Отряд рукокрылых
Московский
государственный университет им. М.В. Ломоносова
Химический
факультет
Кафедра
физической химии
Лаборатория
газовой электронографии
Изучение
равновесия между таутомерными формами молекулы нитрогуанидина с помощью
квантово-химических расчетов
Выполнил студент
408 группы
Научный
руководитель: в. н. с., д. х. н.
Преподаватель:
доц., к. п. н.
Москва 2012
Содержание
Введение. Обзор литературы
.1 Полуэмпирические методы расчета
.2 Неэмпирические методы расчета
.3 Базисные наборы, используемые при
расчетах. Результаты расчетов и их обсуждение
Выводы
Список литературы
Введение
Нитрогуанидин и его производные представляют
большой практический интерес как энергоемкие вещества и находят применение в
качестве компонентов порохов, твердых ракетных топлив и взрывчатых составов.
Поэтому изучение термодинамических свойств нитрогуанидина и родственных ему
соединений и установление закономерностей, связывающих эти свойства со
строением нитрогуанидина, является актуальной задачей.
Кроме того, определение молекулярной структуры
нитрогуанидинов представляет особый интерес для структурной химии из-за
специфического ароматического строения подобных соединений (так называемая Y
-ароматичность). Накопление структурных данных помогает установить количественные
закономерности «структура - свойство» и сделать прогноз новых гипотетических
энергоемких нитриминных структур с требуемым комплексом характеристик и др.
Как известно, молекулярное строение соединения
можно изучать опираясь либо на классическую теорию химического строения, либо
на квантовую.
Классическая теория химического строения
позволяет сказать, пусть только на основе феноменологических построений, какова
может быть структура химического соединения, каковы особенности структуры и
свойств рассматриваемого соединения сравнительно с другими соединениями, каков
должен быть набор химических и физико-химических свойств, присущий этому
соединению. Однако иногда необходима более детальная информация о строении
вещества, которую не может предоставить классическая теория, например, свойства
возбужденных состояний и др. Квантовая теория, благодаря быстрому развитию
вычислительной техники и разработке новых квантово-химических методов, дает
возможность рассчитывать равновесные межъядерные расстояния, валентные и
двугранные углы, оценивать высоту барьеров внутреннего вращения, энергии
образования и диссоциации, энергии активации, сечения и константы скорости
простейших химических реакций и др. [1].
Итак, целью курсовой работы является
квантово-химический расчет термодинамических данных при полной оптимизации
геометрии и оценка количественного содержания наиболее стабильных таутомерных
форм нитрогуанидина при стандартных условиях в газовой фазе с помощью программы
GAUSSIAN-03 [2]. Кроме
того, в работе рассмотрены методы и результаты ранее выполненных исследований в
данном направлении [3 и др.].
I.
Обзор литературы
.1 Полуэмпирические методы расчета
Более ранние полуэмпирические
методы CNDO, INDO
и NDDO были разработаны
Дж. Поплом и его группой в то время, когда вычислительные машины могли
выполнять неэмпирические расчеты лишь для самых простых молекул. Эти методы
ориентированы на корректное воспроизведение электронных характеристик, таких,
как дипольный момент, а не теплот образования и геометрических параметров
молекул. В наиболее простом из них, методе CNDO
(Complete
Neglect of
Differential
Overlap), при расчете
интегралов электрон-электронного отталкивания атомные орбитали рассматриваются
как сферически симметричные. Ориентация р-орбиталей учитывается только в
одноэлектронных резонансных интегралах, величина которых зависит также от
размеров орбиталей, расстояний между центрами и значений констант, определяющих
тип связи. В более сложном приближении INDO
(Intermediate
Neglect of
Differential
Overlap) проводится расчет
одноцентровых интегралов отталкивания между атомными орбиталями для одного и
того же атома. Впервые ориентация р-орбиталей при расчете интегралов
отталкивания учитывалась в следующем по сложности приближении - NDDO
(Neglect of
Diatomic
Differential
Overlap). В этом методе
учитывались трех- и четырехцентровые интегралы, которые ответственны за
перекрывание атомных орбиталей одного и того же атома.
Метод MINDO/3
представляет собой модифицированный вариант приближения INDO,
в котором одноцентровые интегралы отталкивания не вычисляются аналитически, как
в исходной версии, а рассматриваются в качестве параметров. Эти параметры, как
и параметры, при помощи которых вычисляются резонансные интегралы, выбираются
так, чтобы по возможности наилучшим образом воспроизводились экспериментальные
данные.
Метод MNDO
-
это независимый метод, основанный на приближении NDDO.
MNDO позволяет избежать
ряда систематических ошибок метода MINDO/3
при расчете таких молекул, как гидразины и полифторалканы, поскольку учет пространственной
направленности р-орбиталей при вычислении двухэлектронных интегралов дает
возможность более корректно описать отталкивание неподеленных электронных пар.
Преимущество методов MINDO/3
и MNDO перед
неэмпирическими методами заключается в том, что полуэмпирические расчеты
требуют машинного времени на несколько порядков меньше (MNDO
работает приблизительно в 1,5 раза медленнее, чем MINDO/3).
Пренебрежение большим числом интегралов не только ускоряет процесс вычислений,
но также существенно уменьшает требуемый объем оперативной и внешней памяти.
Однако это безусловно приводит к потере точности.
Последние 15 лет довольно
широкое распространение в квантовой химии получили методы Density
Functional
Theory, основанные на
теории функционала плотности. DFT
методы учитывают корреляцию электронов с помощью функционалов электронной
плотности. Эти функционалы отличаются различным представлением
обменно-корреляционных потенциалов и содержат то или иное число эмпирических
параметров. Наиболее известным из DFT
методов является B3LYP.
В методе B3LYP
расчеты ведутся по теории функционала плотности (DFT)
с использованием гибридного обменно-корреляционного функционала B3LYP (Беке,
Ли, Янга, Пара) [6-8].
1.2 Неэмпирические методы расчета
Неэмпирические методы расчета в квантовой химии
являются наиболее точными. Они дают различные приближенные решения уравнения
Шрёдингера, не используя при этом эмпирических параметров (за исключением
фундаментальных физических постоянных).
Одним из приближений среди неэмпирических
расчетов является метод Хартри-Фока, позволяющий получать оценки структурных
параметров и частот колебаний молекул. Недостатком данного метода является
пренебрежение корреляцией электронов, что приводит к недостаточной точности
метода для моделирования энергетики химических реакций.
К методам, учитывающим электронную корреляцию и
основанным на теории возмущений, относятся методы Мёллера-Плессе (MPn).
Наименее затратным является метод MP2,
достаточно точно предсказывающий геометрию молекул. Кроме того, MP2
дает существенно более точные значения энергетических величин, по сравнению с
методом Хартри-Фока.
Для учета корреляции на более высоком уровне
можно использовать методы Мёллера-Плессе более высокого порядка. Наиболее
широкое применение получил метод МР4, который дает значительно лучшие
результаты, чем метод МР2. Однако расчеты по методу МР4 занимают довольно много
времени и требуют высоких параметров компьютера. Метод МР3 дает незначительное
улучшение точности по сравнению с МР2, являясь при этом более затратным, поэтому
почти не используется. По этой же причине редко используют и методы более
высокого порядка, чем МР4.
Методами, учитывающими
корреляцию электронов на более высоком уровне, чем методы МРn,
являются методы связанных (объединенных) кластеров (coupled
cluster, СС),
квадратичного конфигурационного взаимодействия (quadratic
configuration
interaction, QCI)
и конфигурационного взаимодействия (configuration
interaction. CI).
Эти методы могут учитывать одно-, двух-, трех- и четырехкратно возбужденные
электронные конфигурации. В настоящее время методы CC,
QCI и CI
при использовании базисов среднего размера применяются только для небольших
молекул.
1.3 Базисные наборы, используемые
при расчетах
Любая квантово-химическая модель характеризуется
базисным набором, представляющим собой математическое описание атомных
орбиталей в виде линейной комбинации гауссовых функций. Рассмотрим некоторые из
базисных наборов, используемых при решении той или иной задачи.
Минимальный базисный набор (Slater-type
orbitals, ST0-3G)
содержит минимальное число базисных функций, необходимых для каждого атома.
Например, для атома углерода - это орбитали Is,
2s, 2рх,
2ру, 2pz.
Минимальный базисный набор использует АО фиксированного размера. "3G"
в названии базиса указывает, что каждая АО аппроксимируется тремя гауссовыми
функциями [3, 4, 5].
Валентно расщепленные базисные
наборы (split
valence
basis sets)
используют две или более отличающиеся по размеру орбитали для каждой валентной
орбитали и только одну для внутренней АО. К таким базисным наборам относятся
3-21G, 6-31G,
6-311G [3, 4,
5].
В базисном наборе 3-21G
каждая внутренняя АО представлена одной базисной функцией, которая является
линейной комбинацией трех гауссовых функций, а валентные АО - двумя базисными
функциями, которые аппроксимируются двумя и одной гауссовыми функциями разного
типа (2 + 1). Базисный набор 6-31G
использует 6 гауссовых функций одного типа для описания внутренних АО и два
типа гауссовых функций (3 + 1) для валентных. 3-21G
и 6-31G являются базисными
наборами с двойным расщеплением (valence
double zeta,
VDZ), поскольку
используют два типа гауссовых функций для аппроксимации валентных орбиталей. В
базисном наборе 6-311G
с тройным расщеплением (valence
triple zeta,
VTZ) каждая валентная
АО аппроксимируется пятью гауссовыми функциями трех типов (3+1 + 1).
Базисные наборы с
поляризационными функциями (polarized
basis sets)
за счет включения дополнительных орбиталей позволяют изменять не только размер,
но и форму орбиталей, увеличивая таким образом размер базиса. В базисном наборе
6-31 G(d)
(другое обозначение 6-31G*)
к базису 6-31G добавляются
поляризационные функции d-типа
на каждый атом от Li
до Са и f-типа на атомы от Sc
до Zn, а в базисном
наборе 6-31G(d,p)
(другое обозначение 6-31G**)
добавляются также и поляризационные функции р-типа на каждый легкий атом (Н и
Не). Базисные наборы еще большего размера получаются при использовании
мультиплетных поляризационных функций. Так, базисный набор 6-311G(3df,2pd)
содержит три поляризационных функции d-типа
и одну f-типа на тяжелых
атомах и две поляризационных функции р-типа и одну d-типа
на атомах водорода [4, 5, 9, 10, 11].
В базисных наборах с диффузными
функциями (diffuse
functions) используются
функций s- и р-типа большого
размера, которые позволяют орбиталям занимать большее пространство. Базисные
наборы с диффузными функциями важны для систем, где имеется значительная
электронная плотность на больших расстояниях от ядер. Чтобы улучшить точность
расчетов для таких соединений, рассмотренные выше базисные наборы дополняются диффузными
функциями. Базисный набор 6-31+G(d)
- это базисный набор 6-31 G(d)
с диффузными функциями, добавленными к каждому тяжелому атому, а в базисном
наборе 6-31++G(d)
диффузные функции добавлены также к атомам водорода. Диффузные функции на
атомах водорода редко приводят к повышению точности расчетов. Базисные наборы с
диффузными функциями используются для описания электронного взаимодействия в
корреляционных методах и обычно не используются в расчетах Хартри-Фока [4, 9].
Корреляционно согласованные базисные
наборы Даннинга созданы для методов, учитывающих корреляцию электронов. Это
базисные наборы cc-pVDZ
(correlation
consistent, polarized
valence double
zeta), cc-pVTZ
(correlation
consistent, polarized
valence triple
zeta), cc-pVQZ,
cc-pV5Z
и cc-pV6Z,
включающие поляризационные функции и имеющие общее обозначение cc-pVXZ,
а также базисные наборы, дополненные диффузными функциями: aug-cc-VDZ
(augmented
correlation
consistent, polarized
valence double
zeta), aug-cc-pVTZ,
aug-cc-pVQZ
и т.д. [5, 9, 10].
II.
Результаты расчетов и их обсуждение
квантовый
химический таутомерный нитрогуанидин
Квантово-химические расчеты
проводились в приближении выбранного метода MP2(full)/6-311G(3df,2p) при помощи
программы GAUSSIAN-03
[2]. Заряд молекулы, мультиплетность и параметры молекулярной структуры (тип
атомов и их взаимное расположение) были заданы с помощью Z-матрицы
путём последовательного задания внутренних координат атомов - длин связей, а
также валентных и двугранных углов.
Ранее были проведены
квантово-химические расчеты методом B3LYP
и найдены наиболее устойчивые формы нитрогуанидина [3], показанные на рисунке
1.

Рис. 1. Наиболее устойчивые
формы нитрогуанидина
В данной работе проведены расчеты молекулярной
структуры, гармонических силовых полей, частот нормальных колебаний и
термохимических свойств всех пяти таутомеров в значительно более высоком
приближении MP2(full)/6-311G(3df,2p). Пространственная структура наиболее
низколежащей нитроиминной формы (2-нитрогуанидин) по данным расчета
MP2(full)/6-311G(3df,2p) приведена на рисунке 2. Характерной особенностью
молекулы является существенное уплощение гуанидинового фрагмента (сумма
валентных углов трехкоординированного атома углерода 360.0о, bС4=0.6о)
и выравнивание длин связей этого атома с атомами азота (1.315-1.364Å)
и укорочение связи N-N
(1.378Å),
образование водородной связи H10…O6 1.842Å
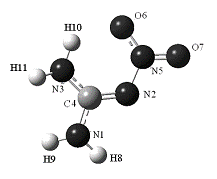
Рис. 2. Равновесная
структура молекулы 2-нитрогуанидина (нитроиминная форма 1). Дана нумерация атомов
при квантово-химическом расчете
Для всех пяти изомерных структур (рис.1)
рассчитанные полные и относительные энергии представлены в таблице 1. Из неё
видно, что наиболее стабильной является нитроиминная форма 1.
Табл. 1. Полные и относительные энергии
устойчивых форм тетранитрогуанидина
|
№
|
Etot,
кДж/моль.
|
ΔE,
ккал/моль
|
|
1
|
-1074794
|
0
|
|
2
|
-1074767
|
6,62175
|
|
3
|
-1074740
|
13,087055
|
|
4
|
-1074759
|
8,599522
|
|
5
|
8,429651
|
Далее на основании полученных в
ходе расчёта значений свободных энергий Гиббса (таблица 2) получили значения
констант равновесий переходов между таутомерными формами 1, 2, 3,
4, 5 (рис. 3(а, б, в)).
Табл. 2. Термодинамические
характеристики для таутомерных форм нитрогуанидина, T=298.15
K
|
Таутомерная
форма
|
|
1
|
2
|
3
|
4
|
5
|
|
ZPE, кДж/моль *
|
209,2
|
209,5
|
208,4
|
209,76
|
208,77
|
|
E0 = Eel + ZPE, кДж/моль
|
-1074585,271
|
-1074557,26
|
-1074531,3
|
-1074548,74
|
-1074550
|
|
E = E0 + Evib + Erot + +Etrans,
кДж/моль
|
-1074567,729
|
-1074539,99
|
-1074513,6
|
-1074531,41
|
-1074533
|
|
H = E + RT, кДж/моль
|
-1074565,251
|
-1074537,52
|
-1074511,2
|
-1074528,93
|
-1074530
|
|
G = H - TS, кДж/моль
|
-1074666,787
|
-1074638,72
|
-1074613,9
|
-1074630,32
|
-1074633
|
|
Cv, Дж/(моль*К)
|
99,43276
|
97,0688
|
97,855392
|
96,637848
|
98,4286
|
|
S, Дж/(моль*К)
|
340,53576
|
339,427
|
344,585872
|
340,050416
|
342,678
|
* Поправка на энергию нулевых
колебаний (zero
potential
energy)

Рис. 3(а). Равновесие между
таутомерными формами нитрогуанидина

Рис. 3(б). Равновесие между
таутомерными формами нитрогуанидина

Рис. 3(в). Равновесие между
таутомерными формами нитрогуанидина
Константы равновесия рассчитывали
следующим образом.
k1=N2/N1,,
k2=N3/N1,
k3=N4/N1,
k4=N5/N1,
k5=N3/N2,
k6=N4/N2,
k7=N5/N2,
k8=N4/N3,
k9=N5/N3,
k10=N5/N4
ΔG1
= RTlnk1,
ΔG2
= RTlnk2,
ΔG3
= RTlnk3,
ΔG4
= RTlnk4,
ΔG5
= RTlnk5,
ΔG6
= RTlnk6,
ΔG7
= RTlnk7,
ΔG8
= RTlnk8,
ΔG9
= RTlnk9,
ΔG10
= RTlnk10
Далее составили систему
уравнений и решили её:
1) X1
+ X2 + X3 + X4 + X5 = 11
= N1/(N1 + N2 + N3 + N4
+ N5)
X2 = N2/(N1
+ N2 + N3 + N4 + N5)3 =
N3/(N1 + N2 + N3 + N4 +
N5)4 = N4/(N1 + N2 + N3
+ N4 + N5)5 = N5/(N1 + N2
+ N3 + N4 + N5)
2) 1/X1
= (N1+
N2+
N3+
N4+
N5)/N1
=1+ k1+ k2+ k3+ k4
1/X2
= (N1+
N2+
N3+
N4+
N5)/N2
=(1/k1)+1+ k5+ k6+ k7
/X3
= (N1+
N2+
N3+
N4+
N5)/N3
=(1/k2)+(1/k5)+1+ k8+
k9
/X4
= (N1+
N2+
N3+
N4+
N5)/N4
==(1/k3)+(1/k6)+(1/k8)+1+
k10
k1
= e-ΔG1/RT,
k2
= e-ΔG2/RT,
k3
= e-ΔG3/RT,
k4
= e-ΔG4/RT,
k5
= e-ΔG5/RT,
k6
= e-ΔG6/RT,
k7
= e-ΔG7/RT,
k8
= e-ΔG8/RT,
k9
= e-ΔG9/RT,
k10
= e-ΔG10/RT
ΔG1
= G(2) - G(1),
ΔG2
= G(3) - G(1),
ΔG3
= G(4) - G(1),
ΔG4
= G(5) - G(1),
ΔG5
= G(3) - G(2),
ΔG6
= G(4) - G(2),
ΔG7
= G(5) - G(2),
ΔG8
= G(4) - G(3)
ΔG9
= G(5)
- G(3),
ΔG10
= G(5)
- G(4)
Затем в полученные формулы
подставляли численные значения соответствующих величин из данных
квантово-химического расчета (табл. 2)
Полученные значения изменений
свободной энергии Гиббса и константы равновесия (Т = 298,15 К) приведены в
таблице 3.
Табл. 3. Изменения свободной
энергии Гиббса и константы равновесия для равновесий рис. 3(а, б, в)
|
ΔG1, Дж/моль
|
28065,573
|
k10
|
1,21018E-05
|
|
ΔG2, Дж/моль
|
52880,591
|
k2
|
5,43508E-10
|
|
ΔG3, Дж/моль
|
36465,027
|
k3
|
4,08553E-07
|
|
ΔG4, Дж/моль
|
34222,722
|
k4
|
1,0095E-06
|
|
ΔG5, Дж/моль
|
24815,018
|
k5
|
4,49112E-05
|
|
ΔG6, Дж/моль
|
8399,4543
|
k6
|
0,033759607
|
|
ΔG7, Дж/моль
|
6157,1492
|
k7
|
0,08341691
|
|
ΔG8, Дж/моль
|
-16415,56
|
k8
|
751,6962379
|
|
ΔG9, Дж/моль
|
-18657,87
|
k9
|
1857,372849
|
|
ΔG10,
Дж/моль
|
1790,752
|
k10
|
0,485575053
|
Соотношение таутомерных форм 1,
2, 3, 4, 5 в равновесной смеси получилось
следующим:
X1
= 99,995% X2
= 0,0042% X3
= 2×10-7%
X4
=0,0001% X5
= 0,0004%
Таким образом, в равновесной
смеси при Т = 298,15 К преобладает таутомерная форма 1. Так же были
рассчитаны электронная, поступательная, вращательная, колебательная, ядерная и
полная молекулярная суммы по состояниям для таутомерных форм 1, 2,
3, 4, 5, представленные в таблице 4.
Табл. 4. Суммы по состояниям
для таутомерных форм 1, 2, 3, 4, 5 нитрогуанидина
(рис. 1) при Т = 298,15 К
|
Таутомерная
форма
|
Q0
|
Qe
|
Qrot
|
Qtr
|
Qvib
|
Q∑
|
|
1
|
4,25E-23
|
1,00E+00
|
1,98E+05
|
4,17E+07
|
1,91E+14
|
6,69E+04
|
|
2
|
3,68E-23
|
1,00E+00
|
4,17E+07
|
1,86E+14
|
5,81E+04
|
|
3
|
9,06E-23
|
1,00E+00
|
2,00E+05
|
4,17E+07
|
2,92E+14
|
2,21E+05
|
|
4
|
3,49E-23
|
1,00E+00
|
2,06E+05
|
4,17E+07
|
1,96E+14
|
5,89E+04
|
|
5
|
6,48E-23
|
1,00E+00
|
2,05E+05
|
4,17E+07
|
2,43E+14
|
1,35E+05
|
Выводы
1. Проделанная работа
показала применимость квантово-химических методов для расчёта равновесий различных
соединений. В процессе выполнения работы освоены основы работы с широко
распространенной квантово-химической программой GAUSSIAN.
2. Рассчитаны
геометрические параметры, характеризующие молекулярную структуру пяти
таутомеров нитрогуанидина. Подтвержден вывод о так называемой Y-ароматичности
нитрогуанидинового фрагмента (выравнивание связей С-N,
уплощение пирамиды связей на атоме С).
3. Получены
термодинамические функции для пяти таутомерных форм нитрогуанидина: свободная
энергия Гиббса, молярная теплопроводность при постоянном объеме, энтальпия,
энтропия и статистические суммы по состояниям.
4. Расчет содержания
таутомерных форм нитрогуанидина в равновесной смеси в газовой фазе при
температуре 298,15 К показал подавляющее преобладание изомера 1, т.е.
нитроиминной формы.
Список литературы
1. Степанов Н. Ф.. Квантовая механика и
квантовая химия. Москва, «Мир», 2001, 3 - 7.
2. Frisch M.J., Trucks G.W.,
Schlegel H.B., Scuseria G.E., Robb M.A., Cheeseman J.R., Montgomery Jr. J.A.,
Vreven T., Kudin K.N., Burant J.C., Millam J.M., Iyengar S.S., Tomasi J.,
Barone V., Mennucci B., Cossi M., Scalmani G., Rega N., Petersson G.A.,
Nakatsuji H., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M.,
Nakajima T., Honda Y., Kitao O., Nakai H., Klene M., Li X., Knox J.E.,
Hratchian H.P., Cross J.B., Adamo C., Jaramillo J., Gomperts R., Stratmann
R.E., Yazyev O., Austin A.J., Cammi R., Pomelli C., Ochterski J.W., Ayala P.Y.,
Morokuma K., Voth G.A., Salvador P., Dannenberg J.J., Zakrzewski V.G., Dapprich
S., Daniels A.D., Strain M.C., Farkas O., Malick D.K., Rabuck A.D.,
Raghavachari K., Foresman J.B., Ortiz J.V., Cui Q., Baboul A.G., Clifford S.,
Cioslowski J., Stefanov B.B., Liu G., Liashenko A., Piskorz P., Komaromi I.,
Martin R.L., Fox D.J., Keith T., Al-Laham M.A., Peng C.Y., Nanayakkara A.,
Challacombe M., Gill P.M.W., Johnson B., Chen W., Wong M.W., Gonzalez C., Pople
J.A. Gaussian 03, Rev.D.01 Pittsburgh (PA): Gaussian Inc., 2003.
3. Хайкин Л.С., Грикина О.Е., Вишневский
Ю.В., Ж. физ. химии, 2007, 81, №1 152 - 154.
. Кларк Т. Компьютерная химия - М.: Мир,
1990.
. Матулис В.Э., Матулис В.Э., Ивашкевич
О.А., Прикладная квантовая химия - Минск, БГУ, 2007
6. Becke
A.D. // Phys. Rev. A, 1988, Vol. 38,
P. 3098-3100
7. Lee
C., Yang W., Parr R.G. // Phys. Rev. B, 1988, Vol. 37, P.
785-789.
8. Becke A.D. // J. Chem. Phys.,
1993, Vol.98, P.5648-5652.
. Moran D., Simmonett A.C.,
Leach F.E., Allen W.D., Schleyer P.v.R., Schaefer H.F., J. Am. Chem. Soc.,
2006, 128, 9342-9343.
10. Fogarasi G., Szalay P.G., J.
Phys. Chem. A, 1997, 101, 1400.
11. Хайкин Л.С.,
Грикина
О.Е.,
Абраменков
А.В.,
Степанов
Н.Ф.,
Ж.
физ.
химии.,
2005, 79, №4., 693-701 (Russ. J. Phys. Chem. 2005, 79,
591).
12. Moeller C., Plesset M.S.
Phys. Rev., 1934, 46, 618.