Важность прецизионного определения структур в неорганической химии
Важность
прецизионного определения структур в неорганической химии
Джереми К. Бердет Перевод д-ра хим. наук С. И.
Троянова
Введение
Неорганическая химия занимается изучением
соединений, которые могут содержать в своем составе фактически любой элемент
периодической системы, и вследствие этого она чрезвычайно разнообразна. Излишне
говорить, что характеристика нового неорганического или металлоорганического
соединения не полна без рентгеноструктурного определения его строения. С другой
стороны, химики-органики нередко удовлетворены уже тогда, когда им удается
провести измерение спектра ЯМР или другое спектроскопическое исследование.
Действительно, сравнение материала настоящей главы и гл. 16 выявляет, ряд
весьма важных различий между этими двумя областями химии. К настоящему времени
органическая химия - гораздо более разработанная область знаний по сравнению с
химией неорганической. В результате появляется относительно меньшее число новых
структурных типов органических соединений по сравнению с новыми типами
координации или координационной геометрии для неорганических или
металлоорганических соединений. Даже в своем несколько несовершенном виде
структурная неорганическая химия - гораздо более широкая область, чем ее
органический аналог. Вследствие этого неорганики и органики используют
прецизионные структурные определения совершенно различным образом. В то время
как химика-органика интересуют довольно малые изменения расстояний в молекуле
при замещении одного или нескольких атомов в исходном соединении, неорганик,
переходя от системы к системе, имеет дело с гораздо более широким диапазоном
изменения длин связей и углов. В неорганической химии метод молекулярных
орбиталей достаточно хорошо разработан для объяснения такого широкого спектра
наблюдений, так что многие эксперименты в действительности проводятся для
проверки существующих теорий.
Мы должны иметь также в виду, что строение
неорганической молекулярной системы, определенное дифракционными методами для
кристалла, может быть несколько отличным от такового для свободной молекулы в
газовой фазе или для сольватированной системы в растворе. Межмолекулярные силы
могут оказывать существенное влияние на тонкие детали строения. Протяженные
твердотельные структуры переходят в раствор обычно лишь за счет образования и
разрыва химических связей. Так, оксиды МО2 и МО3 переходных металлов начала
периодов растворяются в воде, давая разнообразные комплексные полиметаллаты,
формулы которых зависят от рН и существенно отличаются от формул исходных
оксидов. В газовой фазе молекулы существуют в виде короткоживущих и
реакционноспособных частиц. Устойчивые фрагменты таких протяженных систем в
газовой фазе обнаруживаются, как правило, только в тех случаях, когда
„оборванная связь" на конце фрагмента замыкается на другие атомы, что,
естественно, изменяет общую стехиометрию. Известный пример - структура
адамантана C10H16,
содержащего фрагмент из десяти атомов углерода, вычлененных из кубической
структуры алмаза с насыщением валентностей углерода за счет присоединения атомов
водорода. К числу впечатляющих примеров из области неорганической химии
относятся молекулы М9O2
и М11O3 (М = Rb,
Cs), содержащие
соответственно два и три октаэдра М6О с общими гранями (см. [72] и ссылки в
этой работе). За исходную родственную структуру можно принять Cs2O
(анти-CdX2), которая
содержит слои из таких октаэдров, имеющих общие ребра. В противоположность
ситуации в органической химии здесь вообще заранее не очевидно, что частицы с
такой стехиометрией должны быть устойчивы. Это показывает, что
химикам-неорганикам необходимы совершенно иные теоретические подходы, способные
обеспечить их рабочими моделями в этой области химии. Эта глава представляет
собой сугубо личный взгляд на некоторые интересные структурные проблемы
неорганической химии, показывающие ее разнообразие и возможности.
1.
Необычные молекулы и твердые тела)
Выше мы уже указывали на структурное
разнообразие неорганической химии, где точные структурные определения служат
надежной опорой для исследователя. Несколько примеров должно продемонстрировать
всю важность того, насколько существенно иметь действительно хорошие и надежные
данные для обоснования существования новых типов молекул и твердых тел.
Например, открытие ферроцена в 1950-х годах опровергло все прежние
представления химиков о возможных типах молекул. Молекула ферроцена содержит
два плоских циклопентадиенильных кольца (Ср), образующих сандвич с атомом
железа, и, разумеется, определение строения было решающим шагом на пути ее
признания химиками [40]. И сегодня еще студенты спрашивают, каково
координационное число атома железа: десять или два. Современный подход в рамках
метода молекулярных орбиталей [1] предполагает, что электронное состояние лучше
всего описывается концепцией шестикоординированного атома железа. В течение многих
лет во всех изученных мономолекулярных частицах МСр2 плоскости колец были
параллельны, за исключением лишь производных таких металлов, как олово или
свинец, имеющих неподелённую пару электронов. На рис. 20.1 показано строение
недавно синтезированного производного самароцена SmCp2
[41], в котором полностью метилированы оба кольца (обозначается как SmCp2*).
В результате кристаллоструктурного исследования установлена молекулярная
структура, в которой в отличие от ферроцена кольца не параллельны (тогда как FeCp2*
содержит параллельные кольца).
Такое угловое строение (угол измеряется между
нормалями к плоскостям обоих колец, см. рис. 20.1) может возникать по
нескольким причинам.
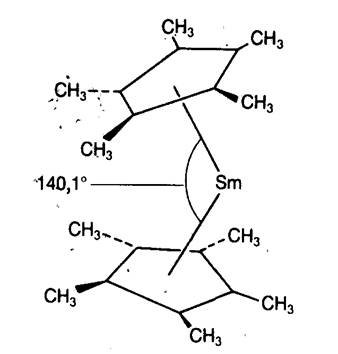
Рис. 20.1. Угловой сандвич в структуре декаметилсамароцена
[41].
Во-первых, соединение может не отвечать формуле SmCp2*
и содержать присоединенный к металлу атом, который не был найден при
структурном определении. Легкий атом (например, водород), координированный
тяжелым атомом самария, вполне может быть пропущен, и, кроме того, не быть
обнаруженным обычным химическим анализом. Известно, например, что в структуре H3NbCp2
имеются три связи М-Н и кольца находятся под углом друг к другу. Однако для
рассматриваемого случая подобная возможность должна быть отвергнута, так как
имелись химические доказательства, что это не HxSmCp*.
Другой причиной могла бы быть “агостическая” связь между атомом металла и одним
или несколькими атомами водорода из органической части молекулы. В дальнейшем
мы опишем этот тип связи, но он может быть исключен из рассмотрения в данном
случае, поскольку расстояния Sm...
Н в целом слишком длинны (наиболее короткое около 2,8 Å)
для того, чтобы этот тип взаимодействия был существенным. Третья возможность
состоит в стереохимической активности f-орбиталей
атома самария (SmII).
Обнаружение нететраэдрических структур у частиц ML4
при электронной конфигурации dn
(n>0) (например,
структура бабочки для молекулы Сг(СО)4) долго рассматривалось как указание на
стереохимическую активность d-орбиталей
(см., например, [1]). Однако f-орбитали
считаются в целом стереохимически неактивными. Одно из доказательств их
незначительной энергетической значимости следует из существенно меньшей ширины
линий в электронных спектрах комплексов с f-орбиталями
по сравнению с шириной линий для d-орбитальных
комплексов. Еще одна возможность состоит в том, что молекулярное строение
определяется так называемыми эффектами упаковки, т. е. равновесием между
внутримолекулярными и межмолекулярными силами в кристалле. Такое объяснение,
однако, исключается, потому что сходное угловое строение было установлено для CaCp2*
и YCp2* в газовой фазе.
И наконец, объяснение, которое кажется наиболее приемлемым, состоит в том, что
кольца сближаются вследствие притяжения за счет сил Ван-дер-Ваальса, т. е. тех
же сил, которые удерживают вместе молекулы углеводородов в кристаллическом
состоянии. Такая модель предполагает, что связи атома металла в значительной
степени ненаправленны. Эта модель интересна тем, что стерические взаимодействия
между лигандами, присоединенными к одному и тому же атому, которые обычно
рассматриваются как отталкивание, в данном случае являются силами притяжения.
На рис. 20.2 показана часть опубликованной
структуры цеолита, которая в свое время привлекла значительное внимание [42],
но впоследствии оказалась ошибочной (см. [71] и список литературы к этой
статье). Цеолиты представляют собой вещества с общей формулой MxS1-xAlxO2,
где металл М (в данном случае одновалентный) необходим для компенсации заряда.
Эту роль могут также играть двух- и трехвалентные металлы. Не содержащий
металла каркас состоит из двухкоординированных атомов кислорода и
приблизительно тетраэдрически координированных атомов кремния или алюминия.
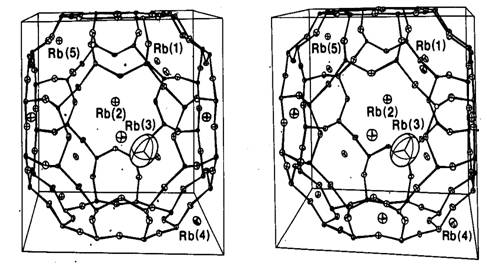
Рис. 20.2. Первоначальное (ошибочное)
представление [42] о „плавающем” ионе
Rb+ (Rb(3))
с „нулевой” координацией в клетке цеолитового каркаса.
Отличительная особенность таких материалов
состоит в наличии в каркасе больших каналов и пустот. Электроположительные
атомы металла всегда координированы атомами кислорода каркаса. Поэтому
представлялось удивительным обнаружение цеолитового каркаса, в котором наряду с
несколькими ионами, координированными атомами кислорода, присутствовал ион, как
бы плавающий в центре клетки каркаса. С позиций электростатики такой ион должен
был бы находиться в положении неустойчивого равновесия, что (если бы это было
правильным) представляло значительный интерес для химии. Однако оказалось, что
частично проблема для этой и некоторых других сходных структур связана с
неправильным выбором пространственной группы при кристаллографическом
уточнении. В цеолитах такого типа атомы кремния и алюминия полностью упорядоченны
в структуре. Если х = 0,5, то, согласно экспериментальным данным, атомы одного
типа не могут находиться в соседних позициях. Этот принцип „самоизбегания”
известен как правило Ловенштейна [51]. Таким образом, различная симметрия
соответствует случаю, когда каркас из атомов неметалла содержит атомы Si/Al
со средней рассеивающей способностью (усредненное статистическое
распределение), или случаю, когда эти атомы упорядоченны. До настоящего времени
не обнаружено ни одной структуры с „нулевой” координацией для ионов этого типа
[65]. Использование неверной пространственной группы представляет распространенную
ошибку в рентгеноструктурных работах. Во многих случаях это не столь важно для
химии, но все же довольно часто различия оказываются существенными (см.,
например, [56]).
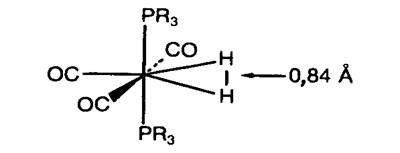
Рис. 20.3. Строение молекулы W(РRз)2(СО)3(Н2),
R = i
Рг (изопропил) [50].
На рис. 20.3 показано строение синтезированной в
1984 г. молекулы, в которой впервые наблюдался новый тип координации диводорода
переходным металлом [50]. Молекула W(PPri3)2(CO)3(H2)
особенно интересна тем, что, находясь в кристалле, она способна обратимо терять
молекулу Н2. Очень похожие комплексы получены с Мо вместо W,
а также с другими фосфинами. Расстояние Н-Н 0,84 Å (без
указания погрешности), полученное методом нейтронографии, близко к значению го=
0,876 Å, установленному по данным
колебательно-вращательной спектроскопии для иона Н3+ (рассчитано из
вращательных постоянных в работе [59]). Экспериментальная величина для молекулы
Н2 равна ге = 0,7417 Å [45].
Эти новые молекулы интересны своим электронным
строением [24], попадая в разряд частиц, где фрагмент Н2 присоединен к
фрагменту (X), имеющему вакантную граничную орбиталь. Рис. 20.4 показывает
электронное состояние для частиц M(PR3)2(CO)3(H2)
(X = M(PR3)2(CO)3)
и для других комплексов, содержащих диводород (X = M(CO)x(NO)3-x),
а также для частиц Н3+ (X = Н+) и CH5+
(X = CH3+).
Последняя частица обнаружена только в масс-спектрометре. Ее строение еще не
установлено экспериментально, и работа в этой области сейчас продолжается (см.,
например, гл. 1, разд. 1.6). Во всех примерах, представленных на рис. 20.4,
частицы X изолобальны (обсуждение принципов изолобальности можно найти в работе
[1]). Однако является ли молекула дигидридом или комплексом с диводородом,
определяется тонким равновесием электронных факторов, зависящих от природы
частицы X [24].
На основе вышевысказанных соображений можно
предположить [22], что должны быть устойчивыми и многие другие частицы Х (Н2).
Например, устойчивость частицы с X = Н+ (т. е. Н3+ ) сразу приводит к
возможному кандидату: FH
(H2), где атом
водорода исходной молекулы HF,
несомненно, должен иметь существенно „протонный” характер. Действительно, такая
молекула была недавно идентифицирована на основании колебательно-вращательных
спектров высокого разрешения [48, 52], также как и родственные ей частицы OН3+(H2)x,
где х = 1 - 3 [62].
 (СО)5(Н2) Со(СО)2(NО)
(Н2) СН3+ H3+
(СО)5(Н2) Со(СО)2(NО)
(Н2) СН3+ H3+
W(РR3)2(СО)3(Н2)
Fе(СО)(NО)2(Н2)
Рис. 20.4. Электронное строение комплексов с Н2.
Рассмотренный тип координации Н2 переходными
металлами теперь уже полностью доказан. Однако в других исследованиях, которые
не выдержали испытания временем, также иногда обнаруживалась необычная
геометрия координированных лигандов. Так, при рентгеноструктурном исследовании
молекулы Сг(СО)5Ру (рис. 20.5,a)
были обнаружены весьма необычные расстояния С-С и C-N
([37]; см. табл. 20.1). Такие расстояния, будь они реальными, указывали бы на
необычный характер химической связи между органической молекулой и металлом.
Однако в процессе уточнения структуры были получены слишком большие тепловые
параметры для атомов лигандов, что должно было вызвать определенное подозрение.
Авторы указывали, что в независимой части элементарной ячейки, содержавшей
необычную молекулу Сг(СО)5Ру, присутствовала еще молекула Сг(СО)5 (пиперидин),
в которой неплоский насыщенный цикл был заменен на плоский ненасыщенный.
Другими словами, обе молекулы в независимой части ячейки имели формулу Сг(СО)5
(пиперидин), но в одной из них гофрированный органический лиганд был
разупорядочен по двум позициям. Поскольку для каждого из шести атомов цикла
имеется два положения, отстоящие на 0,5 Å или
менее друг от друга, уточнение автоматически привело к плоской геометрии (рис.
20.5,6). Таким образом, стали объяснимы необычные расстояния и большие
эффективные коэффициенты теплового движения.
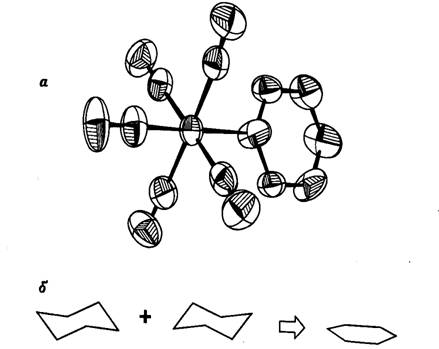
Рис. 20.5. Предполагаемое строение молекулы
Сг(СО)5(пиридин) с плоским гетероциклическим лигандом (а) [37]. Результирующая
плоская геометрия при неадекватном уточнении разупорядоченной молекулы
Сг(СО)5(пиперидин), содержащей гофрированный цикл (б).
Таблица 20.1. Геометрические параметры
пиридинового цикла в комплексе Сг(СО)5 (пиридин) по данным двух
рентгеноструктурных исследований [37, 38]
|
r(C-C)средн., Å
|
r(C-N)средн., Å
|
Литература
|
|
1,442
|
1,386
|
37
|
|
1,366
|
1,345
|
68
|
|
Ожидаемое
значение 1,37
|
1,34
|
|
Во многих случаях кристаллографические проблемы
не настолько отчетливо выражены. Следующий пример взят из координационной химии
молекулярного азота. В то время как в органической химии хорошо известны и
концевой (end-on)
и боковой (sideways-on)
способы координации N2 (например,
в диазометане и диазирине соответственно), в неорганической химии надёжно
охарактеризован лишь первый из них. Однако появилось сообщение [28] о боковой
координации N2 в молекуле RhCl(N2)(Pi
Ргз)2. Повторное исследование при низкой температуре [73] показало, что,
вероятно, диазотный лиганд в действительности координирован по концевому типу,
но из-за разупорядоченности в кристалле некоторые из расстояний во фрагменте M-N2
сильно искажены (те же проблемы явились причиной неверной интерпретации и в
первоначальном исследовании). Расстояние N-N
для комплекса (0,958(5) Å) в
последнем исследовании было короче, чем в свободной молекуле азота (ге = 1,094
Å)
[45].
Структурные исследования доказали свою важность
для понимания нового типа связывания недавно охарактеризованного,
„агостического” атома водорода в химии переходных металлов [13]. В структурном
отношении свойства таких связей имеют много общего с водородной связью. С
позиций электронной структуры эти связи можно объяснить примерно следующим
образом. Соединения многих переходных металлов, где металлический центр имеет
меньше восемнадцати электронов, являются формально ненасыщенными. Один из путей
устранения этой ненасыщенности заключается в увеличении координационного числа
атома переходного металла за счет искажения координированного органического
лиганда таким образом, что атом водорода лиганда приближается к металлическому
центру. Такая геометрическая ситуация и обусловила название для
рассматриваемого атома водорода (agostic
происходит от греческого - прикрепляться, держаться). Рис. 20.6 показывает
некоторые примеры структур, межатомные расстояния для которых приведены в табл.
20.2. Заметим, что в последнем из примеров геометрия двух независимых молекул
немного различается. Данные нейтронографических исследований, приведенные в
табл. 20.2, показывают, что агостические расстояния С-Н и М-Н несколько
длиннее, чем обычные связи С-Н и М-Н. Как уже отмечалось, этот тип
взаимодействия по некоторым характеристикам напоминает водородную связь, в
частности, сила связывания металл-водород, отражающаяся на величине межатомного
расстояния, коррелирует с изменением соответствующего расстояния С-Н в лиганде.
Однако имеется и существенное отличие. В водородной связи атом водорода
притягивается электроотрицательным атомом, тогда как в случае агостического
взаимодействия эффект притяжения оказывается более сильно выраженным для более
электроположительного металла. Весьма вероятно, что такого рода взаимодействия
существенны в реакциях типа полимеризации Циглера - Натта в присутствии
лигандов, координированных на атоме переходного металла начала периода, хотя
пока еще нет достаточно определенных доказательств реального механизма.

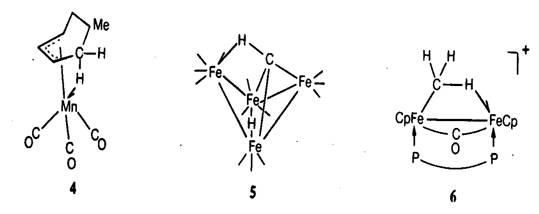
Рис. 20.6. Некоторые примеры „агостических”
атомов водорода [13]. Важнейшие длины связей приведены в табл. 20.2.
Таблица 20.2. Некоторые межатомные расстояния (Å)
для „агостического” атома водорода
|
Молекулаa
|
г(М
- Н)
|
г(С
- Н)
|
г(М...С)
|
Литература
|
|
1б
|
-
|
-
|
2,3
|
69
|
|
2
|
2,27(8)
|
0,97(8)
|
3,055(7)
|
36
|
|
3в
|
1,874(3)
|
1,164(3)
|
2,384(4)
|
17
|
|
4в
|
1,84(1)
|
1,19(1)
|
2,34(1)
|
70
|
|
5
|
1,00(4)
|
1,926(5)
|
9
|
|
6r
|
1,64(4)
|
1,06(4)
|
2,108(3)
|
39
|
|
1,78(3)
|
0,83(4)
|
2,118(3)
|
|
См. рис. 20.6.
6Агостические атомы водорода не локализованы.
вРезультаты нейтронографического исследования.
гВ независимой части элементарной ячейки
присутствуют две независимые молекулы.
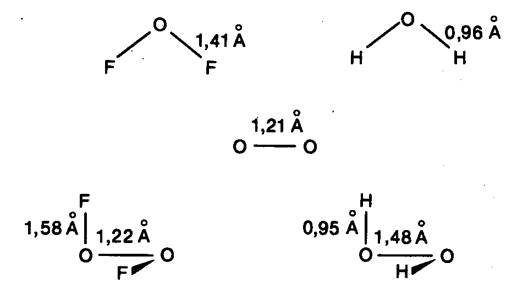
Рис. 20.7. Длины связей в некоторых фторидах и
гидридах [23].
Приведенные примеры наглядно показывают роль
точных структурных определений для этой области, а именно для выявления новых
типов молекул. Другой важной задачей является использование результатов для
детального сравнения длин связей и углов со значениями для других молекул с
целью дать адекватную структурно-электронную характеристику типа молекулы.
Иногда наблюдаемые расстояния сильно различаются для молекул, которые
предполагались весьма сходными. Пример проблемы такого рода дает геометрия
молекулы O2F2
[47]. На рис. 20.7 приведены расстояния в этой молекуле, определенные методом
микроволновой спектроскопии, наряду с некоторыми другими „типичными”
расстояниями О-О, О-Н и О-F
для молекул в газовой фазе: Следует обратить внимание на резкое различие
расстояний О-О в O2F2
и в структурно близкой молекуле O2Н2,
но в то же время близость к расстоянию в молекуле кислорода. Расстояние О-F
гораздо длиннее ожидаемого, что видно при сопоставлении с расстоянием в OF2.
Структурные особенности O2F2
находят отражение в химии этого соединения, являющегося сильным фторирующим
агентом. Понимание некоторых особенностей соединения возможно в рамках простой
теоретической модели [23].
неорганический химия молекула тело
2. Неорганические продукты естественного
происхождения
Точно так же, как химику-органику предоставлена
возможность изучения огромного изобилия природных соединений, химик-неорганик
имеет интереснейший природный материал в виде горных пород, слагающих земную
кору. Многие проблемы взаимосвязи строения и свойств, с которыми в наши дни
сталкиваются химия твердого тела и материаловедение, занимали геохимиков в
продолжение длительного времени. Возможно, первая из проблем - это нахождение
хорошего монокристалла для рентгеноструктурного исследования. В настоящее время
часто оказывается невозможным получить за время, сравнимое с продолжительностью
человеческой жизни, кристалл вещества, подобный тем, которое природа создавала
миллионами лет. Многие из таких материалов имеют очень сложное строение, из-за
чего возникают даже проблемы геометрического описания, с которыми редко
сталкивается химик, изучающий молекулы. Одна из главных проблем для веществ
этой группы часто состоит в большом числе атомов в независимой части ячейки. С
учетом часто реализующейся сложности химического состава, а также того, что
минеральные вещества обычно представляют собой твердые растворы по крайней мере
двух компонентов, становится очевидной вся сложность кристаллографических
исследований природных объектов. Нередко одна позиция в структуре может быть
частично заселена несколькими различными частицами (обычно катионами). В таких
случаях полученная структурная формула является лишь приближенной, так как в
ней суммируются погрешности химического анализа и определения значений заселенности
позиций в процессе кристаллографических вычислений.
Важным аспектом строения этих
материалов является способ упорядочения атомов по возможным позициям структуры.
Для цеолита А с отношением Si/Al, равным
единице, теперь надежно установлено, что в противоположность прежним
представлениям два атома кремния или два атома алюминия не могут занимать
соседние позиции в структуре (правило Ловенштейна, см. обсуждение и литературу
в [32]). Упорядочение атомов кремния и алюминия в полевых шпатах послужило
предметом интенсивных исследований в течение ряда лет. Это представляет очень
интересную кристаллографическую задачу, так как мотив упорядочения ионов иногда
оказывается несоразмерным с трансляционной периодичностью структуры оксидной
фазы [53]. Характер проблем хорошо иллюстрируется на примере полевого шпата -
плагиоклаза, одного из наиболее распространенных минералов горных пород. Одно
время считалось, что между двумя крайними членами - анортитом (CaAl2Si2O8) и
альбитом (NaAlSi3O8) -
реализуется непрерывный ряд твердых растворов. Теперь стало ясно, что при
низких температурах ситуация более сложная. Сверхструктурные пики на
дифрактограммах промежуточных составов не могут быть индицированы с
использованием параметров элементарной ячейки плагиоклаза. Это является надежным
свидетельством присутствия несоразмерной модулированной фазы. Согласно [44]
поведение этих систем описывается в рамках новой концепции структурного
резонанса между двумя упорядоченными фазами, образующимися в результате строго
согласованного замещения Na Са и SiА1. Общая
картина усложняется из-за того, что изменения в каркасе (атомы Si и А1)
накладываются в ней на изменения в соотношении некаркасных катионов Na+ и Са+.
Са и SiА1. Общая
картина усложняется из-за того, что изменения в каркасе (атомы Si и А1)
накладываются в ней на изменения в соотношении некаркасных катионов Na+ и Са+.
. Кристаллографические исследования при низкой
температуре
Химики часто хотят сравнить результаты
теоретических расчетов и эксперимента. Как правило, при таком сравнении
рассчитанные расстояния получаются из гипотетической модели, соответствующей
„замороженному” (0 К) статическому (в отсутствие колебательного движения)
состоянию вещества. Поэтому для сравнения лучше всего подходят параметры,
полученные из экспериментов при низких температурах. Однако при этом возникает
ряд проблем. Например, вещество может претерпевать при охлаждении фазовый
переход. Особенно часто происходит превращение типа металл-изолятор, связанный
либо с твердофазным эквивалентом эффекта Яна - Теллера (эффект Пайерлса), либо
с переходом из высокоспинового в низкоспиновое состояние в молекулах (см.,
например, [1]). Даже в отсутствие фазового перехода при охлаждении часто
происходят существенные изменения структуры, дающие ценную информацию о
веществе. Приведем только два примера.
Известно, что в химии силикатов изменение
валентных углов при атоме кислорода соответствует очень пологому потенциалу,
т.е. энергетические затраты при изменении угла (например) от 120° до 180°
невелики. Благодаря такой структурной нежёсткости (см. [43, 57]) и способно
реализоваться столь огромное число структур силикатов. Существуют два модельных
подхода к этой проблеме. Первый из них [61] предполагает, что из-за короткого
расстояния Si-О
(поскольку кислород-элемент второго периода) атомы кремния могут оказаться на
довольно коротких расстояниях друг от друга, испытывая значительное
отталкивание в случае слишком малых углов Si-O-Si.
Таким образом, энергетика изменения углов определяется балансом между
распределением электронов у атома кислорода (для четырех пар электронов следует
ожидать приблизительно тетраэдрических углов) и стерическими факторами
(способствующими увеличению углов).
Другая модель предполагает π-связывание
между атомами кремния и кислорода, ответственное за стабилизацию линейной
геометрии (относительно новой интерпретации этой модели см. [1, 21]). На
основании сходных аргументов ранее объяснялось плоское строение N(SiH3)3,
а в последнее время - плоское координационное окружение атома кислорода в
оксидных фазах со структурным типом рутила. Мы предпочитаем эту вторую модель.
Молекула субоксида углерода С3О2, строение которой соответствует записи
(ОС)С(СО), как и ее фосфиновый аналог (R3Р)С(РR3),
где R - алкильная
группа, характеризуется мягкой модой деформационных колебаний у центрального
атома углерода. Хотя оба лиганда СО и PR3
являются π-акцепторами, лишь
последний из них имеет объемные концевые группы. И все же молекула С3О2
линейна, тогда как ее фосфиновый аналог имеет угловое строение, что не
соответствует относительному объему, занимаемому лигандами. При любой
интерпретации мы должны были бы из-за наличия колебательного движения ожидать
неких существенных изменений структурных параметров при изменении температуры.
Однако оказалось, что учет колебательного движения в ангармоническом
приближении, особенно при небольшой силовой постоянной (как это необходимо для
рассматриваемых систем), не является легкой задачей, В табл. 20.3 приведены
некоторые из последних результатов для кристобалита (одной из полиморфных
модификаций SiO2), причем
одна и та же процедура уточнения была использована для данных по дифракции нейтронов
для одного и того же порошкообразного образца, полученных при высокой и низкой
температуре [66]. Хорошо виден различный характер изменения расстояний при
изменении температуры.
Мы уже упоминали о том, что лучше всего,
по-видимому, сравнивать с теорией результаты исследований при низкой
температуре. На рис. 20.8 показаны результаты неэмпирических МО-расчетов для
фрагментов структур силикатов [43, 57], использовавшихся для моделирования
свойств силикатных материалов.
Таблица 20.3. Длины связей (Å)
и валентные углы (град.) в кристобалите (SiO2)
по данным нейтронографии порошка при двух температурах [66]
|
Параметр
|
|
Значение
|
|
|
10К
|
|
473
К
|
|
г(Si-О(l))
|
1,602(1)
|
|
1,605(2)
|
|
г(Si -О(2))
|
1,617(1)
|
|
1,590(2)
|
|
<Si-O-Si
|
144,7(1)
|
|
148,4(1)
|
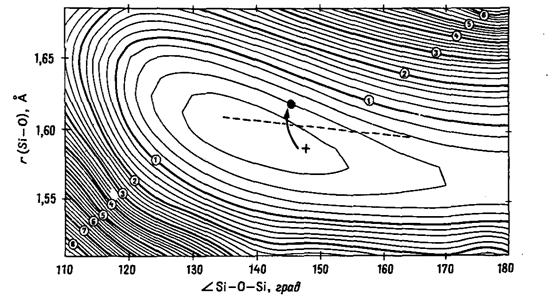
Рис. 20.8. Рассчитанная поверхность
потенциальной энергии в координатах угол Si-O-Si/длина
связи Si-O
для структур силикатов (в качестве модельной системы взят Si2О(OH)6)
[43]. Энергия показана в виде контурной диаграммы с шагом 0,001 а.е. (2,6
кДж/моль); более толстые линии проведены с интервалом 0,005 а.е. (13,1
кДж/моль), они отмечены цифрами от 1 до 8 в порядке увеличения энергии.
Прерывистая линия соответствует корреляции между указанными геометрическими
параметрами, наблюдаемой для многих кристаллических структур. Отмеченные точки
обсуждаются в тексте.
Штриховой линией представлена корреляция длина
связи - валентный угол, полученная из данных структурных определений для
множества силикатов при комнатной температуре. Заметим, что она не
располагается точно в долине, определяемой рассчитанными величинами энергии
искажения. На рисунке также показано изменение одного из расстояний Si-O,
приведенных в табл. 20.3, при изменении температуры. Следует обратить внимание
на движение точки, соответствующей данным для высокой (крестик) и низкой
(кружок) температуры, сопровождающееся уменьшением межатомного расстояния, что
приводит к менее удовлетворительному согласию с теорией (однако необходимо
соблюдать осторожность в выводах на основании данных только для одной точки!).
Изменение другого расстояния Si-O
из табл. 20.3 менее существенно.
Структура рутила ТіО2
(рис. 20.9) относится к таким,
в которых интересные температурные изменения происходят не с самими межатомными
расстояниями, а с другими кристаллографическими параметрами. Атомы металла
имеют формально октаэдрическое окружение, но в этой структуре два аксиальных и
четыре экваториальных расстояния Ti-О
симметрически неэквивалентны. Экспериментально были определены два более длинных
и четыре более коротких расстояния как при комнатной температуре, так и при 15
К.
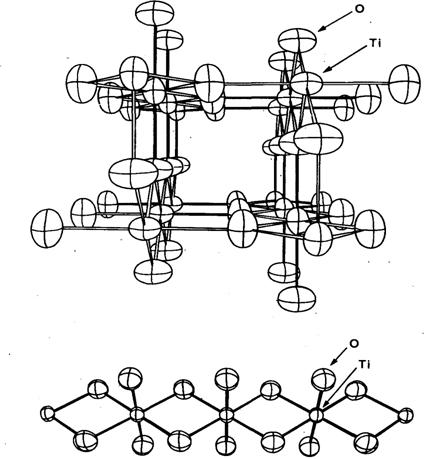
Рис. 20.9.
Две проекции структуры рутила TiO2.
На нижнем рисунке показана цепь из октаэдров ТіО6,
сочленённых по ребрам.
Одна из загадочных особенностей этой структуры
заключается в том, что, согласно расчетам зонной модели для рутила [20], две
аксиальные связи должны быть прочнее и, следовательно, короче, чем
экваториальные, что противоречит экспериментальным данным. Поскольку расчеты
такого рода, как правило, дают правильные предсказания соотношений между
длинами связей, полученный результат казался поразительным. Он по существу
означал, что соотношение межатомных расстояний в этом веществе определяется не
непосредственным взаимодействием Ti-O,
а взаимодействием между атомами кислорода (химики, имеющие дело с молекулярными
структурами, назвали бы это стерическими затруднениями). Однако в MgF2,
также имеющем структуру рутила, относительные расстояния находятся в
соответствии с теорией. Поскольку атомы фтора по размеру меньше, чем атомы
кислорода, а расстояния металл - фтор короче обычных расстояний металл -
кислород, мы вправе ожидать уменьшения отталкивания, обусловленного
невалентными взаимодействиями. Доказательство того, что атомы металла в TiO2
находятся в каких-то особых (напряженных) положениях, размеры которых
определяются матрицей из атомов кислорода, следует из изучения температурной
зависимости тепловых параметров, полученной при нейтронографическом
исследовании порошка (табл. 20.4; [25]). Сходные результаты получены и при
использовании рентгенографии. Обычно более тяжелый атом металла характеризуется
менее интенсивным тепловым движением, чем более легкий атом кислорода, но
оказалось, что при комнатной температуре оба атома имеют близкие значения
анизотропных параметров смещения. Лишь при 15 К эти параметры имеют ожидаемые
значения. В противоположность этому в опубликованной структуре MgF2
не наблюдается каких-либо особенностей теплового движения [7]. Здесь благодаря
меньшему размеру иона F-
геометрия окружения атома металла определяется прямыми взаимодействиями
катион-анион.
4. Интеркалированные материалы
Модификация свойств материалов за счет тонкого
изменения их электронной структуры путем интеркаляции донорными или акцепторными
группами представляет популярное синтетическое направление для
химиков-неоргаников. На рис. 20.10 представлена в упрощенном виде зонная
структура графита [75]. Важно отметить, что уровень Ферми находится на границе
между связывающей и разрыхляющей зонами т-электронов С-С-связи. Поэтому
добавление электронов приведет к заселению разрыхляющей части зоны и ослабит
С-С-связывание [49]. В конечном счёте
добавление одного электрона на каждый атом углерода вызовет гофрировку слоя
наподобие слоям в структуре мышьяка, но нас сейчас будут интересовать лишь
небольшие изменения концентрации
электронов. Рис. 20.11 показывает, как в соответствии с этими простыми
представлениями изменяется расстояние С-С при добавлении электроположительных
атомов калия [58]. Обычно более слабые связи являются и более длинными
(заметим, что ситуация может оказаться более сложной, если интеркалянт является
акцептором электронов, см. [49]).
На рис. 20.12 представлен другой эффект,
обнаруженный в графите (и еще в некоторых материалах) и заключающийся в
последовательном, “ступенчатом” образовании нескольких соединений (см.,
например, [4]). Добавление интеркалянта вызывает появление весьма интересных
локальных особенностей структуры. По-видимому, слои интеркалируемого материала
не имеют тенденции располагаться в стопках непосредственно над и под другой
молекулой интеркалянта, если только это не требуется стехиометрией материала.
Причины такого явления еще до конца не ясны. По-видимому, некоторые из этих
особенностей объяснимы в рамках модели, включающей отталкивание между частицами
интеркалянта в соседних слоях, но притяжение между частицами в том же слое. До
сих пор для интерпретации использовали модель диполь-дипольных взаимодействий,
но необходимо также попробовать применить и простые модели химического
связывания.
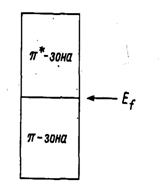
Рис. 20.10. Схема зонной структуры графита,
показывающая соприкосновение π-
и π*-зон
на уровне Ферми в случае чистого материала. При увеличении электронной
концентрации, заселяются π*-уровни и
расстояния С-С в плоскости возрастают.
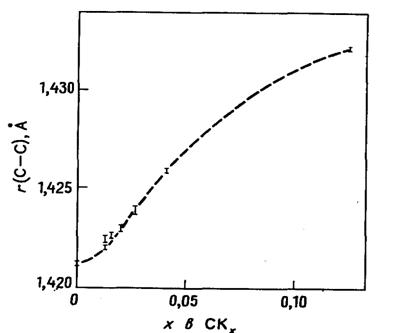
Рис. 20.11. Наблюдаемое изменение расстояний С-С
в графите при интеркаляции калия согласно [58].
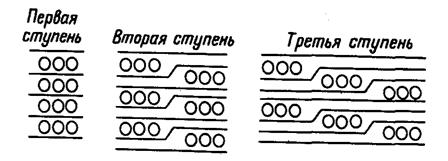

Рис. 20.12. Схема, объясняющая явление
“ступенчатого” образования соединений.
Несмотря на то,
что после интеркаляции двумерная упорядоченность может быть достаточно хорошо
выражена, детали трехмерной структуры не вполне ясны, поскольку положение одной
пары слоев вместе с их содержимым относительно другой часто оказывается
статистически разупорядоченным. Это очевидное следствие слабости сил,
связывающих соседние пакеты между собой. В других интеркалированных материалах
иногда возникают иные проблемы, затрудняющие точное определение структуры.
Например, н-бутиллитий - широко применяемый реагент в этой области, поскольку
атомы лития могут быть внедрены в твердое вещество с одновременным образованием
газообразного углеводорода, который легко удалить. В этой методике интеркаляции
лития используется WO3
[30] в виде прекрасного монокристалла, а для продукта состава Li0,36WО3
монокристалла получить не удалось и для характеристики вещества пришлось
использовать рентгенографию порошка. Хотя для описания подобных систем и используется
термин “интеркалированные материалы”, следует иметь в виду, что это часто
совершенно новые вещества со всеми своими особенностями. В анатаз (модификация TiO2)
может быть интеркалирован литий с образованием нестехиометрического соединения LiхTiO2
со структурной основой исходного анатаза. Однако при х = 0,5 происходит фазовое
превращение и образующийся материал имеет простую структуру шпинели, что можно
было бы и предположить, записав его формулу в виде LiTiO2.
. Правила Полинга
Для химиков-неоргаников особый интерес
представляют длины связей в молекулах и твердых телах, потому что в принципе
они дают важные сведения о способе связывания на основе электронных
представлений. Одно из очень полезных эмпирических соотношений, применимое к
длинам связей во многих кристаллических структурах, носит название правила
суммы валентных усилий (см.[15]), являющееся развитием второго правила Полинга
[64]. Для кристаллических структур, которые не относятся к металлическим,
металлоорганическим или вандерваальсовым (т.е. к таким, где имеется
делокализация, либо некоторые или все межатомные взаимодействия являются
слабыми), это правило вводит представление о валентном усилии s,
определяемом для данной пары атомов лишь расстоянием г между ними. В настоящее
время используется одно из двух выражений [16]:
= (r/ro)-N
(20.1)
s = ехр[(ro
- r)/В] (20.2)
где г0 и N или г0 и В - константы для данной
пары атомов. При использовании этих определений обнаруживается, что сумма
валентных усилий для данного аниона или катиона (i),
направленных к координированным ими соответственно катионам или анионам (j),
почти постоянна. По традиции эта сумма приравнивается значению валентности
атома (Vi):
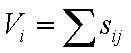 (20.3)
(20.3)
Уравнение (20.2) находит более широкое
применение, чем уравнение (20.1), так как значение В оказывается приблизительно
постоянным (0,37 Å) для многих
систем. В этом случае остается лишь одна переменная (ro),
которую можно выразить, используя аддитивные параметры для катиона и аниона
[16]. Отметим, что здесь не говорится ничего конкретного относительно самой
валентности. Это лишь нормирующий параметр при расчете валентного усилия.
Поэтому уравнение (20.3) определяет способ корреляции вклада отдельных
межатомных расстояний для данного атомного центра. Увеличение одного из
валентных усилий должно приводить к уменьшению всех остальных или некоторых из
них. Это дает также меру относительной силы для связей различного типа.
Шестикоординированный натрий образует связи с валентным усилием 1/6, а
четырёхкоординированный азот (рассматриваемый, как N3-)
дает связи с валентными усилиями, равными 3/4. Эти числа („валентность”,
деленная на координационное число) называют электростатическими усилиями связи,
чтобы избежать путаницы при использовании термина „валентное усилие”.
Соотношение (20.3) очень важно и, хотя основания для его справедливости не
очень ясны, в целом оно устанавливает постоянство общего усилия всех связей
атома. Таким образом, оно определяет и знак силовых констант для взаимодействий
между связями, если отсутствуют другие электронные факторы.
Это правило очень важно в кристаллохимии. Для
рентгеноструктурного анализа это, возможно, пока еще лучший способ отличить гидроксильную
группу от атома кислорода без связанного с ним атома водорода. Атом кислорода
гидроксильной группы будет иметь меньшую сумму валентных усилий, если
учитываются только контакты с уже найденными атомами.
Разумеется, было бы заманчивым использовать эти
соотношения для предсказания кристаллических структур, но, к сожалению,
уравнения (20.3) недостаточно для выбора топологии структуры. Все же Браун [14]
предложил другое правило, которое в сочетании с предыдущим позволяет
продвинуться в понимании распределения связей подлине. Его можно сформулировать
двояким образом: 1) индивидуальные валентные усилия вокруг любого центра имеют
тенденцию к выравниванию; 2) сумма валентных усилий вокруг любой замкнутой
группировки в структуре должна быть равна нулю (если принять знак усилия от
аниона к катиону положительным, а от катиона к аниону - отрицательным). Сходный
метод проверки выполнимости правила суммы валентных усилий был развит в работах
Баура [5]. Чтобы дать представление о точности таких оценок, в табл. 20.5
представлены значения наблюдаемых и предсказанных расстояний в структуре
диопсида CaMgSi2O6
с использованием методов Брауна [14] и Баура. Одно лишь задание связанности в
структуре в сочетании с этими двумя правилами (разумеется, при наличии еще
необходимых параметров для участвующих атомов) приводит к вполне приемлемой
оценке длин связей. Сходные идеи были использованы для поиска метода
систематической генерации возможных кристаллических структур [2]. И хотя общий
алгоритм еще не найден, его разработка оказалась бы весьма плодотворной для
неорганической химии. Развитие работ в этом направлении, без сомнения, основано
на большом числе точных структурных данных. Дальнейшее совершенствование теории
и использование этих методов в повседневной практике в значительной степени
зависят от достигаемой точности структурных определений.
Таблица 20.5. Предсказание длин связей в
диопсиде CaMgSi2O6
согласно работе Брауна [14]
|
Связь
|
Длина
связи
|
|
|
Предсказ.
[14]
|
Предсказ.
[5]
|
Наблюд.
[33]
|
|
Si-O(l)
|
1,07
|
1,60
|
1,62
|
1,60
|
|
Si-О(2)
|
1,17
|
1,56
|
1,59
|
1,58
|
|
Si-О(3)
|
0,88
|
1,67
|
1,67
|
1,66
|
|
Si-О(3)
|
0,88
|
1,67
|
1,67
|
1,69
|
|
σ
(Si - О)
|
|
0,01
|
0,01
|
|
|
Ca-O(l) (x2)
|
0,32
|
2,35
|
2,43
|
2,36
|
|
Ca-C-(2) (x2)
|
0,42
|
2,24
|
2,32
|
2,35
|
|
Са-О(З)
(x2)
|
0,12
|
2,80
|
2,62
|
2,56
|
|
Са-О(З)
(x2)
|
0,12
|
2,80
|
2,62
|
2,72
|
|
σ
(Ca
-
O)
|
|
0,14
|
0,07
|
|
|
Mg-O(l) (x2)
|
0,30
|
2,15
|
2,10
|
2,12
|
|
Mg-O(l) (x2)
|
0,30
|
2,15
|
2,10
|
2,06
|
|
Mg-О(2) (x2)
|
0,40
|
2,01
|
2,06
|
2,05
|
|
σ
(Mg
-
O)
|
|
0,06
|
0,02
|
|
. Высокотемпературные сверхпроводники
год был свидетелем открытия материалов,
обладающих сверхпроводимостью при температурах около 90 К [76]. В последующие
годы были достигнуты еще более высокие температуры перехода в сверхпроводящее
состояние. Критическая температура, ниже которой электрическое сопротивление
падает до нуля, обозначается Тс. Поразительная особенность сверхпроводящих
материалов заключается в их идеальном диамагнетизме (эффект Мейснера), и
научная общественность уже привыкла к фотографиям маленьких магнитов, парящих
над керамическим образцом, охлаждаемым жидким азотом. В отличие от большинства
известных ранее сверхпроводников новые материалы относятся к классу оксидов
металлов, которые несколько лет назад рассматривались физиками как весьма
маловероятные кандидаты в сверхпроводники. С момента их открытия была проделана
значительная экспериментальная работа по исследованию этих сложных оксидов, и
рентгено- и нейтроноструктурный методы сыграли и все еще играют важную роль при
определении строения сверхпроводящих фаз (например, [29, 30, 38]). Ключевой
проблемой здесь оказалось то, что эти материалы имеют нестехиометрический
состав и нестехиометрия оказывает весьма важное влияние на проводимость. Это
свидетельствует о необходимости точного определения их структуры.
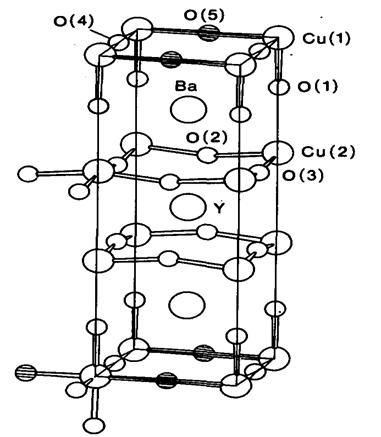
Рис. 20.13. Строение высокотемпературного
сверхпроводника YBа2Сu3О6+x
где 0 < х < 1 [67]. При х < 1 позиции O(4)
и O(5) заселены только
частично.
Интереснейшие данные по этим оксидным системам
свидетельствуют о подвижности некоторых атомов кислорода и об их влиянии на
сверхпроводимость. Тc падает,
когда х становится меньше единицы, а при х, несколько меньшем 0,5,
сверхпроводимость исчезает. Из данных порошковой и монокристальной
нейтронографии при комнатной и более низкой температурах следует, что позиция O(5)
оказывается, как правило, заселенной и при х < 1. В зависимости от равенства
или различия заселенностей позиций O(5)
и O(4) реализуется
тетрагональная или ромбическая структура.
В одном из структурных исследований установлено,
что переход между двумя типами осуществляется вблизи х = 0,5, хотя и не ясно,
является ли этот переход определяющим для сверхпроводимости. Разумеется, все
эти структурные исследования дают лишь усредненную заселенность позиций O(4)
и O(5), которая также
зависит от температуры, что следует учитывать при синтезе этих материалов.
Изучение образца с х = 0,7 [54] показало, что заселенность позиции O(5)
возрастает при повышении температуры. Для области c
0,3<x<0,7 детальные
структурные данные отсутствуют, хотя рентгенодифракционные исследования и
показывают, что индивидуальные фазы могут быть получены либо нагреванием
образцов с высоким содержанием кислорода, либо закаливанием от высоких
температур.
Как мы уже указывали, важно отчетливо
представлять, что в силу особенностей метода такие исследования дают лишь
усредненную картину строения материала. Особенности локального упорядочения
вакансий оказывают решающее влияние на поверхность Ферми материала, как это
было показано ранее в наших работах [26]. Зададимся теперь вопросом, как можно
получить более детальную информацию, выходящую за рамки индивидуальных
структурных определений? Исследование зависимости состава (х) от геометрических
параметров оказалось весьма информативным [67]. Кривые на рис. 20.14 показывают
изменение расстояний (вдоль оси с) различных атомов в структуре YBа2Сu3О6+x
от плоскости, проходящей через атомы Сu(1),
согласно результатам имеющихся структурных определений при комнатной
температуре. Приведенные структурные изменения можно представить происходящими
вследствие изменения степени окисления одной трети атомов меди
(сопровождающегося превращением цепочек в гантели) и движения
электроположительных атомов бария как отклик на появление кислородных вакансий.
В куприте (Сu2О) каждый атом
меди имеет координацию в виде гантели с расстоянием Cu-O
1,85 Å
[77], хотя здесь атомы кислорода окружены четырьмя соседними атомами меди. В YBа2Сu3О6
координационное число атомов O(1)
лишь немного превышает единицу, и следует ожидать существенного укорочения
расстояния Сu(1)-O(1)
по сравнению с таковым в куприте. При уменьшении расстояния Сu(1)-O(1)
расстояние Сu(2)-O(1)
должно увеличиваться для сохранения суммы валентных усилий у атома кислорода O(1),
и оно в действительности удлиняется весьма существенно. Под влиянием удлинения
связи Си(2)-O(1) квадратные
пирамиды при Сu(2) становятся
более растянутыми при х = 0, чем при х = 1. Это следует из рис. 20.14, так как
изменение положения атома Сu(2)
оказывается большим, чем смещение связанных с ним атомов кислорода O(2)
и O(3). Наибольшее
смещение испытывают атомы бария. Видно, что эти атомы отходят от плоскости
атомов O(4) и O(5),
удаляемых из структуры по мере перехода*от
х = 1 к х = 0, что и понятно с позиций обычной
электростатики.
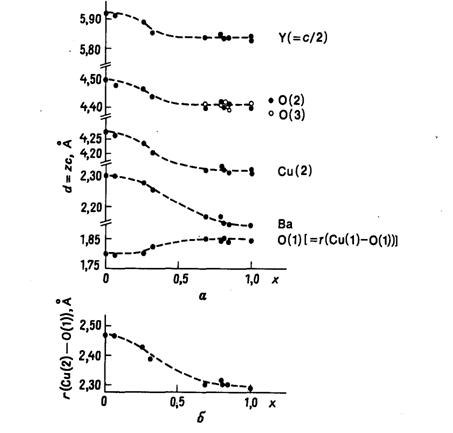
Рис. 20.14. Изменение структурных параметров в YBа2Сu3О6+x
как функция от х [67]. а - расстояние различных атомов от плоскости атомов Сu(1);
б-расстояние Сu(2)-O(1).
Особый интерес представляет, однако, форма этих
кривых. Видно, что зависимость структурных параметров от x
не является линейной, как можно было бы ожидать исходя из модели твердых
растворов, для которых параметры обычно изменяются линейно в зависимости от
стехиометрии или концентрации. На самом деле имеются две сравнительно плоские
области при 0,69<х<1 и при 0 <х<0,31, связанные кривой с гораздо
более крутым наклоном. Такое поведение типично для двухфазных систем, где
зависимость параметров от состава определяется взаимодействиями, обусловленными
притяжением подобного к подобному. Таким образом, из этих данных можно
заключить, что при х>0 дефекты не распределяются равномерно, но
упорядочиваются, образуя области, содержащие атомы СuII,
отделенные от областей с атомами СuI.
Из этой простой модели следует, что более крутой наклон при низких х (высокая
концентрация СuI) по
сравнению с наклоном при больших х (высокая концентрация СиIII)
означает, что энергия кластерообразования для СuIII
выше, чем для СuI.
Следует отметить, что использование кривых типа
показанных на рис. 20.14 позволяет извлечь больше информации, касающейся
микроструктуры, чем из отдельных структурных исследований. Сделанные выводы
находятся в соответствии с результатами исследования методом дифракции
электронов [74] и просвечивающей электронной микроскопии высокого разрешения
[63], которые доказывают, что кислородные вакансии локально упорядочены. Этого
следовало также ожидать, исходя из известной устойчивой геометрии различных
состояний окисления меди. Если бы вакансии были статистически разупорядочены,
то для высоких значений х оба атома меди, связанные с одной и той же
кислородной вакансией, имели бы Т-образную координацию. Простые электронные
соображения показывают [18, 26], что их электронная конфигурация должна быть d10,
а это плохо согласуется с предполагаемой геометрией окружения. Известен только
один пример Т-образного окружения - для молекулярного комплекса Rh1
с низкоспиновой конфигурацией d8
[78]. СuII, однако,
часто имеет линейную координацию в виде гантели. Если же дефекты упорядочиваются
таким образом, что у атома меди оказываются уже две кислородные вакансии, то
такая линейная координация для состояния d10
вполне приемлема. В таком случае реальный процесс упорядочения дефектов должен
минимизировать число атомов меди с Т-образным координационным окружением (на
концах цепочек). Это представляет весьма привлекательную область исследования,
причем прецизионные определения структур совершенно необходимы прежде, чем
могут быть поняты электрофизические свойства этих материалов.
. Значение теории
В неорганической химии теория на количественном
уровне намного хуже развита, чем в органической химии. Имеется лишь очень
немного достаточно глубоких исследований типа описанного, например, в гл. 16.
Частично это является следствием больших размеров рассматриваемых молекул. В то
время как для небольшой органической молекулы, состоящей из нескольких атомов
углерода и водорода, вполне можно выполнить теоретические расчеты высокого
уровня, совсем другое дело провести с такой же точностью вычисления для металлоорганических
молекул, содержащих один или несколько атомов переходного металла. Однако
качественные молекулярно-орбитальные подходы, применимые ко всему набору
элементов периодической системы, развиты достаточно хорошо (см., например, [1,
18]), и имеется множество примеров, когда изменения длин связей и общей
геометрии при изменении числа электронов могут быть сравнительно легко поняты
на качественном уровне при использовании достаточно простых
молекулярно-орбитальных моделей. Например, зависимость расстояний металл -
лиганд от числа электронов в комплексах переходных металлов - известная
двугорбая кривая - вполне объяснима в рамках чрезвычайно простой
молекулярно-орбитальной модели [18]. (Кстати, следует сказать, что,
по-видимому, нужно отказаться от часто используемой теории кристаллического
поля. Зачем использовать для переходных металлов подход, отличающийся от того,
который применяется для всей остальной химии?) Таким же образом на основании
простой схемы подсчета электронов становятся понятными структуры кластеров и
каркасных соединений как непереходных, так и переходных элементов. В этой главе
мы использовали теоретические идеи еще и для того, чтобы рассмотреть
перспективы, касающиеся конкретной структурной проблемы. Довольно часто
теоретические выводы стимулируют постановку эксперимента с целью проверки
правильности необычных предсказаний. Предсказания теории могут служить стимулом
и для другого типа тщательно выполненного эксперимента, ставящего своей задачей
получение молекул или твердых тел, нарушающих установившиеся представления. В
данном разделе будут описаны эксперименты первого типа.
Во многих системах, исследованных в связи с
изучением эффекта Яна - Теллера, наблюдается тетрагональное искажение
октаэдрической координации. Часто возникающий вопрос состоит в том, каков будет
тип искажения для данной системы: две короткие связи и четыре длинные (2S
+ 4L, тип 1) или,
наоборот, две длинные и четыре короткие (2L
+ 4S, тип 2)? Этот
вопрос может быть легко рассмотрен на теоретическом уровне, а предсказания
сделаны в зависимости от числа d-электронов.
Изменение энергии d-орбиталей
(в основном) переходного металла для случая π-донорных
лигандов (например, координированных через атом кислорода) показано на рис.
20.15. В рамках модели углового перекрывания [18] приведены схемы для трех t2g-орбиталей
в случае двух типов (1 и 2) искажения изолированного октаэдра МХ6. Легко
понять, каким образом характер расщепления энергетических уровней зависит от
формы d-орбиталей. Для
описания влияния искажения на взаимодействие π-типа
использован параметр δ.
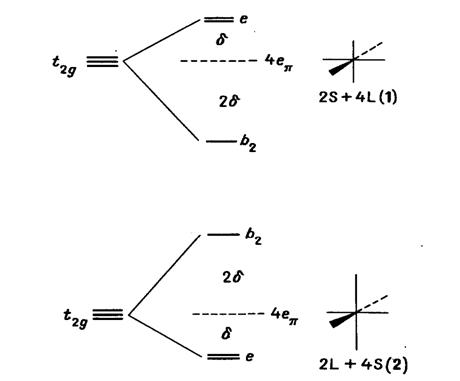
Рис. 20.15. Схема расщепления t2g-орбиталей
d-уровня в случае
двух типов (1 и 2) тетрагонального искажения октаэдра [27]. При искажении
„центр тяжести” уровней энергии остается постоянным. δ
- энергетический параметр для данного типа искажения.
Напомним, что из-за независимости характера
расщепления пары е8-орбиталей от типа искажения, предсказание возможно лишь в
случае асимметричных t2g
-конфигураций. Последние приведены в табл. 20.6. В случае асимметричных еg-конфигураций
необходимо исследовать d-s-смешивание,
обусловленное эффектом Яна -Теллера второго порядка [19], чтобы, например,
понять почти полную универсальность типа 2 в химии СuII.
Если лиганды являются π-акцепторами,
то предсказания на оснований табл. 20.6 следует изменить на противоположные. И
действительно, экспериментально определенное искажение для низкоспинового d5-комплекса
V(CO)6
соответствует типу 1 [8]. Отметим, что надежность предсказаний, а также их
взаимно противоположный характер для π-доноров
и π-акцепторов
при использовании модели углового перекрывания выгодно отличаются от модели
точечных зарядов в теории кристаллического поля.
СгО2 изоструктурен рутилу (рис. 20.9) и
представляет высокоспиновую d2-систему.
Для данной электронной конфигурации предпочтительный тип искажения, согласно
табл. 20.6, отвечает двум более длинным и четырем более коротким расстояниям
Сг-O (тип 2).
Таблица 20.6. Общая энергия d-орбиталей
для t2g-конфигураций
в случае октаэдрической и тетрагонально искаженной октаэдрической координации
на основании молекулярной модели [27]a
|
Конфигурация
d-оболочкиб
|
Правильный
октаэдр
|
Искаженный
октаэдрв
|
|
|
2S + 4L (1)
|
2L + 4S (2)
|
|
d°
|
0
|
0
|
0
|
|
d1
|
4еπ
|
4еπ - 2δ*
|
4еπ - δ
|
|
d2(нc)
|
8еπ
|
8еπ - 4δ*
|
8еπ -
2δ
|
|
d2(вс)
|
8еπ
|
8еπ -
δ
|
8еπ -
2δ*
|
|
d3(нс)
|
12еπ
|
12еπ - 3δ
|
12еπ - 3δ
|
|
d3(вс)
|
12еπ
|
12еπ
|
12еπ
|
|
d4 (нс)
|
16еπ
|
16еπ - 2δ
|
16еπ - 4δ*
|
|
d4 (пс)
|
16еπ
|
16еπ - 2δ*
|
16еπ - δ
|
|
d5(нс)
|
20еπ
|
20еπ - δ
|
20еπ - 2δ*
|
|
d6(нс)
|
24еπ
|
24еπ
|
24еπ
|
а Приведенные энергии отнесены к параметру
модели углового перекрывания , отражающему изменение энергии d-орбитали
π-типа
при координации одного π-лиганда.
Обсуждение этой орбитальной модели и ее связь с моделью точечных зарядов в
кристаллическом поле см. в [18].
бнс - низкоспиновая; вс - высокоспиновая
конфигурация; пс - конфигурация промежуточного типа.
в* указывает самый
низкий уровень энергии в случае π-донорного
лиганда.
Однако рентгеноструктурный эксперимент [34]
выявил противоположный характер искажения, причем такой результат не
ограничивается только оксидными системами. В табл. 20.7 представлены данные по
длинам связей для ряда оксидов и фторидов со структурным типом рутила. Следует
заметить, что, хотя строение FeF2
предсказано правильно, этого нельзя сказать о структуре CoF2
(хотя искажение октаэдра и невелико). В чем здесь дело: неверна теория или
ошибочны экспериментальные данные? Это потребовало повторного определения
структуры СгO2 методом
порошковой нейтронографии при комнатной температуре, 173 К и 10 К, а также
доработки теоретической модели [27]. Оказалось, что при всех трех температурах
характер координации атома хрома сохраняется (значения в табл. 20.7 относятся к
эксперименту при 10 К). В этом случае оказывается неверной теория. После
некоторых раздумий оказалось, что неудача объяснения характера искажения в
кристаллическом СгО2 и ряде других оксидов и фторидов, приведенных в табл.
20.7, не так уж и удивительна. В бесконечно протяженных структурах поле
лигандов на атомах переходного металла часто в значительной степени
определяется требованиями трансляционной симметрии, причем нередко важными
оказываются взаимодействия как через связь, так и через пространство. Первый из
этих подходов основан на идее сверхобмена. Таким образом, в дальнейшем
необходимо использовать теоретическую модель, учитывающую зонное строение
бесконечных кристаллических структур.
Таблица 20.7. Координация атома переходного
металла в оксидах и фторидах со структурным типом рутила
|
Соединение
|
d-электронная конфигурацияа
|
Тип
искажения
|
Литература
|
|
МgF2
|
do
|
1,9968(1)
1,9798(2) -
|
1
|
6
|
|
VF2
|
d3(вс)
|
2,091(3)в
2,074(3)в -
|
1
|
35
|
|
СгF2г
|
d4 (вс)
|
2,01;1,98д
2,43д +
|
2
|
46
|
|
МnF2
|
d5 (вс)
|
2,131(6)
2,104(9) -
|
1
|
7
|
|
FеF2
|
d6 (вс)
|
2,118(4)
1,998(6) -
|
1
|
7
|
|
СоF2
|
d7 (вс)
|
2,049(3)
2,027(5) -
|
1
|
7
|
|
NiF2
|
d8 (вс)
|
2,022(6)
1,981(9) -
|
1
|
7
|
|
СuF2г
|
d9
|
1,93(3)е
2,27(3) +
|
2
|
10
|
|
ZnF2
|
d10
|
2,046(7)
2,012(10) -
|
1
|
7
|
|
ТiO2
|
d0
|
1,9459(3)ж1,9764(4)ж
+
|
2
|
25
|
|
VO2
|
d1
|
1,921(1)
1,933(1) +
|
2
|
55
|
|
СrO2
|
d2 (вс)
|
1,9113(3)з
1,8877(5)з -
|
1
|
27
|
|
RuO2
|
d4 (нс)
|
1,984(6)
1,942(10) -
|
1
|
11
|
|
OsO2
|
d4 (нс)
|
2,006(8)
1,962(13) -
|
12
|
|
|
|
|
|
|
анс - низкий спин; вс - высокий спин.
r2/r4
- отношение величин осевых расстояний к экваториальным при тетрагональном
искажении октаэдра.
вРассчитано по параметрам элементарной ячейки и
координатам, приведенным в первоисточнике.
гМоноклинная структура (три пары расстояний).
дВ первоисточнике погрешность не указана.
еНайдено, что две пары экваториальных расстояний
имеют одинаковую величину.
ж При 15 К.
зПри 10 К.
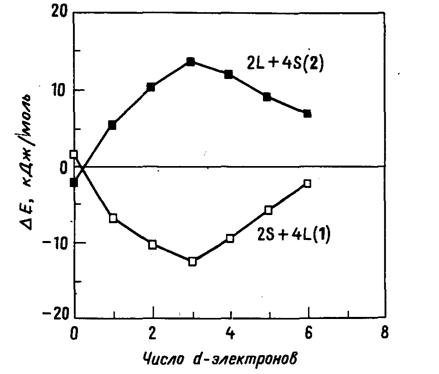
Рис. 20.16. Рассчитанные кривые разности энергии
в зависимости от числа d-электронов
для искажений координационного окружения атома металла (1 и 2) в структуре
рутила с сохранением тетрагональной симметрии [27]. Кривые получены расширенным
методом Хюккеля для тесно связанных зон и соответствуют низкоспиновым
электронным конфигурациям.
В действительности встречаются три различных
типа искажения, отмеченные в табл. 20.7. В рамках пространственной группы
структурного типа рутила отсутствует условие равенства между собой двух наборов
симметрически неэквивалентных расстояний, и, действительно, они всегда
оказываются различными. Искажения типов 1 и 2, представленные в таблице,
возможны в рамках высокосимметричной пространственной группы P42/mnm.
Искажение длин связей М-Х вдоль цепи вызывает существенное понижение симметрии
(до моноклинной), что приводит к трем парам различных расстояний М-Х. Имеются
два примера такого искажения, связанного с вырождением уровня eg,
а именно CuF2 и CrF2.
Здесь нас будут интересовать лишь искажения, сохраняющие тетрагональную
симметрию. На рис. 20.16 для двух типов искажения показаны две кривые разности
энергии в зависимости от числа d-электронов,
полученные при расчете, учитывающем зонную структуру бесконечного твердого
тела. Эти результаты хорошо согласуются с большинством известных структур
оксидов и фторидов переходных металлов, принадлежащих к структурному типу
рутила. При этом превалирующий тип искажения (1) соответствует тетрагонально
сжатому октаэдру (см. табл. 20.7). Немногочисленные исключения ограничены
конфигурацией d° и
высокотемпературной тетрагональной модификацией VO2.
Выше мы уже высказали соображение, что характер искажения в самом рутиле ТiO2
определяется отталкиванием типа анион - анион (см. разд. 20.4). Обратившись к
табл. 20.6, можно заметить, что при новом подходе отсутствует зависимость от
типа электронной конфигурации; иными словами, в фазах со структурой рутила
эффект Яна - Теллера первого порядка уже не влияет на геометрию координационного
окружения атома переходного металла.
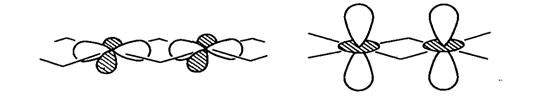
Рис. 20.17. Орбитали симметрии а1, которые могут
взаимоперекрываться (и действительно перекрываются) в реальной электронной
структуре рутила, что опровергает выводы, сделанные исходя из схемы орбиталей
для изолированных октаэдров. Данные работы [27].

Рис. 20.18. а - кривые разности энергии для двух
типов искажения (1 и 2), полученные с использованием молекулярной модели,
показанной на рис. 20.15. б- кривые, полученные из модели, предполагающей
смешивание орбиталей (рис. 20.17); Низкоспиновые конфигурации. Следует обратить
внимание на сходство кривых б с кривыми на рис. 20.16. Данные работы [27].
В действительности за ход обеих кривых
оказывается ответствен второй порядок смешивания орбиталей. Поскольку группа
любой произвольно взятой точки в одномерной зоне Бриллюэна твердого тела
изоморфна С2v, один из наборов еg-орбиталей
и t2g-орбиталей
должен иметь одинаковую симметрию a1
(рис. 20.17). Согласно теории возмущений, энергия стабилизации естаб него
уровня в рамках модели углового перекрывания составляет естаб ≈ 4еπеσ/(4еπ
+ 3еσ).
Рис. 20.18 показывает кривые рассчитанной разности в энергии для обоих типов
искажения с использованием принятой модели для случаев молекулярной структуры
(табл. 20.6) и для бесконечного твердого тела (с учетом этого дополнительного
взаимодействия). В последнем случае наблюдается превосходное согласие с
результатами количественных расчетов, представленными на рис. 20.16. Общий
вывод состоит в том, что даже для типично „ионных" материалов нельзя
недооценивать эффект „ковалентности” (более строго „орбитальные” эффекты),
ответственный за дополнительное связывание соседних центров.
Литература
1. Albright
Т.
A., Burdett J. К.,
Whangbo М.-Н.,
Orbital interactions in chemistry. Wiley-Interscience, New York (1985).
2. Altermatt
D., Brown I. D., Acta Crystallogr. (1985) B41,
240-244.
3. Andersen
R. A., Boncella J. M., Burns C. J., Blom R., Haaland A., Volden H. V., J. Organomet.
Chem. (1986)
312, C49-C52.
4. Bartlett
N., McQuillan B. W. Graphite chemistry. - In: Intercalation chemistry (ed. M.
S. Whittingham, A. J. Jacobson), pp. 19-53. Academic Press, New York (1982).
5. Baur
W. #., Trans. Am. Crystallogr. Assoc. (1970) 6, 129-155.
6. Baur
W. H~., Acta Crystallogr. (1976) B32,
2200-2204.
7. Baur
W. H., Khan A. A., Acta Crystallogr. (1971) B27,
2133-2139.
8. Bellard
S., Rubinson K. A., Sheldrick G. M., Acta Crystallogr. (1979)
B35, 271-274.
9. Beno
M. A., Williams J. M., Tachikawa M., Muetterties E. L., J. Am. Chem. Soc. (1980)
102, 4542-4544.
10. Billy
C, Haendler H. M., J. Am. Chem. Soc. (1957) 79, 1049-1051.
11. Boman
C.-E., Acta Chem. Scand. (1970) 24, 116-122.
. Boman
C.-E., Acta Chem. Scand. (1970) 24, 123-128.
13. Brookhart
M., Green M. L. H., J. Organomet. Chem. (1983) 250, 395408.
. Brown
I. D., Acta Crystallogr. (1977) B33, 1305-1310.
. Brown
I. D., The bond-valence method: An empirical approach to chemical structure and
bonding. - In: Structure and bonding in crystals, Vol. 2 (ed. M. O'Keeffe, A.
Navrotsky), pp. 1-30. Academic Press, New York (1981).
. Brown
/. D., Altermatt D., Acta Crystallogr. (1985) B41, 244-247.
. Brown
R. K., Williams J. M., Schultz A. J., Stucky G. D., Ittel S. D., Harlow R. L.,
J. Am. Chem. Soc. (1980) 102, 981-987.
. Burdett
J. K., Molecular shapes: Theoretical models of inorganic stereochemistry.
Wiley, New York (1980).
. Burdett
J. K., Inorg. Chem. (1981) 20, 1959-1962.
20. Burdett
J. K., Inorg. Chem. (1985) 24, 2244-2453.
21. Burdett
J. K., Caneva D. C., Inorg. Chem. (1985) 24, 3866-3873.
22. Burdett
J. K., Pourian M. R., Inorg. Chem. (1988) 27, 4445-4450.
23. Burdett
J. K., Lawrence N. J., Turner J. J., Inorg. Chem. (1984) 23, 2419-2428.
24. Burdett
J. K., Phillips J. R., Pourian M. R., PoliakoffM., Turner J. J., Upmacis Д.,
Inorg. Chem. (1987) 26, 3054-3063.
. Burdett
J. K., Hughbanks Т.,
Miller G. J., Richardson Jr., J. W., Smith J. V., 3. Am. Chem. Soc. (1987) 10»,
3639-3646.
. Burdett
J. K., Kulkarni G. V., Levin K., Inorg. Chem. (1987) 26, 3650-3652.
. Burdett
J. K., Miller G. J., Richardson Jr., J. W., Smith J. V., 3. Am. Chem. Soc.
(1988) 110, 8064-8071.
. Busetto
C., D:'Alfonso A., Maspero F., Perego G., Zazzetta A., J. Chem. Soc. Dalton
Trans. (1977) 1828-1834.
29. Calestani
G., Rizzoli G, Nature (1987) 328, 606-607.
. Cava
R. J., Santoro A., Murphy D. W., Zahurak S. M„ Roth R. S., J. Solid State Chem.
(1983) 50, 121-128.
31. Cava
R. J., Batlogg В.,
van Dover R. В., Murphy D. W.,
Sunshine S., Siegrist Т.,
Remeika J. P., Rietman E. A., Zahurak S., Espinosa G. P., Phys. Rev. Lett.
(1987) 58, 1676-1679.
. Cheetham
A. K., Fyje C. A., Smith J. V., Thomas J. M., 3. Chem. Soc. Chem. Commun.
(1982) 823-825.
33. Clark
J. R., Appleman D. E., Papike J. J., Mineral. Soc. Am. Spec. Papers. (1969) 2,
31-50.
. Cloud
W. H., Schreiber D. S., Babcock K. R., 3. Appl. Phys. (1962) 33, 1193-1194.
. Costa
M. M. R., De Almeida M. J. M., Acta Crystallogr. (1987) B43, 346-352.
. Cotton
F. A., LaCour Т., Stanislowski A.
G., J. Am. Chem. Soc. (1974) 96, 754-760.
37. Cotton
F. A., Darensbourg D. J., Fang A., Kolthammer B. W. S., Reed D., Thompson J.
L., Inorg. Chem. (1981) 20, 4090-4096.
38. David
W. I. F., Harrison W. T. A., Ibberson R. M., Weller M. Т.,
Grasmeder J. R., Lanchester P., Nature (1987) 328, 328-329.
. Dawkins
G. M., Green M., Orpen A. G., Stone F. G. A., J. Chem. Soc. Chem. Commun. (1982)
41-43.
40. Dunitz
J. D., Orgel L. E., Rich A., Acta Crystallogr. (1956) 9,
373-375.
41. Evans
W. J., Hughes L. A., Hanusa T. P., 3. Am. Chem. Soc. (1984)
106, 4270-4272.
42. Firor
R. L., Seff K„ 3. Am. Chem. Soc. (1976) 08,
5031-5133.
43. Gibbs
G. V., Meagher E. P., Newton M. D., Swanson D. К.,
A comparison of experimental and theoretical bond length and angle variations
for minerals, inorganic solids, and molecules. - In: Structure and bonding in
crystals, Vol. 1 (ed. M. O'Keeffe, A. Navrotsky), pp. 195225.
Academic Press, New York (1981).
44. Heine
V., McConnell J. D. C, J. Phys. C. (Solid State Phys.) (1984)
17, 1199-1220.
45. Herzberg
G., Molecular spectra and molecular structure, Vol. 1, Spectra of diatomic
molecules (2nd edn). Van Nostrand-Reinhold, New York (1950).
46. Jack
К.
H., Maitland Д., Proc. Chem. Soc.
(1957)
232.
47. Jackson
R. H., J. Chem. Soc. (1962) 4585-4592.
48. Jucks
K. W., Miller R. E., J. Chem. Phys. (1987) 87,
5629-5633.
49. Kertesz
M., Vonderviszt F., Hoffmann Д.,
Change of carbon-carbon bond length in layers of graphite upon charge transfer.
- In: Intercalated graphite, Materials Research Society Symposia Proceedings,
Vol. 20 (ed. M. S. Dresselhaus, G. Dresselhaus, J. E. Fischer, M. J. Moran),
pp. 141143. North-Holland, New York (1983).
50. Kubas
G. J., Ryan R. Д.,
Swanson В.
I., Vergamini P. J., Wasserman H. J., J. Am. Chem. Soc. (1984)
106, 451-452.
51. Loewenstein
W., Am. Mineral. (1954) 39, 92-96.
52. Lovejoy
С.
M., Nelson Jr., D. D., Nesbitt D. J., J. Chem. Phys. (1987)
87, 5621-5628.
54. Mclntyre
G. J., Renault A., Collin G., Phys. B. R. Condens. Matter (1988)
37, 5148-5157.
55. McWhan
D. В.,
Marezio M., Remeika J. P., Dernier P. D., Phys. Rev. B, Solid State (1974)
10, 490-495.
56. Marsh
R. E., Schomaker V., Inorg. Chem. (1979) 18,
2331-2336.
57. Newton
M. D. Theoretical probes of bonding in the disiloxy group. - In: Structure and
bonding in crystals, Vol. 1 (ed. M. O'Keeffe, A. Navrotsky), pp. 175-193.
Academic Press, New York (1981).
58. Nixon
D. E., Parry G. S., J. Phys. С (Solid
State Phys.) (1969) 2, 1732-1741.
59. Oka
Т.,
Phys. Rev. Lett. (1980) 45, 531-534.
60. O'Keeffe
M., Hyde B. G., The role of nonbonded forces in crystals. - In: Structure and
bonding in crystals, Vol. 1 (ed. M. O'Keeffe, A. Navrotsky), pp. 227-254.
Academic Press, New York (1981).
. O'Keeffe
M., Hyde B. G., J. Solid State Chem. (1982) 44, 24-31.
62. Okumura
M., Thesis, University of California, Berkeley, USA (1986).
63. Ourmazd
A., Spence J. С. H., Nature (1987)
329, 425-427.
. Pauling
L., J. Am. Chem. Soc. (1929) 51, 1010-1026.
65. Pluth
J. J., Smith J. V., J. Am. Chem. Soc. (1983) 105,
1192-1195.
66. Pluth
J. J., Smith J. V., Faber J., Jr., J. Appl. Phys. (1985)
57, 10451049.
67. Renault
A., Burdett J. K., Pouget J.-P., J. Solid State Chem. (1987)
71, 587-590.
68. Ries
W., Bernal I., Quasi M., Albright T. A., Inorg. Chim. Acta (1984) 83, 5015.
69. Дое
D. M., Bailey P. M., Moseley K., Maitlis P. M., J. Chem. Soc. Chem. Commun. (1972)
1273-1274.
70. Schultz
A. J., Teller R. G., Beno M. A., Williams J. M., Brookhart M., Lamanna W.,
Humphrey M. В., Science (1983)
220, 197-199.
. Seff
K., Mellum M. D., J. Phys. Chem. (1984) 88,
3560-3563.
72. Simon
A., Alkali metal suboxides: A kind of anti-cluster compounds. - In: Crystal
structure and chemical bonding in inorganic chemistry (ed. C. J. M. Rooymans,
A. Rabenau), pp. 47-67. North-Holland, Amsterdam (1975).
73. Thirn
D. L., Tulip Т. H., Ibers J. A.,
J. Chem. Soc. Dalton Trans. (1979) 2022-2025.
74. van
Tendeloo G., Zandbergen H. W., Amelinckx S., Solid State Commun. (1987)
63, 389-393, 603-606.
75. Whangbo
M.-H., Hoffmann Д.,
Woodward R. В., Proc. Roy. Soc. (1979)
A366, 23-46.
76. Wu
M. K., Ashburn J. R., Torng C. J., Ног
P. H., Meng R. L, Gao L., Huang Z. J., Wang Y. Q., Chu C. W., Phys. Rev. Lett. (1987)
58, 908-910.
77. Wyckoff
R. W. G., Crystal structures (2nd edn), Vol. 1, p. 331. Wiley-Interscience, New
York (1963).
78. Yared
Y. W., Miles S. L., Bau Д.,
Reed C. A., J. Am. Chem. Soc. (1977) 99, 7076-7078.