Краун-соединения
Краун-соединения
Применение коронандов (краун-эфиров),
криптандов, подандов в качестве комплексообразователей катионов, анионов и
нейтральных низкомолекулярных веществ вызывает значительный научный и
практический интерес. Химия краун-соединений интенсивно развивается. В
настоящее время постоянно синтезируются новые молекулы со свойствами
краун-эфиров и открываются новые области их применения [1].
Краун-соединения получили такое название
благодаря своей химической структуре и форме комплекса, который образуется при
внедрении ионов металла в их полость. До сих пор нет строгого определения
понятия краун-соединений, но, в общем, их описывают как макроциклические
соединения, имеющие в своих циклических структурах в качестве электронодонорных
атомов гетероатомы, такие, как кислород, азот или сера, и обладающие
способностью включать в свою полость катионы [2].
Мультидентатные моноциклические лиганды с любым
типом донорных атомов называются коронандами (краун-соединениями), а термин
“краун-эфиры” следует оставить лишь за циклическими олигоэфирами, содержащими в
качестве донорных атомов исключительно атомы кислорода. В настоящее время
лиганды в соответствии с топологией их молекул подразделяют на классические
циклические олигоэфиры (краун-эфиры), моноциклические коронанды,
олигоциклические сферические криптанды и ациклические поданды.
Для того чтобы различать свободные краун-эфирные
лиганды и их комплексы с ионами металлов, были предложены названия коронанд для
свободного соединения и коронат для комплекса. Аналогично применяются названия
криптанд/криптат, поданд/подат [2].
Металлокомплексы с краун-соединениями -
уникальный тип координационных соединений, обладающих рядом интересных свойств,
в том числе имитирующих свойства природных веществ. В качестве краун-лигандов
выступают циклические и гетероциклические полиэфиры. Первыми представителями
таких соединений являются краун-эфиры с числом кислородных атомов не менее
трех, которые соединены между собой этиленовыми мостиками [3].
К краун-лигандам относятся также многочисленные
аналоги краун-эфиров и их производные - азакраун-эфиры, тиакраун-эфиры,
криптанды, сферанды, каликсарены, цилиндраны, поданды, лариаты и т.д. Они
являются синтетическими аналогами природных мембраноактивных антибиотиков [4],
подобно им захватывают в макроциклическую полость катионы и удерживают их за
счет ион-дипольных взаимодействий.
Первыми представителями аналогов краун-эфиров
можно считать диэфиры олигоэтиленгликолей, которые были синтезированы задолго
до получения краун-эфиров. Эти открытоцепные аналоги краун-эфиров получили
общее название - поданды [5]. Известна высокая комплексообразующая активность
подандов по отношению к ионам щелочных, щелочно-земельных [6] и редкоземельных
[7] металлов, радиоактивных элементов [8].
Поданды, имеющие на концах достаточно длинных
основных цепей функциональные группы и донорные центры, способны окружать
катионы уникальным образом. Весь скелет лиганда удерживается в конформации
поданда лишь за счет взаимодействий донорных атомов с катионом [2]. В
соответствии с принципом, определяющим, что высокая устойчивость комплекса не
означает одновременно большой селективности при комплексообразовании, поданды
проявляют селективность к различным катионам. Эта селективность определяется
такими специфическими чертами лиганда, как природа и число донорных центров,
жесткость цепи лиганда и особенно тип концевых групп. Следует подчеркнуть, что,
благодаря наличию сильных электронодонорных концевых групп, по своей
комплексообразующей способности поданды не только сравнимы с макроциклическими
соединениями, но в ряде случаев даже превосходят их [9]. При этом самые
распространенные поданды - фосфорилсодержащие [10]:

Рис. 1 - Фосфорилсодержащие поданды
Применение краун-соединений
Многие уникальные свойства
краун-соединений уже нашли свое практическое приложение. Здесь описаны новые
возможности и перспективы применения этих соединений [11].
Высокая растворимость
металлокомплексов с краун-лигандами в неполярных растворителях означает, что
противоионы существуют в виде «обнаженных» анионов, т.е. в несольватированном
или слабосольватированном состоянии. Поэтому гомогенные реакции с участием
труднорастворимых неорганических солей могут протекать в присутствии
краун-соединений с высокой скоростью, что позволяет проводить реакции, ранее
считавшиеся неосуществимыми. Неудивительно, что среди различных областей
применения краун-соединений наибольший прогресс достигнут в области
органического синтеза [1].
Краун-соединения способны
действовать в качестве межфазных катализаторов не только в обычных системах
жидкость - жидкость, но и на границе твердой и жидкой фаз. Такой межфазный
катализ, в котором нет необходимости использовать водный раствор неорганической
соли, является особого типа процессом, свойственным только краун-соединениям
[2].
Благодаря липофильности и малой
растворимости в воде краун-соединения весьма перспективны в качестве
экстрагентов для селективного извлечения, разделения и концентрирования
металлов. Однако трудности технологического характера не позволяют внедрить их
в эту область. Более значительные успехи достигнуты в создании практически
реализуемых конкурентоспособных высокоэффективных экстракционных систем для
разделения изотопов [11].
Результаты исследований показали,
что на коэффициент разделения заметное влияние оказывают тип аниона и природа
растворителя. При этом влияние аниона значительно усиливается в случае
малополярных растворителей, тогда как для полярных растворителей влиянием
аниона можно пренебречь.
Несмотря на кажущееся обилие данных
по изотопным эффектам, делать обобщающие выводы о влиянии различных факторов на
экстрагирующую способность систем на основе краун-соединений затруднительно.
Имеются противоречивые данные по одним и тем же системам. Разночтение чаще
всего вызвано тем, что данные ограничиваются измерением однократных эффектов,
что приводит к неоднозначным, невоспроизводимым и неточным величинам. Более
надежные результаты получаются при использовании различных способов
многократного умножения эффекта [11].
Наибольшее число исследований
посвящено разделению изотопов лития, хотя весьма обнадеживающие результаты
получены также при изучении процессов разделения изотопов водорода, натрия,
калия, магния, кальция, стронция, бария, цинка, церия, урана.
Для изотопов лития, магния и кальция
разработаны физико-химические основы технологических процессов, которые
позволяют уже сейчас достаточно эффективно решать технологические задачи,
связанные с многократным умножением однократных эффектов.
Показана принципиальная возможность
использования фосфорилподандов в качестве экстрагентов в обменных системах
разделения изотопов. Хотя фосфорилподанды и уступают краун-эфирам в
эффективности, но их доступность, а также широкие возможности модифицирования
позволяют проводить целенаправленный поиск экстрагентов среди этого класса
комплексов.
Большие успехи достигнуты в области
ионометрии с применением краун-соединений. Разработаны высокоэффективные
ионселективные электроды для определения различных ионов [11].
Благодаря фотосенсибилизирующим
свойствам комплексов с краунзамещенными фталоцианинами, наряду со способностью
некоторых из них хорошо растворяться в воде, открывается перспективность
использования данного класса соединений для фотодинамической терапии рака.
Выявлены также свойства, указывающие на потенциальную возможность применения
краун-соединений для избирательного удаления опасных для организма человека
ионов, регулирования ионного баланса, переноса кислорода, электронного
переноса, катализа при фотосинтезе, для решения задач охраны окружающей среды,
металлургии, электрохимии, атомной промышленности, электроники и других
областей науки и техники.
Структурная реорганизация
краун-молекул
В комплексах металлов с
краун-эфирами нет ни типично ковалентных, ни типично координационных связей
металл-лиганд. Несмотря на это краун-эфиры образуют удивительно устойчивые
комплексы с ионами щелочных металлов, которые до недавнего времени не считались
металлами-комплексообразователями.
Согласно принципу геометрического
соответствия, сформулированному Ч. Педерсеном [12], селективность краун-эфиров
при комплексообразовании зависит от соответствия размеров и форм
координируемого гостя (ионов) размерам и формам полости хозяина (макроцикла).
Следование этому принципу значительно упрощает задачу поиска наиболее селективных
реагентов и порождает надежды на возможность направленного конструирования
металлокомплексов и регулирования их свойств по заданному сценарию. Однако, как
показал накопленный опыт в области химии комплексов на основе макроциклических
лигандов, данный принцип неадекватно описывает и прогнозирует
комплексообразующие, ионселективные, экстракционные и другие свойства.
Например, в ацетонитриле устойчивость комплексов лития с 12-краун- 4 больше,
чем аналогичных комплексов натрия, тогда как в пропиленкарбонате и метаноле
картина меняется на противоположную. Заметное влияние на устойчивость
комплексов оказывает также тип аниона. Таким образом, кроме соответствия
размеров, факторами, определяющими устойчивость комплексов и селективность
краун-соединений, являются природа растворителя, тип аниона, состав образуемых
комплексов и др. [13]
Тем не менее, принцип соответствия
полезен, в частности при классификации комплексов по типам [14]. Например, по
классификации Даллея комплексы краун-соединений со щелочными металлами в
зависимости от соответствия размеров катиона-гостя и молекулярных полостей
лиганда разделяются на четыре типа (рис. 2):
Размеры катиона близки к размерам
макроциклических полостей, ионы расположены посередине плоских многоугольников,
образованных донорными атомами;
Размеры катионов больше размеров
лигандных полостей, в этом случае образуются сэндвичевые структуры или
наблюдается значительное смещение ионов из средней плоскости донорных атомов;
Размеры катионов меньше размеров
полостей, лиганды «обволакивают» один катион или в состав комплекса входят два
катиона;
Катионы металлов не координируют
лиганды или лиганды связаны не всеми донорными атомами.
Предложена и более детальная
классификация Пуниа [11], которая учитывает помимо принципа геометрического
соответствия характер катион-анионного взаимодействия:
Комплексы состава М:L = 1:1, в
которых катион включен в полость краун-эфира. Донорные атомы макрокольца
расположены практически планарно, а анион или молекула растворителя (если
таковая имеется) занимают аксиальные позиции координационного многогранника.
Так как в процессе комплексообразования ионная пара катион-анион остается
контактной, первый тип назван Пуниа инкапсулированными ионными парами;
Комплексы, в которых захваченный
полностью катион образует сольватно разделенную ионную пару, т.е. когда, кроме
донорных атомов лиганда, металл координирован молекулой (или молекулами)
растворителя, а анион находится во внешней координационной сфере (размер
катиона меньше полости краун-эфира). Пуниа выделил такие комплексы в отдельный
тип - неполностью инкапсулированные;
Комплексы, в которых катион
полностью изолирован и окружен только донорными атомами кислорода. Этот тип
координации назван обволакивающей инкапсуляцией;
Комплексы с соотношением М:L = 2:1.
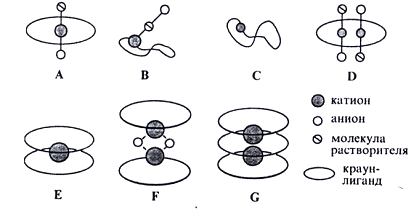
Рис. 2 - Различные типы комплексов
металлов с краун-лигандами (классификация по Пуниа): А - инкапсулированная
ионная пара (пара катион-анион остается контактной, M:L = 1:1), В - не
полностью инкапсулированная ионная пара (катион образует с анионом сольватно
разделенную пару, M:L = 1:1), С - обволакивающая инкапсуляция (M:L = 1:1), D -
соединение с соотношением M:L = 2:1, E - соединение “сэндвичевого” типа с
соотношением M:L = 1:2, F - димеры, связанные мостиковыми лигандами
(соотношение M:L = 1:1), G - трехпалубное “сэндвичевое” соединение (соотношение
M:L = 2:3)
Комплексы со стехиометрией 1:2 и
образование соединений “сэндвичевого типа” отмечены в случае, когда размеры
катиона превышает диаметр полости макроцикла. Комплексы с “обволакивающей
инкапсуляцией” и “сэндвичи” могут быть объединены в класс “полностью
инкапсулированных”.
Краун-соединения в свободном
состоянии в координационном отношении дезорганизованы. При вхождении иона
металла в полость макроцикла донорные атомы четко располагаются вокруг этого
иона и число возможных конформаций макромолекулы резко уменьшается. Наличие
бициклической структуры в криптандах и накопление жестких фрагментов в
гемисферандах еще более ограничивает число конформаций. Однако при этом не
исключаются конформации с малой или нулевой способностью к
комплексообразованию. Только конформации свободных сферандов и комплексов на их
основе практически одинаковые [11].
Развитие конформационного анализа и
особенно спектрально-конформационного анализа позволило на примере большого
числа комплексов (несколько сотен) проанализировать конформационные изменения,
происходящие в краун-лигандах под влиянием тех или иных факторов (например, под
влиянием периферийных заместителей, металла-комплексообразователя, анионов,
растворителя и др.). В результате был предложен принципиально новый
конформационный подход к модификации свойств макрогетероциклических соединений,
в основе которого лежит направленное изменение положения конформационного
равновесия, а, следовательно, комплексообразующих и ионселективных свойств
краун-лигандов. Так возникла идея закрепления с помощью периферийных
заместителей тех конформаций краун-эфиров, которые наиболее подготовлены к
селективному взаимодействию с заданным ионом металла. Такая предварительная
организация лиганда позволяет направленно выбирать нужные для селективного
связывания заданного катиона краун-соединения. Вместе с тем, жесткое
закрепление конформации краун-эфиров лишает их гибкости, которая позволяет этим
соединениям удивительно эффективно адаптироваться к возникшим условиям. Поэтому
при подборе боковых заместителей необходимо добиваться баланса между жесткостью
и гибкостью образующихся лигандов.
Разнообразие конформационных форм
краун-эфиров объясняет многочисленные примеры невыполнения принципа
геометрического соответствия Педерсена. Этот принцип нарушается в том случае,
когда под влиянием аниона, растворителя и межмолекулярных взаимодействий
вынужденно реализуется такой тип конформации лиганда, при котором более
выгодной становится координация с катионом, имеющим больший или меньший размер,
чем это можно ожидать из принципа геометрического соответствия.
Природа аномального алкильного
эффекта
В 1970-х гг. был описан эффект
«аномального арильного упрочнения», который заключался в увеличении
эффективности метилендифосфиндиоксидов R2P(O)CH2P(O)R2 как экстрагентов при
замене алкильных радикалов при атоме фосфора на более электроотрицательные
фенильные [15]. Это противоречило закономерности уменьшения эффективности монодентатных
экстрагентов с ростом электроотрицательности заместителей при атоме фосфора.
Аномальный эффект был обнаружен А.М.Розеном и соавторами при экстракции
актиноидов и лантаноидов из кислых сред диоксидами метилендифосфинов и
карбамоилметилфосфиноксидами. Трактовка аномального эффекта связывается с
бидентатной координацией катиона металла и объясняет увеличение константы
экстракции повышением стабильности извлекаемого шестичленного хелатного
комплекса с тетраарилметилендифосфиндиоксидом за счет подачи электронной
плотности с арильных колец на цикл комплекса [15].
Ранее было описано явление,
родственное аномальному арильному упрочнению. Оно было названо аномальным
алкильным эффектом [16]. Эффект был обнаружен при изучении комплексообразующей
способности фосфорилсодержащих моноподандов по отношению к катионам щелочных
металлов в смеси растворителей ТГФ-CHCl3 (4:1 по объему). Комплексообразующая
способность моноподандов с алкильными заместителями при атоме фосфора оказалась
ниже, чем у аналогичных соединений с фенильными и даже алкоксильными
радикалами.
Аномальный эффект характерен для
монофосфорильных бидентатных аналогов концевых групп моноподандов L1, L2, L3,
L4.

 L2
L2

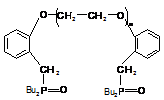 L4
L4
Объяснение, предложенное для эффекта аномального
арильного упрочнения, жестко привязано к структуре метилендиоксидов, поэтому
оно не может быть использовано в случае моноподандов с удаленными фосфорильными
группами.
Предлагается трактовка для аномального
алкильного эффекта, учитывающая различия в свободной энергии “предорганизации”
лигандов с алкильными и фенильными радикалами при атоме фосфора, что может быть
обусловлено различиями как в конформационном строении концевых групп этих
подандов, так и в барьерах вращения фенил- или алкилсодержащих групп R2P(O)
вокруг связи P-C.
Конформации свободного полидентатного лиганда
(например, поданда или краун-эфира) и лиганда, связанного в комплекс,
различаются [2]. На них построена концепция «предорганизации» свободного
лиганда, предложенная Д.Крамом, согласно которой образование комплекса
облегчается в случае «предварительно организованных» лигандов, имеющих
какие-либо структурные элементы жесткости. Даже частичная предорганизация
полиэфирных моноподандов, возникающая при переходе от глимовых к
тетрагидропирановым структурам, существенно повышает комплексообразующие
свойства по отношению к катионам щелочных металлов. Чем больше степень
структурной организации лиганда, тем меньше затраты свободной энергии на
предорганизацию лиганда при образовании комплекса и тем больше экспериментально
определяемая константа устойчивости комплекса в соответствии с выражением:
∆G0эксп = -RT lnK,
где K - экспериментально определяемая константа
устойчивости комплекса;
∆G0эксп - свободная энергия
комплексообразования;- температура;- универсальная газовая постоянная.
Свободная энергия комплексообразования является
интегральной величиной и может быть теоретически описана интегральными схемами,
с той или иной степенью полноты отражающими различные процессы при образовании
комплекса (сольватация и десольватация катиона, лиганда, комплекса и т.д.).
Отличие монодентатной координации от би- или полидентатной должно определяться
составляющей, учитывающей свободную энергию конформационной перестройки или
предорганизации полидентатного лиганда, которая предшествует или сопутствует
образованию комплекса. С этой точки зрения ∆G0эксп может быть описана как
разность двух составляющих - гипотетической «истинной» свободной энергии
образования комплекса би- или полидентатным лигандом в уже сформированной
комплементарной «гостю» конформации (∆G0компл) и указанной выше свободной
энергии предорганизации лиганда (∆G0предорг).
∆G0эксп = ∆G0компл - ∆G0предорг
Заместители при атоме фосфора будут оказывать
влияние на первый член (∆G0компл) через нуклеофильность фосфорильной
группы. На второй фактор (∆G0предорг) будет влиять не столько электронный
эффект заместителей, сколько их пространственное строение. При анализе влияния
природы радикалов при атоме фосфора на процесс комплексообразования двух
соединений в рамках одной реакционной серии необходимо использовать разность
величин ∆G0эксп для сравниваемых лигандов L' и L", которая
пропорциональна разности соответствующих значений lg K.
∆G0эксп(L') - ∆G0эксп(L") = ∆∆G0эксп
= ∆∆G0компл - ∆∆G0предорг
∆∆G0эксп ~ ∆ lg K = lg K(L') -
lg K (L")
В зависимости от соотношения величин ∆G0компл
и ∆G0предорг могут реализовываться различные варианты суммарного влияния
заместителей при атоме фосфора на константы устойчивости би- или полидентатных
лигандов.
∆∆G0компл > ∆∆G0предорг.
В этом случае закономерности влияния заместителей при атоме фосфора в поли- и
монодентатных лигандах будут аналогичны.
∆∆G0компл < ∆∆G0предорг.
Область аномальных явлений, которые не укладываются в рамки шкал электронных
эффектов. Заместители при атоме фосфора оказывают воздействие через
нуклеофильность атома кислорода фосфорильной группы на величину ∆G0компл,
с одной стороны, и косвенно влияют через энергию конформеров и барьеры вращения
вокруг связи Р-С на значение ∆G0предорг, с другой, причем последнее
влияние доминирует.
∆∆G0компл ≈ ∆∆G0предорг.
В этом случае для одного ряда лигандов может осуществляться как нормальный, так
и аномальный или смешанный типы влияния заместителей при атоме фосфора в
зависимости от природы катиона и растворителя. Последний является мощным
фактором, влияющим на процесс комплексообразования, и на энергию
конформационной перестройки.
Влияние заместителей при атоме фосфора на
комплексообразующую способность моноподандов
Для того чтобы охарактеризовать величину
аномального алкильного эффекта, была найдена при помощи эксперимента величина ∆lg
K для соединений L1 и L2 в интервале n=1-5 равная [15]:
∆lg K = lg K(Bu) - lg K(Ph).
Таблица 1 - Разность логарифмов
констант устойчивости (∆lg K) комплексов М+ L моноподандов L1 и L2
|
R
|
N
|
∆lg
K
|
Na
|
K
|
Rb
|
Cs
|
|
Bu
|
1
|
-1,8
|
-1,8
|
-1,4
|
-1,5
|
-1,3
|
|
Bu
|
2
|
-1,3
|
-2,5
|
-2,4
|
-2,2
|
-1,8
|
|
Bu
|
3
|
-2,4
|
-2,9
|
-2,7
|
-2,7
|
-2,4
|
|
Bu
|
4
|
-1,3
|
-2,6
|
-3
|
-3
|
-2,7
|
|
Bu
|
5
|
-0,9
|
-2,5
|
-3
|
-3
|
-2,6
|
Из результатов эксперимента (таблица
1) видно, что все величины ∆lg K имеют знак минус, что символизирует
аномальный эффект. Этот случай относится ко второму варианту (см. выше)
соотношения величин ∆∆G0 комплексообразования и предорганизации:∆∆G0компл
˂ ∆∆G0предорг. Наименьшие по абсолютной величине значения ∆lg
K характерны для комплексообразования коротких тетрадентатных соединений L1 и
L2 при n=1 со всеми катионами (первая строка таблицы 1). Относительно слабый
аномальный эффект обусловлен повышенной нуклеофильностью групп Р(О) бутильного
моноподанда L2 при n=1 по сравнению с его фенильным аналогом L1 при n=1. Это
должно приводить к увеличению ∆G0компл для соединения L2 при n=1
(поскольку «сильные» донорные центры составляют наполовину всех
комплексообразующих центров лиганда) и соответственно - к уменьшению
абсолютного значения ∆lg K. При n>1, наоборот, относительный вклад
«сильных» донорных центров в величину ∆G0компл для лигандов L2
уменьшается, что и сопровождается ростом абсолютных значений ∆lg K, т.е.
усилением аномального эффекта.
Лучше всего ряд наименьших
абсолютных величин ∆lg K характеризуют аномальные эффекты в случае
катиона лития. Катион Li+ обладает высокой плотностью заряда, поэтому он в
наибольшей степени чувствителен к «силе» координирующих центров лиганда. С этой
точки зрения именно повышенная нуклеофильность групп Р(О) бутилзамещенных
моноподандов L2 должна приводить к увеличению значения ∆G0компл в случае
катиона Li+ и, следовательно, к снижению аномального эффекта, что и отражается
в эксперименте. Наименьший эффект в ряду n=1-5 наблюдается для лигандов L1 и L2
при n=5. Гибкость этих моноподандов с длинной цепью обеспечивает максимальное
взаимодействие групп Р(О) с катионом Li+. Относительно малая роль групп Р(О)
обнаруживается в наиболее эффективных по комплексообразующей способности
лигандах L1 и L2 при n=3 (первый столбец таблицы 1), где в сильном
кооперативном взаимодействии с катионом Li+ велика относительная роль «слабых»
донорных центров (максимальный аномальный эффект в этом ряду).
Особенности конформационного
строения некоторых фосфорильных лигандов и их комплексов
Для бензольного сольвата моноподанда
L1 при n=2 методом рентгеноструктурного анализа установлены существенные
различия в конформационном строении как полиэтиленгликолевой цепи, так и
концевой фосфорилсодержащей группы. Если в поданде L1 при n=2 атомы кислорода
фосфорильной и о-оксифенильной групп в максимальной степени разобщены
(трансоидная конформация А), то в комплексе «гость-хозяин» фосфорильная группа
ориентирована внутрь псевдомакроциклической полости комплекса (конформация Б).
Таким образом, в процессе образования комплекса имеет место конформационный
переход А→Б с поворотом о-оксифенильных групп вокруг связи Р−С(о-оксифенил)
на ~180° (рассматривается конформационный переход только в концевых группах)
[15].
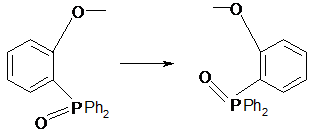
А Б
Аналогичные трансоидные конформации А имеются и
у орто-фосфорилзамещенных анизолов R2P(O)C6H4OMe-o (R = Ph, Bu).
Аномальный алкильный эффект связан с изменением ∆∆G0предорг
вследствие влияния заместителей при атоме фосфора на конформационное строение
концевых групп и барьеры вращения вокруг связи Р−С (о-оксифенил). Для
окисей диалкиларилфосфинов характерна биссекторная конформация В с компланарным
расположением фосфорильной группы и ароматического кольца.
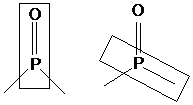
В Г
Бензольные кольца в
трифенилфосфиноксиде имеют различные торсионные углы - от положения, почти
компланарного фосфорильной группе, до 76°, причем угол поворота бензольного
кольца зависит даже от типа кристаллической модификации этого соединения. Такие
же неодинаковые углы поворота арильных колец вокруг связей Р−С (арил) в
моноподанде L1, однако о-оксифенильный радикал отклоняется от компланарности в
наименьшей степени.
Разнообразие ротамеров в
кристаллическом состояии является следствием малых энергитических различий
между ними. Несмотря на что в некоторых полифенильных соединений фосфора
отдельные арильные кольца почти компланарны фосфорильной группе, в среднем
отклонение кольца от плоскости С−Р=О составляет большую величину, чем в
случае биссекторного конформера В диалкиларилфосфиноксидов. В растворе между
ротамерами устанавливается равновесие, которое должно оказывать влияние на интегральную
величину ∆G0предорг. Условно равновесную смесь ротамеров можно
охарактеризовать усредненной гош-конформацией Г. Лиганд в такой конформации
находится в состоянии большей предорганизации (по Д. Краму) и переходит в
комплексообразющую форму Б с меньшей затратой энергии. Большую легкость
изомеризации Г→Б можно трактовать как следствие меньшего барьера
внутреннего вращения вокруг связи Р−С (о-оксифенил) дифенилфосфинильной
группы по сравнению с диалкилфосфинильной (большая вероятность перехода одного
конформера в другой). Последнее может быть обусловлено большими
пространственными препятствиями, создаваемыми такому вращению связями α- и β-С−Н-алкильных
радикалов при атоме фосфора по сравнению с орто-протонами фенильных
заместителей.
Поданды линейной и разветвленной
структуры
В работе [17] проводилось
исследование комплексообразования ионов щелочных металлов с полидентатными
лигандами линейной и разветвленной структуры. Изученные электронейтральные
фосфорорганические соединения (LI, LII и LIII) содержат нуклеофильные
фосфорильные (Р==О) и эфирные (С-О-С) группировки, способные взаимодействовать
с ионом металла за счет неподеленных электронных пар атомов кислорода.
Потенциально возможная максимальная
дентатность лигандов изменяется в ряду LI, LII, LIII от 3 до 7; количество
фосфорильных групп увеличивается от 2 до 4; структура лигандов усложняется от
условно цепочечной до разветвленной «триподандной».
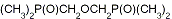
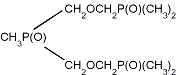
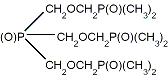
Рис. 3 - Примеры подандов линейной и
разветвленной структуры
Таблица 2 - Логарифмы эффективных
констант устойчивости комплексов ML+ щелочных металлов с лигандами LI, LII и
LIII
|
Лиганд
|
Li
|
Na
|
|
LI
|
2,2
|
1,3
|
|
LII
|
2,6
|
1,6
|
|
LIII
|
2,9
|
Данные таблицы показывают, что наиболее
устойчивые комплексы образуются с лигандом LIII, содержащим наибольшее число
донорных центров.
Согласно результатам работы [25] удлинение
полиэтиленгликолевой цепочки приводит к увеличению комплексообразующей
способности поданда по отношению к РЗЭ.
Квантово-химические методы расчета
Квантовая химия рассматривает молекулу как
образование из точечных ядер и электронов с целочисленными массами и зарядами.
Энергия молекулы имеет составляющие, связанные с кинетическими энергиями
каждого электрона и ядра и попарными энергиями их кулоновских взаимодействий.
Это должно быть надлежащим образом отражено в математическом формализме,
описывающем строение и свойства молекул [18].
Основное уравнение молекулярной квантовой химии
- независящее от времени нерелятивистское уравнение Шредингера:
НY({r, R})=ЕY({r,
R}),
где H-молекулярный гамильтониан;полная энергия
молекулы;
Y({r,R}) - молекулярная волновая функция, с
помощью которой в принципе могут быть рассчитаны химические свойства молекулы;
{r, R} - совокупность координат всех электронов
и ядер.
Иерархия методов квантовой химии
Приближенное решение электронного уравнения
Шредингера для молекулы может быть получено методом Хартри-Фока или одним из
методов, учитывающих электронную корреляцию, на основе физических и
математических законов. Для этого необходимо знать лишь фундаментальные
физические константы, число и сорт атомных ядер. Однако это недопустимо сложный
путь для массовых расчетов и на практике поступают иначе. Прежде всего,
используя приближение Борна-Оппенгеймера, задают структуру молекулы в виде
координат ядер. Затем, как правило, прибегают к приближению МО ЛКАО
(молекулярная орбиталь - линейная комбинация атомных орбиталей) и выбирают
аналитические функции, которыми будут аппроксимироваться АО (атомные орбитали).
Эти функции называются базисными (или просто базисом). Этим «внешняя»
информация и ограничивается и далее проводится строгий самосогласованный расчет
с вычислением всех необходимых интегралов (если возможно - с учетом симметрии молекулы).
Такой способ вычисления МО (молекулярных орбиталей) называется неэмпирическим
или ab initio, т.е. из первых принципов [19].
Степень строгости ССП-расчета, а точнее - его
сложности, может быть различной:в вычисления с разной степенью полноты можно
включить возбужденные электронные конфигурации, а можно ограничиться и методом
Хартри-Фока (т.е. однодетерминантным приближением). Число базисных функций,
используемых при неэмпирическом расчете, также может быть различным. Все
определяется целью расчета и имеющимися компьютерами. Чем более высокого уровня
расчет, тем более точные результаты могут быть получены с его помощью. Следует,
однако, четко сказать, что для многих целей достаточно ограничиться весьма
умеренным уровнем расчета. Более того, в случаях, когда исследуются ряды
соединений и важны лишь относительные, а не абсолютные значения энергии и
других характеристик, можно не вычислять интегралы, возникающие в схеме
расчета, а оценивать их значения на основании экспериментальной информации. При
этом оказывается, что значительную часть интегралов, считая их малыми, можно
приравнять нулю, соответствующим образом эффективно изменив величины
параметров. Такие методы называются полуэмпирическими:они значительно проще и
быстрее неэмпирических методов, а подчас дают и лучшие результаты. Следует
понимать, что это достигается за счет удачной параметризации и одновременно
определяет основной недостаток полуэмпирических методов - их слабую
переносимость от одного класса соединений к другому.
Имеется большое количество квантово-химических
программ, реализующих те или иные расчетные методы. Наиболее полное их собрание
существует в Фонде Квантово-химических Программ при химическом факультете
Университета Индиана (США).
Неэмпирические методы расчета
Неэмпирический метод Хартри-Фока и его
расширение за счет различных способов учета электронной корреляции реализован в
нескольких компьютерных программах, нашедших повсеместное распространение. Это,
прежде всего программы GAUSSIAN, GAMESS и CADPAC. Они ориентированы на работу с
мощными современными UNIX- станциями, хотя имеются и версии для IBM-совместимых
компьютеров. Практическое проведение расчетов по этим программам несложно и
проводится в диалоговом режиме. Необходимо указать метод расчета, задать
координаты ядер молекулы (это обычно делается с помощью, так называемой
Z-матрицы) и число электронов, а также выбрать базис, в котором будет
произведен расчет. Последнее играет важную роль:результаты, время, а также
стоимость расчета зависят от выбранного базиса [20].
Полуэмпирические методы расчета
Принципиально иное направление, сыгравшее
огромную роль в современном развитии химии, состоит в отказе от вычисления
одноэлектронных и двуэлектронных интегралов, фигурирующих в методе Хартри-Фока.
Вместо точного оператора Фока используется приближенный, элементы которого
получают из эмпирических данных. Соответствующие параметры подбирают для
каждого атома (иногда - с учетом конкретного окружения) и для пар атомов:таким
образом, они являются либо фиксированными числами, либо зависят от расстояния
между атомами. При этом часто (но не обязательно) предполагается, что
многоэлектронная волновая функция является однодетерминантной, а базисные
функции - симметричными ортогональными комбинациями ОСТ. Такие комбинации
получаются из исходных ОСТ с помощью преобразования. Расчет МО проводится
обычным итерационным путем.
Полуэмпирические методы обычно на много порядков
быстрее, чем неэмпирические, применимы к большим (часто к очень большим,
например - биологическим) системам и для некоторых классов соединений они дают
более точные результаты. Однако это достигается за счет специально подобранных
параметров, справедливых лишь в пределах узкого класса соединений. При переносе
на другие классы, те же методы могут дать абсолютно неверные результаты. Кроме
того, параметры расчета часто подбираются таким образом, чтобы воспроизводить
те или иные молекулярные свойства, поэтому придавать физический смысл отдельным
параметрам не следует.
С точки зрения подхода к решению молекулярного
уравнения Шредингера принципиально полуэмпирические и неэмпирические методы не
различаются. Для полуэмпирических методов характерна та же общая схема расчета,
что и для методов аb initio. Разница состоит в том, что каждая стадия расчета
существенно упрощается.
Указанные отличия определяют как достоинства,
так и недостатки полуэмпирических методов Основные преимущества по сравнению с
методами аb initio заключаются в следующем:
Скорость расчета увеличивается на несколько
порядков. В результате становится возможным расчет крупных органических
молекул, содержащих 100-200 атомов.
Для отдельных классов химических соединений (в
основном органических) точность расчета некоторых характеристик молекул
полуэмпирическими методами может оказаться не ниже и даже выше, чем методами аb
initio. Это связано с тем, что параметризация полуэмпирических методов
проводится по экспериментальным значениям определенных характеристик реальных
веществ и, естественно, эти значения воспроизводятся с высокой точностью.
Ограничения полуэмпирических методов имеют те же
корни, что и их достоинства. Такая особенность характерна для всех расчетных
методов квантовой химии и обусловлена невозможностью однозначного выбора
наиболее оптимального соотношения в параметрах «точность расчета» - «скорость
расчета» Для всех полуэмпирических методов характерны следующие недостатки:
точность расчета полуэмпирическими методами
ниже, чем методами ab initio в расширенном (биэкспоненциальном или более
сложном) базисе;
круг объектов, а также набор их физических
характеристик, которые могут быть изучены данным полуэмпирическим методом с
удовлетворительной точностью, ограничен особенностями использованной в этом
методе схемы параметризации. Обычно полуэмпирические методы используют для
расчетов органических веществ. Рассмотрение металлоорганических, в том числе
комплексных соединений требует специальных схем параметризации;
предыдущий недостаток, а также «нефизичность»
многих приближений полуэмпирических методов является причиной того, что в
рамках этих методов затруднительно, а часто и невозможно предсказать и
объяснить существование аномалий в свойствах, а также появление новых свойств,
не характерных для соединений рассматриваемого ряда [18].
Точность квантово-химических
расчетов химических свойств молекул
Точность неэмпирического расчета
Ошибки всех неэмпирических квантово-химических
методов возрастают при использовании «коротких» базисных наборов. Следующие
оценки точности расчета методами ХФ или МР2 справедливы для органических
молекул при базисе, не хуже чем DZP или 6-31G*:
Длины связей определяются с точностью 0.01 -
0.02 А ( для элементо- и металлоорганических соединений несколько хуже);
Электронная плотность - 10%, длины связей и
валентные углы - ~ 1%;
Энергии конформационных переходов (вращение и
барьеры инверсии) - < 2 ккал/моль (использование широкого базиса
обязательно);
Частоты колебаний для большинства ковалентных
связей, следующие из метода ХФ, систематически завышены на 10-12 % из-за
пренебрежения электронной корреляцией и ангармонизмом;
Энтропии ~ 0.5 энтропийной единицы (ккал/К
моль);
Реакции атомизации и гомолитического разрыва
связи описываются с большой ошибкой (25-40 ккал/моль);
Барьеры активации реакций также имеют большие
ошибки [19].
Точность полуэмпирических методов
Полуэмпирические методы параметризуются таким
образом, чтобы воспроизводить те или иные свойства молекул. Поэтому расчет всех
свойств на основании одного набора параметров безусловно надежным признан быть
не может. Этот общий и наиболее существенный недостаток полуэмпирической
квантовой химии.
Для молекул с закрытыми оболочками метод MNDO с
параметризациями AM1 и PM3 вполне пригоден, т. к. дает хорошие результаты для
молекулярной структуры и теплоты образования. Метод PM3 параметризован для
большего числа элементов, однако, параметры получены по очень ограниченному
набору экспериментальных данных. Средняя абсолютная ошибка для неводородных
атомов в PM3 составляет 0,036 А; она несколько больше в AM1. Ошибки в валентных
углах равны 3-4 градуса. Это хуже, чем в неэмпирических расчетах даже низкого
уровня, но время расчета несоизмеримо меньше. Ошибка PM3 - расчета энергии
образования органических молекул и переходных состояний органических реакций
составляет менее 5 ккал/моль [19].
Расчеты в HYPERCHEM
Пакет программ HyperChem позволяет проводить
неэмпирические и полуэмпирические расчеты электронных, спектральных и магнитных
характеристик молекул и межмолекулярных комплексов, а также вычислять энергию
переходных состояний комплексов, характеристики гидратной или сольватной
оболочки, производить простейшие расчеты характеристик кристаллов, расчеты
электронных и колебательных спектров.
Последняя версия 8.0 содержит, как и предыдущие
версии, графический редактор, большую базу данных по строению нуклеотидов,
полимеров, элементарных ячеек и способна считать методом функционала плотности
(DFT) с использованием разных обменно-корреляционных потенциалов.
Достаточно большой набор различных методов
молекулярной механики, полуэмпирических методов и всевозможные типы базисов,
используемые в ab initio расчетах, включая расщепленные и поляризованные,
обеспечивают широкий спектр вычислений. Программа написана под Windows, но, к
сожалению, ab initio расчеты требуют больших ресурсов оперативной и общей
памяти машины, большой мощности процессора. К тому же, по окончанию расчетов
электронного спектра с многократным КВ на выходе не указана структура
возбужденных состояний и провести отнесение подобно расчетам с однократным КВ
не представляется возможным. Несомненным преимуществом программы HYPERCHEM
является возможность наглядного изображения графической структуры молекулы и
изменение геометрических параметров при оптимизации химической системы, а также
визуализация полученных в результате расчетов молекулярных орбиталей,
относительной интенсивности электронных 0-0 переходов, потенциалов в двумерном
и трехмерном изображении и анимация колебательных мод. Большая база данных
позволяет построить белки, полимеры, фрагменты ДНК, кластеры металлов,
современные системы металлоорганических соединений.
Расчеты, проведенные с использованием ППП
HyperChem, позволяют исследовать поверхность потенциальной энергии посредством
точечных расчетов, оптимизации и молекулярно-динамического моделирования.
Энергии и производные энергии, такие как силы, действующие на атомы, необходимы
для конструирования поверхности потенциальной энергии [21].
Как молекулярно-механические, так и
квантово-механические методы основываются на приближении Борна-Оппенгеймера. В
квантовой механике уравнение Шредингера (1) дает волновую функцию и энергию
молекулы.
HΨ=EΨ(1)
где Н - молекулярный Гамильтониан, Ψ
- волновая
функция, Е - энергия. Молекулярный Гамильтониан состоит из следующих
операторов:кинетической энергии ядер (N) и электронов (Е), ядерного-ядерного
(NN) и электрон-электронного (ЕЕ) отталкивания и притяжения между ядрами и
электронами (NE).
Ядра значительно больше по массе, чем электроны.
В течение очень небольшого времени, когда движение тяжелых ядер незначительно,
электроны двигаются так, что их распределение равномерно. Это ведет к
приближению, заключающемуся в том, что распределение электронов зависит только
от фиксированных позиций ядер, а не от скоростей ядер. Это приближение
позволяет сделать два упрощения Гамильтониана. Выражение ядерной кинетической
энергии исключается. Так как ядерно-ядерное отталкивание постоянно для
фиксированной конфигурации атомов, это выражение тоже исключается. Гамильтониан
становится исключительно электронным.
Нэлектронный=(кинетическая
энергия)Е+(отталкивание) ЕЕ+ (притяжение) NE (2)
После решения электронного уравнения Шредингера
(2) с целью вычисления поверхности потенциальной энергии необходимо обратно
ввести ядерно-ядерное отталкивание (3).
ППЭ=Еэлектрона+(отталкивание)NN(3)
Получение поверхности потенциальной энергии
(ППЭ) с использованием этого уравнения требует решения для многих конфигураций
ядер. В молекулярной механике электронная энергия в явном виде не вычисляется.
Эти методы получают поверхность потенциальной энергии с использованием
уравнений силового поля. Уравнения силового поля представляют силовую энергию
косвенным образом, через параметризацию.
Методы молекулярной механики (ММ+, AMBER,
BIO+,OPLS)
Молекулярно-механическое силовое поле использует
уравнения классической механики для описания поверхности потенциальной энергии
и физических свойств молекул. Молекула описывается как набор атомов, которые
взаимодействуют друг с другом по простым аналитическим функциям. Это описание
называется силовое поле. Одним компонентом силового поля является энергия,
возникающая из сжатия и растяжения связей. Этот компонент часто аппроксимируется
как гармонический осциллятор и может быть рассчитан с использованием закона
Гука (7).
алкильный эффект молекула моноподанд
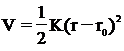 (7)
(7)
Связь между двумя атомами аналогична
пружине, соединяющей две массы. Используя эту аналогию, уравнение (7) дает
потенциальную энергию системы масс, V, и силовую константу пружины, K,
равновесное и смещенное расстояния атомов в связи r0 и r. Как K, так и r0
являются константами для специфической пары атомов, соединенных определенной
пружиной, они являются параметрами силового поля.
Потенциальная энергия молекулярной
системы в силовом поле является суммой индивидуальных компонентов потенциала,
таких как связевые, угловые и ван-дер-ваальсовы потенциалы. Энергия
индивидуальных связевых компонент (связи, углы, пространственные углы) являются
функцией отклонения молекулы от гипотетического соединения, которое
характеризуется минимальными величинами связевых взаимодействий. Абсолютная
энергия молекулы в молекулярной механике не имеет физического смысла, ее можно
использовать только для сравнения между молекулами. Энергии, полученные из
расчетов для данной структуры, относятся к энтальпиям молекул. Однако они не
являются энтальпиями из-за того, что тепловое движение и температурно-зависимые
вклады отсутствуют в выражении для энергии.
Молекулярно-механические расчеты не
могут описывать образование или разрыв связи, системы, в которых происходит
делокализация электронов или молекулярно-орбитальное взаимодействие играет
основную роль в определении геометрии или свойств.
ППП HyperChem предлагает четыре
молекулярно-механических силовых поля:MM+, AMBER, BIO+ и OPLS. Для запуска
молекулярно-механического моделирования нужно в первую очередь выбрать силовое
поле.
Уравнения силового поля для MM+,
AMBER, BIO+ и OPLS подобны в типах членов, которые они содержат:связные,
угловые, диэдрические, ван-дер-ваальсовы и электростатические члены. Существуют
некоторые отличия в форме уравнений, которые могут влиять на выбор силового
поля.
Метод MM+ разрабатывался для
органических молекул. Он учитывает потенциальные поля, формируемые всеми
атомами рассчитываемой системы, и позволяет гибко модифицировать параметры
расчета в зависимости от конкретной задачи, что делает его, с одной стороны,
наиболее общим, а с другой - резко увеличивает необходимые ресурсы по сравнению
с другими методами молекулярной механики. Ряд возможностей для изменения
параметров этого метода можно получить, выбрав кнопку Options в пункте выбора
Силового поля (Force field).
ММ+ является уникальным среди
силовых полей по тому, как оно обрабатывает связи и углы. Как связные, так и
угловые члены могут содержать выражения более высокого порядка, чем стандартные
квадратичные. Эти связные и угловые потенциалы выражают гармоническое движение
лучше, чем гармонический потенциал.
ММ+ также содержит член
растягивание-сгибание, называемый член Юри-Брэдли (Urey-Bradley). Другие
силовые поля в ППП HyperChem обычно оценивают несвязные взаимодействия для
атомов, разделенных тремя или большим количеством связей (1-4 и большие
взаимодействия). Член Юри-Брэдли включает 1-3 взаимодействия, которые являются
необходимыми для точного моделирования молекулы.
ППП HyperChem дополняет стандартное
силовое поле ММ2 посредством дополнительных параметров (силовых констант),
используя две альтернативные схемы. Это расширяет ряд химических соединений,
которые охватывает ММ+. ММ+ также обеспечивает ограничениями для расчета
несвязных взаимодействий и периодических граничных условий.
Метод AMBER разрабатывался для
белков и нуклеиновых кислот. В нем существует возможность выбрать опцию либо
учета всех атомов по отдельности, либо опцию объединенного атома, под которым
подразумевается группа эквивалентных атомов с одинаковыми свойствами. В
последнем случае несколько атомов, либо их групп, обрабатываются как один атом
с одним типом.+ разрабатывался для биологических макромолекул и во многом
повторяет AMBER.разработан для белков и нуклеиновых кислот. Он подобен AMBER,
но более точно обрабатывает не ковалентные взаимодействия.
Расчет методами квантовой химии с
предварительной оптимизацией методом молекулярной механики
При расчете химической системы,
программа HyperChem выбранную часть системы может рассчитывать каким - либо
методом квантовой химии, а остальную методом молекулярной механики. Программа
позволяет проводить подобные расчеты с использованием всех полуэмпирических и
неэмпирических квантово-химических методов. Необходимо помнить, что при
оптимизации геометрии системы только выделенная часть атомов будет менять свои
координаты в ходе оптимизации. Остальные атомы будут вносить свой вклад лишь
как некое статическое поле, генерируемое зарядами на атомах, вычисленными или
присвоенными им ранее.
Чтобы производить расчеты в
смешанном режиме, необходимо выделить ту часть атомов молекулы, которая будет
считаться как квантово- механическая. В случае, если некоторые из молекул лишь
частично выделены, необходимо убедится, что граничные атомы связаны с остальной
частью sp3-связями. Убедится в этом, можно используя параметр Extend to sp3 в
меню Select, до того, как запускать полуэмпирический расчет. Эта опция
распространит выделение рассчитываемой области по всем направлениям до тех пор,
пока выделенная часть не достигнет конечного атома молекулы, либо не найдет
sp3-sp3 связь.
ПП HostDesigner
Алгоритм LINKER
Алгоритм LINKER используется для
соединения трех различных молекулярных фрагментов вместе и делает
предварительный вывод, насколько хорошо результирующая структура свяжет
определенный пользователем гость. Два определенных пользователем фрагмента
комплекса соединяются связывающим фрагментом, молекулярной группой, взятой из
файла LIBRARY. Алгоритм LINKER может генерировать и оценивать на настольном
компьютере миллион структур в минуту и создавать файлы, содержащие описание и
координаты наиболее перспективных кандидатов. Этот процесс является первым
этапом дизайна новых молекул-хозяев, за которым следует более детальный анализ
отобранных структур другими методами [24].
Молекула-хозяин состоит из группы
связывающих участков. Таким образом, любая полидентатная молекула-хозяин может
быть разделена на два или более структурных фрагмента, которые мы обозначим как
компоненты молекулы-хозяина. Например, хорошо известный макроцикл 18-краун-6
может быть разделен на два компонента триглима, три компонента диглима или 6
диметилэфирных компонентов. Возможно определить структуру фрагмента комплекса,
другими словами, часть комплекса гость-хозяин соединением компонента хозяина с
гостем. При конструировании фрагмента комплекса гость может быть расположен
относительно компонента хозяина с получением комплементарной геометрии, т.е.
такой геометрии, которая дает наибольшее взаимодействие между связывающими
участками компонента-хозяина и гостем в действительном комплексе.
Алгоритм LINKER основывается на следующих
допущениях:
Существует оптимальная геометрия для
взаимодействия между каждым связывающим участком молекулы-хозяина и гостем.
Эта оптимальная геометрия независима
от других связывающих участков, которые могут присутствовать в
молекуле-хозяине.
Алгоритм LINKER строит новую
структуру молекул-хозяев соединением двух фрагментов комплекса связывающими
фрагментами, которые берутся из библиотеки.
Хотя утверждение, что расстояние
гость-хозяин с увеличением координационного числа увеличивается, справедливо,
предпочтительная ориентация компонента молекулы-хозяина по отношению к гостю
остается в основном неизменным. Оптимальные углы и двугранные углы могут быть
получены в результате изучения экспериментальной геометрии комплексов
гость-хозяин или через осторожное использование расчетов электронной структуры.
С учетом заданного координационного числа оптимальное расстояние гость-хозяин
может быть получено такими же методами. Таким образом, возможно определить
оптимальную геометрию для взаимодействия гостя с любым компонентом хозяина.
Отбор структур, построенных с
использованием алгоритма LINKER
Для отбора структур, полученных с
использованием алгоритма LINKER, используются два метода. Первый метод отбирает
структуры, учитывая их комплементарность, рассчитанную с использованием
геометрических параметров. Второй метод отбирает структуры, учитывая их
преорганизацию, основанную на расчете конформационной энергии структуры
молекулы-хозяина.
В ходе конструирования каждого
фрагмента комплекса гость располагается относительно компонента
молекулы-хозяина с учетом комплементарной геометрии связывающих участков этого
компонента. Когда два фрагмента комплекса соединяются, степень наложения двух
атомов-гостей является простым критерием для быстрого оценивания степени
комплементарности новой молекулы-хозяина. Оптимальная комплементарность
получается, когда среднее квадратичное отклонение (root mean squared deviation,
RMSD) расстояния между эквивалентными парами атомов двух гостей равно нулю,
другими словами, когда два гостя, реализуя оптимальную ориентацию связей по
отношению к каждому компоненту молекулы-хозяина, точно совпадают.
Алгоритм OVERLAY
Алгоритм OVERLAY реализует другой
подход, чем тот, который был использован в алгоритме LINKER. Он формирует
молекулу-хозяина путем объединения созданного пользователем фрагмента комплекса
связывающим фрагментом. Алгоритм строит новую структуру молекулы-хозяина в
результате формирования двух связей между одним фрагментом комплекса и одним
связывающим фрагментом.
Оценивание структур при использовании
алгоритма OVERLAY
Для оценивания структур, полученных
при использовании алгоритма OVERLAY, используются два метода. Первый метод
ранжирует структуры, основываясь на том, как хорошо связывающий фрагмент
подходит к фрагменту комплекса. Второй метод ранжирует структуры в терминах
преорганизации, основываясь на расчете конформационной энергии структуры
молекулы-хозяина. Степень соответствия измеряется средним квадратичным
отклонением несовпадения, RMSD, четырех точек фрагмента комплекса с четырьмя
точками связывающего фрагмента, где в каждом случае четыре точки являются
концами связывающих векторов. В дополнение к ранжированию структур при помощи
RMSD, второй метод используется для ранжирования структур посредством
преорганизации, основанной на расчете относительной конформационной свободной
энергии молекулы-хозяина. Этот подход идентичен подходу, используемому в случае
алгоритма LINKER.
Соединения РЗЭ с азот- и
фосфорсодержащими подандами
На основе работ [25, 26],
посвященных исследованию экстракции редкоземельных элементов из различных
растворов, можно выявить основные особенности лигандов, ориентированных на
комплексообразование с редкими землями.
Известно, что оксиды
(диалкилкарбамоилметил)диарилфосфинов (КМФО) являются эффективными реагентами
для извлечения редкоземельных элементов из азотнокислых растворов [27]. Было
показано, что введение в метиленовый мостик молекулы КМФО дополнительных
координирующих центров, таких как Ph2P(O)CH2- или Bu2NC(O)CH2-, не приводит к
увеличению коэффициентов распределения РЗЭ(III) при экстракции из азотнокислых
растворов и сорбции полимерными сорбентами, импрегнированными этими реагентами.
Экстракционная способность оксида (дибутилкарбамоилбутил)дифенилфосфина по
отношению к Am(III) и Eu(III) в азотнокислых средах не отличается от таковой
1,7-бис(дибутилкарбамоил)-1,7-бис(дифенилфосфинил)гептана, молекула которого
содержит две координирующие группы Ph2P(O)CHC(O)NBu2, соединенные через
метиленовые группы пентаметиленовой цепочкой [28].
Между тем объединение в одной
молекуле двух бидентатных Ph2P(O)CH2C(O)NH- фрагментов через амидный атом азота
приводит к заметному увеличению способности такого экстрагента извлекать из
азотнокислых сред ионы Am(III) и Eu(III) по сравнению с таковой оксида
(диизобутилкарбамоилметил)октилфенилфосфина [29]. Исследованы некоторые
закономерности межфазного распределения нитратов РЗЭ(III) между водными
растворами HNO3 и растворами в дихлорэтане фосфорсодержащих подандов I и II в
сравнении с их моноаналогом - оксидом (дибутилкарбамоилметил)дифенилфосфина III
[26].
Ph2P(O)CH2C(O)NH(CH2CH2O)nCH2CH2NHC(O)CH2P(O)Ph2,
n=1 (I), n=2 (II);P(O)CH2C(O)NBu2 (III).
Полученные данные показали, что
исследованные P,N-содержащие поданды с двумя бидентатными координирующими
фрагментами, соединенными через амидную группу полиэтиленгликолевой цепочкой,
проявляют более высокую экстракционную способность по отношению к ионам
металлов в азотнокислых растворах по сравнению с таковой оксида
(дибутилкарбамоилметил)дифенилфосфина III.
Функция желательности
Задачу оптимизации процессов,
характеризующихся несколькими откликами, обычно сводят к задаче оптимизации по
одному критерию с ограничениями в виде равенств или неравенств. В зависимости
от вида поверхности отклика и характера ограничений, для оптимизации
предлагается использовать методы неопределенных множителей Лагранжа, линейного
и нелинейного программирования, ридж-анализ и др. К недостаткам этих способов
решения задачи оптимизации следует отнести вычислительные трудности. В
частности, при описании поверхности отклика полиномами второго порядка решение
задачи на условный экстремум с применением неопределенных множителей Лагранжа
приводит к необходимости решать систему нелинейных уравнений.Поэтому одним из
наиболее удачных способов решения задачи оптимизации процессов с большим
количеством откликов является использование предложенной Харрингтоном [30] в
качестве обобщенного критерия оптимизации так называемой обобщенной функции
желательности D.
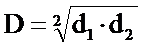
Для построения обобщенной функции
желательности предлагается преобразовать измеренные значения откликов в
безразмерную шкалу желательности d. Построение шкалы желательности, которая
устанавливает соотношение между значением у и соответствующим ему значением d,
является в своей основе субъективным, отражающим отношение исследователя к
отдельным откликам [31].
Для построения шкалы желательности
удобно использовать метод количественных оценок с интервалом значений
желательности от нуля до единицы, хотя возможны и другие варианты шкалы.
Значение d=0 соответствует абсолютно неприемлемому значению данного отклика, а
d=1 - самому лучшему значению отклика, причем дальнейшее улучшение его или
невозможно, или не представляет интереса.
Проведение расчета
Исходные данные
Перед началом моделирования необходимо решить
две задачи:определить строение будущего лиганда и подобрать донорные центры.
Из литературных данных известно, что
наиболее прочные комплексы РЗЭ образуют с подандами, сочетающими в себе
фосфорильные и аминные группы (Рис. 4). Это обусловлено несколькими причинами.
Во-первых, редкие земли имеют высокое сродство к азоту. Во-вторых, на атоме
кислорода фосфорильной группы сосредоточена достаточно высокая электронная
плотность, которая может быть еще увеличена посредством введения фенильных
заместителей. Кроме того, использование именно этих донорных центров дает
возможность для образования внутримолекулярной водородной связи, что в свою
очередь обеспечивает необходимую предорганизацию молекулы.
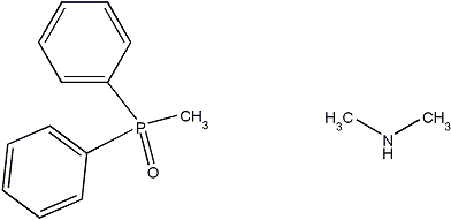
Рис. 4 - Фосфорильная и аминная
донорные группы
Начальные геометрические данные для
донорных групп были взяты из кембриджской базы данных ConQuest ver.1.13.
Использовались соединения с кодами:для аминной группы - HOJJIA [32], для
фосфорильной - UHAYOR [33]. Для аминной группы это длина связи Eu-N (2,586 Å) и валентный
угол C-Eu-N (110,59º).
Для
фосфорильной группы - длина связи Eu-O (2,435 Å), валентный
угол Eu-O-P (145,43º).
При сравнении подандов линейной и
разветвленной структуры можно сделать вывод, что наиболее устойчивые комплексы
образуются с лигандом, содержащим наибольшее число активных центров. Учитывая,
что координационное число европия равно девяти, в данной работе было отдано
предпочтение триподандам с десятью донорными центрами. Расположение активных
центров также выбиралось исходя из возможности образования внутримолекулярных
водородных связей, три фосфорильные группы на одной ветке и по три аминные
группы на двух оставшихся ветках. В качестве центрального атома выступает атом
азота.
Рис. 5 - Форма моделируемого лиганда
- триподанд
Используя представленные данные,
можно переходить к непосредственному подбору мостиков между донорными группами.
При этом необходимо получить две группы мостиков:
между двумя аминными группами;
между двумя фосфорильными группами.
Подбор мостиков
Подбор углеводородных мостиков между донорными
группами осуществлялся при помощи программного продукта HostDesigner. Так как
алгоритм Linker позволяет задавать геометрические характеристики только для донорных
центров и не дает возможности настраивать расположение центров друг
относительно друга, расчеты проводились с использованием обоих алгоритмов.
Рис. 6 - Подбор мостика между
донорными группами с применением алгоритма Linker
При подборе мостиков алгоритмом
Overlay в пределах 5 % варьировались длины связей Eu-O и Eu-N. Начальные
значения углов N-O-N, P-O-P и N-O-P приравнивались 50 градусам, что более или
менее соответствует взаимному расположению десяти активных групп вокруг
центрального атома европия, и изменялись программой HostDesigner в пределах 10
%.
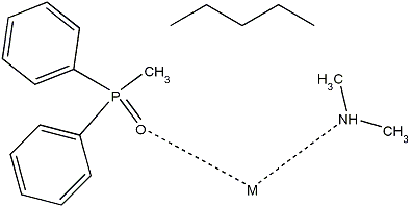
Рис. 7 - Подбор мостика между
донорными группами с применением алгоритма Overlay
Результаты расчетов
В результате работы программы
HostDesigner было получено несколько сотен мостиков, отсортированных по
значениям RMSD и энергии конформации в следующем виде:
1:RMSD=0.209,nrot=2,E=_0.620_cis-bicyclo[4.4.0]decane
:RMSD=0.212,nrot=2,E=_5.292_bicyclo[3.3.1]non-2-ylidine
:RMSD=0.214,nrot=2,E=_4.232_bicyclo[3.3.1]nonane
:RMSD=0.215,nrot=2,E=_1.060_trans-bicyclo[4.4.0]decane
:RMSD=0.216,nrot=2,E=_1.060_1,1-dimethylcyclohexane
:RMSD=0.217,nrot=2,E=_1.060_trans-1,3-dimethylcyclohexane
:RMSD=0.218,nrot=2,E=_1.060_trans-bicyclo[4.3.0]nonane
:RMSD=0.218,nrot=3,E=_1.150_propane
:RMSD=0.220,nrot=2,E=_0.840_cyclohexene
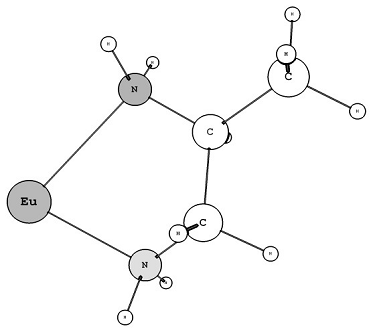
Рис. 8 - Пример пропилового мостика
между двумя аминными группами
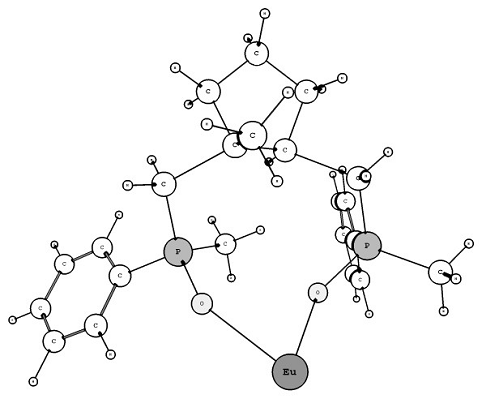
Рис. 9 - Пример
метилциклопентилового мостика между двумя фосфорильными группами
Рис. 10 - Пример комплекса с учетом водородной
связи (этиленовый мостик между фосфорильными группами и бутиленовый - между
аминными группами)
Для уменьшения количества полученных результатов
в первую очередь были отсеяны непредельные углеводороды и слишком экзотичные
структуры. Затем мостики отбирались по принципу наименьших значений энергии
конформации и среднеквадратичного значения несоответствия RMSD. С этой целью
применялась функция желательности, которая позволяет учесть оба этих параметра.
Таблица 3 - Реперные точки для
построения шкалы желательности
|
Удовлетворительное
значение (d=0,8)
|
Неудовлетворительное
значение (d=0,2)
|
|
RMSD
|
0,2
|
0,8
|
0,62
|
4,00
|
Значения обобщенного критерия желательности
рассчитывались отдельно для каждого типа мостиков:между двумя фенильными
группами, между двумя аминными группами, между центральным атомом азота и
фенильной группой. После этого полученные значения обобщенного критерия
желательности принимались за частные критерии желательности и служили исходными
данными для расчета полной функции желательности, сочетающей в себе все
геометрические и энергетические параметры всех рассчитанных мостиков.
Таблица 4 - Наилучшие значения
обобщенных критериев желательности для различных комбинаций мостиков
|
Мостик
между аминными группами
|
Мостик
между фосфорильными группами
|
D
|
|
пропан
|
этан
|
0,71
|
|
циклопентан
|
этан
|
0,71
|
|
бутан
|
этан
|
0,69
|
|
пропан
|
метилциклопентан
|
0,63
|
|
циклопентан
|
метилциклопентан
|
0,63
|
|
бутан
|
метилциклопентан
|
0,62
|
В виду имеющихся ограничений в программе
HostDesigner на присоединение мостиков к атому фосфора, дополнительно были
построены мостики с меньшим количеством атомов углерода, чем можно получить,
используя ПП HostDesigner.
Для дальнейших расчетов использовалась программа
HyperChem. С ее помощью методом молекулярной механики ММ+ были определены
энергии полученных комплексов и рассчитаны энергии предорганизации
соответствующих лигандов. Энергия предорганизации рассчитана методом
SinglePoint. Структуры отсортированы по величине энергии с учетом и без учета
водородной связи.
Таблица 5 - Значения энергий
предорганизации для различных комбинаций мостиков
|
Мостик
между аминными группами
|
Мостик
между фосфорильными группами
|
Энергия
предорганизации, кДж/моль
|
|
1*
|
1
|
55,5
|
|
этан
|
1
|
59,3
|
|
этан
|
2
|
87,1
|
|
пропан
|
этан
|
98,2
|
|
этан
|
этан
|
98,8
|
|
этан
|
3
|
101,5
|
*цифра указывает на количество атомов углерода
непосредственно между двумя атомами фосфора и двумя атомами азота
Таблица 6 - Значения энергий
предорганизации с учетом водородных связей для различных комбинаций мостиков
|
Мостик
между аминными группами
|
Мостик
между фосфорильными группами
|
Энергия
предорганизации, кДж/моль
|
|
бутан
|
этан
|
-84,3
|
|
циклопентан
|
этан
|
-27,8
|
|
этан
|
1
|
-27,7
|
|
1
|
1
|
-20,5
|
|
этан
|
пропан
|
-19,4
|
|
пропан
|
5,1
|
Для проверки расчетов, проведенных методом
молекулярной механики, были выполнены дополнительные расчеты полуэмпирическим
методом PM-3. Полученные данные пропорциональны результатам
молекулярно-механических расчетов.
Таблица 7 - Значения энергий предорганизации,
рассчитанных методом PM-3
|
Мостик
между аминными группами
|
Мостик
между фосфорильными группами
|
Энергия
предорганизации, кДж/моль
|
|
бутан
|
этан
|
638
|
|
циклопентан
|
этан
|
565
|
|
этан
|
1
|
296
|
|
1
|
1
|
278
|
|
этан
|
пропан
|
586
|
|
этан
|
2
|
495
|
Полученные структуры
После объединения всех полученных результатов были
выделены шесть структур, имеющих лучшие значения RMSD, энергии конформации и
энергии предорганизации, как с учетом, так и без учета водородных связей.
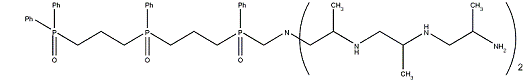



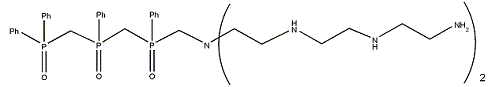

Выводы
В качестве активных центров выбраны фосфорильные
и аминные донорные группы;
Расчетными методами найдены наиболее подходящие
для комплексообразования с европием мостики;
Произведен отбор подандов на основании их
энергий предорганизации;
Получены структуры перспективных
комплексообразователей для атома европия.
Литература
1. Хираока
М. Краун-соединения. М.:Мир, 1974, 363 с.
. Химия
комплексов “гость-хозяин”. Синтез, структуры и применения. Под ред. Ф.Фегтле,
Э.Вебер. М.: Мир, 1988, 511 с.
3. Pedersen
C.J., J. Amer. Chem. Soc.
1970, v.92, p. 391.
4. Овчинников
Ю.А., Иванов В.Т., Шкроб А.М. Мембрано-активные комплексы. М.:Наука, 1974.
. Маркович
И.С., Дзиомко В.М. Ж. Всес. Хим. Общ. Им. Д.И. Менделеева, 1985, т.30, №5, с.
562-570.
. Цивадзе
А.Ю., Левкин А.В., Бондарева С.В. и др. Журнал неорганической химии. 1993.
Т.38, №7 с.1351; Reinoso-Garsia M.M., Dijkman A., Verboom W. et al. European
Journal of Organic Chemistry. 2005. №10. P. 2131.
. Туранов
А.Н., Карандашев В.К., Бондаренко Н.А. Радиохимия. 2006. Т.48, №2. С. 158.
. Туранов
А.Н., Карандашев В.К., Баулин В.Е. Радиохимия. 2007. Т.49, №3. С. 226.
. Еврейнов
В.И., Вострокнутова З.Н., Баулин В.Е. Журнал общей химии. 1989. Т.59, №1. С.
67.
. Буслаева
Т.М., Крылова Е.А., Волчкова Е.В., Громов С.П., Сидоренко Н.И. Известия вузов.
Цветная металлургия. 2008. №6. С. 30.
. Цивадзе
А.Ю. Рос. хим. журнал им. Д.И. Менделеева, т. 40, №4-5, с. 43-54, 1996.
12. Pedersen
C.J. Journal of American Chemistry Society. 1967, v.89, p.7017.
. Цивадзе
А.Ю., Варнек А.А., Хуторский В.Е. Координационные соединения металлов с
краун-лигандами. М.:Наука, 1991, 398 с.
. Цивадзе
А.Ю. Успехи химии, т. 73, №1, с. 6-25, 2004.
. Цветков
Е.Н., Евреинов В.И., Бондаренко Н.А., Сафронова З.В. Журн. общ. хим., т. 66,
вып. 7, с.1081-1092, 1996.
. Евреинов
В.И., Баулин В.Е., Вострокнутова З.Н. и др. Журн. общ. химии, т. 65, вып. 2, с.
223-231, 1995.
. Синявская
Э.И., Яцимирский К.Б., Цветков Е.Н., Крон Т.Е. Журнал неорганической химии.
1982. Т.27. №6. С. 1387.
. Блатов
В.А., Шевченко А.П., Пересыпкина Е.В. Полуэмпирические расчетные методы
квантовой химии. Учебное пособие. Самара:Универс-групп. Издание второе, 2005.
. Минкин
В.И., Симкин Б.Я., Миняев Р.М. Квантовая химия органических соединений.
М.:Химия, 1986.
. Минкин
В.И., Симкин Б.Я., Миняев Р.М. Теория строения молекул. М.:Высшая школа, 1979.
. Семенов
С.А. Пакет прикладных программ HyperChem. Квантовая механика. Учебное пособие.
М.:МИТХТ им. Ломоносова, 2006, 30 с.
. Hay
B.P.; Firman T.K. HostDesigner:a program for the de novo structure-based design
of molecular receptors with binding sites that complement metal ion guests.
Inorg. Chem. 2002, 41, 5502-5512.
23. HostDesigner is available at
no charge from the following website:<http://hostdesigner.emsl.pnl.gov>.
24. Семенов
С.А. Молекулярный дизайн комплексообразователей редких и рассеянных элементов с
использованием программного продукта HostDesigner (часть 1). Учебное пособие.
М.:МИТХТ им. М.В.Ломоносова, 2011 - 77 с.
. Туранов
А.Н., Карандашев В.К., Бондаренко Н.А. Журнал неорганической химии. 2008. Т.
53, №11. С. 1923-1931.
. Туранов
А.Н., Карандашев В.К., Бондаренко Н.А. Радиохимия. 2006. Т.48. №2. с. 158-163.
. Чмутова
М.К., Литвина М.Н., Прибылова Г.А. и др. Радиохимия. 1999. Т. 41, №4. С. 331-335.
. Кочетков
Н.Е., Койро О.Э., Нестерова Н.П. и др. Радиохимия. 1986. Т.28, №3. С. 338-345.
. Arnaud-Neu
F., Bohmer V., Dozol J.-F. et.al. Journal of Chemistry Society, Perkin Trans.
2. 1996. P.1175-1182.
. Harrington
E.C. Industrial Quality Control. 1965. V.21. N 10. P.494-498.
. Семенов
С.А. Планирование эксперимента в химии и химической технологии (часть 2).
Учебное пособие. М.:МИТХТ им. М.В.Ломоносова, 2005, 29 с.
. Ai-Mei
Zhu, Qin-Yan Jin, Ding-Xian Jia et.al. European Journal of Inorganic Chemistry.
2008. P.4756.