Технический анализ углей
Министерство науки и образования
Республики Казахстан
Карагандинский государственный
университет
им. Е.А. Букетова
Химический факультет
Кафедра химической технологии и
экологии
Курсовая работа
на тему:
Технический анализ углей
Подготовила: Завгородняя Мария
студ. гр. ХТОВ-42
Проверила: к. х. н., доцент Халикова З.С.
Караганда 2012
Содержание
Введение
1. Методы исследования углей
1.1 Определение влажности
1.2 Определение зольности
1.2.1 Определение содержания минеральных примесей
1.3 Определение выхода летучих веществ
1.4 Определение спекаемости
1.5 Определение теплоты сгорания углей
Заключение
Список использованных источников
Введение
Уголь является одним из главных источников получения тепловой
энергии, а также представляет собой ценное сырье для химической переработки с
целью получения необходимых для промышленности продуктов.
В мировой добыче всех видов горючих ископаемых, достигающей
примерно 6 млрд. т в год (в пересчете на условное топливо) на твердые горючие
ископаемые (уголь, торф, горючие сланцы) приходится 48%, около 35% добывается
нефти и 17% природных горючих газов.
В балансе мировых запасов горючих ископаемых доля угля и
горючих сланцев составляет 90%, торфа - 5%, нефти и природных газов - 5%.
Большая часть добываемого угля используется для
энергетических целей, а наиболее ценные угли подвергаются термической
переработке - полукоксованию, коксованию, газификации и гидрогенизации.
В настоящее время насчитывается свыше 350 ценных продуктов
различных наименований, получаемых из угля, используемых промышленностью и
сельским хозяйством. В коксохимической промышленности, при получении
синтетических топлив, углеродистых материалов и ряда химических соединений
качество целевой продукции, а иногда и сама возможность ее получения всецело зависят
от вещественного состава и свойств перерабатываемых углей. В связи с этим
изучению качества и разработке методов оценки пригодности углей для различных
отраслей промышленности придается огромное значение.
Для обеспечения разных отраслей промышленности углями
соответствующего качества последние обычно подвергаются предварительной
механической обработке - обогащению для удаления из угля минеральных примесей,
брикетированию, классификации по крупности.
В рациональную схему переработки углей должно входить не
только полное использование всех компонентов органической массы угля, но также
должны быть использованы и минеральные примеси в угле, включая редкие и
рассеянные элементы (всего в составе углей насчитывается более 70 ценных
химических элементов, входящих в те или иные соединения). Существенно важным
для практики вопросом является также извлечение путем обогащения углей
содержащихся в них сульфидов железа (FeS2,), используемых для производства серной
кислоты.
В углях содержатся также элементы, пригодные для нужд
сельского хозяйства. Ценными как стимуляторы роста растений являются
микроэлементы - молибден, цинк, марганец, медь и др., а щелочные золы из угля
являются весьма полезными добавками для кислых почв, повышающими урожайность
бобовых и других культур.
1. Методы
исследования углей
Методы исследования углей - (петрографические, химические и
физические) - включают различный комплекс характеристик в зависимости от задач
исследования. При любой направленности последних первым этапом является
углепетрографическое и частично физическое изучение, по данным которых вместе с
результатами технического и элементарного анализа устанавливаются степень
углефикации и генетический тип угля. Эти данные ориентируют относительно
возможностей промышленного использования угля. Для бурых и низших стадий
каменных углей определяют теплоту сгорания, выход и состав продуктов
полукоксования, а в случае землистых бурых углей - также выход и состав
монтанвоска. Для каменных углей - от газовых до отощенно-спекающихся определяются
спекаемость, вспучиваемость, пластометрические показатели, свойства кокса. Для
тощих углей, полуантрацитов и антрацитов основные технологические
характеристики - теплота сгорания и электропроводность (для антрацитов).
Теоретические исследования угольного вещества проводятся методами
углепетрографии, углехимии и физики. В части химических помимо основных
перечисленных выше показателей для бурых углей включаются определение
содержания и характеристика гуминовых кислот и остаточного угля, для бурых и
каменных углей - определение функциональных групп, группового состава, изучение
с помощью различных органических растворителей и при различных температурах, с
помощью гидролитического расщепления; применяется характеристика углей методами
рентгеноскопии, инфракрасной и ультрафиолетовой спектроскопии, люминесцентного
анализа, электронной микроскопии, парамагнитного и ядерно-магнитного резонанса,
термографии, окисления и гидрогенизации. Перечисленные химические и физические
методы исследования угля позволяют осветить молекулярную структуру угольного
вещества.
Для первичной характеристики горючих ископаемых (ГИ) с целью
определения качества их как товарного продукта необходимы данные исследования
различными методами. Свойства ГИ можно подразделить по некоторым общим признакам,
связанным с методами их определения, на несколько групп:
а) техническая характеристика;
б) элементный состав;
в) физические свойства,
II Технический анализ углей
Все виды твердых горючих ископаемых объединяют в себе две
составляющие: органическое вещество и минеральную компоненту, которую прежде
рассматривали как балласт, но теперь все чаще считают источником ценного
минерального сырья, в частности редких и рассеянных элементов. Для оценки
возможностей и режимов переработки горючих ископаемых применяют технический
анализ, позволяющий определить направления использования их как энергетического
и химического сырья. Под техническим анализом понимается определение
показателей, предусмотренных техническими требованиями на качество угля. В
технический анализ обычно объединяются методы, предназначенные для определения
в углях и горючих сланцах зольности, содержания влаги, серы и фосфора, выхода
летучих веществ, теплоты сгорания, спекаемости и некоторых других характеристик
качества и технологических свойств. Полный технический анализ проводится не
всегда, часто бывает достаточно провести сокращенный технический анализ,
состоящий в определении влажности, зольности и выхода летучих веществ.
1.1
Определение влажности
При анализе угля определяются следующие виды влаги:
Wtr - влага общая (рабочая), %. Определяется по
специальной лабораторной пробе на влагу, обработанной и доставленной в
лабораторию с соответствующими предосторожностями, чтобы сохранить в пробе всю
первоначальную влагу, содержащуюся в угле до самого момента ее определения.
Изменение первоначального веса пробы, указанного в сопроводительных документах,
не должно превышать 0,5 % веса пробы. С момента приготовления пробы до
поступления ее в лабораторию должно пройти не более 12 ч.
Wa - влага аналитическая, %. Определяется по
аналитической пробе. Служит для пересчета показателей качества угля на рабочую,
сухую и условную горючую массы угля;
Wл - влага лабораторная, %. Определяется по лабораторной пробе
для общего анализа;
Wех - влага внешняя или свободная,%. Определяется как разность
по убыли в весе пробы при доведении ее подсушиванием до воздушно-сухого
состояния;
Wh - влага воздушно - сухого состояния, %.
Представляет остающуюся в пробе влагу после подсушивания ее до воздушно-сухого
состояния (при температуре окружающего воздуха 200 С и относительной влажности
воздуха 65%). Ее называют еще влагой связанной или внутренней, также
гигроскопической. Эту влагу определяют по аналитической пробе, доведенной до
воздушно-сухого состояния в указанных условиях лишь в отдельных случаях,
например когда требуется установить гигроскопическую способность угля. Для
массовых определений влагу воздушно-сухого состояния определяют по
аналитической пробе, доведенной в течение 24 ч до воздушносухого состояния в
комнатных и, следовательно, не точно обусловленных условиях температуры и
влажности воздуха. Поскольку для практической оценки угля эти условия обычно
близки в различных лабораториях, то данные о содержании определенной таким
образом влаги могут служить для общей характеристики угля.
Взаимосвязь между влагой рабочей, лабораторной, внешней и
воздушно-сухого состояния определяется следующими соотношениями:
а) в случае отсутствия потери веса пробы при доставке ее в
лабораторию
= Wл; Wtr = Wех + Wh
б) в случае потери веса пробы (до 0,5%) при доставке ее в
лабораторию
=W п + Wn 100 - Wп/100
где Wп - потеря веса пробы, %.
Содержание влаги в углях определяют прямыми и косвенными
методами.
Прямые методы заключаются в отгонке воды из навески угля в
замкнутой системе, улавливании выделяющихся при этом паров воды и определении,
путем непосредственного замера, веса или объема воды в навеске.
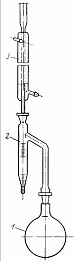
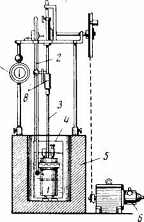
Рис 1. Прибор определения содержания влаги в угле
объемным методом: 1 - колба для перегонки; 2 - приемник; 3 - холодильник
Косвенными методами содержание влаги определяется по
косвенным показателям, например по убыли в весе навески угля после высушивания
ее в сушильном шкафу до постоянного веса, по реакциям, в которые вступает влага
угля при тех или иных на нее воздействиях, по изменению свойств угля в
зависимости от содержания в нем влаги.
Прямые методы определения влаги обеспечивают наиболее точные
результаты анализа, но они трудоемки и поэтому их применяют преимущественно в
тех случаях, когда возникают повышенные требования к точности определения
влаги.
Определение влаги прямыми методами
При определении содержания влаги производится отгонка воды из
навески угля в токе азота или с парами какой-либо жидкости, например толуола
или ксилола, не смешивающейся с водой.
Объемное определение влаги
Объемное содержание влаги в угле определяют методом
дистилляции в приборе, показанном на рис.1. Сущность метода заключается в
нагревании навески угля с толуолом, отгонке выделяющихся из угля паров воды
вместе с парами толуола и замере сконденсировавшейся воды.
Предварительно производят калибровку прибора с толуолом путем
перегонки точно измеренных объемов воды в количествах, соответствующих
возможному содержанию влаги в подлежащих анализу навесках угля, и составляют
график зависимости между количеством воды, взятой для перегонки, и количеством
воды, полученным в приемнике при перегонке.
Во избежание прилипания капелек воды к стенкам колбы,
холодильника и приемника их перед началом опыта обрабатывают раствором
двухромовокислого кальция в серной кислоте, промывают дистиллированной водой и
высушивают.
В течение 1 5 - 20 мин постепенно на песчаной бане нагревают
колбу до кипения, а потом поддерживают кипение таким образом, чтобы
испаряющаяся вода, охлаждаясь, стекала из холодильника в приемник со скоростью
до четырех капель конденсата в секунду. Перегонку ведут до тех пор, пока не
прекратится увеличение объема воды в приемнике в течение 10 - 1 5 мин. Смывают
горячими парами толуола капли воды, оставшиеся на внутренней поверхности
холодильника и верхней части приемника. Возобновляют на некоторое время (5 - 1
0 мин) нагрев жидкости в колбе, так как часть воды со смывом могла попасть
обратно в колбу. Воде в приемнике дают отстояться и охладиться до комнатной
температуры. Замеряют (по нижнему мениску) объем полученной воды в приемнике и
вносят и нему поправку по составленному ранее графику.
Содержание влаги в аналитической пробе угля вычисляют по
формуле: Wa=Vn×100/G, %, где Vn - объем воды в
приемнике, исправленный по графику, мл (считая плотность воды равной 1 г/см3,
можно не делать пересчета с объемных единиц на весовые); G - навеска угля, г.
Допускаемые расхождения результатов приведены в табл.7
Таблица 1
Допускаемые расхождения результатов параллельных определений
содержания влаги
|
Угли
|
Допускаемые
расхождения, %
|
|
|
в одной
лаборатории
|
в разных
лабораториях
|
|
Каменные
|
До 10
|
0,3 абс.
|
0,5 абс.
|
|
Более 10
|
3,0 отн.
|
5,0 отн.
|
|
Бурые
|
До 20
|
0,4 абс.
|
0,8 абс.
|
|
Более 20
|
2,0 отн.
|
4,0 отн.
|
Весовое определение содержания влаги.
уголь влажность зольность сгорание

Рис. 2
Весовое определение влаги в угле производят на установке
(рис.2) путем отгонки воды из навески угля в токе азота при температуре 105 -
1100 С, улавливания и определения количества испарившейся влаги. В качестве
поглотителя влаги используют хлорнокислый магний или серную кислоту.
Установка состоит из системы для подачи азота, поглотительной
системы для улавливания паров воды, выделяющихся при нагревании навески испытуемого
угля, и сушильного шкафа.
Система для подачи азота имеет баллон 1, заполненный азотом,
содержащим не более 0,5% кислорода, редуктор 2, моностат 3 для поддержания
постоянного давления в системе, осушительную колонку 4 и U-образную трубку 5,
заполненные хлорнокислым магнием, реометр 6 для измерения расхода азота.
Поглотительная система состоит из основной и контрольной U-образных трубок 7, 8,
заполненных хлорнокислым магнием, и гуська 9 с серной кислотой, не позволяющей
влаге из воздуха попасть в систему. Если в качестве поглотителя влаги применяют
серную кислоту, то вместо U-образных трубок используют калиаппарат и вместо осушительной
колонки - склянку Дрекселя.
Сушильный шкаф 10 с электрическим или газовым обогревом имеет
устойчивую температуру нагрева 105 - 1100 С. На задней стенке шкафа делают
отверстия 11 и подставку для помещения в шкаф пробирок с навесками угля. В
шкафу имеется гнездо для термометра 13.
Установка рассчитана для проведения одновременно анализа двух
навесок угля.
Содержание влаги в аналитической пробе угля вычисляют по
формуле
Wa= (G1-G2/G)
100, %,
где G1 - увеличение веса основной поглотительной трубки при нагревании
навески угля, г; G2
- увеличение веса контрольной поглотительной трубки, г; G - навеска угля, г.
Вычисление результатов анализа производят с точностью до
0,01%. Допускаемое расхождение результатов определений содержания влаги
составляет 0,3%.
Определение влаги косвенными методами путем высушивания
навесок угля
Сущность метода определения влаги заключается в высушивании
навесок угля в сушильном шкафу при заданной температуре и вычислении содержания
влаги в угле по убыли веса навески при высушивании.
Содержание влаги рабочей, лабораторной и аналитической (Wp, Wл,Wa) вычисляют по формуле:
=G1×100/G, %
где G - навеска угля, г; G1 - убыль веса навески при сушке, г.
Таблица 2
Допускаемые расхождения результатов определений содержания
влаги
|
Содержание
влаги, %
|
Вид влаги
|
Допускаемые
расхождения, %
|
|
|
в одной
лаборатории
|
в разных
лабораториях
|
|
До 10
|
Wa, wji
|
0,3 абс.
|
0,4 абс.
|
|
Wa, Wh
|
0,2 абс.
|
|
|
Более 10
|
wa, wji
|
3,0 Отн.
|
5,0 отн.
|
|
Wa, Wh
|
0,3 абс.
|
|
1.2
Определение зольности
Определение зольности углей производят прямыми и косвенными
методами.
Прямые методы определения зольности заключаются в озолении
навески угля, помещаемой в фарфоровой лодочке или в небольшом противне в
муфельную печь, и прокаливании зольного остатка. Выход зольного остатка в
процентах к первоначальному весу навески принимают за зольность угля.
Точность определения зольности может изменяться в зависимости
от конечной температуры, достигаемой в печи, от скорости нагрева и расположения
навесок, от атмосферы, которая может быть создана в печи (например,
прокаливание в токе кислорода или в токе инертных газов, предварительное
смачивание навески концентрированной соляной или азотной кислотой и т.д.).
От изменения указанных факторов изменяются и условия
определения зольности угля. Большое значение для условий определения имеет и
требуемая точность анализа, в соответствии с чем различают медленное и
ускоренное озоление навески угля.
Примерами косвенных методов являются определения зольности,
основанные на различной проницаемости угля и минеральных примесей
рентгеновскими лучами, радиоактивными изотопами и т.д. Эти методы дают менее
точные по сравнению с прямыми методами результаты определений зольности угля.
Но для их применения необходимо иметь дорогую и требующую квалифицированного
обслуживания аппаратуру.
Для расчетов угольных предприятий с потребителями за качество
зольность угля и продуктов обогащения в расчетно-аналитических товарных пробах
углехимические лаборатории определяют, применяя прямые методы медленного и
ускоренного озоления навески угля (по ГОСТ 11022 - 64).
Для озоления навески применяют муфельные печи с максимальной
температурой нагрева 900' С. При ускоренном определении зольности печь должна
иметь вытяжную трубу диаметром 20 мм, расположенную в задней стенке печи.
Определение зольности
Вычисление зольности аналитической пробы угля и пересчет
зольности на сухую и рабочую массы угля производят по формулам:
=G1×Q100/G Ad = Аа (100/100-W3) Ar
= Aa (100 - Wrt /100-W3)
где G1 - вес зольного остатка, г; G - навеска угля, г; Wa, Wrt - влага аналитическая и
рабочая, %.
Все вычисления ведут с точностью до 0,01%, округляя
окончательные результаты до десятых долей процента (табл. 9).
Таблица 3
Допускаемые расхождения результатов параллельных определений
зольности
|
Зольность угля,
%
|
|
в одной
лаборатории
|
в разных
лабораториях
|
|
От 12 до 25
|
0,3
|
0,5
|
|
Более 25
|
0,5
|
0,7
|
1.2.1
Определение содержания минеральных примесей
Зола, получаемая при озолении навески угля и после
прокаливания ее, отличается по содержанию от минеральных примесей, находившихся
в исходном угле. Последние под действием высоких температур претерпевают
различные превращения: из алюмосиликатов удаляется кристаллизационная вода, в
связи с чем их вес уменьшается примерно на 8 %, из карбонатов выделяется
двуокись углерода и вес их уменьшается до 40 - 50%; серный колчедан окисляется
с образованием окиси железа и двуокиси серы, которая может улетучиться или
вступить во взаимодействие с имеющимися в минеральной части угля окислами
кальция и магния. Улетучиваются также и некоторые хлориды.
В процессе озоления угля в его минеральной части возможны,
помимо потерь кристаллизационной воды, следующие основные реакции:
=CaO+CO2;
Fe0+02= 2Fe2O3;
FeS2+1102=2Fe2O3+8SO2;
SO2+2CaO+02=2CaSO4;4×2H2O=CaSO4+ 2H2O.
Могут быть и другие превращения минералов.
Приближенно истинное содержание в угле минеральных примесей
определяют по зольности, введя в нее поправочный коэффициент 1,08 - 1,10. Для
более точных подсчетов содержание минеральных примесей определяют химическим
путем.
Сущность метода заключается в частичном извлечении
минеральных примесей путем обработки навески угля соляной и фтористоводородной
кислотами в условиях, при которых вещество угля не изменяется. Вычисляют убыль
в весе навески в результате обработки ее кислотами, а затем навеску озоляют и в
зольном остатке определяют содержание железа. По содержанию железа в золе
вычисляют содержание серного колчедана в угле. Определяют также количество
соляной кислоты, поглощенной углем.
Для испытания берут навеску угля 6 г и переносят ее в
химический стакан из кислотоупорного материала (политена или поливинилхлорида)
вместимостью 200 мл, добавляют в стакан 40 мл 5 н. соляной кислоты. Стакан
помещают на водяную баню, в которой поддерживают температуру 55 - 600 С, и
выдерживают на ней 45 мин, перемешивая через каждые 5 мин содержание стакана,
остальное время стакан должен быть закрыт.
После отстаивания взвеси угля в стакане в течение 10 мин
раствор декантируют, сливая его на фильтр, куда переносят и уголь, который
промывают водой и затем смывают (не более 10 мл воды) опять в стакан. Добавляют
в стакан 40 мл фтористоводородной кислоты (плотность 1,13 г/см) и нагревают на
водяной бане так же, как и нагревали с соляной кислотой. Взвесь в стакане
отстаивают и декантируют раствор через тот же фильтр. Уголь в стакане промывают
3 раза водой, сливая ее на фильтр, а затем переносят на фильтр весь уголь (при помощи
палочки с резиновым наконечником) и промывают его 20 раз порциями горячей воды
по 25 мл. В течение 5 - 1 0 мин просушивают уголь на воздухе, затем фильтр с
углем помещают в вакуумный шкаф, где его сушат в течение 1,5 ч при температуре
500 С. Высушенный уголь выдерживают на воздухе в течение 1 ч и взвешивают.
Возможно полнее переносят уголь в склянку с притертой пробкой и взвешивают
очищенный фильтр. Вес угля определяют по разности между весом фильтра с углем и
без угля.
Тщательно перемешивают обработанный кислотами уголь и
определяют в нем содержание влаги, хлора, а также зольность и содержание железа
в зольном остатке. По содержанию хлора вычисляют эквивалентное ему содержание
соляной кислоты, а по содержанию железа в зольном остатке - эквивалентное ему
содержание серного колчедана в угле. Все результаты пересчитывают на сухую
массу угля (содержание влаги в испытуемой навеске угля определяют
предварительно).
Содержание минеральных примесей в угле вычисляют по формуле
= (G-G1+K+HCl+1,1A) 100/G, %,
где G - навеска угля, г;
G1 - вес навески после обработки ее соляной и
фтористоводородной кислотами г;
К - вес серного колчедана, содержащегося в навеске, г;
HCl - вес соляной кислоты, содержащейся в навеске,
г;
А - вес зольного остатка за вычетом веса окиси железа, г
В настоящее время разработан целый ряд физических методов для
определения содержания в ТГИ минеральных компонентов: а) микроскопическое
определение содержания минеральных компонентов по их рельефу, цвету, степени
блеска, т.е. по оптическим признакам; б) рентгеноскопический, использующий
особенности рассеивания рентгеновских лучей различными минеральными веществами;
в) радиоизотопный, основанный на взаимодействии атомов минеральных примесей с
радиоактивным излучением изотопов.
Степень минерализации ТГИ зависит от многих генетических
факторов. Зольность торфа колеблется в зависимости от степени минерализации
питающих вод и типа торфа. Зольность низинного торфа изменяется от 6 до 18 %,
переходного - от 4 до 6 %, верхового - от 2 до 4 % при средних величинах 7,6;
4,7 и 2,4 %. Компонентный состав минеральной части торфов определяется
минеральным режимом торфообразования. В торфах верхового типа преобладают
оксиды кремния (47 % от массы золы). В низинных торфах накапливаются соли
кальция и железа.
Зольность углей зависит не только от состава исходного
растительного материала, условий их накопления и первичного превращения, но и
от горно-геологических условий формирования угольных пластов. Зольность углей
может формироваться под влиянием факторов, действующих на разных стадиях его
образования. В соответствии с этим различают зольность: а) внутреннюю,
связанную с содержанием в первичном материале золообразующих элементов (в
основном щелочных металлов); б) внешнюю первичную, обусловленную накоплением в
торфянике растворимых солей из подстилающей почвы, внесением минеральных
примесей грунтовыми водами и ветром; в) внешнюю вторичную, обусловленную
проникновением минеральных веществ в пласты органических накоплений при
погружении торфяника в недра земли и на дальнейших стадиях их генезиса; г)
случайную, связанную с добычей ТГИ, особенно в условиях применения механизации,
за счет вовлечения в товарный продукт боковой породы из кровли, почвы и
природных прослоек.
Зольность ТГИ играет исключительно большую роль как показатель
их качества, является балластом и приводит к значительным транспортным расходам
при перевозке ТГИ. Высокая их зольность при использовании в качестве топлива
снижает показатели работы энергетических установок. Для производства кокса
используют угли с зольностью 7 - 10 %. Высокая зольность углей ухудшает
качество кокса и показатели работы доменных печей: увеличивается расход кокса
на 1 т производимого чугуна и снижается их производительность. Для уменьшения
содержания минеральных примесей в углях и сланцах их подвергают обогащению
различными методами: гравитационным или флотационным, а также путем
обеззоливания химическими реагентами. В результате получают угольный
концентрат, промежуточный продукт и отходы, т.е. в основном минеральную часть.
1.3 Определение
выхода летучих веществ
Летучими веществами считаются паро- и газообразные продукты
термического разложения органической массы угля и некоторых минеральных его
примесей, выделяющиеся из угля при нагревании без доступа воздуха.
Сюда не входит влага, содержащаяся в угле и испаряющаяся при
его нагревании.
Выход летучих веществ и их состав в значительной степени
зависят от температурных условий и времени нагревания угля.
Поэтому сравнимые результаты определений выхода летучих
веществ могут быть получены лишь при строго регламентированных температурном
режиме и времени нагревания угля.
Определяют весовой и объемный выходы летучих веществ.
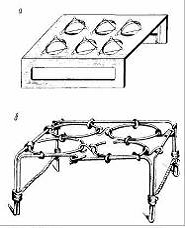
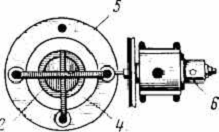
Рис.3. Подставка для тиглей: из а-жаростойкой
стали; б - из нихромовой проволоки
При определении выхода летучих веществ в каменных углях и
антрацитах для их классификации зольность проб не должна быть более 10%. В
противном случае их обогащают расслоением в четыреххлористом углероде
плотностью 1,4 г/см3 (каменные угли) или в хлористом цинке плотностью 1,8 г/см3
(антрациты).
Весовой выход летучих веществ определяют (по ГОСТ 6382 - 80)
нагреванием навески аналитической пробы угля. За выход летучих веществ
принимают разность между общей убылью веса навески после нагревания, выраженной
в процентах к первоначальному весу, и убылью ее веса за счет испарившейся
влаги.
Определение весового выхода летучих веществ
Навески аналитической пробы угля берут на различной глубине
из двух-трех мест в предварительно взвешенные фарфоровые или кварцевые высокой
формы тигли с притертыми крышками.
Навески бурых углей (а также окисленных каменных углей) перед
испытанием брикетируют в лабораторном брикетном прессе. Брикеты вынимают из
пресса и взвешивают в тиглях, уточняя взятую для испытания навеску угля.
Для нагревания навесок угля применяют электрическую муфельную
печь с температурой нагрева до 1 0000 С и с автоматическим регулированием
температуры. В передней стенке печь должна иметь отверстия для выпуска летучих
веществ, а в задней стенке - отверстия для установки термопары. В печи должен
быть участок с устойчивой температурой 850 ± 100 С, площадь которого
определяется предварительным замером температуры в закрытом муфеле.
Термопару располагают так в муфельной печи, чтобы горячий
спай ее находился в пространстве на расстоянии 1 0 - 20 мм от пода печи по
середине зоны с устойчивой температурой.
Тигли с навесками, закрытые крышками, вводят в печь, нагретую
до 8600 С, на специальной подставке на шесть, четыре или два тигля (рис.1 2) из
нихромовой проволоки или из жаростойкой стали в зону устойчивой температуры, с
таким расчетом, чтобы под печи был от дна тигля на расстоянии 20 мм.

Рис.4. Схема установки тиглей в муфельной печи
Схема установки тиглей в печи показана на рис.4.
В закрытой печи выдерживают тигли в течение 7 мин.
Температура, понизившаяся при установке тиглей в печь, должна снова достичь 850
□ 10° С не более чем за 3 мин. В противном случае испытание повторяют.
После 7-минутного пребывания тиглей в печи их вынимают оттуда
и охлаждают сначала в течение 5 мин на воздухе (не снимая крышек), а затем до
комнатной температуры в эксикаторе, после чего взвешивают и освобождают от
нелетучего остатка.
Вычисление выхода летучих веществ аналитической пробы угля
производят по формулам
= (G1×100) /G-Wa; Va=
(G1×100) /G-Wa - [ (C02) ak],
где G1 - убыль веса навески после выделения летучих веществ и испарения
влаги, г;
G - навеска угля, г;
Wa - влага аналитическая, %;
[ (C02) ak] - разность между содержанием двуокиси углерода карбонатов в
исходной пробе испытуемого угля и в нелетучем остатке после определения выхода
летучих веществ, %.
При содержании в аналитической пробе более 2% двуокиси
углерода карбонатов (C02) ak выход летучих веществ рассчитывают с поправкой на двуокись углерода.
Выход летучих веществ в аналитической пробе пересчитывают на
условную горючую массу угля:
= Va×100/ (100-Wa -
Va)= Va×100/ (100-Wa-Va - [ (C02)
ak],
Твердый нелетучий остаток в тигле, получаемый после
определения выхода летучих веществ, переносят путем опрокидывания и легкого
встряхивания тигля на фарфоровую или стеклянную пластинку.
В зависимости от внешнего вида и прочности нелетучего остатка
его характеризуют по следующей классификации: порошкообразный; слипшийся - при
легком нажиме пальцем рассыпается в порошок; Допускаемые расхождения между
результатами двух параллельных
Таблица 4
|
Выход летучих
веществ, %
|
Допускаемые
расхождения, %
|
|
в одной
лаборатории
|
в разных
лабораториях
|
|
Менее 9
|
0,3
|
0,5
|
0,5
|
1,0
|
|
45 и более
|
1,0
|
1,5
|
Определение объемного выхода летучих веществ
Сущность метода определения объемного выхода летучих веществ
заключается в нагревании пробирки с навеской аналитической пробы
угля в трубчатой печи при температуре 900 ±10 С в течение 15
мин и определении объема выделившихся из навески летучих веществ. Установка для
проведения анализа показана на рис.5.

Рис.5. Установка для определения объемного выхода
летучих веществ: I - гальванометр; 2 - термопара; 3 - трубка хлоркальциевая; 4
- отводная трубка; 5 - пробирка кварцевая; 6 - трубчатая печь; 7 - навеска
угля; 8, 9, 14, 16 - краны; 1 0 - манометр; 11 - термометр; 12 - аспиратор; 13
- уравнительный сосуд; 1 5 - тройник; 1 7 - мерный цилиндр
Аспиратор наполняют насыщенным раствором хлористого натрия,
слегка подкисленным соляной кислотой и подкрашенным метиловым оранжевым индикатором.
Закрывают пробкой открытый конец манометра и открывают все краны, кроме крана
на уравнительном сосуде. При этом из аспиратора вытекает раствор хлористого
натрия в мерный цилиндр, вследствие чего в системе создается разрежение. Если
система герметична, то ток раствора через 1,0-1,5 мин прекращается.
Определяют величину поправки на расширение воздуха в пробирке
при ее нагревании.
Пробирку (без навески) закрывают пробкой с отводной трубкой,
присоединяемой к системе, и опускают на глубину 145 мм в печь, нагретую до
температуры 910 - 9150 С, где выдерживают в течение 15 мин, поддерживая
температуру нагрева 900 ± 10 С. Объем жидкости, вытекшей из аспиратора за это
время, принимают за постоянную величину.
Для определения выхода летучих веществ берут навеску
аналитической пробы угля.
Навеску помещают в кварцевую пробирку диаметром 11 мм, длиной
210 мм, соединяют с установкой через отводную трубку и опускают в нагретую до
температуры 910 - 9150 С трубчатую печь на глубину 145 мм. Краны, соединяющие
пробирку с манометром и аспиратором, должны быть при этом открыты.
Понизившаяся при установке пробирки температура в печи должна
подняться до 900 +100 С не более чем за 3 мин, в противном случае испытание
повторяют. Последняя температура поддерживается в печи за весь период
испытания. Ток раствора хлористого натрия из аспиратора регулируют так, чтобы
давление в системе поддерживалось постоянным.
Нагревание пробирки с навеской производят в течение 15 мин,
записывая через каждые 3 мин температуру газа в аспираторе. Затем закрывают
вначале краны у манометра и мерного цилиндра, а затем кран, соединяющий
пробирку с аспиратором, вынимают пробирку из печи. В мерном цилиндре отмечают
объем вытекшей в него жидкости.
Объемный выход летучих веществ в навеске аналитической пробы
рассчитывают для нормальных условий по формуле
об= (V1 - V2) ×293P/ (273+t) ×760, мл/г
где V1 - объем раствора хлористого натрия, вытекший из аспиратора, мл;
V2 - объем воздуха, выделившийся из пробирки при нагревании,
мл;
- нормальная температура воздуха (200 С), 0К (градусы
абсолютной шкалы);
Р - барометрическое давление во время испытания с учетом
разрежения в аспираторе (Р = Р1 - Р), где Р1 - барометрическое давление, р -
разрежение), мм рт. ст.;
t - средняя температура газа в аспираторе, 0 С;
+ t - то же, 0 К;
- нормальное атмосферное давление, мм рт. ст.
Объемный выход летучих веществ на 1 г горючей массы
пересчитывают по формуле
6= Vao6×100/100-Wa-Aa, мг/г
где - содержание влаги в аналитической пробе, %,
Аа - зольность аналитической пробы, %.
При анализе углей новых месторождений в них определяют
содержание двуокиси углерода карбонатов. Если содержание ее более 2 %, то
вводят поправку на объемный выход летучих веществ. Содержание двуокиси углерода
карбонатов определяют в навеске 1 г и в нелетучем остатке (для анализа берут
весь остаток) после определения объемного выхода летучих веществ. Разница между
результатами этих определений представляет собой количество двуокиси углерода
карбонатов, перешедшей в газ при анализе на объемный выход летучих веществ.
Объем двуокиси углерода карбонатов, перешедшей в газ,
вычисляют по формуле
VaCO2=g (22400×293P) / (44 (273+t) 760), мл
где g - вес двуокиси углерода, перешедшей в газ.
Остальные значения букв - см. предыдущие формулы.
С учетом двуокиси углерода карбонатов объемный выход летучих
веществ в навеске антрацита составит
=Vao61 - VaCO2, мл/г.
Допускаемые расхождения в результатах параллельных
определений выхода летучих веществ Vaoб для одной лаборатории - 6 % по отношению к
меньшему результату определения; Vгoб для разных лабораторий - 8 % отн.
Определение весового содержания двуокиси углерода,
карбонатов.
Определение двуокиси углерода карбонатов производят (по гост
13455 - 68) путем воздействия на навеску аналитической пробы угля соляной
кислотой и весового определения выделяющейся при этом в результате разложения
карбонатов двуокиси углерода, поглощаемой едким кали, и натронной известью.
1.4
Определение спекаемости
Способность углей спекаться является одним из важнейших их
свойств, определяющих возможность получения из угля прочного кускового кокса.
О спекающей и коксующей способности угля первое качественное
представление получают по внешнему виду коксового королька (нелетучего
остатка), образующегося при анализе угля на выход летучих веществ путем нагревания
навески аналитической пробы угля без доступа воздуха и ее прокаливания.
Например, коксовыми считают угли, образующие сплавленный и нормально вспученный
королек. Угли неспекающиеся для производства кокса непригодны.
Численное выражение для спекаемости углей было впервые
предложено английским ученым Кампредоном (1895 г.). Максимальное весовое
количество песка, спекаемого 1 г измельченного угля при нагревании смеси без
доступа воздуха, было принято за число, выражающее спекающую способность угля. Для
неспекающихся углей это число получилось равным нулю, для хорошо спекающихся
коксовых углей - оно достигало 17.
Этот принцип определения спекаемости угля по количеству
спекаемого им инертного материала положен в основу и некоторых современных
методов исследования углей на спекаемость.
Важнейшей характеристикой коксового королька, определяющего
спекаемость угля, является степень его вспученности. Это вспучивание происходит
в период пластического состояния угля, когда идет процесс его разложения с
образованием газообразных и парообразных продуктов. Газы и пары стремятся
удалиться из пластической массы. Встречая на своем пути сопротивление массы,
они распирают ее, в результате чего происходит вспучивание королька при
затвердевании массы. В зависимости от вязкости пластической массы вспучивание
королька будет тем больше, чем больше ее вязкость, и наоборот, чем меньше
вязкость массы, тем меньше вспучиваемость королька.
Таким образом, численное выражение степени вспучивания
коксового королька может также служить характеристикой спекаемости углей.
Современные углехимические лаборатории характеризуют
спекаемость углей следующими числовыми показателями: спекающей способностью
угля (по ГОСТ 2013 - 49); числами вспучивания - дилатометрическими показателями
в приборе ИГИ - ДМеТИ (по ГОСТ 14056 - 68); индексом свободного вспучивания
королька, индексом Рога (по ГОСТ 9318 - 79).
Последние два метода определения спекаемости применяют при
международной классификации углей.
Определение спекаемости
Определение спекающей способности угля (ускоренное)
Сущность метода определения спекающей способности заключается
в нагревании смеси испытуемого угля с песком при установленном температурном
режиме и определении спекающей способности угля по степени пластичности
размягченной угольной массы.
Для характеристики этого параметра определяют способность
проникновения размягченных угольных зерен под воздействием внешней силы в
пустоты между частицами угля и смешанного с ним мелкозернистого кварцевого
песка.

Рис. 6
Спекающую способность определяют в аппарате ИГИ (рис.6),,
состоящем из печи и съемной части. Основная рабочая часть аппарата состоит из
стаканчика (цилиндра), помещаемого в печь, рычага с грузом для воздействия на
нагрузку и шкалы для отсчета показателей. Съемная часть аппарата монтируется на
станине 1, имеющей ручку 11 и ножки 21. На стойках 10 и укреплены шкала 5 с
отвесом 2 и рычаг 7 с грузом 19 и кулисой 6. С помощью винтов 3 шкалу можно
передвигать. Рычаг штифтами 8 шарнирно соединен со стойкой и штемпелем 9. К
съемной части относятся также и стаканчик 12, и трубка 1 8 со вставным
донышком, куда помещается смесь испытуемого угля с песком. Трубка обогревается
электропечью, состоящей из цилиндра 1 5 с обмоткой из нихромовой проволоки,
уложенной на изоляционную прокладку из асбеста, установленного на подставке 16.
Печь помещена в кожух 1 4 с асбестовой набивкой, покоящейся на подставке 1 7,
имеющей установочные винты 1 8. Температура печи контролируется с помощью
термопары, помещаемой в углубление 20. Регулирование температуры осуществляется
ползунковым реостатом.
Для испытаний угля на спекаемость можно применять любой
чистый кварцевый песок с окатанными зернами. Подготовку песка осуществляют,
освобождая его, прежде всего, от примесей глинистых веществ (путем
отмучивания), а затем обрабатывают 10% - ным раствором соляной кислоты с
последующей промывкой, высушиванием и прокаливанием. Чистый песок просеивают на
стандартных ситах. Песок, проходящий через сито № 60 (класса 0,21 - 0,28 мм),
собирают отдельно от песка, проходящего через сито № 80 и остающегося на сите №
100 (класс 0,16 - 0,21 мм).
Для работы оставляют песок, состоящий на 60,5% из класса 0,21
- 0,28 мм и 40 - 35% из класса 0,16 - 0,21 мм.
В навеску для испытания берут 5,86 мл песка (в указанном
объеме стандартный песок весит 8,5 г).
Чтобы проверить эти величины, в сухую бюретку объемом 25 мл
засасывают 15 мл воды, всыпают и нее немного песка и легким постукиванием по
бюретке выравнивают уровень песка по одному из нижних делений бюретки, после
чего добавляют в 6юретку 8,5 г испытуемого песка (взвешенного с точностью до
0,0002 г). Через 2 - 3 ч, когда песок осядет, отмечают верхний и нижний уровни
взятой навески, получая объем V, занимаемый 8,5 г испытуемого песка. Вес песка G, соответствующий 5,86
мл, будет
=8,5×5,86/V, г.
Отвешивают на технических весах 1,8 г (с точностью до 0,002
г) измельченного до крупности 0,28 мм воздушно-сухого угля и смешивают в
фарфоровой чашке с песком.
С помощью шаблона устанавливают стрелку рычага на нулевое
деление шкалы, вынимают трубку, извлекают из нее шаблон и через воронку всыпают
приготовленную смесь (площадь сечения трубки в этом случае должна быть точно в
пределах 1,12 - 1,18 см). Трубку опускают в стаканчик и вставляют в нее
штемпель.
Печь разогревают и поддерживают в ней не менее 5 мин
температуру 500 - 5050 С, после чего съемная часть аппарата устанавливается на
печи в рабочем положении (трубка с навеской смеси угля и песка опущена в
стакан).
При достижении температуры размягчения угля происходит, ввиду
пластической деформации его и проникновения размягченной угольной массы в
пустоты между частицами песка, уменьшение объема углепесочной смеси в трубке и
штемпель с рычагом постепенно опускаются. Испытание продолжают до прекращения
движения рычага, примерно 1 5 мин.
Отмечая начальное Н и конечное К деления по шкале, определяют
спекающую способность угля:
=К - Н.
При расхождении результатов двух параллельных определений
спекающей способности угля более 10% опыт повторяют.
Антрациты и тощие угли практически почти не обладают
спекающей способностью. Определенная этим методом спекающая способность
коксующихся углей обычно колеблется в пределах 13 - 30 единиц.
Для определения спекающей способности многозольных углей (А
> 15%) их предварительно обогащают в растворе хлористого цинка, затем
промывают и сушат до воздушно-сухого состояния.
1.5
Определение теплоты сгорания углей
Теплота сгорания углей является одной из важнейших его
энергетических характеристик. Основными горючими веществами в угле являются
углерод, водород и отчасти сера (органическая и колчеданная). При горении
углерода по реакции С+О2=СО2 выделяется 8137 ккал/кг тепла.
Соединения водорода с углеродом дают примерно такие же
количества тепла, но водород, связанный с кислородом (Н2О), в тепловом
отношении является не только потерянным, но даже вредной составной частью угля
(затрачивается тепло на испарение). Процентное содержание связанного водорода в
угле упрощено, его представляют равным м 8 '
Сера выделяет при сгорании в SO2 2187 ккал/кг тепла.
Если допустить, что элементы, входящие в состав угольного
вещества, дают при сгорании столько же тепла, сколько и в свободном состоянии,
то приближенно по составу угля его высшую теплоту сгорания можно выразить так:
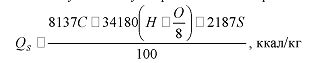
Д.И. Менделеев дает упрощенную формулу (округление
коэффициентов, уменьшение числа составных частей) для определения теплоты
сгорания угля:
=81C+300H-26 (O-S), ккал/кг,
где C, H,
O, S - содержание в угле
соответственно углерода, водорода, кислорода и серы, %.
При определении теплоты сгорания надо учитывать фазовое
состояние, как сжигаемого вещества, так и продуктов реакции. Если в результате
реакции получается газ, то определенная опытом теплота сгорания при постоянном
объеме Qv отличается от теплоты сгорания при постоянном давлении Qp на величину работы,
которую могут совершить образующиеся газы:
=Qp+RT (n'
- n),
где n' - число молей газа до реакции;
n - число молей газа после реакции;
R - универсальная газовая постоянная;
Т - температура, 0К.
Экспериментально теплоту сгорания угля определяют (по ГОСТ
147 - 74) путем сжигания навески аналитической пробы угля в калориметрической
бомбе (при постоянном объеме) в среде сжатого кислорода, насыщенного водяным
паром, и в определении количества теплоты, выделившейся при сгорании угля, а
также при образовании и растворении в воде серной и азотной кислот в условиях
испытания угля.
Навеску угля помещают в чашечку, располагаемую внутри
калориметрической бомбы, заполненной кислородом под давлением 25 - 30 кг/см2 и
соединенной с запалом тонкой (диаметр 0,1 - 0,2 мм) проволокой, с помощью
которой, пропуская электрический ток, поджигают навеску.
Зная вес сожженного угля и количество воды в
калориметрическом сосуде, наблюдая повышение ее температуры, вычисляют теплоту
сгорания Qas угля.
Следует учитывать, что при сжигании угля в топках
содержащаяся в нем вода переходит в пар, удаляющийся вместе с топочными газами,
унося с собой скрытую теплоту парообразования, в бомбе же пар конденсируется.
Содержащиеся в угле сера и азот в бомбе окисляются и образуют
серную и азотную кислоты с выделением некоторого количества тепла. В топках
этого не происходит - горючие соединения серы в угле образуют сернистый газ,
уходящий вместе с продуктами сгорания угля.
Азот же не окисляется и удаляется из топки в виде инертного
газа. Поэтому в полученные результаты анализа вносят поправки на скрытую
теплоту парообразования в топках и теплоту образования кислот в бомбе.
Калориметрическая установка
а б
Рис.7. Калориметрические бомбы: а -
самоуплотняющаяся; б - несамоуплотняющаяся
Калориметрическая установка для определения теплоты сгорания
состоит из собственно бомбы, калориметрического сосуда с мешалкой, оболочки для
тепловой изоляции, контрольно-измерительной аппаратуры и вспомогательных
приборов. Применяют самоуплотняющиеся и несамоуплотняющиеся бомбы.
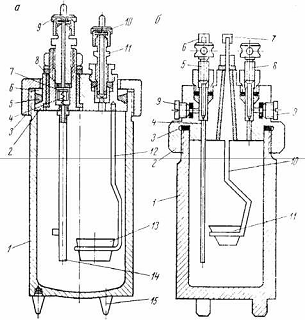
Самоуплотняющаяся бомба (рис.7, а) состоит из стакана 1,
крышки 2, накидной гайки 3 с накаткой, металлического кольца
и уплотняющего резинового кольца 5. Крышка с кольцами
завинчивается от руки накидной гайкой, чем достигается предварительное
уплотнение бомбы. При подаче в бомбу сжатого кислорода избыточное давление его
приподнимает крышку вверх, сжимая резиновое кольцо. Этим достигается хорошее
самоуплотнение бомбы.
Входной штуцер бомбы закрывается самоуплотняющимся клапаном
7, прижимаемым давлением кислорода в бомбе к эластичной кольцевой прокладке 8.
Пружина 4 служит для предварительного уплотнения клапана. Снизу клапан
заканчивается газопроводной трубкой 1 4, служащей одновременно одним из
электродов. Второй электрод 1 2, ввинченный в крышку бомбы, заканчивается
кольцом, в которое помещают чашечку 13 для сжигаемой навески угля.
Выходной штуцер 11 бомбы выполнен в виде игольчатого вентиля.
Оба штуцера (входной и выходной) снабжены колпачками 9 и 10 с эластичными
прокладками. Колпачки имеют штифты для подведения тока. Стакан бомбы стоит на
ножках 15.

а б в
Рис.8. Калориметрический сосуд с вертикальной
мешалкой: 1 - бомба; 2 - термометр; 3 - мешалка; 4 - калориметрический сосуд; 5
- двухстенный кожух; 6 - электродвигатель; 7 - секундомер; 8 - лупа
Несамоуплотняющаяся бомба (рис.7, б) представляет собой
цилиндрический стакан 1 емкостью около 300 мл. В нижней части стакана имеются
три ножки, служащие для укрепления бомбы в специальной подставке. Бомбу
закрывают навинчивающейся на нее по резьбе крышкой 2, где сделан кольцевой
желоб, куда для герметизации закладывают свинцовую прокладку 8. Крышку
завинчивают двуплечим ключом, который накладывают на выступающий прямоугольный
выступ, на этот же выступ сверху ввинчивают контргайки с запорными вентилями 5
- для подачи в бомбу кислорода и вентилями 8 - для выпуска газов после
испытания. Под контргайки для герметичности укладывают набивку из баббита или
фибры. На выступе крышки имеются два горизонтальных канала, закрываемые
резьбовыми пробками 9. При наполнении бомбы кислородом пробку 9 вывинчивают, а
вместо нее вставляют штуцер соединительной трубки для манометра. Трубка 4
служит для подачи в бомбу кислорода и одновременно является токоведущим
стержнем для воспламенения запальной проволоки. Контактная проволока - второй
электрически изолированный токоведущий стержень 10 из жароупорной стали,
который имеет загнутый в виде кольца нижний конец, куда вставляется чашечка 11
с навеской испытуемого угля. К штифтам 6 и 7 присоединяют концы электрической
схемы зажигания.
Для сохранения герметичности бомб их надо содержать в чистоте
и промывать водой после каждого опыта, а обращение с вентилями, гайками и
прокладками должно быть осторожным. Если замечено, что бомбы негерметичны, надо
проверить и подтянуть контргайки, сменить набивку и т.д. Бомбы ежегодно
опрессовывают гидравлическим давлением на 100 кг/см, после чего тщательно
промывают бензином, спиртом и эфиром.
Калориметрический сосуд (рис.8) представляет собой сделанный
из латуни никелированный цилиндр, заполненный дистиллированной водой, в него
погружают калориметрическую бомбу. Сосуд устанавливают в кожух, подкладывают
под него подставку из теплоизоляционной пластмассы (эбонита, текстолита и др.)
или из стекла.
В сосуд опускают мешалку (рис.8), состоящую из стержня а с поперечной
планкой б, к концам которой прикреплены две вертикальные планки в, несущие три
серповидные пластинки с отверстиями, перемешивающие при вращении воду около
бомбы. Для вращения мешалки установлен электродвигатель мощностью 50 - 100 вт.
Скорость вращения регулируют с помощью реостата. Калориметрический сосуд
защищен от внешних тепловых воздействий кожухом, называемым оболочкой
калориметра, состоящей из двух стенок, сделанных из латуни с двойным дном.
Сосуд покрыт текстолитовой или деревянной лакированной крышкой, состоящей из
двух половинок и имеющей отверстия для термометра, стержня мешалки и шнура для
запала навески угля. Пространство между стенками калориметрического сосуда
заполняется воздухом или водой. Для предохранения воды от порчи в нее подливают
несколько капель фенола. Для наблюдения за уровнем ртути в термометре имеется
двухлинзовая лупа или короткофокусная зрительная труба с фокусным расстоянием
0,5 - 1,0 мм.
Проведение калориметрического испытания.
Предварительно для каждого калориметра должна быть определена
его фактическая теплоемкость - количество тепла, необходимое для нагревания на
1 0 С калориметрической системы, состоящей из калориметрической бомбы,
помещенной в сосуд с водой, мешалки и термометра при данной глубине его
погружения.
Фактическую теплоемкость калориметра определяют сжиганием в
бомбе навески эталонной бензойной кислоты, теплота сгорания которой заранее
известна (6320 кал/г), и замером изменяющейся при этом температуры
калориметрической системы.
В калориметрический сосуд наливают дистиллированную воду в
таком количестве, чтобы несамоуплотняющаяся бомба после установки на дно сосуда
была погружена в воду до 2/3 высоты контргаек вентилей. Самоуплотняющаяся бомба
должна быть покрыта водой до нижнего края колпачка.
Количество воды в калориметрическом сосуде при последующих
определениях теплоты сгорания угля должно быть тем же самым, что и при
определении фактической теплоемкости калориметра.
Испытания проводят в следующем порядке. Берут навеску 1 □
0,1 г бензойной кислоты, спрессовывают ее и взвешивают с точностью до 0,0002 г
в чашечке для сжигания (вес которой предварительно определен). Поместив чашечку
с брикетом в кольцо токоведущего стержня, один конец запальной проволоки
присоединяют к трубке 12, а другой к токоведущему стержню 18. Наливают в стакан
бомбы 1 мл дистиллированной воды, закрывают и завинчивают бомбу крышкой.
Осторожно наполняют бомбу кислородом, до давления 25 - 30
кг/см не вытесняя из нее воздуха, наблюдая за равномерностью подачи кислорода
по манометру, установленному на редукторе кислородного баллона.
Заполнив бомбу кислородом, отключают подводящую кислород
трубку и отверстия в крышке бомбы закрывают резьбовыми пробками. Устанавливают
калориметрический сосуд на изоляционную подставку. Чтобы иметь возможно меньшую
поправку на радиацию, температура воды в сосуде должна быть ниже температуры
воды в оболочке на 5 - 1,50 С. Для получения требуемой температуры воду
разбавляют соответственно более теплой или более холодной водой при непрерывном
ее помешивании (термометром). Бомбу погружают в калориметрический сосуд (не
касаясь пальцами воды). В случае выделения из бомбы пузырьков кислорода
испытание отменяют и проверяют герметичность вентилей и прокладки. Если бомба
герметична, включают подводку тока запальной цепи напряжением 1 2 в и
устанавливают мешалку и термометр. Проверяют правильность сборки отдельных
частей калориметрической установки (термометра и стенок сосуда; положения
ртутного резервуара термометра, середина которого должна находиться на высоте
половины части бомбы, погруженной в воду; перемешивающие части мешалки должны
быть в воде). Осторожно закрывают сосуд крышкой так, чтобы не внести каких-либо
нарушений в положение отдельных частей установки.
В процессе самого испытания можно отметить три периода:
начальный, предшествующий сжиганию навески, когда
производится учет теплообмена данной калориметрической установки с окружающей
средой при начальной температуре испытания;
главный, во время которого сгорает навеска и при передаче
тепла калориметрической системы происходит выравнивание температуры всех ее
частей;
конечный, служащий для учета теплообмена калориметрической
установки при заключительной температуре испытания.
Испытание начинают через 5 - 10 мин после начала
перемешивания воды в калориметрическом сосуде, когда выровняется температура
всех частей калориметрической установки. Отсчеты температуры (по шкале
термометра) делают в 1 - м периоде каждую минуту, всего 5 отсчетов за 5 мин
(точностью до 0,0010 С). Моменты отсчетов определяют по секундомеру.
Тысячные доли градуса отсчитывают при помощи лупы или
короткофокусной трубы, деля "на глаз” промежутки между двумя соседними
делениями термометра на 10 равных частей. Чтобы преодолеть действие капиллярных
сил, препятствующих равномерному перемещению мениска ртути, перед отсчетом
температуры легонько постукивают по термометру тонкой палочкой с резиновым или
пробковым наконечником. В момент последнего отсчета начального периода замыкают
ток запальной цепи, присоединенной к зажимам бомбы, в результате чего запал и
навеска бензойной кислоты сгорают.
Во 2-м периоде отсчеты производят каждые полминуты и считают
этот период законченным при поступлении равномерного изменения температуры воды
в сосуде в полуминутные промежутки. Если возникают сомнения в окончании главного
периода, увеличивают продолжительность относимых к нему отсчетов на 1 - 2 мин.
Отсчеты в главном периоде производят с точностью до 0,10 С
при повышении температуры за полминуты больше, чем на 0,50 С, с точностью до
0,010 С при повышении температуры за этот же период от 0,1 до 0,50 С и с
точностью до 0,0010 С при повышении температуры за полминуты менее чем на 0,10
С.
В 3-м конечном периоде производят через каждые полминуты 1 0
отсчетов при равномерном изменении температуры с точностью до 0,0010 С.
По окончании испытания выключают мешалку, отсоединяют
провода, приподнимают и вытирают термометр, вынимают и вытирают бомбу и,
осторожно открыв вентиль, выпускают в течение 5 мин газы. Отвинчивают и снимают
крышку с бомбы, осматривают ее и при наличии сажистого налета опыт бракуют. При
отсутствии сажистого налета тщательно промывают дистиллированной горячей водой
крышку и стакан бомбы, чашечку и все внутренние части бомбы, оба вентиля и
трубку для впуска кислорода, собирая промывочные воды (150 - 200 мл) в стакан.
Последний накрывают часовым стеклом и кипятят воду в течение 5 мин. Затем
приливают 2 капли в стакан фенолфталеина, титруют содержимое стакана 0,1 н
раствором едкого натра до появления неисчезающего розового окрашивания
жидкости, определяя содержание образовавшейся во время опыта азотной кислоты (1
мл израсходованного на титрование едкого натра соответствует 1,43 кал тепла,
выделившегося при образовании и растворении азотной кислоты).
Стакан и крышку бомбы вытирают изнутри чистым сухим
полотенцем. Вентили до следующего испытания оставляют открытыми и в конце
рабочего дня продувают их подогретым воздухом.
Сжигание навески угля в бомбе для определения теплоты
сгорания проводят так же, как сжигание бензойной кислоты для определения
фактической теплоемкости калориметра. Дно чашечки для сжигания навески
покрывают слоем прокаленного волокнистого асбеста.
Перед взятием навески аналитическую пробу угля перемешивают в
открытой банке шпателем или ложечкой по возможности на полную глубину. Затем из
двух-трех мест пробы берут навеску для испытания 0,8 - 1 - 5 г. Для
многозольного угля навеску надо брать более 1 г, для малозольного - менее 1 г.
Перед сжиганием уголь брикетируют (впрессовывая в брикет запальную проволочку).
Уголь с зольностью более 35%, а также антрациты и другие небрикетирующиеся угли
сжигают в виде порошка.
Если после сжигания наблюдаются частицы угля на дне бомбы, то
делают повторное испытание.
По окончании опыта смыв бомбы доводят до 150 - 200 мл.
Вычисление теплоты сгорания
Общий подсчет результатов калориметрического испытания
аналитической пробы угля производят по формуле

где S - теплота сгорания в бомбе аналитической пробы угля, кал/г;
G - навеска испытуемого угля, г
где ε - коэффициент,
учитывающий теплоту разложения карбонатов, равный 40 кДж/кг на 1 % диоксида
углерода карбонатов;
 - массовая доля диоксида углерода карбонатов, не
разложившихся при сжигании навески сланца в бомбе, %.
- массовая доля диоксида углерода карбонатов, не
разложившихся при сжигании навески сланца в бомбе, %.
Заключение
Ископаемые угли (гумусовые) в большинстве своем слагаются из
отдельных петрографических компонентов (ингредиентов или мацералов),
представляющих собой блестящие, полублестящие, матовые и волокнистые
разновидности угольного вещества, образующие в угольном пласте чередующиеся
слои, линзы или агрегатные включения. Петрографический состав углей оказывает
большое влияние на их технологические свойства и имеет важное значение при
решении вопроса об использовании углей.
При оценке свойств и качества углей используют результаты
технического и петрографического анализов. Основными показателями качества
углей являются: влажность (W), зольность (A), выход летучих веществ (V), теплота сгорания (Q), элементный состав (C, Н, О, N), содержание серы (S) и фосфора (P), петрографический
состав углей и отражательная способность витринита (R). Кроме этих показателей
определяют:
для каменных углей - показатель (индекс) РОГА (RI), показатель ГРЕЙ-КИНГА
(GK),
толщина пластического слоя (Y); для бурых углей -
выход смолы полукоксования (TSK), содержание битума (В) и гуминовых кислот (НА);
для антрацитов - анизотропия отражения витринита (AR);
Определение показателей проводят по аналитическим пробам и по
специальным формулам пересчитывают для различных состояний топлива. Обозначение
показателей твердого топлива состоит из:
символа, характеризующего основной показатель топлива;
нижнего индекса символа, дополняющего характеристику
основного показателя;
верхнего индекса символа, уточняющего состояние топлива, к
которому относится соответствующее свойство.
Список
использованных источников
1.
Митронов Д.В. Введение в химию и технологию переработки угля: Учебное пособие.
- Якутск, 2011. - 172с.
.
Глущенко И.М. Теоретические основы горючих ископаемых: Учебник для вузов. - М.:
Металлургия, 1990. - 296с.
.
Химическая технология твердых горючих ископаемых: Учебник для вузов/ од ред.
Г.Н. Макарова и Г.Д. харламповича. - М.: Химия. 1986. - 496с.
.
М.И. Байкенов, В.А. Хрупов, К.А. Жубанов Синтетические продукты из твердого
углеводородсодержащего сырья: Учебное пособие. - Караганды: Изд-во КарГУ, 2004.
- 126с.
.
Халикова З.С. Химия и физика нефти, газа и угля: Учебное пособие. Караганда:
Изд-во КарГУ, 2010. - 143с.
6. Е.М. Тайц, И.А. Андреева. Методы анализа и испытания углей. М.:
Недра, 1983. - 302 с.
. Менковский М.А., Флодин А.А. Аналитическая химия и технический
анализ углей. М.: Недра, 1973. - 367 с.