прежде всего это
связано с тем, что на начальной стадии элюирования адсорбент поглощает из
обогащенной водным буфером подвижной фазы загрязняющие компоненты, которые в
дальнейшем элюируются и проявляются на хроматограмме в виде "горбов",
"порогов" и отдельных пиков, сильно затрудняющих выделение полезных
сигналов аналитов.
Наиболее чистые растворители требуются для
проведения групповых определений следовых количеств веществ в режиме
градиентного элюирования.
Для выполнения определений в градиентном
режиме элюирования, а также прецизионных определений в изократическом режиме,
подвижная фаза должна применяться однократно, то есть элюат должен сбрасываться
или утилизироваться.
При изократическом элюировании, если нет
особенных проблем с чувствительностью, отработанный элюент можно применять
повторно. Система, в которой элюат после прохождения через детектор поступает
обратно в емкость с подвижной фазой, называется "системой с
рециклом". Особенно полезной такая система оказывается в случае проведения
большого числа рутинных изократических определений на стандартных колонках
(250х4.6, 150х4.6) при объемной скорости порядка 1 мл/мин. В этих случаях
система с рециклом обеспечивает экономию до 200-300 мл органического
растворителя в день. Такая экономная система позволяет применять для анализа
очень чистые, дорогие растворители. Вопрос экономии дорогих растворителей стоит
менее остро в случае применения микроколонок (80х2, 100х2), поскольку для
проведения разделения требуется на порядок меньший объем подвижной фазы.
. Дегазация растворителей. Растворители,
применяющиеся в хроматографии для приготовления подвижных фаз, обычно содержат
растворенный воздух. Особенно много воздуха содержит вода.
При работе на недегазированных элюентах
пузырьки воздуха попадают в различные узлы жидкостной системы: насос, колонку,
капилляры, детектор. При попадании воздуха в жидкостную систему на
хроматограмме появляются высокие периодические шумы, вызванные колебанием
давления в жидкостной системе. Это приводит к резкому уменьшению
чувствительности анализа.
Для удаления воздуха их элюента проводят
его дегазацию. Как правило, дегазируют только элюенты для обращенно-фазовых
разделений - поскольку водноорганические смеси содержат значительные количества
растворенного воздуха. Особенно тщательно дегазация должна осуществляться в
случае градиентного элюирования, а также при применении флуориметрического
детектирования.
В ходе градиентного элюирования в
обращенно-фазовом режиме происходит смешивание двух элюентов -
водноорганических смесей различного состава. Смешивание недегазированных
элюентов приводит к интенсивному выделению растворенного воздуха, что критично
для проведения определения в целом (на хроматограмме пузырьки воздуха
регистрируются в виде резких "выбросов" на нулевой линии).
Чувствительность флуориметрического
детектирования уменьшается при высоком содержании растворенного воздуха в
подвижной фазе (происходит тушение флуоресценции). Таким образом, при
применении флуориметрического детектирования дегазации элюента должно уделяться
особое внимание.
Существует три основных способа дегазации
подвижных фаз для жидкостной хроматографии.
а. Дегазация вакуумом - элюент выдерживают
в колбе Кляйзена под вакуумом водоструйного насоса в течение нескольких минут.
При проведении дегазации следует избегать кипения элюента.
б. Термическая дегазация применяется для
дегазации водноорганических элюентов с высокой долей воды. Подвижную фазу
помещают в колбу, которую не герметично закрывают пробкой, и оставляют в водной
бане при температуре около 50ºС. Через 10-15 минут
колбу закрывают пробкой герметично и охлаждают ее под струей воды до комнатной
температуры.
в. Дегазация ультразвуком. Подвижную фазу
несколько минут обрабатывают ультразвуком, а затем дают ей отстояться в течение
10-15 минут. Этот способ часто бывает недостаточно эффективен для дегазации
водноорганических элюентов.
Современные насосные системы для
жидкостной хроматографии комплектуются системами автоматической дегазации. Тем
не менее, при проведении градиентных анализов обе подвижные фазы лучше
дегазировать предварительно и "вручную", по одному из приведенных методов.
. Фильтрация подвижной фазы. Для
обеспечения бесперебойной работы насоса подвижную фазу желательно фильтровать
под вакуумом с применением мембранного фильтра.
. Промывка колонки и узлов жидкостной
системы. После работы с водноорганическими подвижными фазами, содержащими соли
и кислоты, всю жидкостную систему (включая колонку) следует промыть
дистиллированной водой с добавлением 5-10% органического растворителя. Такая
промывка делается для того, чтобы в нерабочее время дополнительно не
изнашивались узлы жидкостной системы хроматографа и сама неподвижная фаза.
Не проведение такой промывки приводит,
прежде всего, к тому, что после остановки насоса на его деталях и стенках
кюветы детектора из элюента осаждаются соли. Это, в свою очередь, приводит к
неустойчивой работе прибора в целом, а также преждевременному износу движущихся
частей насоса. Регулярное отсутствие промывки системы от содержащих соли и
кислоты элюентов может привести к сокращению времени жизни неподвижной фазы.
Добавка к промывочной воде некоторого
количества органического растворителя необходима для предотвращения
биологического загрязнения жидкостной системы.
. Переход на новую подвижную фазу, которая
не смешивается с предыдущей. Такой переход осуществляется через промежуточный
растворитель, неограниченно смешивающийся с обеими подвижными фазами - обычно,
через изопропанол или ацетон.
Для перехода с водного элюента на
неполярный элюент жидкостную систему следует промыть водой с добавкой
органического растворителя, затем снять хроматографическую колонку, промыть
систему изопропанолом (ацетоном), промыть систему неполярным элюентом,
установить новую колонку.
Для обратного перехода снимают
хроматографическую колонку, промывают жидкостную систему изопропанолом
(ацетоном), затем водным элюентом, затем ставят новую колонку.
При переходе от водного элюента к
неполярному следует заранее убедиться, что материал уплотнений насоса рассчитан
на работу с неполярными растворителями.
. Фильтрация пробы. Если анализируемая
проба содержит нерастворенную взвесь, то ее желательно отфильтровывать,
пропуская пробу через мембранный фильтр, соединенный со шприцем. К сожалению,
если пробы мало, менее миллилитра, то профильтровать ее таким способом
становится практически невозможно.
При регулярном анализе проб, содержащих
взвеси, входной фильтр на колонке (фрит) может засориться, что приведет прежде
всего к увеличению давления в системе. В этом случае входной фильтр лучше
заменить, а при отсутствии замены - промыть его в органическом растворителе с
обработкой ультразвуком в течение 10-15 минут.
Наиболее оптимальным решением проблемы
является применение ин-лайн фильтра перед колонкой. Ин-лайн фильтр содержит
сменный фрит - такой же, как и на колонке. Замена фрита на ин-лайн фильтре
является рутинной операцией, которая может проводиться достаточно часто.
. Применение предколонок. При регулярном
анализе "грязных" проб хроматографическая колонка достаточно быстро
загрязняется и теряет свою разделительную способность. Известной альтернативой
тщательной пробоподготовки в этом случае является применение предколонки,
защищающей основную колонку от загрязнения.
Иногда пробоподготовку целесообразно
вообще не проводить, а поставить в линию перед основной колонкой ин-лайн фильтр
и предколонку. Преимуществами этой схемы являются простота и экспрессность
анализов при меньшей затрате труда и реагентов.
. Консервация хроматографических колонок.
Перед достаточно длительным хранением хроматографические колонки промываются и
заполняются растворителем, вполне определенным для каждого типа неподвижной
фазы.
Так, хроматографические колонки для работы
в нормально-фазовых системах обычно заполняются высококипящим углеводородом,
например, изооктаном. Обращенные фазы промывают водой и заполняют
ацетонитрилом, или на небольшой скорости подачи - изопропанолом. Фазы,
предназначенные для работы с водными буферами, заполняются водой с небольшой
добавкой азида натрия (бактериостатика).
Инструкции по хранению колонки могут
указываться в ее паспорте.
. Хранение водных буферов. В случае
проведения рутинных определений бывает достаточно удобно готовить сразу большой
объем водного буфера для приготовления подвижной фазы. К сожалению, водный
буфер не может храниться больше нескольких дней, если не добавить в него азид
натрия, бактериостатик. Очень плохо хранятся подвижные фазы на основе
фосфатного буфера.
Иногда большой объем водного буфера
готовят для того, чтобы "повысить воспроизводимость анализа". Вообще
говоря, при таком подходе вопроизводимость анализа не повышается, а вот
проблемы с хранением буфера появляются неизбежные.
Вообще же говоря, ответ на вопрос -
готовить ли водный буфер на неделю или на один день? - определяется
исключительно принципом удобства.
. Регулярность проведения калибровки. Как
правило, калибровку по стандарту проводят каждый день, или же каждый раз при
приготовлении нового элюента.
Калибровка проводится при достижении
стационарного состояния хроматографической системы; считываемыми параметрами
являются время удерживания пика стандарта, его площадь (при
спектрофотометрическом детектировании - на опорной длине волны), спектральные
отношения (при применении сканирующего или диодно-матричного
спектрофотометрического детектора).
В начале работы стандарт может быть
проанализирован дважды - для подтверждения воспроизводимости времени
удерживания.
1.
Определение компонентов препарата "БИЦИЛЛИН-3" методом ВЭЖХ
Бициллин-3 является пенициллином пролонгированного действия и
представляет собой смесь натриевой, новокаиновой и бензатиновой солей
бензилпенициллина (БП). По действующей ВФС 42-3034-98 определение БП в
препарате проводят с помощью ВЭЖХ, новокаин определяют спектрофотометрически, а
бензатин (N,N1-дибензилэтилендиамин) экстрагируют эфиром из водного раствора,
насыщенного хлоридом натрия. После выпаривания эфира бензатин определяют
титрованием хлорной кислотой.
В Европейской Фармакопее содержание БП и бензатина в
бензатиновой соли БП определяют с помощью градиентной ВЭЖХ в смеси метанола с
раствором фосфата натрия при рН 3,5.
Цель работы - разработка метода ВЭЖХ в изократическом режиме
для определения компонентов в бициллине-3.
Экспериментальная часть
Использовали бициллин-3 производства АКО "Синтез"
(Курган). Исследование проводили на хроматографе фирмы "Waters" (США)
с насосом модели 510, УФ-детектором модели 481 и инжектором модели 7125 (Rheodyne)
с дозирующей петлей вместимостью 50 мкл. Для детектирования использовали длину
волны 214 нм, при которой хорошо детектируются все анализируемые соединения.
Регистрацию хроматограмм и расчет площадей пиков и основных параметров
удерживания проводили с помощью персонального компьютера с аналого-цифровым
преобразователем и программой "Мультихром".
Был изучен обращеннофазный вариант метода ВЭЖХ на колонке
"Luna C18 (2)" размером 250 х 4,6 мм фирмы "Phenomenex"
(США), поскольку колонка зарекомендовала себя ранее как относительно дешевая с
улучшенной симметрией выхода пиков органических аминов. С этой же целью в
качестве подвижной фазы использовали смесь ацетонитрила с буферным раствором,
содержащим в качестве одного из компонетов триэтиламин, имеющим pH 5,0.
Исходный раствор для приготовления подвижной фазы - 2,5 М
раствор фосфорной кислоты, который титровали триэтиламином до рН 5,0. Буферный
раствор для ВЭЖХ получали разбавлением исходного раствора водой в 10 раз.750 мл
полученного буферного раствора смешивали с 250 мл ацетонитрила. При этом
значение кажущегося рН подвижной фазы повышалось до 5,7. Скорость подвижной
фазы 1 мл/мин. Хроматографирование проводили при комнатной температуре. Время
анализа 20 мин.
Поскольку входящие в состав препарата компоненты различаются
по кислотно-основным свойствам - БП является кислотой, а новокаин и бензатин -
основаниями, при увеличении рН их времена удерживания в интервале рН, где их
ионизация меняется, сдвигаются в разные стороны. Поэтому путем изменения рН
легко подобрать удобное удерживание анализируемых компонентов. Однако
увеличение рН приводит к заметному ухудшению формы пика бензатина, а уменьшение
- к недостаточному разрешению новокаина и продуктов гидролиза БП. Разделение
компонентов бициллина-3 при указанных выше условиях представлено на рисунке.
Времена удерживания новокаина, бензатина и БП составляли 4,2, 11,6 и 14,8 мин
соответственно.
Существенным является выход пика новокаина между 2 пиками,
представляющими собой продукты гидролиза БП. В связи с этим для лучшего
разделения компонентов рекомендуется добавлять в подвижную фазу небольшие
количества 2,5 М растворов фосфорной кислоты или триэтиламина и контролировать
разделение хроматографированием смеси новокаина и БП, раствор которого хранился
около суток при комнатной температуре.
Для количественного определения 20-25 мг бициллина-3 вносили
в мерную колбу вместимостью 100 мл и растворяли в 20% водном растворе
ацетонитрила. Использование для растворения метанола или его растворов вело к
частичному метилированию БП. Увеличение концентрации ацетонитрила приводило к
уширению пика новокаина. Верхний предел концентрации лекарственного вещества
ограничен его растворимостью. Калибровочные графики для БП и бензатина получали
с использованием натриевой соли БП и диацетата бензатина после соответствующего
пересчета. Калибровочный график для БП линеен в области 0,1-0,5 мг/мл, для
бензатина и новокаина - в области 0,01-0,05 мг/мл. Результаты определения
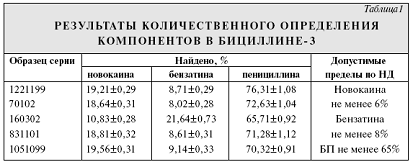
компонентов в 5 сериях препарата представлены в табл.1, где каждое значение
представляет собой среднее из 5 определений. Относительное среднее квадратичное
отклонение составило 1,6% для новокаина, 3,4% для бензатина и 1,4% для БП.
Из табл.1 следует, что результаты количественного определения
с помощью ВЭЖХ укладываются в допустимые пределы, регламентируемые НД.
Для подтверждения правильности предложенной методики были
проанализированы компоненты бициллина-3 в модельных смесях, приготовленных
смешением натриевой соли БП, бензатина диацетата и новокаина. Результаты
приведены в табл.2. Результаты пересчитаны на исходные компоненты.
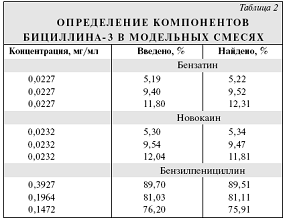
Каждое значение в графе "Найдено" табл.2 - средний
результат 3 определений. Средняя величина относительного отклонения составила
2,2 % для бензатина, 0,9 % для новокаина и 0,8 % для БП, что коррелирует с
относительными средними стандартными отклонениями, найденными при анализе
компонентов в реальных пробах. Для бензатина разброс результатов несколько
выше, чем для остальных компонентов, что объясняется низкой высотой и
неправильной формой пика и соответственно большей ошибкой интегрирования.
Другой причиной относительно больших ошибок при определении бензатина может
явиться эффект памяти инжектора при анализе сильно адсорбирующихся веществ.
Однако и такой разброс, несколько превышающий принятый для анализов с помощью
метода ВЭЖХ, вполне допустим для определения бензатина.
Выводы
1. Разработана методика обнаружения и количественного
определения компонентов в препарате "Бициллин-3".
. Методика проверена на ряде серий препарата и подтверждена
анализом модельных смесей известного состава.
2. ВЭЖХ в
анализе препаратов, содержащих пропифеназон
Пропифеназон (4-изопропил-2,3-диметил-1-фенил-3-пиразолин-5-он;
изопропилантипирин) относится к ненаркотическим анальгетикам пиразолонового
ряда и входит в состав комбинированных безрецептурных препаратов. В настоящее
время в лечебной практике широко используются таблетки каффетин (состав:
пропифеназона-0,21 г, парацетамола-0,25 г, кофеина-0,05 г, кодеина фосфата-0,01
г) и саридон (парацетамола-0,25 г, пропифеназона-0,15 г, кофеина-0,05 г).
Согласно существующей нормативной документации для подтверждения подлинности
таблеток каффетина используется хроматография в тонком слое сорбента.
Количественное определение предложено проводить в отдельных навесках препарата
разными для каждого компонента методами: с помощью спектрофотометрии,
титриметрии, сочетание тонкослойной хроматографии и спектрофотометрии. В
нормативной документации на саридон парацетамол, кофеин и пропифеназон
определяют методом ВЭЖХ на колонках длиной 12,5 см с сорбентом Merck
Lichrospher C18 и подвижной фазой состава: метанол-0,01 М фосфорная кислота в
соотношении 30: 70.
Цель настоящей работы - разработка методик обнаружения и
количественного определения компонентов таблеток каффетина и саридона с помощью
ВЭЖХ.
В работе использовали таблетки каффетина производства
"Алкалоид Скопье" (Республика Македония), саридона производства
"Лаборатория Рош Николас С. А.", Гайярд (Франция) и субстанции
компонентов, входящих в их состав. Исследование проводили на отечественном
микроколоночном жидкостном хроматографе "Милихром-4" с
УФ-спектрофотометрическим детектором, колонкой длиной 8 см с обращенно-фазовым
сорбентом Сепарон-С18 в качестве неподвижной фазы. Полярный характер
анализируемых соединений, их хорошая растворимость в воде и ацетонитриле
обусловили выбор водно-ацетонитрильных смесей в разных соотношениях в качестве
подвижной фазы. Были испытаны подвижные фазы ацетонитрил - вода в соотношениях
9: 1; 7: 3; 6: 4; 8: 2 и ацетонитрил-вода-диэтиламин (3: 2: 0,2). Избегали
использовать подвижные фазы с объемной долей органического растворителя более
80%, чтобы исключить нормально-фазовые взаимодействия, затрудняющие дальнейшее
регулирование состава подвижной фазы. Объемная доля растворителя менее 5%
приводит к функциональной неустойчивости подвижной фазы и невоспроизводимости
времен удерживания. Введение в состав подвижной фазы диэтиламина в качестве
модификатора позволило добиться разделения всех 4 компонентов таблеток
каффетин. Известно, что на поверхности октадецилсиликагеля находится
значительное количество остаточных силанольных групп, способных к ионообменному
взаимодействию. Диэтиламин исключает из хроматографического процесса
силанольные группы, улучшает форму пиков, сокращает время анализа и регулирует
рН на поверхности силикагеля. Измерение проводили в следующих условиях: масштаб
регистрации 2,0, время удерживания 0,8 с, скорость расхода элюента 50 мкл/мин,
объем вводимой пробы 3 мкл. Детекцию пиков осуществляли при 2 длинах волн - 238
и 276 нм.
Идентификацию проводили по параметрам удерживания, которые
определяли предварительно на стандартных растворах исследуемых веществ.
Компоненты таблеток каффетина разделены при использовании
подвижной фазы ацетонитрил-вода-диэтиламин (3: 2,2: 0,2). Время удерживания
парацетамола составило 3,08 мин, пропифеназона - 5,73 мин, кофеина - 4,0 мин,
кодеина - 4,67 мин.
Компоненты саридона можно также разделить, используя
подвижную фазу ацетонитрил-вода (8: 2). Время удерживания для парацетамола -
3,9 мин, пропифеназона - 5,11 мин, кофеина - 4,44 мин.
Для количественного определения применяли метод абсолютной
калибровки. Прямая пропорциональная зависимость концентрации вещества от высоты
пика наблюдалась для парацетамола в диапазоне 50-200 мкг/мл, для пропифеназона
- 25-128 мкг/мл, кофеина - 20-50 мкг/мл, кодеина - 59-234 мкг/мл.
Метод ВЭЖХ имеет некоторые ограничения в анализе сложных
смесей. При одновременном присутствии в смеси веществ в макро - и
микроколичествах возникает перегрузка колонки, что оказывает влияние на
качество разделения и форму выходящих пиков. В каффетине содержание кодеина
фосфата по отношению к парацетамолу и пропифеназону в 21-25 раз меньше, поэтому
рекомендуется жидкостная экстракция для отделения кодеина от остальных
компонентов таблеток. Предварительно мы установили, что парацетамол,
пропифеназон и кофеин извлекаются при однократной экстракции этилацетатом из
водных растворов при рН 2,0 в количестве соответственно 87,43, 87,29 и 87,84%,
а кодеин полностью остается в водном растворе и для его извлечения и
концентрирования необходимо использовать хлороформ при рН 9,0-10,0.

Методика количественного определения
парацетамола, пропифеназона и кофеина в таблетках саридона и каффетина.20 таблеток растирают в
ступке в мелкий однородный порошок, отвешивают около 0,01 г (точная навеска)
порошка растертых таблеток и помещают в мерную колбу вместимостью 25 мл,
прибавляют 10 мл ацетонитрила и тщательно перемешивают.
Содержимое колбы доводят до метки ацетонитрилом, перемешивают
и фильтруют. Раствор вводят в колонку хроматографа в объеме 3,0 мкл. Содержание
парацетамола, пропифеназона и кофеина определяют методом абсолютной калибровки.
Результаты определения приведены в табл.1 и 2, из которых видно, что полученные
данные укладываются в допустимые пределы содержания по нормативной документации
(НД).

Методика количественного определения кодеина
фосфата в таблетках каффетина. Около 0,2 г растертых таблеток (точная навеска)
растворяют в 20 мл воды, хорошо перемешивают до получения однородного раствора,
фильтруют через фильтр для мелких и самых мелких осадков, фильтр промывают 10
мл очищенной воды. Раствор подкисляют 10% раствором серной кислоты до рН 2,0.
Экстрагируют трижды этилацетатом порциями по 10 мл. Экстракты отбрасывают. К
водному раствору добавляют 25% раствор аммиака до рН 9,0-10,0. Экстрагируют
трижды хлороформом порциями по 10 мл. Объединенные хлороформные вытяжки
помещают в фарфоровые чашки и испаряют при комнатной температуре. Сухие остатки
растворяют в ацетонитриле, переносят в мерную колбу вместимостью 25 мл и
доводят до метки тем же растворителем.3 мкл полученного раствора вводят в
колонку хроматографа и определяют кодеин в описанных условиях. Результаты
определения приведены в табл.3.

Для оценки точности предлагаемых методик и проверки
воспроизводимости результатов были приготовлены и исследованы модельные смеси.
Данные на примере таблеток саридона приведены в табл.4. Как видно из табл.4,
относительная погрешность определения не превышает для парацетамола ±1, 19%,
пропифеназона ±1,16%, кофеина ±1,63%.
Методика количественного определения компонентов
таблеток саридона в модельных смесях. Отвешивают точные навески парацетамола (около
0,08 г), пропифеназона (около 0,05 г) и кофеина (около 0,016 г), переносят в
мерную колбу вместимостью 50 мл, растворяют в небольшом объеме ацетонитрила и
доводят до метки тем же растворителем. Отбирают аликвоту 2,5 мл и переносят в
мерную колбу вместимостью 25 мл, объем до метки доводят тем же растворителем,
перемешивают и фильтруют. Раствор вводят в колонку хроматографа в объеме 3 мкл.

Выводы
1. Разработана методика обнаружения компонентов таблеток
каффетина и саридона с помощью ВЭЖХ.
Время удерживания для парацетамола составило 3,08 мин, для
пропифеназона - 5,73 мин, кофеина - 4 мин и кодеина - 4,67 мин.
. Предложен метод ВЭЖХ для количественного определения
компонентов таблеток каффетина и саридона.
Относительная ошибка определения составила для парацетамола
±1, 19-1,21%, пропифеназона ±1,16-1,71%, кофеина ±1,22-1,63% и для кодеина
±2,95%.
.
Стандартизация препарата "Аданол"
Фармацевтической фирмой "Полисан" разработан ряд
комплексных лекарственных препаратов метаболического действия, стимулирующих
обменные процессы головного мозга, в том числе "Цитофлавин"
(инъекции, таблетки) и "Аданол". "Аданол" обладает
выраженными антигипоксическими и противоишемическими свойствами и является
перспективным лекарственным средством для лечения больных с последствиями
инсульта. Он представляет собой таблетированную лекарственную форму, покрытую
кишечно-растворимой оболочкой.
В его состав входят янтарная кислота (ЯК), пирацетам (Пц),
рибоксин (Рб), никотинамид (НА), пиридоксина гидрохлорид (ПГ), рибофлавина
мононуклеотид (РФ).
Цель работы - разработка метода качественного и
количественного определения ЯК в сложных многокомпонентных смесях на примере
препарата "Аданол".
В работе использовали жидкостный хроматограф высокого
давления фирмы "Shimadzu" (Япония) с УФ-детектором и колонкой
Hypersil BDS C18 фирмы "Supelco Inc." зернением 5 мкм, длиной 250 мм
с внутренним диаметром 4,6 мм. Подвижная фаза - водно-органическая фаза на
основе фосфатного буфера (рН 2,6-7,0). Длина волны детектирования 206 нм. Режим
анализа изократический, скорость элюирования 500 мкл/мин; объем проб 20 мкл.
УФ-спектры сняты на спектрофотометре UV mini-1240 фирмы "Shimadzu".
Для количественного определения большинства субстанций,
входящих в состав препарата, предложены спектрофотометрические методы анализа.
Однако сравнение спектральных характеристик компонентов препарата показало, что
области поглощения ЯК, ПГ, НА, Рб и Пц в УФ-зоне перекрывают друг друга
(рис.1).
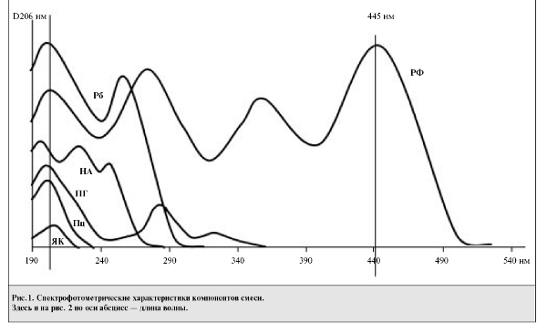
В связи с этим содержание их в смеси нельзя определять
методом прямой спектрофотометрии и в таком случае целесообразно использовать
метод ВЭЖХ. Только РФ имеет специфическую область поглощения более 350 нм (λmax=373, Е1% 1см=202 и λmax=445 нм, Е1% 1см=243),
поэтому для него предложен качественный и количественный анализ
спектрофотометрическим методом.
На основе полученных данных выбрана оптимальная рабочая длина
волны для анализа 5 субстанций, составляющая λопт=206 нм (см. рис.1).
Это значение является максимумом УФ-спектра ЯК, которая обладает наименьшим
удельным поглощением (λmax=206 нм Е1% 1см= 5,8) по
сравнению с остальными компонентами смеси (см. таблицу).
Поскольку все вещества, входящие в препарат, ионогенны, для
их анализа целесообразно применять обращенно-фазную хроматографию с
использованием неполярной неподвижной и полярной подвижной фаз. При
обращенно-фазной хроматографии ионогенных соединений значение водородного
показателя - один из факторов, который существенно влияет на эффективность
разделения индивидуальных веществ. При работе с современными обращенно-фазными
сорбентами в составе подвижной фазы обычно используют буферные растворы рН от
2,0 до 8,0. Так как выбранное оптимальное значение рабочей длины волны равно
206 нм, то лучше всего применять фосфатный буфер, потому что он не имеет
положения в УФ-диапазоне длин волн более 200 нм.
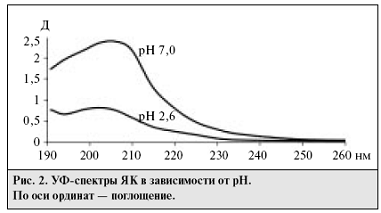
Для изучения поведения ЯК при различных значениях рН были
сняты ее спектры в фосфатных буферных растворах рН 7,0 и 2,6 (рис.2). При рН
2,6 наблюдается гипохромный эффект молекулярной формы ЯК - поглощение снижается
в 3 раза по сравнению со спектром при рН 7,0 (при этом значении рН диссоциация
ЯК полностью подавлена). С учетом этого оптимальным значением рН подвижной фазы
является 7,0. Далее было изучено влияние состава и рН подвижной фазы на
эффективность разделения компонентов препарата. При рН 2,6 не произошло полного
разделения смеси на индивидуальные пики - Пц, На и ПГ не делятся и выходят
первыми одним пиком, за ними следуют ЯК и Рб в виде индивидуальных пиков. При
рН 5,5 также не было полного разделения компонентов. При рН 7,0 произошло
полное разделение компонентов смеси. Все вещества выходили в виде отдельных пиков
в следующей последовательности: ЯК, Пц, ПГ, НА и Рб. Однако процесс разделения
длителен - 45-50 мин.
Для обеспечения большей элюирующей силы подвижной фазы и
ускорения процесса разделения необходимо ввести в ее состав менее полярный
органический растворитель. Из раство рителей, наиболее часто употребляемых в
качестве элюирующих агентов в ВЭЖХ, по пределу прозрачности в УФ-свете при
выбранной рабочей длине волны (λопт = 206 нм) мы могли
применить ацетонитрил и метанол, у которых пределы прозрачности равны 195 и 205
нм соответственно.
При введении в состав подвижной фазы 2% метанола время
процесса сократилось, однако пик НА имел высокую асимметрию и пик Рб
накладывался на хвост пика НА. После снижения концентрации метанола до 1% пики
Рб и НА не перекрывались, однако асимметрия пика НА увеличилась. С целью ее
снижения в подвижную фазу, содержащую 1% метанола, вводили ацетонитрил.
В результате экспериментов была подобрана подвижная фаза -
водно-органическая фаза, состоящая из фосфатного буфера рН 7,0, содержащего 1%
метанола и 0,5% ацетонитрила, которая обеспечила эффективное разделение всех 5
компонентов (рис.3). Общее время хроматографирования в этой системе составило
35 мин, а времена удерживания компонентов приблизительно (в мин): у ЯК - 5,3,
Пц - 15, ПГ - 19,3, НА - 26, Рб - 31.
Количественное определение проведено методом внешнего
стандарта с использованием растворов стандартных образцов индивидуальных
компонентов.
Метрологические характеристики предлагаемой методики
количественного определения изучены на модельных смесях в 5 повторностях и
представлены в таблице.
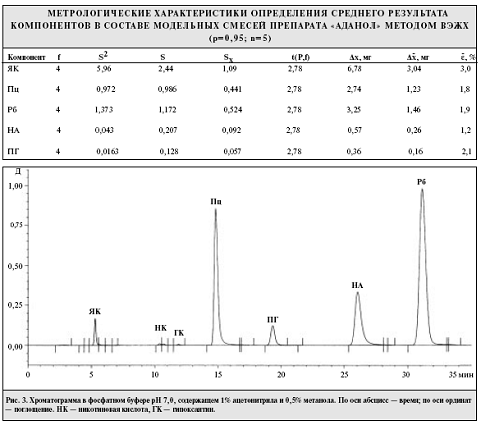
Как следует из таблицы, с помощью разработанной методики в
препарате "Аданол" можно определять все 5 компонентов с относительной
ошибкой не более 3% с доверительной вероятностью 95%.
Помимо пиков основных компонентов препарата, на
хроматограмме, полученной в вышеописанных условиях, идентифицированы пики
гипоксантина и никотиновой кислоты, присутствующих в исходных субстанциях, а
также образующихся в процессе гидролиза из Рб и НА соответственно. Таким
образом, разработанная методика позволяет качественно и количественно
определить эти посторонние примеси в препарате.
Выводы
1. Определены оптимальные условия количественного анализа
янтарной кислоты с применением метода ВЭЖХ в многокомпонентной смеси.
. Разработана методика качественного и количественного
анализа компонентов препарата "Аданол", включая примеси, с
относительной ошибкой определения не более 3%.
Список
использованной литературы
1. М.А.
Казьмин, А.В. Михалев, А.П. Арзамасцев "Определение компонентов препарата
"БИЦИЛЛИН-3" методом ВЭЖХ" // Фармация - №5 - 2002 - с.5-6.
2. Т.Х.
Вергейчик, Н.С. Онегова "ВЭЖХ в анализе препаратов, содержащих
пропифеназон" // Фармация - №6 - 2002 - с.13-16.
. А.Ю.
Петров, С.А. Дмитриченко, А.Л. Коваленко, Л.Е. Алексеева "Стандартизация
препарата "Аданол" // Фармация - №5 - 2002 - с.11-13.
Дополнительная
. Барам
Г.И., Федорова Г.А. // Применение хроматографии в пищевой, микробиологической и
медицинской промышленности: Мат. Всес. Конф. - Геленджик, 1990 г. - С.43-44.
2. Кричковская
Л.В., Черненькая Л.А. // Применение хроматографии в пищевой, микробиологической
и медицинской промышленности: Мат. Всес. Конф. - Геленджик, 8-12 октября 1990
г., М., 1990. - С.49.
. Хроматография:
Практическое приложение метода: В 2-х частях. Ч.2 - М.: Мир, 1986. - 422 с.