Исследование термодинамических функций малоразмерных наночастиц при использовании квантово-химических методов
Балтийский
Государственный Технический Университет «ВОЕНМЕХ» им. Д.Ф. Устинова
Кафедра
Космических аппаратов и двигателей
Курсовая
работа
на
тему: Исследование термодинамических функций малоразмерных наночастиц при
использовании квантово-химических методов
Санкт-Петербург,
2011
Введение
В настоящее время большой интерес представляет
изучение наноразмерных структур. Частицы данных размеров обладают избытком
поверхностной энергии, что позволяет рассматривать возможность применения их в
качестве компонента топлив, обеспечивающего рост их энергетического потенциала.
Однако данная особенность влечет за собой ряд проблем, связанных с сохранением
данного вида энергии в течение всего процесса изготовления и хранения
энергетических конденсированных систем. Рост удельной энтальпии образования
частиц, очевидно, ведет к росту их химической активности и, как следствие, к
образованию соединений, обладающих меньшим запасом энергии.
Сейчас активно рассматривается идея решения этой
задачи при помощи покрытия наноструктур атомами других элементов. Рассмотренные
в более ранних работах пути предотвращения коагуляции путем ионизации
поверхностного слоя кластера [1] не оправдали ожиданий, поскольку силы
отталкивания превалировали над силами притяжения лишь на определенных,
достаточно больших расстояниях, но при меньших дистанциях не могли
препятствовать сближению частиц. Исследование термодинамических функций частиц
позволит оценить их энергетический ресурс, а так же помочь решить задачу
предотвращения взаимодействия кластеров с окружающей средой и их коагуляции,
поскольку осуществление данных процессов будет приводить к снижению ресурса
энергии и, в конечном счете, сведет на нет сам смысл использования этих
структур.
1. Общая характеристика
малоразмерных наночастиц (кластеров)
Наноразмерные объекты занимают промежуточное
положение между объемными материалами и атомами (или молекулами). В современном
научном мире под наноразмерными структурами принято понимать структуры, размеры
которых не превышают 100 нм, по крайней мере, в одном направлении. При таких
величинах начинают проявляться размерные эффекты, т. е. наночастицы
демонстрируют свойства, отличные как от свойств твердого тела, так и от свойств
микрочастиц, заключающиеся в изменении свойств и характера взаимодействия
частиц между собой или же с другими элементами. Когда размер зерен становится
менее 10 нм - так называемые малоразмерные наночастицы - размерные эффекты
становятся наиболее отчетливыми. Число микрочастиц в таких кластерах не
превышает нескольких десятков, что приводит к существенному избытку
поверхностной энергии частиц. Причиной этого является особое строение
наночастиц.
В отличие от классической дисперсной фазы, в
которой, не смотря на достаточно маленькие размеры самих частиц, подавляющее
большинство атомов находится внутри зерна и взаимодействует одновременно с
большим количеством окружающих их атомов, в кластерах число микрочастиц
поверхностного слоя и микрочастиц, находящихся внутри зерна - величины одного
порядка. К примеру, в наночастице, состоящей из 13 атомов, лишь один атом
располагается в центре, а все остальные (92% от общего объема) находятся на
поверхности. Атомы, находящиеся на границе имеют оборванные связи, в силу чего
возникает поверхностная (граничная) энергия, дополнительная к свободной энергии
объема кластера, которая и придает этим структурам уникальные особенности,
отсутствующие в масштабах «ньютоновской физики». Поверхностная энергия
обуславливает значительное уменьшение потенциальной энергии каждой микрочастицы
в сравнении с кристаллической фазой, что приводит к росту энтальпии образования
наночастиц. Естественным следствием высоких значений энтальпии будет
возрастание свободной энергии Гиббса и химического потенциала, непосредственно
связанного с химической активностью кластеров. Таким образом, с увеличением
доли атомов поверхностного слоя (уменьшения числа микрочастиц в составе
кластера) увеличивается удельная величина поверхностной энергии и,
следовательно, повышается химическая активность наночастиц.
2. Методы расчетного определения
характеристик наночастиц
К настоящему времени разработаны два
принципиально различных подхода к описанию наночастиц, которые в общем случае
можно рассматривать как достаточно большие молекулы: квантово-химический и
подход, базирующийся на классической механике и на использовании
экспериментальных данных о взаимодействии между микроскопическими частицами.
Рассмотрим эти подходы.
В рамках методов, базирующихся на классической
механике, электроны в явном виде не рассматриваются, изучается взаимное
положение ядер атомов [2]. При этом закономерности поведения данных объектов
соответствуют классической механике. Наиболее существенной проблемой данной
группы методов является корректное описание межмолекулярного (межатомного)
взаимодействия. Чаще всего это осуществляется при помощи потенциалов
взаимодействия - аппроксимаций действительного взаимодействия между частицами.
Это описание базируется на экспериментальной информации, получение которой
представляет собой значительные сложности. Поэтому экспериментальные данные
имеются для ограниченного числа элементов.
Метод молекулярной механики обеспечивает
определение строения и энергии молекулярной системы. Поверхность потенциальной
энергии, которая в квантово-химических моделях подлежит прямому расчету, здесь
аппроксимируется определенными эмпирическими функциями разной степени
сложности, чаще всего это суммы парных потенциалов взаимодействия атомов. Эти
потенциальные функции, определяющие так называемое силовое поле молекулы,
содержат некоторые параметры, численное значение которых выбирается в согласии
с экспериментально полученными данными. В простейшем случае параметрами
являются равновесные длины связей и валентные углы, а также силовые постоянные,
то есть коэффициенты жесткости упругих сил, связывающих пары атомов [3].
Простейшие модели молекулярной механики
учитывают растяжения связей, деформацию валентных и торсионных углов,
взаимодействие валентно несвязанных атомов, называемое также ван-дер-ваальсовым
взаимодействием, электростатические вклады и т.д.
U =  Uраст + Uдеф + Uторс +
Uраст + Uдеф + Uторс +  Uвдв + Uэл-стат (1)
Uвдв + Uэл-стат (1)
где: Uраст - энергия растяжения связей в
наночастице; Uдеф - энергия деформации валентных углов; Uторс - энергия
деформации двугранных узлов; Uвдв - энергия, учитывающая Ван-дер-Ваальсово
взаимодействие атомов в частице; Uэл-стат - энергия электростатического
взаимодействия (кулоновская энергия).
Для каждого слагаемого записывается определенное
аналитическое выражение (например, энергия электростатического вклада
Uэл-стат., описывается кулоновской функцией, но, быть может, с нецелыми
зарядами в качестве параметров) и параметры соответствующих функций подгоняются
по каким-либо свойствам базовых молекул. Сумма всех перечисленных вкладов
определяет энергию молекулы как функцию геометрической конфигурации ядер. Для
нахождения равновесной геометрической конфигурации исследуемой молекулы
необходимо определить ее минимум. Данная модель позволяет моделировать
достаточно сложные молекулы даже в отсутствие значительных вычислительных
мощностей
Метод молекулярной динамики (МД) обеспечивает
моделирование детальной картины внутренней подвижности системы, состоящей из
молекул. В его основе лежит расчет классических (ньютоновских) траекторий
движения взаимодействующих классических частиц в фазовом пространстве их
координат и импульсов.
Атомы и молекулы могут рассматриваться как
объекты классической механики. Необходимо сравнивать длину волны де Бройля с
размером атома, чтобы определить, можно ли трактовать его как классический
объект. Основу метода составляет численное решение классических уравнений
Ньютона для системы взаимодействующих частиц.
 (2)
(2)

где:
N -
количество частиц;
 - масса i-той
частицы;
- масса i-той
частицы;
 - соответствующая проекция суммарной
силы, действующей на частицу со стороны остальных частиц:
- соответствующая проекция суммарной
силы, действующей на частицу со стороны остальных частиц:
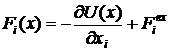 ;
;
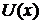 - потенциал взаимодействия;
- потенциал взаимодействия;
 - сила взаимодействия частиц с
окружающее средой.
- сила взаимодействия частиц с
окружающее средой.
Наиболее существенными проблемами
использования метода МД является корректное определение потенциала
взаимодействия и способ решения системы уравнений (2). На каждом временном шаге
необходимо вычислять  сил
взаимодействия. Учитывая, что типичный МД-шаг по времени составляет порядок
фемтосекунд (10-15 с), очевидны существенные затраты машинного времени на
реализацию метода. Расчеты проводятся, как правило, при постоянной температуре.
сил
взаимодействия. Учитывая, что типичный МД-шаг по времени составляет порядок
фемтосекунд (10-15 с), очевидны существенные затраты машинного времени на
реализацию метода. Расчеты проводятся, как правило, при постоянной температуре.
По-видимому, как разновидность
МД-метода можно рассматривать метод Монте-Карло. В рамках этого метода каждая
конфигурация системы определяется не посредством решения уравнений Ньютона, а с
использованием случайных процессов.
Метод основан на выборе случайных процессов
вместо детерминированных, которые позволяют получить набор конфигураций. Этот
метод не может дать временных зависимостей, однако обеспечивает определение
термодинамических функций молекулярных систем за меньшие времена счета в
сравнении с МД-методом.
Еще один подход к описанию взаимодействия
кластеров с окружающей средой - метод нанотермодинамики. Сущность метода
заключается в поиске строения наночастиц, обеспечивающего в состоянии
равновесия минимальное значение соответствующей термодинамической функции, и
использовании для описания их взаимодействия с окружающей средой и между собой
[2, 4]. В качестве исследуемой термодинамической функции определяется
потенциальная энергия частицы для условно твердого состояния или энергия
Гельмгольца для условно жидкого.
Как и в методе молекулярной механики,
потенциальная энергия частицы задается в виде суммы парных полуэмпирических
потенциалов. Варьируя конфигурацию наночастицы или ее размеры (при «жидком»
агрегатном состоянии), можно получить значение целевой функции, а затем в
рамках идеологии «надмолекул» [5-6], а также с использованием аппарата
классической и статистической термодинамики определить и другие интересующие
термодинамические функции кластера.
При наличии сведений о структуре
наночастицы и свойствах образующих ее микрочастиц может быть получено значение
ее химического потенциала 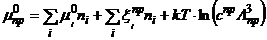 , где
, где  химический
потенциал наночастицы;
химический
потенциал наночастицы; 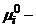 химический
потенциал микрочастицы i-того типа с неподвижным центом масс
в вакууме; ni -
количество микрочастиц i-того типа;
химический
потенциал микрочастицы i-того типа с неподвижным центом масс
в вакууме; ni -
количество микрочастиц i-того типа; 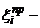 работа
переноса микрочастицы i-того типа из вакуума в наночастицу;
cnp -
концентрация наночастиц;
работа
переноса микрочастицы i-того типа из вакуума в наночастицу;
cnp -
концентрация наночастиц;  длина волны
де Бройля наночастицы; k - постоянная Больцмана; Т -
температура. [4]
длина волны
де Бройля наночастицы; k - постоянная Больцмана; Т -
температура. [4]
Определенная в каждом случае
комбинация химических потенциалов образует химическое сродство процесса:  , где mi, mj -
химический потенциал i-того и j-того
веществ при данных значениях температуры и давления. Химическое сродство
является универсальным «инструментом» для определения наличия взаимодействий,
включая химические реакции и их скорости. [7]
, где mi, mj -
химический потенциал i-того и j-того
веществ при данных значениях температуры и давления. Химическое сродство
является универсальным «инструментом» для определения наличия взаимодействий,
включая химические реакции и их скорости. [7]
В квантово-химических методах
состояние частицы задается волновой функцией  , являющейся аналогом уравнения
движения в классической механике. Однако в отличие от классического
представления движения тел в микромире принципиально невозможно определить
точные координаты частицы. Ее движение подчиняется лишь вероятностным законам,
зная которые, возможно получить средние значения интересующих величин.
, являющейся аналогом уравнения
движения в классической механике. Однако в отличие от классического
представления движения тел в микромире принципиально невозможно определить
точные координаты частицы. Ее движение подчиняется лишь вероятностным законам,
зная которые, возможно получить средние значения интересующих величин.
Фундаментальное уравнение квантовой
механики - волновое уравнение Шрёдингера, описывает движение квантовой частицы
в заданном потенциальном поле.
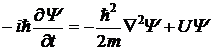 (3)
(3)
где:  , h -
постоянная Планка; m - масса частицы, U - внешняя
по отношению к частице потенциальная энергия в точке
, h -
постоянная Планка; m - масса частицы, U - внешняя
по отношению к частице потенциальная энергия в точке 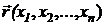 .
.
Решение этого уравнения дает
комплексную волновую функцию , квадрат величины которой  определяет
вероятность нахождения частицы в момент времени t в объеме dV. Точное
решение уравнения Шредингера принципиально может быть получено только для
водородоподобных частиц. При нахождении в рассматриваемой системе двух и более
электронов, в уравнении возникает дополнительный член, учитывающий
межэлектронное отталкивание, величина которого зависит от координат всех
электронов. А также при рассмотрении многоатомной системы возникают члены,
учитывающие электростатическое отталкивание ядер и их кинетическую энергию.
Таким образом, решение уравнения методом разделения переменных становится
невозможным ни в одной координатной системе. Тем не менее, существует множество
методов, позволяющих с той или иной точностью описывать многоэлектронные атомы
и атомные системы.
определяет
вероятность нахождения частицы в момент времени t в объеме dV. Точное
решение уравнения Шредингера принципиально может быть получено только для
водородоподобных частиц. При нахождении в рассматриваемой системе двух и более
электронов, в уравнении возникает дополнительный член, учитывающий
межэлектронное отталкивание, величина которого зависит от координат всех
электронов. А также при рассмотрении многоатомной системы возникают члены,
учитывающие электростатическое отталкивание ядер и их кинетическую энергию.
Таким образом, решение уравнения методом разделения переменных становится
невозможным ни в одной координатной системе. Тем не менее, существует множество
методов, позволяющих с той или иной точностью описывать многоэлектронные атомы
и атомные системы.
В 1927 г. был разработан метод
Хартри-Фока, согласно которому движение электрона происходит в усредненном
поле, создаваемом всеми зарядами системы. Это позволяет не рассматривать
взаимодействие электронов между собой, заменив его взаимодействием электрона с
усредненным полем, т. е. представить в виде функции, зависящей от координат
только одного электрона. Тогда полная волновая функция записывается в виде
произведения волновых функций всех электронов:  . В конечном виде уравнения
Хартри-Фока выглядят следующим образом.
. В конечном виде уравнения
Хартри-Фока выглядят следующим образом.
 (4) i=1,2…n
(4) i=1,2…n
где:
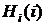 - гамильтониан Хартри для i-го
электрона.
- гамильтониан Хартри для i-го
электрона.
Они представляют собой точный
гамильтониан электрона, в котором член, учитывающий электростатическое
отталкивание электронов в атоме, заменен эффективным сферически симметричным
потенциалом (выражение под знаком суммы). Эффективный потенциал отражает
усредненное взаимодействие i-го электрона со всеми остальными
электронами, а также включает поправку Фока, учитывающую наличие обменной
энергии [2]. Величина  описывает
энергию электрона на i-й орбитали гамильтониана
Хартри-Фока. Данные уравнения решаются численными методами.
описывает
энергию электрона на i-й орбитали гамильтониана
Хартри-Фока. Данные уравнения решаются численными методами.
Другой метод - метод функционала
плотности является следствием принципа неразличимости тождественных частиц,
согласно которому элементарные частицы принципиально не могут быть распознаны и
отличены одна от другой. Электронная плотность - плотность вероятности распределения электронов
в квантовой системе, задается как функция радиус-вектора любого электрона.
Теоретическое обоснование теории
функционала плотности было сформулировано в 1964г. Хоэнбергом и Коном в виде
двух теорем:
1. Электронная плотность основного
состояния однозначно соответствует многоэлектронной волновой функции основного
состояния.
2. Полная энергия основного состояния
многоэлектронной системы может быть рассчитана как функционал электронной
плотности.
На основании этих утверждении были получены
уравнения Кона-Шэма, согласно которым каждой системе взаимодействующих
электронов, движущихся во внешнем поле V0(r), можно поставить в соответствие
"невзаимодействующую" систему с локальным потенциалом Vs(r), таким,
что в основном состоянии электронные плотности ρ(r)
и ρs(r)
для обеих систем будут равны:
 (5)
(5)
 (6)
(6)
где:
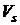 -эффективный
одноэлектронный потенциал;
-эффективный
одноэлектронный потенциал;
 -
обменно-корреляционный потенциал, функционально зависящий от полного
распределения электронной плотности ρs(r); r- радиус-вектор точки
пространства, в которой находится электрон.
-
обменно-корреляционный потенциал, функционально зависящий от полного
распределения электронной плотности ρs(r); r- радиус-вектор точки
пространства, в которой находится электрон.
Второе слагаемое в выражении для
эффективного одноэлектронного потенциала учитывает кулоновское отталкивание
электронов, а последнее слагаемое учитывает обменно-корелляционные
взаимодействия между ними.
3. Анализ возможностей
квантово-химических методов
Широкое применение квантово-химических методов
на практике стало возможно только с появлением высокопроизводительных ЭВМ. Были
разработаны программные пакеты, позволяющие получить решение уравнения
Шредингера при помощи численных методов.
На сегодняшний день данные пакеты обладают
возможностями представленными ниже:
. Возможность ввода координат различных типов -
декартовые, координаты Хильдербрандта, Z-матрицы в стандарте MOPAC и GAUSSIAN,
специальные случаи линейных молекул.
. Возможность расчета молекулярных волновых
функций методом самосогласованного поля в приближении ограниченного метода
Хартри-Фока RHF, неограниченного метода Хартри-Фока UHF, ограниченного метода
Хартри-Фока для систем с открытыми электронными оболочками ROHF, обобщенного
метода валентных связей GVB и многоконфигурационного самосогласованного поля
MCSCF;
. Расчет энергии системы при заданной
конфигурации, поиск ее градиента и вторых производных
. Учет энергии электронной корреляции на основе
теории возмущений, конфигурационного взаимодействия MP2,
связанных кластеров и функционала плотности DFT;
. Возможность выполнения полуэмпирических
расчетов методами MNDO, AM1 и PM3;
. Автоматическая оптимизация геометрии, поиск
переходных состояний с использованием аналитических градиентов;
. Исследование седловых точек поверхности
потенциальной энергии.
. Вычисление нормальных колебательных частот и
интенсивности инфракрасных (IR) спектров.
. Определение пути прохождения реакции при
помощи расчета внутренней координаты реакции
Таким образом, основные функции различных
программных пакетов схожи, однако их реализации в различных программах могут
отличаться.
4. Описание программных средств
В рамках данной работы квантово-химические
расчеты были проведены в программном комплексе PC
GAMESS/Firefly
v.7.1G.
[10] Данный пакет разработан группой во главе с Александром Грановским на
химическом факультете МГУ на базе пакета программ GAMESS (US). Автором было
переписано 60-70 % программ. Наибольшие изменения коснулись платформозависимых
частей программы (выделение памяти, дисковый ввод-вывод, параллельный запуск),
математических функций (например, матричные операции) и квантовохимических
методов (метод Хартри-Фока, теория Мёллера-Плессе, теория функционала
плотности).
Данный программный продукт не имеет
пользовательского интерфейса и работает в режиме командной строки. Все исходные
данные задаются в соответствующем входном текстовом файле.
Пример входного файла для расчета
оптимизированной структуры кластера Al13:
$CONTRL SCFTYP=ROHF
RUNTYP=OPTIMIZE
MAXIT=300 MULT=2
$END $SYSTEM
TIMLIM=525600 MEMORY=1000000
$END $BASIS
GBASIS=N21
NGAUSS=3 $END
$SCF DIRSCF=.TRUE.
$END $STATPT
OPTTOL=0.0001 NSTEP=20
$END $DATA
cluster 13.0 -0.276366065695649 0.0625902070045782 -0.198625995009080 13.0
-1.79978592768002 0.859280968982388 2.33388869786128 13.0
-1.09517338991369 -2.10445089183915 2.03560131424229 13.0
-1.06653343961383 -0.638488149412364 4.90063505647716 13.0
-4.74654653688141 1.60806242569331 1.98021341642658 13.0
-3.36954645295062 -0.716006321810898
0.230681509766823
13.0 -2.53303837800308 2.35704968833590
-0.232858068084099
13.0 1.14697469927196 0.110499152485722 2.68756361366965 13.0
-3.85789715037053 -1.14929618891116 3.38219852358965 13.0
-2.50439806671642 3.82301249242038 2.63217575168369 13.0
-0.230025015109394 2.43456798566201 4.43709548646525 13.0
0.258325371603761 2.86785770668611 1.28557844379604
Al 13.0
-3.32320589900943 1.65597166462748 4.86640372345958 $END
Входной файл должен содержать вид расчета, метод
расчета, мультиплетность исследуемого соединения (группа $CONTRL);
системные параметры, такие как максимальное время счета и выделяемый объем
памяти (группа $SYSTEM);
сведения о выбранном базисном наборе (группа $BASIS);
группу симметрии (с1); а также структуру исследуемого соединения с указанием ее
заряда и координат образующих ее атомов.
5. Расчет характеристик металлических
кластеров
Расчет характеристик кластера Al13
произведен ограниченным методом Хартри-Фока для систем с открытыми электронными
оболочками (ROHF) с
использованием базисных наборов STO-3G,
3-N21, 6-N31,
DZV.
STO-3G
один из простейших базисных наборов представленных в PC
GAMESS. В данном базисе
осуществляется представление атомных орбиталей слетеровского типа (STO) в виде
комбинации трех гауссовых функций.
Валентно-расщепленные базисные наборы 3-N21
и 6-N31 представляют
атомные орбитали валентных электронов в виде двух валентных функций одинаковой
симметрии. Одна из них является более сжатой, другая - размытой, диффузной.
Такое представление атомных орбиталей обеспечивает большую гибкость базисного
набора в зависимости от химического окружения атомов, на которых локализованы
данные валентные электроны, и обеспечивает увеличение точности расчетов.
Дальнейшее увеличение базисных наборов позволяет
учитывать поляризацию соединений, заряд ионов, коррелированное движение
электронов, однако при этом сильно возрастает объем вычислений, что требует
использования больших вычислительных ресурсов.
Структура и запас энергии.
Поскольку точные данные о строении кластеров
отсутствуют, необходимо произвести оптимизацию структуры наночастицы, а затем
рассчитать полную энергию, соответствующую данной геометрии. Для решения задачи
нахождения оптимальной геометрии соединения необходим выбор начального
приближения (структуры соединения). В общем случае начальная структура
соответствует неустойчивой конфигурации молекулы. В данной работе в качестве
начального приближения, задаваемого в исходном файле, были использованы
координаты атомов в составе кластера, полученные при помощи аппарата
нанотермодинамики.
Все программы расчетов по методу
самосогласованного поля используют одинаковые подходы для поиска волновой
функции оптимизированной молекулы, поскольку решение этой задачи требует
проведение двух обязательных процедур. Одна из этих процедур состоит в
нахождении наилучшей волновой функции для фиксированного набора геометрических
параметров молекулы. Для этого осуществляется процедура самосогласования поля.
Волновые функции молекулярных орбиталей, входящие в выражения для полной
энергии молекулы, представляют собой линейные комбинации атомных орбиталей,
каждая из которых умножена на неизвестный коэффициент разложения. Процедура
самосогласования как раз и заключается в поиске этих коэффициентов и нахождении
полной энергии соединения для заданной конфигурации.
Далее начинается вторая процедура - процедура
непосредственной оптимизации, в которой для найденой полной энергии
рассчитывают градиенты по всем независимым координатам молекулы. Величина
градиента определяет шаг изменения i-ой координаты, а знак - направление
(«минус» - увеличение, «плюс» - уменьшение). Совокупность градиентов
характеризует направление (вектор) спуска, в соответствии с которым программа
рассчитывает новые координаты. Далее опять решается уравнение Шредингера,
находятся энергия, градиенты и т.д. Оптимизационный цикл завершается на n-ном
шаге, когда разница энергий соединения на шаге n
и n-1 не превышает
некоторого малого значения, отвечающего сходимости.
На выходе мы получаем значение энергии
соединения, соответствующее оптимизированной структуре, и координаты атомов в
соединении. Энергия рассчитывается в Хартри, атомных единицах энергии.
(1Хартри=627.5096ккал/моль=2625.5 кДж/моль)
Выходные данные расчета оптимизации структуры
кластера Al13.
ENERGYELECTRON ENERGY =
-8226.9207189553ELECTRON ENERGY = 3163.3165889373REPULSION ENERGY =
1935.8942577394ENERGY = -3127.7098722786 ELECTRON-ELECTRON POTENTIAL ENERGY =
3163.3165889373 NUCLEUS-ELECTRON POTENTIAL ENERGY = -11350.1649457820
NUCLEUS-NUCLEUS POTENTIAL ENERGY = 1935.8942577394POTENTIAL ENERGY =
-6250.9540991052KINETIC ENERGY = 3123.2442268267RATIO (V/T) = 2.0014298099OF
ALL ATOMS ARE (ANGS) ATOM CHARGE X Y Z AL 13.0
-0.4346287384 0.1303403608 -0.0349581387 AL 13.0 -1.7997779966 0.8592780340
2.3338973142 AL 13.0 -1.1127938464 -1.8751718772 2.0953313194 AL 13.0
-1.0917220705 -0.5036943579 4.7017608181 AL 13.0 -4.5140760085 1.5434665826
1.9715632859 AL 13.0 -3.2659973256 -0.5629044235 0.3860230080 AL 13.0
-2.5078436844 2.2223362378 -0.0340857882 AL 13.0 0.9143837662 0.1751321073
2.6962400394 AL 13.0 -3.7332928312 -0.9857061663 3.2632734131 AL 13.0
-2.4867322124 3.5937598365 2.5725002340 AL
13.0 -0.3336136723 2.2813700095 4.2816816222 AL
13.0 0.1337520111 2.7042389247 1.4045650412 AL
13.0 -3.1648736424 1.5882054715 4.7027593057
При помощи различных программ-интерпретаторов
можно визуализировать полученные результаты. Данная визуализация получена в
программе ChemCraft.
Обозначения связей присваиваются исходя из расстояния между атомами.

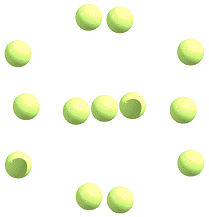

С использованием полученных выходных данных
получаем энергию связи атомов в кластере алюминия как разницу полных энергий
кластера Al13 и
тринадцати невзаимодействующих друг с другом атомов алюминия.
Термодинамические функции.
Для проверки соответствия оптимизированной
структуры точке минимума на поверхности потенциальной энергии производится
расчёт гессиана (матрицы вторых производных энергии по координатам ядер). Если
в диагонализированной матрице отсутствуют мнимые силовые постоянные (частоты
колебаний), значит структура отвечает минимуму на поверхности потенциальной
энергии. Фактически также происходит расчёт ИК-спектра. Ввиду малой затратности
методами статистической термодинамики (ТД) также производится расчёт энтропии
(S), теплоемкости при постоянном объеме (CV)
и давлении (CP),
термохимических поправок (относительно полной энергии E) к энтальпии (H),
энергии Гиббса (G), полной внутренней энергии (абсолютной внутренней энергии, не
учитывающей энергию ядер (E)).
наночастица кластер малоразмерный металлический
Определение термодинамических свойств методами
статистической механики базируется на определении статистической суммы.
Предположим, что имеется подчиняющаяся законам
термодинамики система, находящаяся в постоянном тепловом контакте со средой,
которая имеет температуру T, а объём системы и количество составляющих её
частиц фиксированы. Обозначим точные состояния, в которых может находиться
система n (n=1,
2, 3..), а полную энергию системы в состоянии n
- En. Как правило, эти
микросостояния можно рассматривать как дискретные квантовые состояния системы.
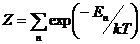 ,
,
=1,38Ч10-23 Дж/К - постоянная
Больцмана, (7) - каноническая сумма по состояниям (статистическая сумма)
системы при квантовомеханическом описании. В классической статистической
механике было бы некорректно определять статистическую сумму в виде суммы
дискретных членов, как в приведённой выше формуле. В классической механике
координаты и импульсы частиц могут меняться непрерывно, и множество
микросостояний несчётно. При этом статистическая сумма принимает вид интеграла.
Например, статистическая сумма газа из N классических частиц равна
Z = 1/N!h3N ∫exp(−
H( p,q) /kT)dpdq, (8)- статистический интеграл системы при классическом
описании. Где H(p,q) - функция Гамильтона - сумма кинетической и потенциальной
энергий системы, записанных как функции обобщенных импульсов р и обобщенных
координат q. Пределы интегрирования в выражении (8) определяются допустимыми
значениями импульсов и координат: проекция импульса частицы может принимать
значения от - ∞ до + ∞, значения координат частиц ограничиваются
геометрическими размерами системы. Множитель 1/N!h3N в выражении
статистического интеграла (8) учитывает неразличимость частиц, в результате
чего число различных микросостояний уменьшается в N! раз, и отражает тот факт,
что минимальный элемент объёма фазового пространства классической системы N
частиц, в силу соотношения неопределенности Гейзенберга, равен h3N [8].
Статистическая сумма является
функцией, в первую очередь, температуры T, а во вторую - энергий микросостояний
E1, E2, E3 и т. д. Энергии микросостояний определяются другими
термодинамическими величинами, такими как число частиц и объём, а также
микроскопическими свойствами, такими как масса частиц. Эта зависимость от
микроскопических свойств является основной в статистической механике. По модели
микроскопических составляющих системы можно рассчитать энергии микросостояний,
а следовательно, и статистическую сумму, которая позволяет рассчитать все
остальные термодинамические свойства системы [11].
Статистическая сумма может быть использована для
расчёта термодинамических величин, поскольку она имеет очень важный статистический
смысл. Вероятность Pn, с которой
система находится в микросостоянии n,
равна
 .
.
F(T,V,N) = - kT lnZ(T,V,N) ),
(9)
где F(T,V,N) - энергия Гельмгольца
системы Формула (9) вытекает из канонического распределения Гиббса и
справедлива для системы, описываемой макроскопическими параметрами T, V, N. При
помощи энергии Гельмгольца термодинамические функции могут быть выражены через
сумму по состояниям системы:
Энтропия
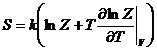
Внутренняя энергия

Энергия Гиббса

Энтальпия

Теплоемкость при постоянном объеме

Так как идеальный газ состоит из
невзаимодействующих молекул, то, согласно свойству мультипликативности Z, сумма
по состояниям системы равна произведению сумм по состояниям Q отдельных
молекул. Следует отметить, что при формулировке свойства мультипликативности Z
подразумевалось, что невзаимодействующие части системы (а в данном случае это
молекулы идеального газа) являются различимыми. В действительности одинаковые
частицы неразличимы. Для учета этого, необходимо разделить полученный результат
на N!. Таким образом, сумма по состояниям Z системы, состоящей из молекул
идеального газа,
 (10),
(10),
где Q - сумма по состояниям
отдельной молекулы. Энтропия идеального газа определяется через сумму по
состояниям молекулы:
Аналогичным образом определяются
теплоемкости при постоянном объеме и давлении, а также термохимические
поправки.
Энергия поступательного движения
молекулы (кинетическая энергия) не зависит от энергии её внутренних степеней
свободы. Поэтому сумма по состояниям молекулы Q равна произведению
поступательной суммы по состояниям Qпост и суммы по состояниям, обусловленной
внутренними степенями свободы молекулы
вн, Q=QвнQпост (11)
Молекула рассматривается как жесткий
ротатор и гармонический осциллятор, таким образом, вращение и колебания
молекулы независимы [9]. С использованием допущения о независимости различных
видов энергии внутренних степеней свободы молекулы, сумма по состояниям Qвн, в
соответствии со свойством мультипликативности, будет равна произведению сумм по
вращательным, колебательным, электронным и ядерным состояниям молекулы,
вн = Qвр·Qкол·Qэл·Qяд. (12)
В химических процессах спин ядер не
меняется. Так как в практических задачах требуется рассчитывать лишь изменение
термодинамических функций, то составляющие ядерного спина сокращаются и на
результат не влияют. Поэтому термодинамические функции рассчитывают без учета
ядерного спина [8]. Термодинамические функции, рассчитанные без учета ядерного
спина, называют практическими. В справочниках приведены именно практические
значения термодинамических величин.
Поступательная сумма по состояниям
отдельной молекулы идеального газа имеет вид
 (13),
(13),
где h=6,626Ч10-34
Дж·с - постоянная Планка, m - масса молекулы.
Для идеального газа, 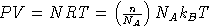 ,
следовательно
,
следовательно  .
.
Колебательная сумма по состояниям:
 , где
, где
ν - частота колебаний, Гц,  = 0, 1, 2,
3, …, ∞ - колебательное квантовое число. Частоты колебаний определяются
при расчете гессиана.
= 0, 1, 2,
3, …, ∞ - колебательное квантовое число. Частоты колебаний определяются
при расчете гессиана.
Так как сумма представляет собой бесконечную
убывающую геометрическую прогрессию, имеем
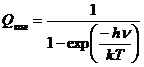 (14).
(14).
Для всех типов нелинейных молекул
справедливо следующее выражение вращательной суммы по состояниям:
 (15),
(15),
где  - число симметрии молекулы, равно
числу эквивалентных положений молекулы при всех возможных вращениях [8].
Моменты инерции молекулы также определяются при расчете гессиана и выводятся в
разделе термохимии выходного файла.
- число симметрии молекулы, равно
числу эквивалентных положений молекулы при всех возможных вращениях [8].
Моменты инерции молекулы также определяются при расчете гессиана и выводятся в
разделе термохимии выходного файла.
В электронной сумме по состояниям необходимо
учитывать либо только одно слагаемое (основное электронное состояние), либо
несколько первых слагаемых (основное и низколежащие возбужденные электронные
состояния):
 (16),
(16),
где g0 - степень вырождения
основного электронного состояния, g1, g2, … - степени вырождения возбужденных
состояний, Еэл1, Еэл2, … - энергии возбужденных состояний.
Поступательная сумма по состояниям
(13) определяется исходя из справочных величин (постоянная планка, постоянная
Больцмана) и задаваемого состава соединения (масса). Для получения величины
колебательной (14) и вращательной (15) сумм по состояниям исползьуются так же
значения частот колебаний рассматриваемой молекулы и ее моменты инерции,
определяемые при помощи аппарата квантовой химии при расчете гессиана, их
значения так же приводятся в выходном файле вычислений в разделе термохимии.
Для электронной суммы по состояниям (16) в firefly учитывается
только основное электронное состояние, которое характеризуется мультиплетностью
молекулы.
Перемножение полученных составляющих
дает в итоге значение суммы по состояниям отдельного кластера и ее логарифма,
которые непосредственно используются в соотношениях определяющих
термодинамические функции наночастиц алюминия.
Раздел термохимии в выходном файле расчета
гессиана кластера Al13:
THERMOCHEMISTRY AT T= 298.15 KIDEAL
GAS, RIGID ROTOR, HARMONIC NORMAL MODE APPROXIMATIONS. P= 1.01325E+05 PASCAL.
ALL FREQUENCIES ARE SCALED BY 1.00000 THE MOMENTS OF INERTIA ARE (IN
AMU*BOHR**2) 6072.55222 6197.56240 6197.84150 THE ROTATIONAL SYMMETRY NUMBER IS
1.0 THE ROTATIONAL CONSTANTS ARE (IN GHZ) 0.29692 0.29094 0.29092 THE HARMONIC
ZERO POINT ENERGY IS (SCALED BY 1.000) 0.012701
HARTREE/MOLECULE 2787.503821 CM**-1/MOLECULE 7.969881 KCAL/MOL 33.345984 KJ/MOL
Q LN Q ELEC. 2.00000E+00 0.693147 TRANS. 2.58207E+08
19.369274 ROT. 5.46711E+06 15.514261 VIB. 4.67199E+25 59.106212 TOT.
1.31904E+41 94.682894 E H G CV CP S KJ/MOL KJ/MOL KJ/MOL J/MOL-K J/MOL-K
J/MOL-K ELEC. 0.000 0.000 -1.718 0.000 0.000
5.763 TRANS. 3.718 6.197 -48.015 12.472 20.786 181.830 ROT. 3.718 3.718 -38.459
12.472 12.472 141.464 VIB. 88.503 88.503 -113.175 253.387 253.387 676.429 TOTAL
95.939 98.418 -201.367 278.330 286.644 1005.486 E H G CV CP S KCAL/MOL KCAL/MOL
KCAL/MOL CAL/MOL-K CAL/MOL-K CAL/MOL-K ELEC. 0.000 0.000 -0.411 0.000 0.000
1.377 TRANS. 0.889 1.481 -11.476 2.981 4.968 43.458 ROT.
0.889 0.889 -9.192 2.981 2.981 33.811 VIB.
21.153 21.153 -27.049 60.561 60.561 161.670 TOTAL
22.930 23.523 -48.128 66.522 68.510 240.317
......END
OF NORMAL
COORDINATE
ANALYSIS......
С использованием значений полной энтропии и
полной энергии кластера возможно получить все интересующие термодинамические
функции. Величина энергии связи атомов в кластере позволяет определить удельную
энтальпию образования наночастиц алюминия. И, далее, с использованием
абсолютного значения энтропии из выходного файла при помощи уравнения
Гиббса-Гельмгольца рассчитать энергию Гиббса
Пример расчета термодинамических функций для Al13
по данным, полученным в базисе 3-N21.
Полная энергия одного атома алюминия
Eal= -240,551
а. е. э.
Полная энергия 13 атомов алюминия
E13al=
-3127,163 а. е. э.
Полная энергия кластера из 13 атомов алюминия
Eal13= -
3127,7098 а. е. э.
Тогда энергия связи одной металлической
наночастицы
 а. е. э.= -2,3839Ч10-18 Дж.
а. е. э.= -2,3839Ч10-18 Дж.
Удельную энергию связи определяем
как
 Дж/кг,
Дж/кг,
где m=582,4498Ч10-27
кг - масса одной наночастицы.
При использовании методов
молекулярной механики эта величина составляет 3,51Ч106Дж/кг.
Из справочных данных удельная
энтальпия образования газообразного алюминия при стандартной температуре
Т=298,15єК: Hfalгаз=329,7
кДж/моль=12,2198Ч106 Дж/кг.
Следовательно, удельная энтальпия
образования кластеров при стандартной температуре
 Дж/кг
Дж/кг
При расчете гессиана для
оптимизированной структуры кластера, в выходном файле получаем значение
абсолютной энтропии:
Sal13=1005
Дж/мольЧК
И далее, пользуясь уравнением
Гиббса-Гельмгольца, определяем:
μ=H-TЧS=2,8507Ч106-298,15Ч1005,486=2,5509Ч106
Дж/моль
Однако необходимо отметить, что расчет
термохимии имеет смысл только в том случае, если мнимые частоты колебаний
отсутствуют, в противном случае полученные результаты могут значительно
отличаться от истинных.
Из-за несовершенства численных методов
квантово-хиических расчетов, реализованных в квантово-химических пакетах
программ, минимум на поверхности потенциальной энергии, найденный посредством
оптимизации геометрии системы, не всегда соответствует минимуму при дальнейшем
расчете гессиана для данной структуры. Так при расчете гессиана с
использованием базисов 6-N31
и DZV не удалось найти
геометрию, при которой бы отсутствовали мнимые частоты колебаний.
Сравнение полученных данных с результатами,
полученными при использовании термодинамического метода.
Результаты проведенных расчетов приведены в
таблице 1 (см. приложение).
Каждый из рассматриваемых в данной работе
методов оценки энергии связи микрочастиц в кластере и термодинамических функции
обладает недостатками, которые влияют на достоверность (accuracy)
получаемых результатов. Так основными источниками неточности в
термодинамическом методе является отсутствие учета многочастичного
взаимодействия атомов в кластере, а так же использование парных потенциалов
взаимодействия, которые, вообще говоря, являются лишь эмпирическими
аппроксимациями реального взаимодействия между частицами и позволяют получать
приемлемое описание только в узкой области расстояний и для ограниченного числа
элементов. Достоверность данных получаемых при расчете квантово-химическими
методами во многом определяется точностью задаваемого описания: начальным
приближением расположения атомов, порогом сходимости, методом расчета и, в
максимальной степени, выбираемым базисным набором функций для описания атомных
орбиталей. Об этом свидетельствуют значительно отличающиеся между собой, в
зависимости от сложности используемого базиса, величины энергий связи атомов.
Однако оценить точность (т.е. отклонение
среднего аналитического результата от некоторой предполагаемой истинной
величины) получаемых в итоге значений термодинамических функций достаточно
проблематично. Для этого необходимо обладать экспериментальными данными,
получение которых само по себе представляет значительные сложности. Более того,
эти данные будут соответствовать каким-то определенным условиям, вероятно
отличающимся от тех, для которых проводились расчеты (изолированный кластер
алюминия при стандартной температуре и давлении). Поэтому в рамках данной работы
мы можем говорить лишь о согласовании или несогласовании между собой данных
полученных в рамках термодинамического и квантово химического подходов.
Переходя к сравнению данных, прежде всего,
необходимо отметить достаточно значимую корреляцию значений энергий связи
атомов в кластере (погрешность не превышает 14%) и, как следствие, значений
удельной энтальпии (отклонение порядка 7%) образования наночастиц, полученных
на основе расчетов из первых принципов, и значений, получаемых на основе
эмпирического подхода, базирующегося на использовании парных потенциалов
взаимодействия. Такое согласование определяемых величин во многом
обеспечивается подбором параметров модельных потенциалов в соответствии с
экспериментальными данными.
С другой стороны, имеет место существенное
отличие получаемых значений энтропий (около 99%). Поскольку энтропия
определяется как разность энтальпии и энергии Гиббса, деленная на температуру,
даже незначительные погрешности при определении энергии связи атомов в
кластере, могут приводить к существенным ошибкам при переходе к значениям
абсолютной энтропии.
Таким образом, причина несогласующихся
результатов по энтропии может лежать как в методе нанотермодинамики, при
аппроксимации реального взаимодействия атомов модельными потенциалами, так и в
квантовой химии, при определении величин (частот колебаний и моментов инерции),
используемых для расчета термохимии.
Выбор того или иного метода при описании
малоразмерных наноструктур представляет собой отдельную задачу, решение которой
зависит от располагаемых технических ресурсов для проведения расчетов,
необходимой точности, начальной информации об интересующей системе, целей
исследования. Так методы, базирующиеся на квантовой теории, реализуют расчеты
без каких-либо эмпирических данных. Данные методы обладают широкими
возможностями, такими как определение пути прохождения реакции при помощи
расчета внутренней координаты реакции. Однако трудности получения результатов
при их использовании связаны с тем, что они всеобъемлющи, так что численное
решение уравнений представляет крайне сложную задачу и требует значительных
вычислительных ресурсов. Кроме того, необходимо отметить, что существующие
алгоритмы квантово-химических расчетов крайне требовательны к заданию
начального приближения. В ряде случаев может не сходиться процедура
самосогласования, или же оптимизированная структура соединения может не
соответствовать теоретически прогнозируемой геометрии. Отчасти это связано с
тем, что реализованный в них градиентный метод поиска минимума обладает значительным
недостатком: при его использовании определяется местоположение некоторого
минимума, который, вообще говоря, не обязательно будет глобальным.
В термодинамическом подходе напротив,
используется большое число параметров, которые должны быть получены на основе
экспериментальных данных, что ограничивает область использования данных
подходов лишь заранее исследованными классами соединений. Данный метод
схематизирует реальное взаимодействие между атомами в соединении, что приводит
к некоторому упрощению исследуемой модели, при этом значительно сокращая
затраты машинного времени. Предварительно по итогам проведенного исследования
можно заключить, что использование метода нанотермодинамики для изучения
наноразмерных структур оказывается вполне оправданным, поскольку при
существенной экономии вычислительных ресурсов, он позволяет получить результаты
сопоставимые с результатами, получаемыми при проведении большого количества
сложных вычислений методами квантовой химии.
Заключение
В рамках данной работы были показаны возможности
методов квантовой химии для описания наночастиц и их термодинамических
характеристик. Получены значения основных термодинамических функций для
кластера Al13 в
стандартных условиях по температуре и давлению и проведен анализ полученных
данных. Выполнено сравнение результатов, полученных различными методами, из
которого был сделан вывод о том, что использование эмпирического метода
нанотермодинамики для изучения кластеров вполне правомерно и дает величины
хорошо кореллирующие значения, определяемые при помощи квантовой химии.
Для построения более полной картины о
термодинамическом состоянии наночастиц необходимо продолжать данные
исследования с использованием других доступных методов, в частности метода
функционала плотности.
Дальнейшее же изучение кластеров, необходимое
для принятия решения о возможности использования их в качестве компонентов
высокоэнергетических топлив, по всей видимости, должно лежать в области
определения энергии активации реакций взаимодействия кластеров друг с другом и
элементами окружающей среды, а также исследования кинетики этих процессов.
Список литературы
1. Бабук В.А., Салимуллин Р.М.,
Зеликов А.Д., Ванеева О.В., Принципы использования малоразмерных наночастиц в
составе высокоэнергетических материалов., Химическая физика и мезоскопия. Т.
13, № 3, С. 356-362, Ижевск, 2011.
. Ибрагимов И. М., Ковшов А. Н.,
Назаров Ю. Ф., Основы компьютерного моделирования наносистем, СПб, «Лань», 2010
.Буркерт У., Аллинжер Н.,
Молекулярная механика. М., Мир, 1986
. Бабук В.А., Зеликов А.Д.,
Салимуллин Р.М. Термодинамический метод определения характеристик наночастиц.
. Алесковский В.Б. Химия
надмолекулярных соединений. СПб: Химия, 1996, 256 с.
. Русанов А.И. Нанотермодинамика:
химический подход // Российский химический журнал. 2006, т. 50, №2, с. 145-151.
. Бабук В.А., Зеликов А.Д.,
Салимуллин Р.М., Проблемы взаимодействия наночастиц с окружающей средой.
Подходы к решению и результаты анализа., Химическая физика и
мезоскопия. Т.12, №2, С. 224-231,
Ижевск, 2010.
. Леванов А. В., Антипенко Э. Е.,
Определение термодинамических свойств статистическими методами. Классический
идеальный газ., МГУ, 2006
. Joseph W. Ochterski, Termochemistry
in Gaussian.,
2000
http://www.gaussian.com/g_whitepap/thermo.htm
.
http://classic.chem.msu.su/gran/gamess/index.html
.
Ландау Л. Д., Лифшиц, Е. М. Статистическая физика. Часть 1 - Издание 5-е. - М.:
Физматлит, 2005. - 616 с.
Приложение
Таблица 1.
|
Al13
|
Al13, а.е. э.
|
13Al, а. е. э.
|
Eсв, а. е. э.
|
Eсв, МДж/кг
|
Н,
МДж/кг
|
Н,
МДж/моль
|
S, Дж/мольЧК
|
μ,
МДж/моль
|
μ, МДж/кг
|
|
ROHF STO-3G
|
-3108,2785
|
-3107,3445
|
-6,9906
|
5,2292
|
1,8342
|
529,804
|
1,6762
|
4,7788
|
|
ROHF 3-N21
|
-3127,7098
|
-3127,163
|
-0,5468
|
-4,0925
|
8,1273
|
2,8507
|
1005,486
|
2,5509
|
7,2725
|
|
ROHF 6-N31
|
-3144,5182
|
-3144,1042
|
-0,414
|
-3,0986
|
9,1212
|
3,1993
|
902,643
|
2,9302
|
8,3538
|
|
DZV
|
-3144,5707
|
-3144,1152
|
-0,4556
|
-3,4090
|
8,8063
|
3,0889
|
601,960
|
2,9094
|
8,2946
|
|
Метод
нанотермодинамики
|
-
|
-
|
-
|
-3,5273
|
8,6898
|
3,0480
|
2002,328
|
2,4510
|
6,9877
|