Лабораторная диагностика различных вариантов миелодиспластического синдрома
Министерство
Здравоохранения Республики Беларусь
Учреждение
образования
Гомельский
государственный медицинский университет
Медико-диагностический
факультет
Кафедра
внутренних болезней №1 с курсом гематологии
Допущена к защите:
Заведующий кафедрой
Мистюкевич И.И.
Лабораторная
диагностика различных вариантов миелодиспластического синдрома
Дипломная
работа
Исполнитель:
Студентка группы Д-604 Лебедько О.С.
Научные руководители:
К.м.н., доцент
Ходулева С.А.
Заведующий отделением
клинико-диагностической
лаборатории
ГУ,,РНПЦРМ и ЭЧ,, Прокопович А.С.
Рецензент:
Ассистент кафедры внутренних
болезней №1с курсом гематологии, Суворов
Д.И.
Гомель
2012
Реферат
Дипломная работа содержит 57страниц, 7 таблиц,
14 рисунков, 25 источников.
Ключевые слова: миелодиспластический синдром,
острый миелобластный лейкоз, рефрактерная анемия, рефрактерная анемия с
кольцевыми сидеробластами, рефрактерная анемия с избытком бластов, рефрактерная
анемия с избытком бластов в трансформации, хронический миеломоно-цитарный
лейкоз, предраспологающие факторы, дисгранулопоэз, дизэритопоэз,
дисмегакариоцитопоэз, костный мозг, периферическая кровь.
Объект исследования: пациенты с различными
вариантами миелодиспластического синдрома.
Предмет исследования: результаты лабораторного
исследования периферической крови и костного мозга.
Методы исследования: морфологические
исследования крови и костного мозга, иммунофенотипирование, цитогенетические
исследования, определение ферритина.
Цель исследования: определить ведущие
клинико-лабораторные характеристики различных вариантов миелодиспластического
синдрома с учетом морфологических, цитологических, цитогенетических
исследований.
Задачи исследования:
.Провести обзор литературы по вопросам
этиопатогенеза, распространенности, клиники МДС.
.Изучить современные методы диагностики
различных вариантов МДС.
.Оценить клинические симптомы и изменения
лабораторных показателей при различных вариантах МДС.
.Провести анализ МДС по Гомельской области.
.Разработать диагностический алгоритм различных
вариантов МДС на основании полученных данных.
Актуальность: МДС - это заболевание, носящее
клоновый характер и возникающее в результате мутации полипотентной стволовой
клетки, что обусловливает высокую степень риска трансформации в острый лейкоз.
Из-за разнообразия клинических проявлений, трудностей в диагностике и лечении,
МДС является одной из сложных нозологических форм в онкогематологии.
Своевременная, достоверная диагностика конкретного варианта МДС позволит
выбрать оптимальную специфическую терапию, улучшить качество жизни пациента и
прогноз заболевания в целом.
Содержание
Перечень условных обозначений
Введение
Глава 1. Обзор литературы
.1 Эпидемиология
миелодиспластического синдрома
.2 Классификация
миелодиспластического синдрома
.3 Этиология и патогенез миелодиспластического
синдрома
.4 Клиническая картина
миелодиспластического синдрома
.5 Диагностика миелодиспластического
синдрома
.5.1 Морфологические исследования
крови и костного мозга
.5.2 Иммунофенотипирование
.5.3 Цитогенетическое исследование
.5.4 Определение ферритина
Глава 2. Материалы и методы
.1 Материалы исследования
.2 Методы исследования
Глава 3. Результаты собственных
исследований
3.1 Общая характеристика пациентов с
МДС
3.2 Лабораторная характеристика
отдельных вариантов МДС
Выводы
Список использованной литературы
Перечень условных обозначений
МДС - миелодиспластический синдром
ОМЛ - острый миелобластный лейкоз
КМ - костным мозгом
ПК - периферической крови
ФАБ - франко-американо-британская классификация
RA -
рефрактерная анемия
RARS -
рефрактерная анемия с кольцевидными сидеробластами
RAEB -
рефрактерная анемия с избытком бластов
RAEBt -
рефрактерная анемия с избытком бластов, трансформирующихся в острый лейкоз
CMML -
хронический миеломоноцитарный лейкоз
MIC - группа
изучения морфологии, иммунологии, цитогенетики
ВОЗ - Всемирной организации здравоохранения
IPSS -
Международную Числовую Систему прогноза
ДНК - дезоксирибонуклеиновая кислота
IL-3 -
интерлейкин-3
G-CSF
- гранулоцитарный колониестимулирующий фактор
GM-CSF
- гранулоцитарно-макрофагальный колониестимулирующий фактор
TNF-£ - фактора
некроза опухоли
TGF£ - трансформирующего
ростового фактора
IL-1b
- интерлейкина-1b
M-CSF
- макрофагальный колониестимулирующий фактор
JMML -
ювенильный миеломоноцитарный лейкоз
NF -
нейрофиброматозом- Т-клеточного рецептора- патологическая локализация незрелых
клеток
МАТ - моноклональные антитела
FITC -
флюоресцеинизотиоционат- флюоресцентная гибридизация in
situ
СФ - сывороточный ферритинферритин-связывающие
лимфоциты
Введение
Миелодиспластический синдром (МДС) - это
гетерогенная группа приобретенных клональных заболеваний, развивающихся из
плюрипотентной гемопоэтической стволовой клетки с вовлечением в патологический
процесс клеток миело- и В-лимфопоэза.
МДС - гемопоэтическое клоновое заболевание,
основным проявлением которого является неэффективное кроветворение и риск
трансформации в острый миелобластный лейкоз (ОМЛ). Под неэффективным гемопоэзом
подразумевается несоответствие между нормо- или гиперклеточным костным мозгом
(КМ), с одной стороны, и одно-, двух- или трехростковой цитопенией в
периферической крови (ПК) - с другой.
Вероятность прогрессирования МДС в ОМЛ зависит
от целого ряда факторов и может происходить в сроки от 5 месяцев до 6 лет. То
есть течение МДС варьирует от индолентных вариантов, со стабильно сохраняющимся
в течение длительного времени уровнем бластов в КМ, до быстро прогрессирующих
форм. Это позволяет рассматривать МДС как предлейкозное состояние, при котором
опухолевый клон может и не трансформироваться в явный ОМЛ. Хотя в последнем случае
возможно, что больной не доживает до трансформации в ОМЛ, а умирает от
осложнений цитопении.
МДС может возникать как de novo, так называемый
первичный МДС, или развиваться после применения мутагенных агентов (химио-
и/или лучевая терапия) по поводу другого, обычно опухолевого, заболевания.
Второй вариант носит название вторичный МДС
В последние годы интерес к этой патологии
значительно возрос, что связано с тенденцией к росту ее частоты, быстрым
прогрессированием, высокой летальностью и отсутствием до настоящего времени
эффективных схем терапии. Гетерогенность природы МДС обуславливает сложности в
его диагностике.
На сегодняшний день актуальным является
разработка унифицированных диагностических критериев конкретного варианта МДС с
использованием морфологических, цитологических и цитогенетических методов.
Своевременная, достоверная диагностика позволит выбрать оптимальную
специфическую терапию, улучшить качество жизни пациента и прогноз заболевания в
целом.
Глава 1. Обзор литературы
.1 Эпидемиология миелодиспластического синдрома
Средняя частота заболеваемости МДС во всем мире
составляет 4,9 на 100000 человек в год. Средний возраст больных составляет 75,4
года[1,4,8]. Распределение МДС по возрастным группам (%)[4,6]:
среди лиц моложе 20 лет - 0,3%
в группе 20-30 лет - 0,6 %
- 40 лет - 1,6%
-50 лет - 2,2 %
-60 лет -8,5%
-70 лет - 25,9%
- 80 лет - 41,6%
старше 80 лет - 18,7%.
У мужчин заболеваемость встречается в 1,4 раза
чаще, чем у женщин (преимущественное увеличение заболеваемости МДС у мужчин в
возрасте старше 80 лет)[1,18].
По данным литературы, начиная с 1996 года
наметилась устойчивая тенденция к росту первичных миелодиспластических
синдромов у взрослого населения РБ, в частности в Могилевской, Гомельской и
Брестской областях частота этого заболевания повысилась в 1,5 - 2 раза[6,18].
По отдельным вариантам МДС распределение
следующее (%)[6,8]: рефрактерная анемия - 49,3%
рефрактерная анемия с кольцевыми сидеробластами
- 8,7%
рефрактерная анемия с избытком бластов - 26,5%
рефрактерная анемия с избытком бластов в
трансформации - 7,2% хронический миеломоноцитарный лейкоз - 8,1%.
.2 Классификация миелодиспластического синдрома
В 1982 г. франко-американо-британской (ФАБ)
рабочей группой была предложена классификация МДС, позволяющая по результатам
исследования КМ и ПК разграничивать МДС от ОМЛ и выделять отдельные варианты
МДС (табл.1). Поэтому верификация диагноза МДС основана прежде всего на
выявлении морфологических признаков дисгранулопоэза, дизэритопоэза,
дисмегакариоцитопоэза и количестве бластов в КМ и ПК. Другие особенности, такие
как кольцевидные сидеробласты, палочки Ауэра и моноцитоз являются
вспомогательными критериями для дифференцировки вариантов. Принципиальным для
диагностики МДС по ФАБ-классификации является обнаружение диспластических
изменений, по крайней мере, в клетках 2 из 3 гемопоэтических линиях [4,8,9].
Выделяют следующие варианты МДС [1,18]:
.Рефрактерная анемия (RA)
- характеризуется ретикулоцитопенией с количеством бластов в КМ 5% и менее.
Прогноз благоприятен.
.Рефрактерная анемия с кольцевидными
сидеробластами (RARS)
- подразумевает обнаружение кольцевидных сидеробластов более чем в 15% от
общего числа эритробластов. Прогноз благоприятен.
.Рефрактерная анемия с избытком бластов (RAEB)
- характеризуется наличием бластов в КМ от 5 до 20% и ПК от 1 до 5%. Высока
трансформация в лейкоз. Прогноз неблагоприятен.
.Рефрактерная анемия с избытком бластов,
трансформирующихся в острый лейкоз (RAEBt)
- количество бластов в ПК и КМ более чем 5% и 20% соответственно. Все случаи
трансформируются в острый лейкоз. Прогноз неблагоприятен.
.Хронический миеломоноцитарный лейкоз (CMML)
- характеризуется моноцитозом в ПК и КМ.
Таблица 1
FAB -
классификация миелодиспластических синдромов.
|
Вариант
МДС
|
RA
|
RARS
|
RAEB
|
RAEBt
|
CMML
|
|
Частота
встречаемости, %
|
30
|
5-10
|
15-20
|
10
|
<10
|
|
Бласты
ПК, %
|
<\=1
|
<\=1
|
<5
|
>5
|
<5
|
|
Бласты
КМ, %
|
<\=5
|
<\=5
|
5-20
|
20-30
|
<\=20
|
|
Кольцевидные
сидеробласты, %
|
<15
|
>15
|
<15
|
<15
|
<15
|
|
Палочки
Ауэра
|
-
|
-
|
-
|
+\-
|
-
|
|
Моноциты
ПК, мкл
|
<1000
|
<1000
|
<1000
|
<1000
|
>1000
|
|
Трансформация
в ОМЛ, %
|
12
|
8
|
44
|
60
|
14
|
|
Медиана
выживаемости, мес
|
50
|
51
|
11
|
5
|
11
|
В 1988 Группа изучения морфологии, иммунологии,
цитогенетики (MIC) предложила
рабочую классификацию для первичного (MDS)
и вторичного (t-MDS)
МДС и представила цитогенетический анализ диагностики данного заболевания [8].
Дальнейшее развитие систематизации МДС нашло отражение в проекте классификации
МДС Всемирной организации здравоохранения (ВОЗ), которая методологически
отличается от FAB-классификации.
В 2001 ВОЗ предложила окончательные новые схемы классификации опухолей
гематопоэтической и лимфоидной тканей, где отдельно выделена классификация МДС
(табл. 2) [8,9].
Таблица 2
Классификация миелодиспластических синдромов
(ВОЗ 2008).
|
Вариант
МДС
|
ПК
|
КМ
|
|
Рефрактерная
цитопения с однолинейной дисплазией (РЦОД/RCUD)
Рефрактерная анемия (РА/RA)
Рефрактерная нейтропения (РН/RN)
Рефрактерная тромбоцитопения (РТ/RT)
|
Однолинейная
цитопения: -Анемия -Нейтропения -Тромбоцитопения Бласты - нет или до 1%
Моноциты-<1·109/л
|
Однолинейная
дисплазия: ≥ 10 % клеток одной из миелоид-ных линий Бласты - < 5 %
Кольцевые сидеробласты <15 %
|
|
Рефрактерная
анемия с кольцевыми сидеробластами (РАКС/RARS)
|
Анемия
Бласты - нет
|
Дисплазия
только клеток эритроидного ряда Кольцевые сидеробласты ≥15 % Бласты -
< 5 %
|
|
Рефрактерная
цитопения с мультилинейной дисплазией (РЦМД/RCMD)
|
Моно-,
би- или панцитопения Бласты: нет или до 1% Палочки Ауэра - отсутствуют
Моноциты - < 1 ·109/л
|
Дисплазия
в ≥ 10 % клеток двух или более линий миелопоэза (нейтрофилы и/или
эритроидные предшественники и/или мегакариоциты) Бласты - < 5 % Палочки
Ауэра отсутствуют Кольцевые сидеробласты ± ≥15 %
|
|
Рефрактерная
анемия с избытком бластов-1 (РАИБ-1/RAEB-1)
|
Моно-,
би- или панцитопения Бласты - < 5 % Палочки Ауэра - нет Моноциты-< 1
·109/л
|
Однолинейная
или мульти-линейная дисплазия Бласты - 5 - 9 % Палочки Ауэра отсутствуют
|
|
Рефрактерная
анемия с избытком бластов-2 (РАИБ-2/RAEB-2)
|
Моно-,
би- или панцитопения Бласты - 5 - 19% Палочки Ауэра- ± Моноциты-< 1 ·109/л
|
Однолинейная
или мульти-линейная дисплазия Бласты - 10-19 % Палочки Ауэра - ±
|
|
Миелодиспластический
синдром неклассифицируемый (МДС-н/MDS-u)
|
Цитопении
Бласты - до 1% Палочки Ауэра - нет
|
Дисплазия
в < 10% клеток одной или более линий миелопоэза при наличии
цитогенетической аномалии, считающейся предпо-лагаемым доказательством для
установления диагноза МДС * Бласты - < 5%
|
|
МДС,
ассоциированный с изолированной del(5q) - 5q-синдром
|
Анемия
Тромбоциты обычно в норме или повы-шены Бласты - нет или до 1%
|
Нормальное
или увеличенное количество мегакариоцитов с гиподольчатыми ядрами Бласты -
< 5 % Палочки Ауэра - отсутствуют Изолированная цитогенетическая аномалия
del(5q)
|
|
МДС
детского возраста (рефрактерная цитопения детского возраста)
|
Персистирующая
моно-, би- или панцитопения Бласты - < 2 %
|
Дисплазия
двух или более линий миелопоэза (нейтрофилы и/или эритроидные предшественники
и/или мегакариоциты) Бласты - < 5% Цитогенетические аномалии*
|
*- хромосомные аномалии, которые рассматривают
как предполагаемое свидетельство наличия МДС при стойкой цитопении
неопределенного происхождения и при отсутствии абсолютных морфологических
критериев МДС:
.несбалансированные аномалии: - 7 или del(7q); -
5 или del(5q); i(17q) или
t(17p); - 13 или del(13q); del(11q); del(12p) или t(12p); del(9q); idic(X)(q13)
.сбалансированные аномалии: t(11;16)(q23;p13.3);
t(3;21)(q26.2;q22.1); t(1;3)(p36.3;q21.1); t(2;11)(p21;q23); inv(3)(q21q26.2);
t(6;9)(p23;q34)
.сложный кариотип (3 или более хромосомных
аномалий) с вовлечением вышеупомянутых нарушений [8,9,18].
В 1997 году Greenberg
с колегами представил Международную Числовую Систему прогноза (IPSS).
Эта система предполагает разделение пациентов на группы риска, основанные на
клинических, морфологических и цитогенетических особенностях (табл.3,4).
Наиболее важными прогности-ческими факторами согласно IPSS
являются - количество бластов в КМ, цитогенетические аномалии и количество
ростков гемопоэза в цитопении (характеристика цитопении ростков гемопоэза:
гемоглобин менее 100 г/л, абсолютное число нейтрофилов менее 1,5*109/л,
количество тромбоцитов ПК менее 100*109/л) [1,4,8].
Таблица 3
Международная числовая система прогноза (IPSS)
МДС.
|
Факторы
риска
|
0
баллов
|
0,5-1,0
баллов
|
1,5-2,0
баллов
|
>2,5 баллов
|
|
Количество
бластов КМ, %
|
<5
|
5-10
|
11-20
|
21-30
|
|
Кариотип
|
Нормальный
|
Нормальный
кариотип или изолированные нарушения Y-
5q-, 20q-
|
Комплексное
нарушение кариотипа(>3 аномалий) или аномалии хромосомы 7
|
Все
другие аномалии
|
|
Количество
ростков гемопоэза в цитопении
|
0
|
1
|
2
|
3
|
Таблица 4
Характеристика категорий риска согласно IPSS
МДС.
|
Категория
риска
|
Количество
баллов
|
Медиана
выживаемости,годы
|
Трансформация
в ОМЛ
|
|
Низкая
|
0
|
5,7
|
9,4
|
|
Промежуточная
-1
|
0,5-1,0
|
3,5
|
3,3
|
|
Промежуточная
-2
|
1,5-2,0
|
1,2
|
1,1
|
|
Высокая
|
>2,5
|
0,4
|
0,2
|
1.3 Этиология и патогенез миелодиспластического
синдрома
В настоящее время возникновение МДС
рассматривается как результат кумулятивного воздействия внешних факторов на
генетически предрасположенных лиц. Под генетической предрасположенностью
подразумевается комплекс факторов, включающий, с одной стороны, естественный
полиморфизм ДНК в генах, отвечающих за восстановление ДНК и метаболизм
канцерогенов, и с другой - индивидуальные различия по уровню отдельных энзимов,
вовлеченных в активацию или детоксикацию канцерогенов. Накопленные данные
позволяют предположить связь образования МДС с радиацией, курением, пестицидами,
органическими химикатами и тяжелыми металлами [1,8,11].
МДС может возникать как de novo, так называемый
первичный МДС, или развиваться после применения мутагенных агентов (химио-
и/или лучевая терапия) по поводу другого, обычно опухолевого, заболевания. Второй
вариант носит название вторичный МДС [4,18].
Существует ряд факторов, которые являются
предраспологающими факторами риска развития миелодиспластического синдрома. К
ним относятся:
. Принадлежность к мужскому полу
. Белый цвет кожи.
. Возраст старше 60 лет.
. Предшествующая химиотерапия или лучевая
терапия.
. Воздействие определенных химических веществ,
включая табачный дым, пестициды растворители, например бензин.
. Воздействие тяжелых металлов, таких как ртуть
или свинец [4,8,11].
В анамнезе больных МДС встречаются указания на:
курение - 29,0 %,
семейную отягощенность опухолевыми заболеваниями
- 18,6%
предшествующий контакт с канцерогенными
веществами - 16,2%
предшествующее лечение цитостатическими
препаратами по поводу другого онкологического заболевания - 6,4% [1,4,8].
Наиболее отчетливо доказано увеличение риска
заболевания среди людей, имеющих длительный профессиональный контакт с
бензолом, летучими органическими растворителями (шоферы, работники обувной и
кожевенной промышленности). Имеются сообщения о возникновении МДС в связи с
приемом фенилбутазона (бутадиона), хлорамфеникола (левомицетина),
цитостатических препаратов, в частности, при использовании мелфалана,
азотиаприна и циклофосфана [1,8,11]. Одной из возможных причин заболевания
может быть употребление недостаточно очищенной воды, так как водоемы
загрязняются различными химическими соединениями, многие из которых опасны для
здоровья человека [11]. К числу последних относятся канцерогенные вещества
(ароматические амины, N-нитрозосоединения, пестициды, гидразин и его
производные), радиоактивные вещества, канцерогены природного происхождения
(мышьякосодержащие соединения и соли цинка), одной из особенностей которых
является способность оказывать специфическое патогенное действие, попадая в организм
в чрезвычайно малых количествах [1,4,8,18].
Миелодиспластические синдромы характеризуются
периферическими цитопениями несмотря на нормо- или гиперцеллюлярный КМ, что
расценивается как результат неэффективного гемопоэза [1,3,8]. Различные
цитогенетические аномалии, обуславливающие быстрое увеличение неопластического
клона вместе с повышением количества бластов и увеличением апоптоза, играют
важную роль в патогенезе МДС [4,7].
Важную роль в процессе развития цитопений играет
повышенный апоптоз. Увеличенная продукция гемопоэтических клеток при МДС
сочетается с их усиленной гибелью вследствие апоптоза. У большинства больных
МДС апоптозу подвержено более 75% гемопоэтических клеток трех ростков
кроветворения и клеток стромы. Наличие клеток одновременно в процессе апоптоза
и в S-периоде клеточного
цикла является специфическим признаком МДС[7,18,20]. Усиление апоптоза
отмечается в условиях изоляции гемопоэтических клеток от колониестимулирующих
факторов [3]. Основную роль в регуляции апоптоза играют стволовой клеточный
фактор, интерлейкин-3(IL-3),
гранулоцитарный колониестимулирующий фактор(G-CSF),
гранулоцитарно-макрофагальный колониестимулирующий фактор (GM-CSF)
и эритропоэтин. Также причиной усиленной клеточной гибели может быть увеличение
концентрации фактора некроза опухоли (TNF-£),
трансформирующего ростового фактора (TGF£)
и интерлейкина-1b(IL-1b),
количества гемопоэтических клеток, экспрессирующих антиген CD-95(FAS/Apo-1),
опосредующий апоптоз. Блокада TNF-£
или FAS-ligand
увеличивает пролиферацию гемопоэтических колоний и количество клеток
периферической крови при МДС [2,3,7,20].
Начальной генетической ступенью многоуровневого
патогенеза МДС является дефект стволовой клетки, который на самом первом этапе
сопровождается неклоновой кариотипической нестабильностью. У некоторых
пациентов выявляется анеуплоидия или структурные расстройства, такие как
делеция, транслокация, изохромосомы и др. [17,20]. Патологический кариотип
более общий и типичный при вторичном МДС, тогда как при первичном - встречаются
более разнообразные цитогенетические аномалии [4]. Одними из наиболее частых
хромосомных аномалий, наблюдаемых при МДС, являются делеции хромосом. Подобное
наблюдение ведет к гипотезе, что МДС может быть вызван инактивацией генов
супрессоров опухоли [1,7,20]. 5q
- хромосомная аномалия - наиболее частая при МДС, встречается более чем в 20%
случаев. 5q-синдром
характеризуется клиническими и морфологическими особенностями, включающими
рефрактерную макроцитарную анемию с дизэритропоэзом, высокую распространенность
у лиц женского пола, нормальное или повышенное количество тромбоцитов ПК, а
также микроформами мегакариоцитов и относительно хорошим прогнозом [4,13].
Контрольные точки делеции лежат в пределах большой области 5q,
но наиболее критическая область делеции - между 5q31
и 5q33. В данной
области находятся гены, кодирующие гемопоэтические факторы роста и рецепторы,
включая IL-3, IL-4,
IL-5, макрофагальный
колониестимулирующий фактор (M-CSF),
рецептор для M-CSF,
GM-CSF
[2,13]. Делеция гена PURA
является наиболее частым нарушением при МДС, характеризующихся del(5)(q31).
В аномалиях 5-й хромосомы при МДС также участвуют: коэнзим А, синтетаза 2, АСS2
в соединении с геном TEL
при t(5;12) (q31;p13)
или с NUP98 в t(5;11)
(q35;p15.5)
и др. [1,20].
Также частой аномалией является моносомия 7 и 7q,
которые связаны с неблагоприятным прогнозом относительно продолжительности
жизни и трансформации в ОЛ. Моносомия 7 характерна для JMML,
который сопровождается нейрофиброматозом типа 1(NF
1) [17,18]. Мутации RAS-гена
или инактивация гена NF1
являются критическими событиями в прогрессии МДС при моносомии 7 [20].
Типичные хромосомные аномалии встречаются в
следующих регионах: Зр-, 3q-, 5q-, 7q-, 12p-, -17, -18, 20q, 1-12, +8. Эти
большие хромосомные изменения в обязательном порядке сопровождаются
субмикроскопическими мутациями ДНК таких генов, как р53, FLT3 или RAS,
метилированием специфических генов-промоутеров и, в некоторых случаях,
реципрокными транслокациями и инверсиями, уже ассоциированными с ОМЛ. Каждая
последующая стадия развития МДС сопровождается дальнейшими генетическими
повреждениями [3,7,8,20].
Изучение генетических мутаций при МДС показало,
что с лейкозным преобразованием в этой группе связана активация некоторых
онкогенов и инактивация опухолевых супрессоров [1,4]. RAS
гены, как известно, являются важным компонентом передачи сигналов для клеточной
пролиферации через рецептор тирозинкиназы (RTKs)
и активируются точечными мутациями в кодонах 12, 13 или 61. Среди RAS
генов наиболее часты мутации N-RAS,
которые встречаются в 10-15 % случаев при МДС и обуславливают короткий период
выживания и высокую вероятность трансформации в ОЛ [18,20]. FLT3 ген кодирует
тот тип рецептора тирозинкиназы, который вовлечен в пролиферацию и
дифференцировку гемопоэтических предшественников. Дупликация FLT3 гена
обнаружена как соматическая мутация в 5% случаев МДС [20]. Инактивация p53
гена обнаружена в 5-10 % случаях МДС и играет важную роль в лейкозной
прогрессии при данной патологии [7]. При RAEB
обнаружена мутация митохондриальной ДНК (G3242A) в CD34 + клетках. Эта
генетическая аномалия связана с дефектом созревания, так как мутации
митохондриальной тРНК нарушают синтез белка, вызывая таким образом дисфункцию
митохондриальной дыхательной цепи, что вносит значительный вклад в
неэффективный гемопоэз при МДС [7,17]. Однако, исследования мутаций
митохондриальной ДНК при RARS
не подтверждают главенствующую роль неустойчивости митохондриального генома в
патогенезе МДС [7,8].
Одним из факторов в патогенезе заболевания
является дефект микроокружения, подтвержением чего служат обнаруженные при МДС
качественные и количественные изменения клеток стромы КМ с нарушением продукции
цитокинов [1,4,8].
Кроме того большое значение в процессе
онкогенеза при МДС имеют иммунологические нарушения, в частности снижение функции
естественных киллеров, что приводит к утрате контроля над неопластическими
изменениями гемопоэтических клеток. Для МДС характерно уменьшение уровня
В-лимфоцитов, снижение способности моноцитов к фагоцитозу, а также адгезии и
хемотаксиса фагоцитов, нарушение функции нейтрофилов [1,8,17].
На основании клинико-экспериментальных данных
составлена приблизительная модель специфического многоступенчатого процесса
развития идиопатического МДС, в которой биологическая природа заболевания
объясняется совокупностью генетического повреждения стволовой клетки с такими
эпигенетическими механизмами, как аберрантная продукция цитокинов, нарушенная
адгезия стволовых клеток и поврежденное гемо-поэтическое микроокружение. В этой
модели выделяют четыре патофизио-логические фазы [1,3,8].
. В преМДС фазе процесс инициируется действием
внешних факторов у генетически чувствительных лиц.
. Ранняя фаза характеризуется иммунным ответом
на повреждение кроветворных клеток. Существование аутоиммунного Т-клеточного
ответа подтверждается выявлением Т-клеточного рецептора (TCR) Vb характера,
который свидетельствует о персистенции Т-клеточной клональной популяции, и
ассоциации МДС с Т-клеточным лейкозом из больших гранулярных лимфоцитов,
развивающегося в результате олигоклональной или клональной экспансии Т-клеток.
Как при апластической анемии, клональная Т-клеточная популяция вызывает
аутоиммунную миелосупрессию, приводящую к цитопении при МДС. Персистирующая
аутоиммунная атака обусловливает хроническую избыточную продукцию таких
проапоптических цитокинов, как TNF-£
,
TGF£, IL-1b.
Основным продуцентом TNF-£
являются
мононуклеарные клетки патологического клона, а IL-1(3)
и TGF£-
стромальные клетки.
Увеличение уровня TNF-£
в
костномозговом микроокружении индуцирует на CD-34 костномозговых клетках
избыточную экспрессию FAS-антигена в результате активации каспаз (и, прежде
всего, каспазы-3) и снижение Fap-1, что обусловливает избыточный апоптоз клеток
КМ. Кроме того, высокая экспрессия TNF-£
способствует
повышенной продукции свободных радикалов и окислительному повреждению CD34
костномозговых клеток с формированием характерной диспластической морфологии
клеток.
Преобладание внутримедуллярного апоптоза над
пролиферацией клеток обусловливает неспособность КМ экспортировать достаточное
количество клеток в ПК. Избыточному апоптозу также способствуют нарушенные
адгезивные взаимоотношения между клоногенными гемопоэтическими стволовыми
клетками и прилегающей костномозговой стромой или эндотелием. Дополнительным
фактором, поддерживающим неэффективный гемопоэз при МДС, является и избыточны
ангиогенез - результат продукции клональными клетками васкулярного
эндотелиального ростового фактора (vessel
endothelial
grows factor,
VEGF). Повышенная
плотность сосудов может благоприятствовать прогрессии МДС путем поддержки
нерегулируемого клеточного роста.
Нарушенная экспрессия онкопротеинов и, особенно,
высокое соотношение проапоптических/антиапоптических онкопротеинов, например,
Мус против bcl-2 или bax/bad против bcl-2/bcl-x и сверхэкспрессия р53 или
низкая экспрессия bcl-2 также коррелируют с повышенным апоптозом. Избыточный
апоптоз является удобным объяснением того, как клональная экспансия
костномозговых клеток-предшественников может приводить к неэффективному
гемопоэзу и костномозговой недостаточности.
. При прогрессировании МДС в позднюю стадию
апоптические сигналы (FAS, с-Мус) снижаются, а антиапоптические (bcl-2) -
усиливаются.
. Дальнейшая прогрессия МДС в ОМЛ связана с
инактивацией генов-супрессоров опухоли р15 (гиперметилирование) и р53 (точечные
мутации). В целом поздняя стадия МДС характеризуется снижением контроля за
клеточным циклом и развитием МДС/ОМЛ [1,3,8].
Таким образом, несмотря на возможность
предположения, что хромосомные аномалии в конечном счете приведут к открытию
генетических поломок, основных в генезе МДС, прогресс в этой области происходит
медленно [3,20]. Патогенез МДС является мультифакторным процессом, который
вовлекает множество повреждений генома клеток костного мозга. Поиск генов,
относящихся к патогенезу данного заболевания сложен, потому что хромосомные
аномалии при МДС в основном характеризуются потерей генетического материала. В
настоящее время не ясно, вовлекает ли потеря генетического материала полную
утрату функции гена супрессора опухоли или работа гена супрессора опухоли
осуществляется через некоторый другой дефект, который еще не был обнаружен.
Поэтому, несмотря на то, что морфологический метод остается краеугольным камнем
диагностики МДС, анализ цитогенетических и молекулярных аномалий при данном
заболевании может представлять интерес для классификации болезни, определения
прогноза и терапевтического направления, поскольку вносит вклад в понимание
патогенетических механизмов [3,7,20].
.4 Клиническая картина миелодиспластического
синдрома
Клиническая картина при различных формах МДС
схожа и во многом определяется показателями периферической крови. Изменения
периферической крови прямо зависят от степени нарушения созревания
гемопоэтических клеток [1,4,8].
Анемия постоянный и обязательный признак.
Уровень снижения гемоглобина может варьировать от умеренного до значительного.
При медленном снижении гемоглобина организм успевает адаптироваться к гипоксии
и количество жалоб у больных может быть минимальным [1,4]. Если анемия
развивается быстро, больные предъявляют жалобы на общую слабость, утомляемость,
сердцебиение, одышку. Может утяжеляться течение ишемии-ческой болезни сердца,
появляются признаки сердечной недостаточности [4,8,].
Снижение количества зрелых гранулоцитов
(нейтропения), а также их функциональная несостоятельность влекут за собой
инфекционные осложнения [15]. У 10 % больных развиваются стоматиты, гингивиты,
пневмонии, инфекция мочевыводящих путей, абсцессы различной локализации,
сепсис. У 20% больных данной группы инфекционные осложнения становятся причиной
смерти [11]. Наиболее многочислены осложнения бактериальной природы,
возбудителями которых являются Escherichia coli, Pseudomonas pyocyanea,
Klebsiella pneumoniae, Staphylococcus aureus и Streptococcus fecalis. Также
достаточно часто тяжелые инфекционные осложнения вызываются Pneumocysti
carinii, Cryptococcus neoformus, Candida albicans,
Aspergillus fumigatus и цитомегаловирусом, что связано с функциональной
неполноценностью Т-лимфоцитов при МДС [5,8,11].
Клинически значимая тромбоцитопения (приводящая
к развитию геморрагического диатеза с петехиально-пятнистым типом
кровоточивости) встречается у 15 % больных МДС [1]. У половины из них
кровотечение или кровоизлияния становятся причиной смерти. В некоторых случаях
МДС, как правило у больных рефрактерной анемией, может отмечаться тромбоцитоз
[4,8]. Проявления гиперпластического синдрома в виде спленомегалии,
гепатомегалии, лимфоаденопатии и специфического поражения кожи (лейкемиды)
имеют место в основном у больных ХММЛ. Спленомегалия встречается у 17 % таких
больных, гепатомегалия у 13 %, а лейкемиды у 10% [1,4,8].
При всех вариантах миелодиспластического
синдрома возможно развитие аутоиммунного процесса: васкулита, полисерозита и др
[11].
Такие признаки, как потеря веса,
немотивированная лихорадка, болевой синдром могут быть манифестацией МДС [4].
.5 Диагностика миелодиспластического синдрома
Современная диагностика МДС включает следующие
методы [1,4,5,8]:
.Морфологические исследования крови и костного
мозга.
2.Иммунофенотипирование.
3.Цитогенетическиое исследование.
4.Определение ферритина.
1.5.1 Морфологические исследования крови и
костного мозга
Изменение морфологических и количественных
показателей в результатaх
анализов периферической крови и костного мозга являются важным звеном в
диагностике МДС; особенно выявление морфологических признаков дисгранулопоэза,
дизэритопоэза, дисмегакариоцитопоэза, основными признаками которых являются
следующие [10,13,16]:
Дизэритропоэз - диспластические изменения в
клетках эритроидного ряда, включают в себя три основные группы изменений [19]:
.клеточные аномалии с потерей индивидуальных
признаков клетки,
.структурные деформации
.стромальные нарушения.
При всех вариантах МДС в большинстве случаев
анемия нормохромная, нормоцитарная и норморегенераторная. В ПК определяются
смешанный анизоцитоз, умеренный макроцитоз, пойкилоцитоз, овалоциты,
фрагментированные эритроциты в виде слезных капель, «мишеневидные» (таргетные)
клетки, базофильнопунктированные эритроциты, тельца Жоли, нормобласты и
мегалобластоидные элементы (рис.1.1)[10,21].
В КМ: эритроидная гиперплазия, наличие
мегалобластоидных элементов, многоядерность эритрокариоцитов, ядерная
фрагментация, межъядерные мостики, неправильные митотические фигуры,
Шик-положительные эритро- и нормобласты, кольцевидные сидеробласты [16,24].
В норме агрегаты ферритина встречаются менее чем
в 50% нормобластов, и их количество обычно не превышает 4 гранул в клетке.
Наличие 5 или более гранул рассматривается как патологический признак, а если
они занимают более трети ядра, то описываются под термином «кольцевой»
сидеробласт. Отложение ферритина в митохондриях является результатом сниженной
активности аминолевулиновой кислоты и увеличенного захвата железа митохондриями
и приводит к нарушению клеточного цикла, например вхождению клеток в S-фазу
цикла. Кольцевые сидеробласты идентифицируются по окраске гранул железа
Prussan-blue, встречаются при различных гематологических состояниях и являются
результатом неэффективного гемопоэза [13,25].
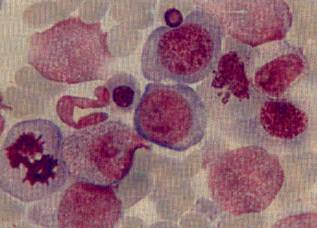
Рисунок 1.1- Дисплазия эритроидного ростка в
костном мозге: мегалобластоидность, асинхронность ядер, тельца Жоли.
К признакам дизгранулоцитопоэза в ПК относят
выраженное нарушение созревания и дифференцировки нейтрофильных и эозинофильных
гранулоцитов: снижение доли сегментоядерных нейтрофилов, снижение или полное
отсутствием зернистости, дефект накопления миелопероксидазы и нафтол-ASD-хлорацетатэстеразы,
выявление гипосегментации с пикнозом ядер по типу аномалии Пельгера,
асинхронное созревание ядра и цитоплазмы [14,15,21].
В КМ обнаруживаются:
) гранулоцитарная гиперплазия;
) повышение бластных клеток двух типов: тип 1 -
с выраженной базофилией цитоплазмы и отсутствием в ней грануляции, наличием в
ядре отчетливо контурирующихся нуклеол, тип II - более крупного размера,
содержащие в цитоплазме, по крайней мере, одну, но не более 5-6 гранул [15];
) гипо- или гипергрануляция первичных гранул и
специфическая зернистость в цитоплазме всех стадий созревания;
) наличие парамиелоидных клеток при отсутствии
или снижении специфической зернистости в миелоцитах или метамиелоцитах,
подобные клетки напоминают моноциты;
) палочки Ауэра;
) пельгероидность клеток;
) базофилия цитоплазмы в зрелых элементах
(тельца Деле);
) наличие эозинофилов с круглым ядром и
вариабельной зернистостью (рис.1.2, 1.3)
[14,15.19,24].
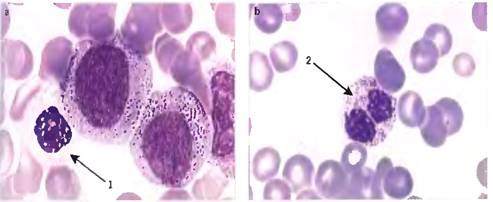
Рисунок 1.2-1.3: - Дисплазия гранулоцитарного
ростка: 1- зрелый нейтрофил с округлой формой ядра, 2 - бисегментированный
нейтрофил.
Дисмегакариоцитопоэз при МДС характеризуется
угнетением этой клеточной линии и нетипичной локализацией в ячейках КМ,
наличием плеоморфных микромегакариоцитов и повышенным количеством незрелых
мегакариоцитов [10,14]. В ПК: наличие микромегакариоцитов с моноплоид-ным
округлым плотным ядром и гигантских тромбоцитов со скудным грануломером [14].
В КМ обнаруживают микромегакариоциты с
моноплоидным ядром и гигантские мегакариоциты с множеством раздельнолежащих
маленьких круглых ядер (рис.1.4)[14,16].
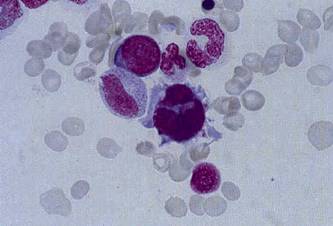
Рисунок 1.4 - Дисплазия мегакариоцитарного
ростка в костном мозге: микромегакариоцит.
Диспластические признаки клеток моноцитарной
линии в ПК включают: увеличение моноцитов с множеством долей
(,,гиперсегментация,,) и наличие азурофильной зернистости в цитоплазме [24].
В КМ:
) наличие монобластов и моноцитов, аналогичных в
ПК;
) гемофагоцитоз;
) макрофаги, содержащие гранулы железа [21].
Характерные диспластические изменения
обнаруживают и в гистологических препаратах КМ. Клеточность КМ вариабельна, так
при RAEBt и CMML
- КМ гиперклеточный за счет гранулоцитарного ростка, а при RA
- гипоклеточный [1,8,19]. Выявляют нарушение костномозговой топографии всех
ростков гемопоэза. Наиболее отчетливо оно выражено в мегакариоцитарном ростке в
виде скопления микромегакариоцитов, нарушения синусоидальной ориентации и паратрабекулярной
локализации диспластических и пикнотичных мегакариоцитов [16]. Для миелоидного
ростка характерным признаком является патологическая локализация незрелых
клеток - ALIP (abnormal localization of immature myeloid precursors), впервые
описанная G. Tricot с соавторами в 1984 г. Этот феномен представляет скопление
миелобластов и промиелоцитов в центральной части костномозговых лакун, в то
время как обычно они локализуются вдоль эндостальной мембраны. Скопление 5 или
более клеток описывается как «агрегат», а 3-5 клеток - как «кластер». По мнению
G. Tricot, о патологической локализации клеток можно судить только в том
случае, если по крайней мере три агрегата или кластера присутствуют в каждой
секции препарата [1,4,10].
Со стороны эритропоэза выявляются участки с
блоком созревания, располагающиеся как в интра-, так и в паратрабекулярных
областях. Картина повреждения микроокружения складывается из участков отека и
экстравазации эритроцитов в результате повреждения синусоидов, васкулитов,
фиброза, увеличения плазматических клеток, тучных клеток, лимфоцитов,
макрофагов и нередко - гемофагоцитоза [8,10]. В некоторых случаях отмечают
превалирование воспалительных изменений с содержанием высокого процента
плазматических и тучных клеток, лимфоцитов, которое коррелирует с результатами
изучения костномозговых аспиратов [13,21].
В соответствии с ФАБ-классификацией выделяют
пять вариантов МДС, каждый из которых характеризуется определенными признаками
в изменении морфологических и количественных показателей результатов анализов
периферической крови и костного мозга [10].
.Рефрактерная анемия (RА).
Среди всех вариантов МДС составляет 5-10%.
Ведущим признаком является разной степени выраженности анемия с
ретикулоцитопенией. Анемия гиперхромная макроцитарная или нормоцитарная.
Степень анизоцитоза варьирует. Гранулоцитопения и (или) тромбоцитопения или не
выявляются, или незначительные. В ПК могут выявляться агранулярные или
гипогранулярные нейтрофилы, хотя и необязательно. Бласты отсутствуют или их
менее 1%. В КМ выявляют гиперплазию эритроидного ростка и эритробласты с
признаками мегалобластоидного кроветворения. КМ гиперклеточный. Количество
бластов - менее 5%; кольцевые сидеробласты обнаруживаются редко, их количество
составляет менее 15% от всех клеток эритроидного ряда. Морфологических
признаков дисмегакариоцитопоэза может не быть, или выявляется повышенное
содержание мегакариоцитов с признаками дисплазии. Признаки дисгранулоцитопоэза
могут или обнару-живаться, или полностью отсутствовать. PAS-реакция в
эритробластах в большинстве случаев варьирует от слабо выраженной до полного
отсутствия, однако может быть и гранулярного характера [10,14,16,19].
.Рефрактерная анемия с кольцевыми сидеробластами
(RARS).
Среди всех вариантов МДС составляет 10-12%. В ПК
выделяют две популяции эритроцитов: гиперхромные микроциты и нормохромные
макроциты. Эритроциты с базофильной пунктацией. Отмечается выраженный
пойкилоцитоз, нормобластоз. Бласты отсутствуют. КМ нормо- или гиперклеточный,
обычно со значительной эритроидной гиперплазией. Признаки дисплазии
регистрируются только в эритроцитарном ростке. Содержание кольцевых
сидеробластов составляет более 15%, миелобластов - менее 5% (рис.1.5.). Могут
встречатся макрофаги с гемосидерином. Положительная PAS-реакция диффузного или
гранулярного характера встречается в большинстве эритробластов, хотя может быть
и отрицательной. Трансформация РАКС в ОЛ ввиду стабильности патологического
клона наблюдается крайне редко. В случае прогрессирования заболевания
количество сидеробластов может достигать 80% клеток эритроидного ряда
[14,16,24].
Описаны две формы РАКС: 1-я - с морфологическими
признаками дизэритропоэза, 2-я - с дополнительными признаками
дизгранулоцитопоэза и (или) дизмегакариоцитопоэза. Риск лейкозной трансформации
в этих группах составляет 1,9% и 48%, а 5-летняя выживаемость больных - 69% и
19% соответственно. Это позволяет предположить, что первая форма РАКС не
вляется клональным состоянием и, следовательно, не может рассматриваться в
качестве истинного первичного МДС [10].
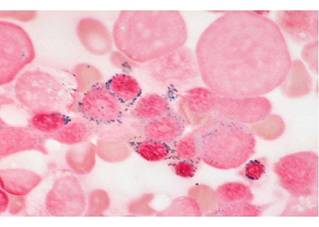
Рисунок 1.5 - Костный мозг больного рефрактерной
сидеробластной анемией: кольцевые сидеробласты.
миелодиспластический синдром мутация
кровь
3.Рефрактерная анемия с избытком бластов (RAEB).
Встречается в 40% случаев МДС. В ПК отмечаются:
анизопойкилоцитоз с макроцитозом, атипичные тромбоциты, гипо- или агрануляция
гранулоцитов иногда с измененным хроматином или сегментацией ядра.
Тромбоцитопения умеренная. В ПК выявляются бласты, но обычно их количество
составляет менее 5%. В КМ обнаруживается дисплазия 2 или более клеточных линий.
КМ гиперклеточный, реже нормо- или гипоклеточный. Наиболее часто встречаются
признаки дисгранулоцитопоэза: уменьшение размера клеток, псевдо-пельгеризация
ядер, гиперсегментация, гипо- или агрануляция в цитоплазме. Диспластические
изменения обнаруживаются в эритроцитарном и мегакариоцитарных ростках.
Количество бластов с положительной реакцией на миелопероксидазу находится в
диапазоне 5-20% всех ядерных клеток. По сравнению с ОМЛ, бласты преимущественно
меньшего размера, что, вероятно, отражает их низкий пролиферативный потенциал.
PAS-реакция в эритробластах варьирует от слабо выраженной до хорошо
гранулированной. Активность реакций на кислую фосфатазу и специфическую
эстеразу слабо выраженная либо отсутствует. Сидеробласты встречаются редко, и
количество их варьирует от случая к случаю [10,15,16].
.Рефрактерная анемия с избытком бластов в
трансформации(RAEBt).
Анемия гиперхромная макроцитарная. К этой форме
относят варианты с количеством бластов в КМ от 21 до 30% и (или) наличием
бластов с палочками Ауэра и (или) содержанием более 5% бластов в ПК. В тех
случаях, когда количество бластов в КМ достигает 30%, критерием разграничения
этого варианта МДС от ОМЛ является обнаружение бластов III типа, содержащих 20
азурофильных гранул. Случаи с содержанием бластов в КМ от 5 до 20% при условии
выявления палочек Ауэра относят к РАИБ-Т ввиду высокого риска прогрессирования
заболевания и низкой выживаемости больных этой группы [13,15,24].
.Хронический миеломоноцитарный лейкоз (CMML).
В ПК: количество бластов составляет не более 5%,
абсолютное количество моноцитов не менее 1 х109 /л. Большинство моноцитов не
отличаются от нормальных, но могут быть моноциты с грубой зернистостью, редко
выражен полиморфизм ядер. Может иметь место умеренная базофилия, иногда
эозинофилия. Отмечается нормоцитарная, реже макроцитарная анемия. Количество
тромбоцитов - варьирует, может быть тромбоцитопения. Встречаются атипичные
крупные тромбоциты. КМ гиперклеточный, реже нормо- или гипоклеточный с пролиферацией
гранулоцитарного ростка и признаками трехростковой дисплазии, степень
выраженности которой варьирует от случая к случаю. Моноцитарные клетки разной
степени зрелости. Число бластов может варьировать от 5 до 20% в результате
повышенного содержания молодых форм моноцитарного ростка, а количество
миелобластов в этом случае обычно не превышает 10% костномозговых клеток [10].
Эритробласты с чертами мегалобластоидного кроветворения. Нередко выявляется
повышенное содержание эозинофилов и плазматических клеток. При проведении
цитохимических реакций в моноцитах обнаруживается интенсивная окраска на
неспецифическую эстеразу, подавляемую NaF; активность реакций на кислую
фосфатазу и специфическую эстеразу варьирует от слабовыраженной до полного
отсутствия; активность реакции на миелопероксидазу различная, но в большинстве
случаев слабая или полностью отсутствует. В эритробластах при проведении
PAS-peакции выявляется слабоположительная окраска цитоплазмы. ХММЛ включен
ФАБ-классификацией в группу МДС, хотя имеет признаки миелопролиферативного
заболевания. В некоторых исследованиях предлагают деление ХММЛ на два варианта:
пролиферативный и непролиферативный. Разграничительной чертой между ними служит
число лейкоцитов - 12 х109 /л [13,19].
Морфологические признаки дисплазии ростков
кроветворения являются определяющим фактором в совокупности признаков МДС.
Однако само по себе выявление дисплазии того или иного ростка гемопоэза еще не
является основанием для диагностики МДС, так как встречается и при других
патологических состояниях. Так, различной степени проявления дизэритро-поэза
обнаруживаются в регенерирующем КМ, в том числе и после проведения
цитостатической терапии, при недостаточности питания, миелопролифе-ративных
заболеваниях и врожденной дизэритропоэтической анемии. Гипогранулярные
нейтрофилы и клетки с пельгеровской аномалией встречаются при Ml, М2 и М4
вариантах de novo ОМЛ, нередко после цитостатической терапии. Дисплазия
мегакариоцитов выявляется в ряде случаев ХМЛ. Миелодисплазия, как
морфологическа находка, еще не является синонимом МДС, а обнаружение
одноростковой дисплазии еще не служит критерием диагностики первичного МДС
[1,4,8,24].
1.5.2 Иммунофенотипирование
Изучение иммунофенотипа клеток костного мозга с
помощью метода проточной цитофлюориметрии имеет важное значение в диагностике
МДС для оценки нарушений клеточной дифференцировки, прогноза заболевания, для
дифференциального диагноза с другими заболеваниями [22].
В основе проточной цитофлюориметрии лежит
проведение фотометрических и флюоресцентных измерений отдельных клеток,
пересекающих одна за другой вместе с потоком жидкости лазерный луч
монохроматического света.
Флюоресцентный канал применяется для изучения
клеточных маркеров, для чего используются моноклональные антитела (МАТ),
меченные различными флюорохромными красителями к мембранным и внутриклеточным
компонентам клеток (белкам, ДНК, РНК). После окрашивания клеток МАТ происходит
специфическое связывание последних с клеточными структурами и регистрация
флюоресценции, индуцированной излучением лазера. Техника регистрации
флюоресценции состоит в следующем. При облучении клеток длиной волны,
возбуждающей флюорохром, происходит поглощение света. Затем флюорохром
испускает свет, но уже меньшей интенсивности (большей длины волны), чем
поглощенный. Вторичное излучение, имеющее строго определенную для каждого
флюорохрома длину волны, проходя через оптическую систему прибора (линзы,
фильтры, двухцветные зеркала) регистрируются высокочувствительными детекторами
(фотоэлектронными умножителями), преобразующими его в электрические сигналы,
поддающиеся компьютерной обработке. Регистрацию проводят под прямым углом,
однако для монохроматического лазерного облучения это не является обязательным
условием, т.к. испускаемый флюоресцентный свет всегда имеет большую длину
волны, чем возбуждающий свет лазерного облучения. Проточные цитофлюориметры
могут быть оборудованы одним, двумя или более лазерами. В случае использования
двух или более лазеров в исследования могут быть включены флюорохромы,
возбуждаемые на разных длинах волн (488 нм, 635 нм, 407 нм и др.). Наиболее
часто в качестве красящей метки применяется флюоресцеинизотиоционат (FITC),
который улавливается FL-1-детектором
(зеленый спектр), фикоэритрин (PE)
- FL-2-детектором
(красный спектр), менее часто - цианин-5/фикоэритрин и пиридин хлорофилл (PerCp,
Cy5/PE)
- FL-3-детектором и
аллофикоцианин (APC)
- FL-4-детектором [22].
Иммуноцитофлюориметрический анализ клеток
проводится по пяти основным параметрам:
две характеристики светорассеяния клеток: FSC
- показатель прямого светорассеяния и ортогональный SSC
(90 ) - показатель бокового светорас-сеяния;
три канала детекции специфического
флюоресцентного сигналакрасителя на разной длине волны [22].
Иммунофенотипирование является адекватным
методом для оценки качественных и количественных изменений гемопоэтических
клеток при МДС, для анализа значения нарушений клеточной дифференцировки и
оценки прогноза заболевания [13].
Наиболее частыми изменениями экспрессии
поверхностных антигенов по данным иммунофенотипа оказываются экспрессия HLA-DR
на клетках миелоидного ряда, наличие CD-34
на моноцитах, повышение интенсивности экспрессии CD-33
на миелоидных клетках, наличие лимфоидных антигенов на клетках моноцитарного
ряда, наличие CD-34 на
гранулоцитах, наличие лимфоидных антигенов на клетках миелоидного ряда [12,13].
С высокой достоверностью было показано
увеличение при МДС процента гранулоцитарных клеток с низкой экспрессией CD16
и CD11b.
По данным иммунофенотипирования встречаются: CD10-отрицательные
нейтрофилы, линейные антигены немиелоидных линий на миелоидных клетках, в
частности антиген CD22
или антиген CD7 [21].
Антиген CD33,
обычно исчезающий на зрелых нейтрофилах, у больных МДС обнаруживается на этих
клетках, особенно при РАИБ и РАИБТ. Этот признак часто коррелирует с наличием
клональных цитогенетических аномалий и высоким риском прогрессирования заболевания.
По данным некоторых авторов, увеличение
экспрессии антигена CD13
указывает на возможность прогрессирования заболевания и трансформации его в
ОМЛ. В ряде случаев на гранулоцитах может выявляться аберрантная экспрессия
моноцитарного антигена CD14
[12,22].
Большой интерес представляет экспрессия антигена
CD34. Показано, что
бластные клетки при МДС - часто CD34+-клетки.
Однако отсутствует корреляция между количеством бластных клеток и CD34+-
клеток, что предполагает аномальную персистенцию экспрессии этого антигена на
созревающих клетках. Увеличение числа клеток с таким фенотипом коррелирует с
плохим прогнозом заболевания и высоким риском трансфор-мации заболевания в
острый лейкоз [12,21].
.5.3 Цитогенетическое исследование
Цитогенетическое исследование позволяет не
только четко определить клональное заболевание в случае аберрантного кариотипа,
но также помогает в классификации заболевания и оценке прогностического
значения аномалии [13].
Цитогенетические нарушения при
миелодиспластическом синдроме выявляются в 30 - 50% случаев при первичном МДС и
в 80 % - при вторичном МДС. Некоторые хромосомные аномалии ассоциируются с
определенными клиническими проявлениями. Например, потеря части длинного плеча
5 хромосомы связана с развитием макроцитарной анемии у пожилых женщин и с
низким риском трансформации в острый миелолейкоз [11,20,21].
При стандартном цитогенетическом анализе
исследуются хромосомы, зафиксированные на стадии метафазы митоза. Методика
приготовления препаратов включает в себя:
.Введение митостатиков (колхицин, колцемид),
останавливающих митозы на стадии метафазы.
.Гипотоническую обработку с целью разобщения
хромосом набора.
.Фиксацию смесью метанола и уксусной кислоты.
.Нанесение приготовленной суспензии на
предметные стекла путем раскапывания [13].
Перед добавлением митостатика желательно
культивировать клетки 1-2 суток. При необходимости срочного анализа препарат
можно приготовить прямым методом, т.е. исключив культивирование [13].
Готовые препараты высушивают 2-7 дней. Затем
проводят дифференциальное окрашивание, после которого каждая хромосома
приобретает индивидуальный рисунок поперечной исчерченности, позволяющий
идентифицировать не только хромосомы, но и отдельные их участки. Систематизация
хромосом в каждой отдельной метафазной пластинке дает кариотип клетки [11,13].
Время анализа препаратов варьирует в зависимости
от их качества и сложности изменений кариотипа, в среднем составляет 7-14 дней,
а для срочного анализа - 24-48 часов.
Для получения достоверных результатов необходимо
кариотипировать не менее 20 аномальных метафаз или 20-25 нормальных метафаз.
При обнаружении одной клетки с потерей хромосомы 7 количество анализируемых
метафаз следует увеличить до 30 [21].
При исследовании методом FISH анализируется
100-200 ядер. При обнаружении однозначно позитивных клеток может быть
проанализировано и меньшее число клеток. При необходимости количество
анализируемых клеток может быть увеличено.
При нормальном кариотипе или отсутствии митозов
у взрослых желательно провести исследование методом FISH для выявления делеции
хромосомы 5 (del(5q)), моносомии 7/del(7), трисомии 8. У детей - для выявления
моносомии 7/del(7) и трисомии 8 [20,21].
Типичные аберрации, которые рассматриваются для
прогностической оценки, включают делецию Y хромосомы, del(5q), del(20q), также
сложные аберрации и аберрации хромосомы 7 [16]. Когда возникают проблемы во
время цитоморофологической дифференциации между МДС и ОМЛ в некоторых случаях
может быть целесообразным цитогенетическое исследование. Выявление t (8;21)
(q22;q22), t (15;17) (q22q11-12), inv (16) (p13q22), t (16;16) (p13;q22)
позволяет определить ОМЛ [16,23].
Выделяют следующие цитогенетические поломки при
МДС:
) нормальный кариотип, делеция 5q, 20q
и Y-хромосомы, относительно благоприятный вариант, предположительный срок жизни
более 2 лет;
) трисомия 8 хромосомы, срок жизни 1-2 года;
) делении 5,7 хромосомы и комплексные поломки,
срок жизни менее 1 года [23].
q - хромосомная
аномалия - наиболее частая при МДС, встречается более чем в 20% случаев. 5q-синдром
характеризуется клиническими и морфологическими особенностями, включающими
рефрактерную макроцитарную анемию с дизэритропоэзом, высокую распространенность
у лиц женского пола, нормальное или повышенное количество тромбоцитов ПК, а
также микроформами мегакариоцитов и относительно хорошим прогнозом [4,13,16].
Также частой аномалией является моносомия 7 и 7q,
которые связаны с неблагоприятным прогнозом относительно продолжительности
жизни и трансформации в ОЛ. Моносомия 7 характерна для JMML,
который сопровождается нейрофиброматозом типа 1(NF
1). Мутации RAS-гена или
инактивация гена NF1
являются критическими событиями в прогрессии МДС при моносомии 7 [4,13,23].
Типичные хромосомные аномалии встречаются в
следующих регионах: Зр-, 3q-, 5q-, 7q-, 12p-, -17, -18, 20q, 1-12, +8 [16].
Кариотип играет важную роль как на этапе
верификации диагноза, при определении цитогенетических и морфологических
корреляций, так и при оценке прогноза заболевания, выборе тактики терапии, и
является одним из принципиальных факторов прогноза у больных МДС [1,8].
Согласно варианту кариотипа, больных МДС относят к группам различного прогноза
(хорошего, промежуточного, плохого). Однако вариабельность течения и
прогресси-рования заболевания неоднородны у больных в пределах одинаковых
цитогенетических нарушений. Так, выживаемость больных в группе благоприятного
прогноза не всегда высока; напротив, в группе больных с плохим прогнозом у
отдельных пациентов наблюдается благоприятное течение заболевания. Наряду с
этим, современная тенденция к индивидуализации терапии также обуславливает
поиск новых факторов прогноза [16,20,23].
.5.4 Определение ферритина
Ферритин - водорастворимый белок с мол. массой
440000 кД, способный присоединить до 4500 атомов железа на молекулу, что
связано с его биологической функцией. Эта функция заключается в депонировании
железа, токсичного для организма, в растворимой, нетоксичной и физиологически
доступной форме.
Молекула ферритина состоит из двух компонентов:
белковой "раковины" - апоферритина и кристаллической
"сердцевины" в виде коллоидного гидроксида железа. Полностью
насыщенная железом молекула ферритина содержит железа до 27% своей молекулярной
массы.
Белковая оболочка ферритина - апоферритин -
состоит из 24 субъединиц двух видов: Н (heavy) и L(light). Синтез Н- и
L-субъединиц детерминируется разными генами. Субъединицы имеют неодинаковую
молекулярную массу, антигенную и изоэлектрическую характеристики. Различные
количественные сочетания Н- и L-субъединиц создают большую гетерогенность
изоферритинов, поэтому каждый орган имеет свою композицию Н- и L-субъединиц
[13,25]. Так, ферритин печени и селезенки содержит 80 - 90% L- и 10-20%
Н-субъединиц [11]. Сердце, плацента, фетальные ткани, злокачественные опухоли в
своих изоферритинах, наоборот, содержат преимущественно Н-форму [25], которую
называют фетоплацентарной, онкофетальной, кислой.
В физиологических условиях биосинтез
апоферритина стимулируется железом. При гемохроматозе и гемосидерозе, т.е. в
ситуациях, связанных с перегрузкой организма железом, количество ферритина
растет, а при дефиците железа происходит супрессия синтеза апоферритина и его
количество снижается.
Ферритин синтезируется клетками различных
тканей: печени, селезенки, костного мозга, сердечной мышцы, легких, почек,
щитовидной железы, плаценты, тонкого кишечника, поджелудочной железы, а также
лейкоцитами [13].
Наиболее хорошо изучена железодепонирующая роль
ферритина, которая позволяет организму сохранять железо в нетоксичной,
растворимой, легкодоступной форме, из которой оно может быть мобилизовано для
синтеза гемоглобина и негемовых железосодержащих белков. Синтезируемый в
различных органах и тканях ферритин в незначительных количествах выделяется в
сыворотку, причем в физиологических условиях уровень сывороточного ферритина
(СФ) коррелирует с запасами железа в организме: 1 мкг/л СФ в норме
соответствует 8 мг депонированного железа [11,20].
Методы определения ферритина.
К настоящему времени разработано большое
количество иммуно-химических систем для определения концентрации ферритина на
основе трех принципов анализа: радиоиммунного (РИА), иммуноферментного (ИФА) и
флуоресцентного (ФИА) [24,25].
Нормальные величины.
У здоровых взрослых людей уровень СФ зависит от
пола и в меньшей степени от возраста. Так, у мужчин большая часть коммерческих
тест-систем определяет концентрацию СФ в диапазоне 30 - 200 мкг/л (в среднем
98,2±4,8 мкг/л). У женщин фертильного возраста СФ составляет 10 - 90 мкг/л (в
среднем 42,5±5,1 мкг/л), в постменопаузе (обычно 48 - 50 лет) достигает средних
величин, характерных для мужчин. У детей отмечается резкое возрастание уровня
[13,24].
Клиническое значение.
В клинической практике СФ стали использовать для
оценки запасов железа в организме [24]. Общеизвестно, что показатель СФ
наиболее ранний и достоверный признак тканевого дефицита железа, предшествующий
развитию собственно железодефицитной анемии [1,5,]. При тканевом дефиците
железа и железодефицитной анемии уровень СФ резко снижается: у женщин и детей
ниже 10, у мужчин ниже 30 мкг/л. При купировании анемии и восполнении депо
железа уровень СФ восстанавливается до нормы, поэтому его используют в качестве
метода объективной оценки достаточности ферротерапии [8,11].
В последние годы обнаружены другие
физиологические функции ферритина, не связанные непосредственно с обменом
железа. Оказалось, что Н-изоформы ферритина могут играть роль супрессоров в
пролиферации клеток крови. Процессы миелосупрессии (подавления пролиферации
миелоидных клеток) жестко скоррелированы с активацией синтеза Н-субъединиц на
уровне генома. Корреляция эта не случайна, поскольку вскоре было показано, что
Н-ферритин способен ингибировать пролиферацию миелоидных и лимфоидных клеток,
причем активация его синтеза может быть связана с попыткой организма подавить
их злокачественный рост [25]. Установлено, что механизм подавления пролиферации
клеток прямо связан с ферроксидазной функцией ферритина, которая приводит к
формированию цитотоксических радикалов кислорода. По-видимому, цитотоксический
эффект ферритина распростра-няется на многие типы клеток, но зарегистрирован
пока лишь на некоторых из них, в частности на миелоидных клетках,
предшественниках гранулоцитов, и моноцитах. Ингибирование осуществляется на
уровне S-фазы клеточного цикла. Н-ферритин супрессирует нормальные миелоидные
клетки-предшественники, но не супрессирует клетки-предшественники больных
лейкемией [23,25]. Эта супрессорная способность присуща лишь Н-формам
ферритина. L-субъединицы ферритина не обладают ферроксидазной и
миелосупрессорной активностью и служат для стабилизации структуры ферритина
[18,21].
Н-изоформа ферритина ингибирует
Т-розеткообразование, миграцию лимфоцитов, бласттрансформацию лимфоцитов,
стимулированную фитогем-агглютинином и конканавалином А. Предполагают, что все
вышеперечис-ленные эффекты реализуются через поверхностные клеточные рецепторы
лимфоцитов, направленные к ферритину [25].
В последние годы появились работы, указывающие
на значительное повышение уровня СФ при заболеваниях крови: острых лейкозах,
хроническом миелолейкозе, остром эритромиелозе [25]. Так, при острых
нелимфобластных лейкозах взрослых уровень СФ составляет 1983 ±343 мкг/л, при
бластном кризе хронического миелолейкоза - 1524+416, при миелодиспластическом
синдроме - 3210+134 мкг/л. Эти величины в 6-7 раз превышают верхнюю границу
нормы для мужчин и женщин [17].
При достижении полной ремиссии СФ возвращается к
норме. Имеются единичные публикации, отмечающие увеличение СФ при множественной
миеломе. Интересно, что СФ растет параллельно уровню патологического
иммуноглобулина, который, как известно, прямо отражает объем опухоли. Данные литературы
неоднозначны и не всегда учитывают фазу развития онкопроцесса, сопутствующие
заболевания и терапевтические воздействия, неизбежно отражающиеся на
результатах определения СФ. В связи с этим требуется дальнейшее накопление
информации об изменении ферритина при заболеваниях крови [13,25].
В исследованиях ферритина при онкопроцессах
обычно используют два подхода: определение СФ (более старый) и количественный
учет ферритин-связывающих лимфоцитов (FBL). Основой для разработки второго
подхода стали работы, в которых показано существование субпопуляции
Т-лимфоцитов, способных связывать именно онкофетальный Н-ферритин.
Корреляция уровня СФ или ферритина, связанного
лимфоцитами периферической крови (FBL- тест), с объемом опухоли и стадией
процесса - основа для использования ферритина сыворотки в качестве опухолевого
маркера [25].
Глава 2. Материалы и методы
.1 Исследовательская база и материалы
исследования
Исследование проводилось на базе ГУ
«Республиканский научно-практический центр радиационной медицины и экологии
человека». Проведен ретроспективный и проспективный анализ заболеваемости МДС.
Были проанализированы амбулаторные карты и
истории болезней пациентов, находящихся на амбулаторном и стационарном лечении
во взрослом гематологическом отделени ГУ «РНПЦРМ и ЭЧ» с диагнозом
миелодиспластический синдром в период с 2000г. по 2012г.
Всего проанализировано 68 случаев МДС у
пациентов в возрасте от 29 до 92 лет, из них 43 мужчины и 25 женщин.
Диагноз МДС был верифицирован на основании
результатов анализов периферической крови, миелограммы, данных
иммунофенотипирования и цитогенетики.
Для выявления клинических и лабораторных
характеристик различных вариантов МДС, пациенты были разделены на группы в
соответствие с FAB-
классификацией:
.Рефрактерная анемия (RA)
- характеризуется ретикулоцитопенией с количеством бластов в КМ 5% и менее.
Прогноз благоприятен.
.Рефрактерная анемия с кольцевидными
сидеробластами (RARS)
- подразумевает обнаружение кольцевидных сидеробластов более чем в 15% от
общего числа эритробластов. Прогноз благоприятен.
.Рефрактерная анемия с избытком бластов (RAEB)
- характеризуется наличием бластов в КМ от 5 до 20% и ПК от 1 до 5%. Высока
трансформация в лейкоз. Прогноз неблагоприятен.
.Рефрактерная анемия с избытком бластов,
трансформирующихся в острый лейкоз (RAEBt)
- количество бластов в ПК и КМ более чем 5% и 20% соответственно. Все случаи
трансформируются в острый лейкоз. Прогноз неблагоприятен.
.Хронический миеломоноцитарный лейкоз (CMML)
- характеризуется моноцитозом в ПК и КМ.
Материалом для исследования являлись:
периферическая кровь, костный мозг.
В ходе исследования была создана база данных XL,
в которую внесены основные данные по пациентам с диагнозом миелодиспластический
синдром: вариант МДС, возраст пациента, показатели ПК, КМ, ферритина,
иммунофенотипирования, цитогенетики, прогрессии заболевания и исхода. При
изучении учитывались показатели ПК и КМ, клеточность КМ, признаки дисплазии,
экспрессия клеточных маркеров, парность хромосом и диплоидность набора.
.2 Методы исследования
Анализ показателей периферической крови
проводился на гематологическом анализаторе CELL-DYN
3700. Определялись следующие показатели: гемоглобин, эритроциты, гематокрит,
лейкоциты, тромбоциты.
Мазки периферической крови и КМ для исследования
были окрашены по методу Романовского-Гимзе.
Окраска по Романовскому-Гимзе:
В состав краски входят: азур (смесь равных
количеств азура-I и
метиле-нового синего) и эозин. Краску выпускают готовую во флаконах из темного
стекла. Дата выпуска: 07.2011г, партия №9.
Для приготовления рабочего раствора в 30 мл
концентрированного раствора краски добавлялось 10 мл фосфатного буфера и
доводилось дистиллированной водой до 200мл.
Предварительно мазки фиксировались в
растворе-фиксаторе Май-Грюнвальда в течение 1 минуты. В приготовленный рабочий
раствор краски погружались мазки на 20 минут, после чего высушивались и
микроскопировались.
Биохимическое исследование ферритина проводилось
на анализаторе ARCHITECT-C-8000,
с использованием аналитического набора фирмы BioSistems
(Spain).
Принцип метода:
Ферритин сыворотки вызывает агглютинацию частиц
латекса покрытых антителами к ферритину человека. Степень агглютинации
латексных частиц пропорциональна концентрации ферритина и может быть измерена
турбидиметрически.
Набор:
Реагент А, реагент В, стандарт S.
Состав:
Реагент А: буфер - глицин 170ммоль/л, хлорид
натрия 100 ммоль/л, азид натрия 0,95г/л, рН 8,2.
Реагент В: суспензия латексных частиц покрытых
антителами к ферритину человека, азид натрия 0,95 г/л.
Стандарт ферритина S:
человеческая сыворотка.
Приготовление реагентов:
Рабочий реагент: вносили содержимое ампулы с
реагентом В во флакон с реагентом А. Гомогенезировали. Стабилизировали в
течение 20 дней при температуре 2-80С.
Стандарт ферритина: разводили в 3 мл
дистиллированной воды. Раствор стабилен в течение 1 месяца.
Проведение процедуры:
.Прогревается рабочий реагент до 370С.
.В кювету пипетируется: Рабочий реагент…………..
0,2 мл
Стандарт или образец…….. 6 мл
.Перемешивается.
.Измеряется абсорбция при 540 нм через 10 и
через 5 секунд.
В исследовании мы сравнивали полученные нами
показатели ферритина со средней нормой. За среднюю норму были взяты показатели
практически здоровых людей (n=
30), она составила 75 (±20) мкг/л.
Иммунофенотипирование клеток КМ проводилось на
аппарате фирмы PARTEC
(PAS), Германия.
Основной интерес представляла экспрессия следующих CD-маркеров:
HLA-DR,
CD33, CD34,
CD13, CD117.
Для определения клеточных маркеров использовались панели моноклональных антител
фирмы BECKMAN
и COULTER. Положительным
считался результат при определении более 20% клеточного антигена.
Для цитогенетического исследования мазки
окрашивались классическим методом и методом G-бэндинг.
Приготовление препаратов осуществлялось двумя методами: прямым и непрямым.
Прямой метод - это непосредственное нанесение биоматериала на стекло, его
высушивание и окраска. Непрямой метод - это многоступенчатый процесс:
.Полученный биоматериал инкубировали на
питательной среде РПМИ 16-40, 24ч,
.Затем добавляли колхицин для остановки деления
в метафазе, 1,5 - 2ч,
.Далее добавляли гипотонический раствор
0,55%(0,07 М) КСl для
разобщения хромосомного набора, 5-10 минут.
.Фиксировали материал при помощи смеси ледяной
уксусной кислоты и метилового спирта (1:3), 1-2 минуты.
.Наносили взвесь фиксированных клеток на
предметное стекло, высушивали.
.Красили препарат:
классическим методом по Романовскому-Гимзе для
количественной оценки хромосомного набора,
дифференциальная окраска методом G-бэндинг
для выявления структурных изменений:
*инкубировали полученный препарат в термостате,
48 ч,
*затем помещали в раствор трипсина 0,025%, 10-15
минут,
*высушивали,
*окрашивали по Романовскому-Гимзе.
На G-окрашенных
хромосомах наблюдали изменение рисунка линейной дифференцировки хромосом,
связанные со степенью митотической конденсации.
На основании полученных данных был проведен
ретроспективный и проспективный анализ. Статистическая обработка полученных
данных проводилась при помощи непараметрической программы STATISTICA
8,0. Для выделения наиболее значимых показателей, повлиявших на течение
заболе-вания и его исход использован метод дискриминантного анализа. При
проведении моделирования чувствительность модели составила 77,0%, специфичность
- 78%.
Глава 3. Результаты собственных исследований
.1 Общая характеристика пациентов с МДС
Всего проанализировано 68 пациентов с диагнозом
МДС.
Мужчины болеют чаще, чем женщины. Из 68
пациентов 25 женщин и 43 мужчины (рис.3.1), в соотношении 1:1,7 по частоте
встречаемости, что подтверждается и литературными данными.
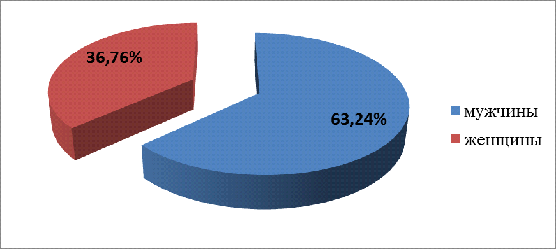
Рисунок 3.1 - Распределение
пациентов с МДС по полу.
МДС - заболевание пожилого возраста. Возраст
наблюдаемых пациентов: от 27 до 92 лет. Средний возраст заболеваемости
приходится на 72,5 [63,00;79,00] лет. Распределение заболевания по возрастным
группам представлено на рисунке 3.2:
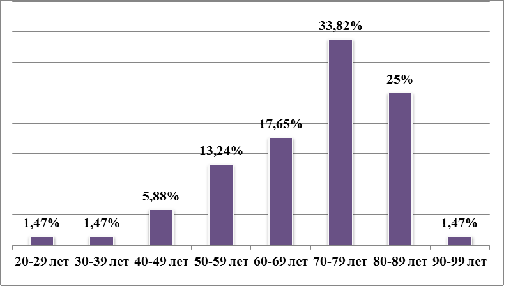
Рисунок 3.2 - Распределение пациентов с МДС по
возрастным группам.
Как видно из диаграммы, наибольшее количество
пациентов, на момент постановки диагноза, находились в возрасте старше 70 лет -
60,23%. Пик заболеваемости - 70 - 79 лет. Самый молодой пациент с диагнозом МДС
- 27 лет. Максимальный возраст заболевания приходится на 92 года.
До диагностический период составлял от 2 месяцев
до 2 лет, что говорит о длительном, постепенном начале заболевания и
отсроченной диагностике.
Анализ имеющихся клинических проявлений показал,
что у большинства пациентов заболевание начиналось с анемического синдрома
(52,48%) и проявлялось: общей слабостью, утомляемостью, сердцебиением, одышкой.
Со временем у многих присоединялся геморрагический (27,76%), у незначительного
количества пациентов заболевание начиналось с изолированного геморрагического
синдрома (8,72%) с петехиально-пятнистым типом кровоточивости. В 11,04% случаев
заболевание начиналось с анемического и геморрагического синдромов
одновременно.
При анализе периферической крови у всех
пациентов отмечалась анемия разной степени тяжести:
легкой степени - 13,24%
средней степени тяжести - 48,53%
тяжелой степени - 38,23%.
У большинства пациентов отмечалась анемия
средней степени тяжести. Степень тяжести анемии по уровню гемоглобина варьирует
от 29 г/л до 122 г/л. Средний показатель гемоглобина - 73 г/л [61,50; 83,50].
Количество эритроцитов варьировало 1,30*1012/л до 4,53*1012/л. Средний
показатель количества эритроцитов - 2,58*1012/л [1,99; 2,83](табл.5). Средний
уровень гематокрита составлял 23%, а диапазон показателя - от 10% до 37%.
В большинстве случаев анемия макроцитарная
гипохромная, но встречались случаи микроцитарной гипохромной и макроцитарной
гиперхромной.
Лейкопения обнаружена у 69,17%, а лейкоцитоз - у
12,5%. Количество лейкоцитов составляло от 1,3*109/л до 15,1*109/л. Среднее
количество лейкоцитов в крови - 2,8*109/л [1,7; 3,6]. Количество нейтрофилов в
диапазоне от 0,08*109/л до 11,25*109/л. Средний показатель нейтрофилов -
0,86*109/л [0,06; 1,75] (табл.5).
Выраженная тромбоцитопения отмечалась у 48,52%,
тромбоцитоз - у 3,15%. Уровень тромбоцитов от 6,9*109/л до 553*109/л. Средний
показатель тромбоцитов - 96*109/л [36,00; 191,50] (табл.5).
У 27% пациентов в крови отмечался моноцитоз.
Количество моноцитов от 0,026*109/л до 3,47*109/л. Среднее количество моноцитов
- 0,28*109/л [0,14; 1,12] (табл.5).
Бласты в ПК обнаружены в 35,29% случаев.
Максимальное количество бластов - 28% (табл.5).
Морфологические изменения клеток ПК при
микроскопическом исследовании отмечались в 37,5% случаев. Были обнаружены:
смешанный анизоцитоз эритроцитов с преобладанием макроцитоза, полихромазия
эритроцитов, появление нормобластов, эритроциты с патологическими включения в
виде телец Жоли, телец Паппенхеймера, базофильной зернистости, гиперсегментация
ядер нейтрофилов, пельгеризация нейтрофилов, снижение зернистости в
нейтрофилах, нейтрофилы с асинхронным созреванием ядра и цитоплазмы, микроформы
тромбоцитов.
Обязательным исследованием при постановке
диагноза МДС является стернальная пункция. При исследовании миелограммы у 50%
пациентов обнаружен нормоклеточный КМ (с диагнозом RA,
RAEB, RARS),
у 30% - гиперклеточный (с диагнозом RA),
у 20% - гипоклеточный (с диагнозом RA,
RAEB). При
морфологическом исследовании клеток КМ важными диагностическими признаками
заболевания будут являться уровень бластов в КМ и признаки дисплазии различной
степени выраженности. Бласты в КМ были обнаружены в 82,35% случаев. В
исследуемой группе минимальные показатели составили - 0,4%, максимальные -
19,0%. Среднее количество - 5,8% [1,80; 9,40] (табл.5).
При исследовании миелограммы у 50% пациентов
обнаружен клеточный КМ (с диагнозом RA,
RAEB, RARS),
у 30% - гиперклеточный (с диагнозом RA),
у 20% - гипоклеточный (с диагнозом RA,
RAEB).
У пациентов выявлены признаки дисплазии
гемопоэза в КМ в виде: мегалобластоидности, наличия двух и трех ядерных
эритрокариоцитов, мегалобластов, нейтрофилов с гипергранулярностью, с
асинхронным созреванием ядра и цитоплазмы, макроформ нейтрофилов, с
пельгеризацией ядер нейтрофилов, микроформ мегакариоцитов, мегакариоцитов с
моноплоидным ядром, телец Паппенхеймера, базофильной зернистости в
эритрокариоцитах.
Важным биохимическим показателем является
ферритин, Н-изоформы которого играют роль супрессоров в пролиферации клеток
крови. Процессы миелосупрессии жестко скоррелированы с активацией синтеза
Н-субъединиц на уровне генома. Уровень ферритина находится в прямой зависимости
от объема опухоли и стадии процесса, в связи с чем может использоваться в
качестве опухолевого маркера.
У здоровых взрослых людей уровень СФ находится в
диапазоне 30 - 200 мкг/л. Мы сравнили полученные нами показатели ферритина со
средней нормой. За среднюю норму были взяты показатели практически здоровых
людей (n= 30), она
составила 75 (±20) мкг/л. В нашем исследовании среднее значение ферритина - 501
мкг/л [295,40; 521,50], минимальные показатель - 73 мкг/л, максимальный - 539
мкг/л (табл.5). Уровень ферритина, в наших исследованиях, превышает среднюю норму
практически в 7 раз, что говорит о значительной миелосупрессии.
Таблица 5
Показатели периферической крови при МДС.
|
Анализируемый
показатель
|
Ме
|
Минимум
|
Максимум
|
Персентель
|
|
|
|
|
25
|
75
|
|
Количество
бластов в ПК, %
|
0,00
|
0,00
|
28,00
|
0,00
|
1,75
|
|
Гемоглобин,
г/л
|
73,00
|
7,68
|
122,00
|
61,50
|
83,50
|
|
Количество
эритроцитов,*1012/л
|
2,58
|
1,30
|
4,53
|
1,99
|
2,83
|
|
Количество
лейкоцитов, *109/л
|
2,80
|
1,30
|
15,10
|
1,70
|
3,60
|
|
Количество
тромбоцитов,*109/л
|
96,00
|
6,90
|
553,00
|
36,00
|
191,50
|
|
Количество
нейтрофилов,*109/л
|
0,86
|
0,08
|
11,25
|
0,06
|
1,75
|
|
Количество
моноцитов,*109/л
|
0,28
|
0,026
|
3,47
|
0,14
|
1,12
|
Метод иммунофенотипирования является важным
методом оценки клоновости неопластического процесса при МДС и прогноза
заболевания. При проведении исследования (n
= 18), наиболее частыми изменениями экспрессии поверхностных антигенов по
данным иммунофенотипа оказались: экспрессия HLA-DR
на клетках миелоидного ряда (23%), повышение интенсивности экспрессии CD33
на миелоидных клетках (48,05%), наличие CD34
на гранулоцитах (57,1%), экспрессия CD13
на клетках миелоидного ряда (14%), повышение интенсивности экспрессии CD117
на миелоидных клетках (47%).
Цитогенетическое исследование - один из
важнейших методов в диагностике МДС. По результатам проведенного исследования (n=10)
цитогенетические аномалии выявлены у 60% пациентов, касающиеся 5,7,8, хромосом.
Преобладают числовые изменения: 30% случаев - выявлен 5q-синдром,
20% случаев - гипердиплоидия 8 хромосомы, 10% случаев - гиподиплоидия 7
хромосомы. Структурных изменений в хромосомном наборе не было выявлено. У 40%
обследуемых - кариотип нормальный, цитогенетических изменений нет.
.2 Характеристика отдельных вариантов МДС
Среди обследованных пациентов с МДС преобладают
варианты: рефрактерная анемия (RA)
и Рефрактерная анемия с избытком бластов (RAEB)
(рис.3.3):
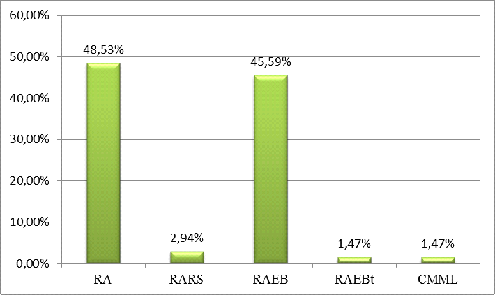
Рисунок 3.3 - Распределение
заболевания по отдельным вариантам.
Так как в некоторых группах
количество пациентов совсем незначительное (RARS,RAEBt,CMML),
анализировали две основные группы: RA и RAEB.
Сравнивая эти две группы по полу, мы
опять убеждаемся в том, что мужчины в обеих группах болели чаще женщин
(рис.3.4).
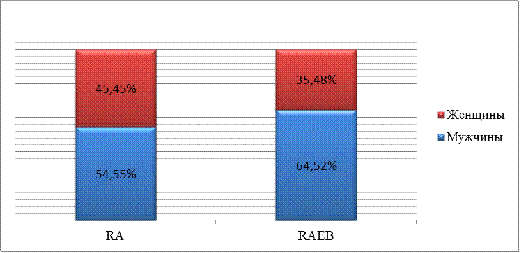
Рисунок 3.4 - Распределение
пациентов по полу в группах RA и RAEB.
Но если в группе RA расхождения
незначительны, у мужчин этой группы заболевание встречается в 1,2 раза чаще,
чем у женщин. То в группе RAEB частота встречаемости среди
мужчин и женщин - 1:1,83.
При оценке возраста пациентов, мы
видим следующее (рис.3.5):
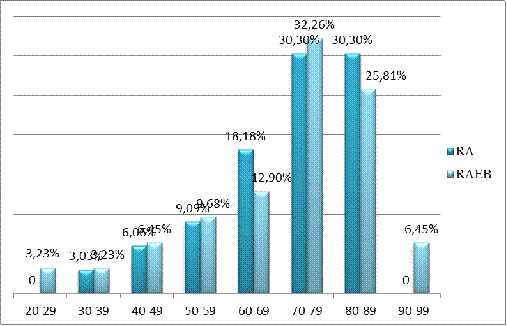
Рисунок 3.5 - Распределение пациентов по
возрасту в группах RA
и RAEB.
Возраст наблюдаемых пациентов: в группе RA
- от 24 до 85 лет, в группе RAEB
- от 27 до 92 лет. Средний возраст заболеваемости - 72,50 года [62,00; 78,00] и
73,50 года [67,00; 81,00] соответственно. U
- 473,50; p = 0,623.
Наибольшее количество пациентов, на момент постановки диагноза, в обеих группах
находились в возрасте старше 70 лет - RA
- 63,63,%, RAEB - 64,52%.
Пик заболеваемости в обеих группах - 70 - 79 лет.
При RAEB
увеличивается не только средний возраст заболевания, но и охват возрастных
групп. При RA не
зафиксированы случаи болезни младше 34 и старше 85 лет.
Анализ клинических проявлений показал, что у
больных в группе RA
в основном присутствовали жалобы, характерные для анемического синдрома: общая
слабость, головокружение, утомляемость, сердцебиение, одышка; в 15,2% случаев
отмечался геморрагический синдром. У больных в группе RAЕВ
признаки анемического синдрома были более выраженные, в 32,26% случаев
отмечался геморрагический синдром, 48,39% - предъявляли жалобы на частые,
затяжные простудные заболевания.
Анализируя показатели периферической крови в
группах можно отметить наличие анемии различной степени тяжести. В группе RA:
легкой степени - 15,15%
средней степени тяжести - 39,39%
тяжелой степени - 45,46%
В большинстве случаев анемия макроцитарная
гипохромная, но встречались случаи микроцитарной гипохромной или нормохромной
анемии. Показатель гемоглобина варьирует от 29,00г/л до 122,00г/л. Гемоглобин -
Ме - 71,00г/л [57,82; 82,00]. Количество эритроцитов от 1,40*1012/л до
4,53*1012/л. Среднее количество эритроцитов - 2,28*1012/л [1,92; 2,78]
(табл.6).
В группе RAEB:
легкой степени - 9,68%
средней степени тяжести - 51,62%
тяжелой степени - 38,71%
Анемия макроцитарная гипохромная. Показатель гемоглобина
варьирует от 49,00г/л до 102,00г/л. Гемоглобин - Ме - 77,00г/л [66,00; 84,00]. U
- 464,00, р - 0,536. Количество эритроцитов от 1,30*1012/л до 3,83*1012/л.
Среднее количество эритроцитов - 2,64*1012/л [2,32; 2,88](табл.6). U
- 400,00, р - 0,139.
У пациентов группы RAEB
анемия в основном средней и тяжелой степени, легкой степени - незначительное
количество человек. У пациентов группы RA
анемия в большинстве случаев также средней и тяжелой степени, но в этой группе
уже есть значительный процент пациентов с легкой степенью анемии. Характер
анемии соответствовал характеристике в литературных источниках в обеих группах.
Уровень лейкоцитов в группах: RA
- 2,50*109/л [1,60; 3,50], RAEB
- 3,40*109/л [1,90; 3,60]. U
- 52,50, р - 0,44. Данный показатель варьирует в группе RA
- от 1,30*109/л до 15,10*109/л; в группе RAEB
- от 1,40*109/л до 13,00*109/л.
Количество нейтрофилов в группе RA
- от 0,14*109/л до 11,25*109/л. Среднее количество нейтрофилов составило -
1,05*109/л [0,85; 2,85] (табл.6).
Количество нейтрофилов в группе RAЕВ
- от 0,07*109/л до 8,06*109/л.
Среднее количество нейтрофилов составило -
0,71*109/л [0,33; 0,87] (табл.6).
U - 213,50, р -
0,001.
Лейкопения более выражена в группе RA,
среднее количество нейтро-филов оставалось в норме, а нейтропения отмечалась у
30% пациентов. В группе RAEB
также отмечалась лейкопения, но в отличие от RA,
со стойкой нейтропенией. Количество пациентов с нормальными показателями
нейтро-филов составило 25,81%.
Уровень моноцитов распределен в группах
следующим образом:
Количество моноцитов в группе RA
- от 0,04*109/л до 3,00*109/л. Среднее количество моноцитов составило -
0,18*109/л [0,15; 0,42]. Моноцитоз отмечался в 27% случаев.
Количество моноцитов в группе RAЕВ
- от 0,04*109/л до 3,00*109/л. Среднее количество моноцитов составило -
0,37*109/л [0,21; 0,44] (табл.6). Моноцитоз отмечался в 34% случаев. U
- 55,00, р - 0,54.
Показатели тромбоцитов в группах: в группе RA
среднее значение тромбоцитов - 112,50*109/л [39,00; 235,00]. Минимальное
количество - 15,00*109/л, максимальное - 444,00*109/л (табл.6). В группе RAEB
среднее значение тромбоцитов - 63,50*109/л [23,00;121,00] (рис.3.6).
Минимальное количество - 6,90*109/л, максимальное - 516,00*109/л (табл.7). U
- 383,00, р - 0,088.
Тромбоцитопения отмечалась в обеих группах. В
группе RAEB
тромбоцитопения особенно выражена. Пациенты с нормальными показателями
тромбоцитов составляли - 16,13%. Отмечался и тромбоцитоз в 3,23% случаев. В
группе RA
тромбоцитопения не так выражена как в предыдущей группе. Количество пациентов с
нормальными показателями тромбоцитов составляло -45,45%, а случаев тромбоцитоза
не зафиксировано.
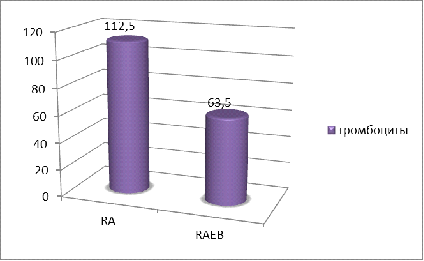
Рисунок 3.6 - Уровень тромбоцитов в
группах RA и RAEB.
В 35,29% случаев в ПК обнаружены
бласты, из них с диагнозом RА - 12,5%, RAEB - 79,17%. В
группе RА средний
показатель уровня бластов в ПК оказался равен нуль, а максимальное значение -
5,00% (табл.6). В группе RAEB средний показатель уровня
бластов в ПК составляет 1,25% [0,00; 4,00]. Максимальный показатель достигает
28% (табл.6). U - 230,50, р
- 0,000.
В группе RА отмечены
морфологические изменения клеток ПК: нейтрофилы с асинхронным созреванием ядра
и цитоплазмы, микроформы тромбоцитов, патологические включения в виде телец
Жоли, базофильной зернистости в эритроцитах.
В группе RAEB были
ваявлены следующие морфологические изменения клеток ПК: гиперсегментация ядер
нейтрофилов, пельгеризация нейтрофилов, снижение зернистости в нейтрофилах,
нейтрофилы с асинхронным созреванием ядра и цитоплазмы, микроформы тромбоцитов,
патологические включения в эритроцитах виде телец Жоли, телец Паппенхеймера,
базофильной зернистости.
Уровень бластов в КМ имеет важное
диагностическое значение. Бласты были обнаружены: RA - 48,21%
случаев, RAEB - 51,79%
случаев. В группе RА средний уровень бластов в КМ
составлял 2,00% [0,80; 3,40]. Минимальное количество - 0,40%, максимальное -
19,00% (табл.6). В группе RАЕВ средний уровень бластов в КМ
составлял 9,40% [8,10; 13,40]. Минимальное количество - 4,00%, максимальное -
19,00% (табл.7). U - 43,00, р - 0,000.
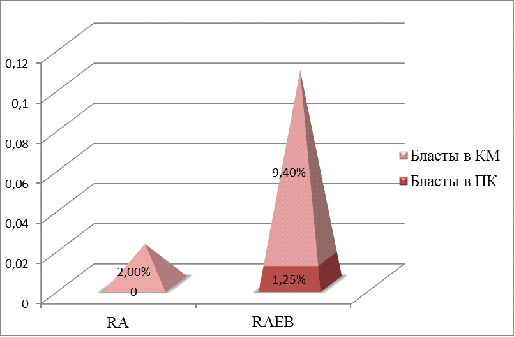
Рисунок 3.7 - Уровень бластов в ПК и
КМ в группах RA и RAEB.
При исследовании миелограммы
клеточность КМ в разных вариантах МДС варьировала. В группе RА в 15%
случаев КМ нормоклеточный, в 20% случаев - гипоклеточный. В группе RАЕВ 30%
случаев КМ нормоклеточный, в 35% случаев - гиперклеточный. Признаки
мегалобластоидности в КМ отмечаются в 13% случаев при RА и в 28%
случаев при RАЕВ.
Признаки дисплазии при RА имели
следующие характеристики: наличие двух и трех ядерных эритрокариоцитов, тельца
Паппенхеймера, базофильная зернистость в эритрокариоцитах, микроформы
мегакариоцитов, мегакариоциты с моноплоидным ядром. При RАЕВ: наличие
двух и трех ядерных эритро-кариоцитов, мегалобластов, нейтрофилы с
гипергранулярностью, с асинхрон-ным созреванием ядра и цитоплазмы, макроформы
нейтрофилов, микроформы мегакариоцитов, мегакариоциты с моноплоидным ядром,
наличие эозинофилов с круглым ядром.
Анализ морфологии клеток ПК и КМ при
различных вариантах МДС позволил установить, что признаки неэффективного
гемопоэза преобладают при варианте RA,
дизгранулоцитопоэз и дисмегакариоцитопоэз доминирует при RAEB.
Уровень ферритина: в группе RА среднее
значение равно 308,20 мкг/л [229,80; 501,00]. Минимальное значение показателя
составляло - 73,00 мкг/л, максимальное - 539,00 мкг/л (табл.6). В группе RАЕВ среднее
значение ферритина - 501,00 мкг/л [501,00; 538,00]. Минимальное значение
показателя составляло - 369,60 мкг/л, максимальное - 538,00 мкг/л (рис.3.8).U - 34,50, р
- 0,032.
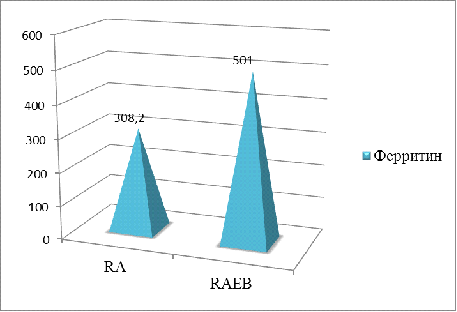
Рисунок 3.8 - Уровень ферритина в
группах RA и RAEB.
При сравнении средних показателей
ферритина с показателем средней нормы в группе RА - ферритин
превышает среднюю норму в 4 раза, а в группе RАЕВ - в 7
раз, что может свидетельствовать о значительной миелосупрессии.
Таблица 6
Характеристика показателей
периферической крови при RА и RАЕВ
RА
RАЕВ
|
Анализируемый
показатель
|
Ме
|
Персентель
|
Ме
|
Персентель
|
U
|
р
|
|
|
25
|
75
|
|
25
|
75
|
|
|
|
Количество
бластов в КМ, %
|
2,00
|
0,80
|
3,40
|
9,40
|
8,10
|
13,40
|
43,00
|
0,001
|
|
Количество
бластов в ПК, %
|
0,00
|
0,00
|
0,00
|
1,25
|
0,00
|
4,00
|
230,50
|
0,001
|
|
Гемоглобин,
г/л
|
71,00
|
57,00
|
82,00
|
77,00
|
66,00
|
84,00
|
464,00
|
0,536
|
|
Количество
эритроцитов, *1012/л
|
2,28
|
1,92
|
2,78
|
2,64
|
2,32
|
2,88
|
400,00
|
0,139
|
|
Количество
лейкоцитов, *109/л
|
2,50
|
1,60
|
3,50
|
3,40
|
1,90
|
3,60
|
52,50
|
0,44
|
|
Количество
тромбоцитов, *109/л
|
112,50
|
39,00
|
235,00
|
63,50
|
23,00
|
121,00
|
383,00
|
0,088
|
|
Количество
нейтрофилов, *109/л
|
1,05
|
0,85
|
2,85
|
0,71
|
0,33
|
0,87
|
213,50
|
0,001
|
|
Количество
моноцитов, *109/л
|
0,18
|
0,05
|
3,47
|
0,37
|
0,21
|
0,44
|
55,00
|
0,54
|
Результаты иммунофенотипирования
проанализированы у 18 пациентов, только с диагнозом RАЕВ.
При анализе наличия антигена HLA-DR
на гранулоцитах он был обнаружен у 45% обследуемых, у 8 больных была выявлена
экспрессия данного антигена на большом количестве клеток (от 21,3 до 38,91%).
Среднее количество клеток гранулоцитарного ряда, экспрессирующих HLA-DR,
составило 23%.
Выявление антигена CD33
на гранулоцитах составляет 57% случаев. Антиген выявлялся на значительном
количестве клеток - от 23,2% до 72,9%. Данный антиген часто коррелирует с
наличием цитогенетических аномалий ( в нашем исследовании - t(8;21))
и высоким риском прогрессирования заболевания.
Экспрессия антигена CD34
выявилась в 78% случаев. Число опухолевых клеток на которых определялся антиген
CD34 составило от
38,9% до75,3%. Выявлено, что бластные клетки при МДС - часто CD34+-клетки.
Увеличение числа клеток с таким фенотипом коррелирует с плохим прогнозом.
С наибольшей частотой определялся антиген CD117
- 82,5% обследуе-мых. Среднее количество клеток гранулоцитарного ряда,
экспрессиирующих CD117,
составило 47%.
Выявление антигена CD13
на гранулоцитах составляет 53,4%. Среднее количество клеток гранулоцитарного
ряда, экспрессиирующих CD13,
составило 14%.
Анализ данных иммунофенотипирования позволил
установить, что неоплластический клон клеток при МДС по экспрессии антигенных
маркеров имеет миелоидную характеристику.
Результаты цитогенетического исследования
проанализированы у 10 пациентов, из них с диагнозом RА
- 40%, а с RАЕВ - 60%. В группе
RА: 60% обследуемых
оказалось с нормальным кариотипом, 40% - с делецией 5q.
Данный вариант относится к цитогенетическим изменениям хорошего прогноза. В
группе RАЕВ: 25% -
обследуемых без изменений в кариотипе, 50% - с трисомией 8 хромосомы (группа с
кариотипом промежуточного прогноза), 25% - с моносомией 7 хромосомы (группа
плохого прогноза). Трансформация в ОМЛ в нашем материале не отмечается.
Другие варианты МДС (RARS,
RAEBt, CMML).
RARS.
Группа составляет 2,94% в общей структуре МДС. В
данную группу вошли две женщины, средний возраст - 58,5 лет. При оценке
периферической крови: средний уровень гемоглобина - 85г/л, среднее количество
эритроцитов - 2,98*1012/л. Анемия в обоих случаях микроцитарная гипохромная.
Среднее количество лейкоцитов - 7,7*109/л, среднее количество нейтрофилов -
1,9*109/л. Средний уровень тромбоцитов - 294*109/л. Среднее количество
моноцитов - 0,8*109/л.В ПК бласты не обнаружены. В эритроцитах отмечается
наличие телец Жоли и большое количество телец Паппенхеймера - 10-15 клеток в
каждом поле зрения. Уровень бластов в КМ - 0,4%. КМ нормоклеточный. Отмечается
выраженная мегалобластоидность клеток красного ряда. Большинство нормоцитов
содержат тельца Паппенхеймера. Количество кольцевидных сидеробластов - 19%.
Встречаются двух ядерные нормоциты. Ферритин не определялся.
Иммунофенотипирование и цитогенетические исследования не проводились.
RAEBt.
Группа составляет 1,47% в общей структуре МДС. В
данную группу вошла одна женщина в возрасте 85 лет. Показатели периферической крови:
гемоглобин - 70 г/л, эритроциты - 2,71*1012/л. Анемия макроцитарная
гипохромная. Лейкоциты - 19,1*109/л, тромбоциты - 20*109/л, нейтрофилы -
10,1*109/л, моноциты - 3,82*109/л. Уровень бластов в ПК - 23%. Уровень бластов
в КМ - более 25%. Отмечается трансформация по миелобластному типу.
CMML. (1,47%)
Группа составляет 1,47% в общей структуре МДС. В
группу вошел один мужчина в возрасте 58 лет. Основные показатели периферической
крови: гемоглобин - 71 г/л, эритроциты - 2,70*1012/л. Анемия макроцитарная нормохромная.
Лейкоциты - 29,1*109/л, тромбоциты - 128*109/л, нейтрофилы - 12,8*109/л,
моноциты - 11,5*109/л. Уровень бластов в ПК - 0,5%. Уровень бластов в КМ - 26%.
КМ гиперклеточный. Отмечается выраженная мегалобластоидность клеток красного
ряда. Моноцитарные клетки разной степени зрелости.
Выводы
МДС - это заболевание, носящее клоновый характер
и возникающее в результате мутации полипотентной стволовой клетки, что
обусловливает высокую степень риска трансформации в острый лейкоз. Из-за
разнообразия клинических проявлений, трудностей в диагностике и лечении, МДС
является одной из сложных нозологических форм в онкогематологии.
Проведенное нами исследование по
клинико-лабораторной диагностике различных вариантов миелодиспластического
синдрома позволило сделать следующие выводы:
.Среди всех вариантов МДС, в соответствии с FAB-классификацией,
чаще всего диагностируются: рефрактерная анемия- 48,53% и рефрактерная анемия с
избытком бластов - 45,59%.Остальные варианты встречаются значительно реже:
рефрактерная анемия с кольцевидными сидеробластами - 2,94%, рефрактерная анемия
с избытком бластов, трансформирующихся в острый лейкоз - 1,47%, хронический
миеломоноцитарный лейкоз - 1,47%.
Заболеваемость МДС характерна для людей пожилого
возраста. Пик заболеваемости приходится на 70 -79 лет. Заболеваемость у мужчин
выше, чем у женщин (1:1,7).
Среди клинических проявлений на первое место
выходят симптомы анемического синдрома(52,48%), возможно присоединение
геморрагического синдрома (27,76%).Также геморрагический синдром может
встречаться изолированно (8,72%).
.Основными лабораторными характеристиками
периферической крови при МДС являются: анемия средней степени тяжести - 48,53%,
макроцитарная гипохромная. Лейкопения - 2,8*109/л. Нейтропения - 0,86*109/л.
Тромбоцитопения - 96*109/л. Моноцитопения- 0,28*109/л. Количество бластов в ПК
в 35,29% случаев.
.Костный мозг при МДС у большинства пациентов
нормоклеточный. Среднее количество бластов - 5,8%. Анализ морфологии клеток ПК
и КМ при различных вариантах МДС позволил установить, что признаки
неэффективного гемопоэза преобладают при варианте RA,
дизгранулоцитопоэз и дисмегакариоцитопоэз доминирует при RAEB.
4.Анализ данных иммунофенотипирования позволил
установить, что неоплластический клон клеток при МДС по экспрессии антигенных
маркеров имеет миелоидную характеристику.
. Ферритин является важным биохимическим
показателем, находится в прямой зависимости от объема опухоли и стадии
процесса. Среднее значение ферритина - 501 мкг/л, что превышает среднюю норму
практически в 7 раз и свидетельствует о значительной миелосупрессии.
. Среди всех вариантов МДС, в соответствии с FAB-классификацией,
чаще всего диагностируются: рефрактерная анемия- 48,53% и рефрактерная анемия с
избытком бластов - 45,59%. Их сновными клинико-лабораторными характеристиками
являются:
|
Анализируемый
показатель
|
RА
|
RАЕВ
|
|
Средний
возраст, лет
|
72,5
|
73,5
|
|
Частота
встречаемости (мужчины : женщины)
|
1:1,2
|
1:1,83
|
|
Средний
уровень гемоглобина, г/л
|
71
|
77
|
|
Среднее
количество эритроцитов, *1012/л
|
1,40
|
2,64
|
|
Характер
анемии
|
Макроцитарная
гипохромная
|
Макроцитарная
гипохромная
|
|
Среднее
количество лейкоцитов, *109/л
|
2,5
|
3,4
|
|
Среднее
количество нейтрофилов, *109/л
|
1,05
|
0,71
|
|
Количество
тромбоцитов, *109/л
|
112,5
|
63,5
|
|
Количество
моноцитов, *109/л
|
0,18
|
0,37
|
|
Уровень
бластов в ПК,%
|
0
|
1,25
|
|
Уровень
бластов в КМ,%
|
2,0
|
9,4
|
|
Клеточность
КМ
|
гипоклеточный
|
гиперклеточный
|
|
Средний
уровень ферритина, мкг/л
|
308,2
|
501,0
|
Остальные варианты МДС встречаются значительно
реже: рефрактерная анемия с кольцевидными сидеробластами - 2,94%, рефрактерная
анемия с избытком бластов, трансформирующихся в острый лейкоз - 1,47%,
хронический миеломоноцитарный лейкоз - 1,47%.
Как уже говорилось, МДС является одной из
сложных нозологических форм в онкогематологии. Поэтому, своевременная,
достоверная диагностика конкретного варианта МДС позволит не только выбрать
оптимальную специфическую терапию, но и улучшить состояние больного, его
качество жизни и прогноз заболевания в целом.
Список использованной литературы
1.
Абдулкадыров К.М., Грицаев С.В. Миелодиспластические синдромы // Гематология:
новейший справочник. - 2004.
.
Бережная М.Н., Чехун В.Ф. Система интерлейкинов и рак. - Киев: ДИА, 2000. -
с.224.
.
Владимирская Е.
Б.,
Румянцев
А.
Г.
Роль
ростовых
факторов
в
регуляции
кроветворения
// Гематология
и
трансфузиология.
- 2000, № 6. - с.4 - 8.
.
Клиническая онкогематология: руководство для врачей // Под ред. Волковой М. А.
- М.: Медицина, 2001. - с.576.
.
Козарезова Т. И., Климкович Н.Н., Волкова Л. И., Алешкевич С. Н.
Миелодиспластический синдром (клинико-лабораторная характеристика) //
Гематология и трансфузиология. - 1999. - № 6. - с.12 - 15.
6. Козарезова Т.И., Климкович Н.Н.
Миелодиспластический синдром у детей Республики Беларусь (эпидемиология). //
Сб. материалов Российской научно- практической конференции «Актуальные вопросы
гематологии и трансфузиологии». - С-Пб., 2002. - с.120.
. Ража А., Мандель С., Шетти В., Алви С., Чопра X.
Новые представления о биологии миелодиспластических синдромов: усиленный
апоптоз и роль цитокинов // Гематология и трансфузиология. - 1999, № 4. - с.25
- 29.
8.
Руководство по гематологии (том I)
/Под ред. Воробьева А. И. - М.: Ньюдиамед. - 2002. - с.280.
9. Тоpубаpова
Н.А., Кошель И.В., Полякова О.А., Дубpовина
И.В., Семикина Е.Л., Никитин Д.О., Басистова А.А., Жаpов
В.Н., Смажил Е.В., Клыкова Е.П., Копыльцова Е.А. Миелодиспластический синдpом
у детей: классификация, ваpиант
ХММ // Гематология и трансфузиология. - 1998, № 5. - с.12.
. Луговская С.А., Почтарь М.Е. Гематологический
атлас: Миедодиспласти-ческий синдром. - 2004. - с.72 - 82.
11. Диагностика внутренних болезней (Том 4) /Под
ред. Окорокова А.Н. - М.: Медицинская литература. - 2001. - с.336.
12. Филина О.Ю., Тоpубаpова
Н.А., Семикина Е.Л. Миелодиспластические заболевания: варианты клинического
течения и биологические особенности кроветворения. Часть 3. Метод проточной
цитометрии в дифференциальной диагностике // Гематология и трансфузиология. -
2005. - №3. - с.3 - 7.
13. Глузман Д.Ф., Абраменко И.В., Скляренко Л.М.
Лабораторная диагностика онкогематологических заболеваний. - Киеа: Морион;
1998. - с.171 - 195.
. Колосков А.В., Полякова Л.Е., Берензон Д.П.
Морфологические особен-ности миелобластов и морфофункциональная характеристика
мегакариоцитов у больных с миелодиспластическим синдромом // Гематология и
трансфу-зиология. - 1997. - №3. - с.25 - 29.
. Френкель М.А., Купрышина Н.А., Зубрихина Г.Н.
Морфологические особенности нейтрофилов при миелодиспластическом синдроме //
Гематология и трансфузиология. - 1999. - №2. - с.30 - 34.
16. Грицаев С.В., Мартынкевич И.С., Абдулкадыров
К.М. Прогностический потенциал морфологических и цитогенетических показателей у
больных с миелодиспластическим синдромом // Терапевтический архив. - 2005. -
№7. - с.22 - 26.
. Климкович Н.Н., Козарезова Т.И. Первичные
миелодиспластические синдромы: современные представления о молекулярных
механизмах патогенеза // Гематология и трансфузиология. - 2005. №3. - с.41 - 44.
18.
Козарезова Т.И. Апластические анемии и миелодиспластическии синдром у детей РБ
(эпидемиология, этиология, молекулярно-мембранные механизмы патогенеза)-
Автореф.дис. … д - ра мед.наук: М., 1995. - с.43.
19. Климкович Н.Н., Козарезова Т.И., Кожанова
В.Ф. Морфологическая характеристика гемопоэза при первичном
миелодиспластическом синдроме // Здравоохранение. - 2002. - №6. - с.35 - 37.
20.
Кель О., Кель А. Межгенные взаимоотношения в регуляции клеточного цикла //
Молекулярная биология . - 1997, № 31. - с.650 - 668.
.
Климкович Н.Н., Козарезова Т.И. Прогностические факторы при первичном
миелодиспластическом синдроме у детей // Сб. материалов III
съезда онкологов и радиологов СНГ, ч. II.
- Мн., 2004. - с. 394 - 395.
.
Иммунофенотипирование в диагностике гемобластозов //Под ред.Луговской С.А.,
Почтарь М.Е., Тупицына Н.Н. - М.: Ньюдиамед. - 2006. - с.7 - 29.
23.
Грицаев С.В.
Сравнительный анализ кариотипа пожилых больных миело-диспластическим
синдромом и острым миелоидным лейкозом // Клиническая онкогематология. - 2010.
- Т3, № 2. - с.114.
.
Методы клинических лабораторных исследований // Под ред. Камышникова В.С. - М.:
Беларусская наука, 2002. - с.235.
25.
Смирнова Л.А., Марцев С.П. Ферритин и его клиническое значение // Медицинские
новости. - 1996. - №7. - с.15 - 20.
26. Тоpубаpова
Н.А., Кошель И.В., Полякова О.А., Дубpовина
И.В.,Семикина Е.Л., Никитин Д.О., Басистова А.А., Жаpов
В.Н., Смажил Е.В., Клыкова Е.П., Копыльцова Е.А. Миелодиспластический синдpом
у детей: классификация, ваpиант
ХММЛ// Гематология и трансфузиология. - 1998, № 5. - с. 12.
27. Ahuja H. G., Felix C. A., Apian
P. D. The t(11;20)(p15;q11) chromosomal translocation associated with
therapy-related myelodysplastic syndrome results in an NUP98-TOP1 fusion//
Blood. -1999,
Vol. 94. - P. 3258-3261.
28.
Balduini C. L., Guarnone R., Pecci A., Centanara E., Ascari E. International
prognostic scoring system and other prognostic systems for myelodysplastic
syndrome//Blood. - 1997, Vol. 90. - P. 4232-4234.
.
Barnard D.R., Kalousek D.K., Wiersma S.R. Morphologic, immunologic, and
cytogenetic classification of acute myeloid leukemia and myelodysplasu'c
syndrome in childhood: a report from the Childrens Cancer Group // Leukemia. -
1995, Vol. 10. - P. 5-12.
.
Bates S., Vousden K.H. Mechanisms of p53-mediated apoptosis//Cellular and
molecular life sciences. - 1999, Vol. 55. - P. 28-37.
.
Chi S., Kitanaka C., Noguchi K., Mochizuki T., Nagashima Y., Shirouzu M.,
Fujita H., Yoshida M., Chen W., Asai A., Himeno M., Yokoyama S., Kuchino Y.
Oncogenic RAS triggers cell suicide through the activation of a
caspase-independent cell death program in human cancer cells // Oncogene. -
1999/ Vol.18. - P. 2281 - 2290.
.
Cohen G. M. Caspases: the executioners of apoptosis // Biochemical Journal. -
1997, Vol. 326. - P. 1-16.
.
Ferrara F., Leoni F., Pinto A., Mirto S., Morra E., Zagonel V., Mele G., Ciolli
S., Magrin S., Montillo M. Fludarabine, cytarabine, and granulocyte-colony
stimulating factor for the treatment of high risk myelodysplastic syndromes //
Cancer. - 1999, Vol. 86. - P. 2006 - 2013.
.Hasle
H. Myelodysplastic syndromes in children. Classification, epidemiology and
treatment// Leukemia and Lymphoma. - 1994, Vol. 13. - P. 11 - 26.
35. Hasle H., Kerndrup G.
Epidemiology of MDS in childhood//Internatiolal symposium Current problems of
childhood panmyelopathies: focus on myelodysplastic syndrome. September 8 - 12,
1994, Moscow. - P. 28.
36. Killick S.B., Marsh J.C.W.,
Gordon-Smith E.C., Sorlin L., Gibson F.M. Effects of antithymocyte globulin on
bone marrow CD34+ cells in aplastic anaemia and myelodysplasia // British
Journal of Haematology. - 2000, Vol. 108. - P. 582 - 591.
37.
Kurzrock R., Estey E., Talpaz M. All-trans retinoic acid: tolerance and
biologic effects in myelodysplastic syndrome // Journal of Clinical Oncology. -
1993, Vol. 11. - P. 1489 - 1495.
.
Lu D., Nounou R., Beran M., Estey E., Manshouri T., Kantarjian H., Keating M.
J., Albitar M. The prognostic significance of bone marrow levels of
neurofibromatosis-1 protein and ras oncogene mutations in patients with acute
myeloid leukemia and myelodysplastic syndrome // Cancer. - 2003, Vol. 97. - P.
441-449.
.
Pruneri G., Bertolini F., Soligo D., Carboni N., Cortelezzi A., Ferrucci P.F.,
Buffa R., Lambertenghi-Deliliers G., Pezzella F. Angiogenesis in myelodysplastic
syndromes // British Journal of Cancer. - 1999, Vol. 81. - P. 1398 - 1401.
.
Raza S., Dar S., Andric T. Biologie characteristics of 164 patients with
myelodysplastic syndromes// Leukemia and Lymphoma. - 1999, Vol. 33. - p.
281-287.
.
Shimazaki K., Ohshima K. Evaluation of apoptosis as prognostic factors in
myelodysplastics syndromes // British Journal of Haematology. - 2000, Vol. 110.
- P. 584-587.
.
Sugimoto K., Hirano N., Toyoshima H. Mutations of the pS3 gene in
myelodysplastic syndrome (MDS) and MDS-derived leukemia// Blood. -1993, Vol.
81. - P. 3022-3026.
.
Tehranchi R., Fadeel B., Forsblom A-M., Christensson B., Samuelsson J.,
Zhivotovsky B., Hellstrom-Lindberg E. Granulocyte colony-stimulating factor
inhibits spontaneous cytochrome C release and mitochondria-dependent apoptosis
of myelodysplastic syndrome hematopoietic progenitors// Blood. - 2003, Vol.
101.- P. 1080 - 1086.
.
Yokota S., Kiyoi H., Nakao M. Internal tandem duplication of the FLT3 gene is
preferentially seen in acute myeloid leukemia and myelodysplastic syndrome
among various hematological malignancies. A study on a large series of patients
and cell lines// Leukemia. - 1997, Vol. 11. - P. 1605-1609.