Особенности фармацевтического анализа
Содержание
Вступление
.
Основные принципы фармацевтического анализа
.1
Критерии фармацевтического анализа
.2
Общие принципы испытаний подлинности лекарственных веществ
.3
Общие требования к испытаниям на чистоту
.
Виды и методы контроля качества
.1
Методы анализа
.2
Виды внутриаптечного контроля
.3
Особенности экспресс-анализа лекарственных форм в условиях аптеки
.
Принципы проведения экспресс-анализа
.1
Качественный экспресс-анализ
.2
Количественный экспресс-анализ
.
Методики экспресс-анализа
.1
Анализ неорганических лекарственных веществ
.2
Анализ органических лекарственных веществ
.
Экспериментальная часть
.1
Методика анализа
.2
Результаты анализа
Выводы
Список
использованной литературы
Вступление
Фармацевтический анализ - это наука о химической
характеристике и измерении биологически активных веществ на всех этапах
производства: от контроля сырья до оценки качества полученного лекарственного
вещества, изучения его стабильности, установления сроков годности и
стандартизации готовой лекарственной формы. Фармацевтический анализ имеет свои
специфические особенности, отличающие его от других видов анализа. Эти
особенности заключаются в том, что анализу подвергают вещества различной
химической природы: неорганические, элементорганические, радиоактивные, органические
соединения от простых алифатических до сложных природных биологически активных
веществ. Чрезвычайно широк диапазон концентраций анализируемых веществ.
Объектами фармацевтического анализа являются не только индивидуальные
лекарственные вещества, но и смеси, содержащие различное число компонентов.
Количество лекарственных средств с каждым годом увеличивается. Это вызывает
необходимость разработки новых способов анализа.
Способы фармацевтического анализа нуждаются в
систематическом совершенствовании в связи с непрерывным повышением требований к
качеству лекарственных средств, причем растут требования как к степени чистоты
лекарственных веществ, так и к количественному содержанию. Поэтому необходимо
широкое использование не только химических, но и более чувствительных
физико-химических методов для оценки качества лекарств.
К фармацевтическому анализу предъявляют высокие
требования. Он должен быть достаточно специфичен и чувствителен, точен по
отношению к нормативам, обусловленным ГФ XI, ВФС, ФС и другой НТД, выполняться
в короткие промежутки времени с использованием минимальных количеств испытуемых
лекарственных препаратов и реактивов.
Фармацевтический анализ в зависимости от
поставленных задач включает различные формы контроля качества лекарств: фармакопейный
анализ, постадийный контроль производства лекарственных средств, анализ
лекарственных форм индивидуального изготовления, экспресс-анализ в условиях
аптеки и биофармацевтический анализ.
Составной частью фармацевтического анализа
является фармакопейный анализ. Он представляет собой совокупность способов
исследования лекарственных препаратов и лекарственных форм, изложенных в
Государственной фармакопее или другой нормативно-технической документации (ВФС,
ФС). На основании результатов, полученных при выполнении фармакопейного
анализа, делается заключение о соответствии лекарственного средства требованиям
ГФ или другой нормативно-технической документации. При отклонении от этих
требований лекарство к применению не допускают.
Заключение о качестве лекарственного средства
можно сделать только на основании анализа пробы (выборки). Порядок ее отбора
указан либо в частной статье, либо в общей статье ГФ XI (вып. 2). Отбор пробы
производят только из неповрежденных укупоренных и упакованных в соответствии с
требованиями НТД упаковочных единиц. При этом должны строго соблюдаться
требования к мерам предосторожности работы с ядовитыми и наркотическими
лекарственными средствами, а также к токсичности, огнеопасности,
взрывоопасности, гигроскопичности и другим свойствам лекарств. Для испытания на
соответствие требованиям НТД проводят многоступенчатый отбор проб. Число
ступеней определяется видом упаковки. На последней ступени (после контроля по
внешнему виду) берут пробу в количестве, необходимом для четырех полных физико-химических
анализов (если проба отбирается для контролирующих организаций, то на шесть
таких анализов).
Из расфасовки "ангро" берут точечные
пробы, взятые в равных количествах из верхнего, среднего и нижнего слоев каждой
упаковочной единицы. После установления однородности все эти пробы смешивают.
Сыпучие и вязкие лекарственные средства отбирают пробоотборником, изготовленным
из инертного материала. Жидкие лекарственные средства перед отбором проб
тщательно перемешивают. Если это делать затруднительно, то отбирают точечные
пробы из разных слоев. Отбор выборок готовых лекарственных средств осуществляют
в соответствии с требованиями частных статей или инструкций по контролю,
утвержденных МЗ РФ.
Выполнение фармакопейного анализа позволяет
установить подлинность лекарственного средства, его чистоту, определить
количественное содержание фармакологически активного вещества или ингредиентов,
входящих в состав лекарственной формы. Несмотря на то, что каждый из этих
этапов имеет свою конкретную цель, их нельзя сматривать изолированно. Они
взаимосвязаны и взаимно дополняют друг друга. Так, например, температура
плавления, растворимость, рН среды водного раствора и т.д. являются критериями
как подлинности, так и чистоты лекарственного вещества.
Необходимость внутриаптечного контроля
обусловлена высокими требованиями к качеству лекарственных форм,
изготавливаемых в аптеках. Поскольку изготовление лекарств в аптеках
ограничивается сжатыми сроками, оценку их качества осуществляют
экспресс-методами. Основные требования, предъявляемые к экспресс-анализу, -
расход минимальных количеств лекарственных форм, простота и быстрота
выполнения, достаточная точность и возможность проведения анализа без изъятия
приготовленного лекарства.
В настоящее время в аптеках широко используют
различные методы как качественного, так и количественного экспресс-анализа. Для
этого применяют различные химические и физико-химические методы.
Целью данной работы является изучение методик
проведения экспресс-анализа.
В экспериментальной части работы проведен анализ
таблеток анальгина 0,5 г.
1. Основные принципы фармацевтического анализа
.1 Критерии фармацевтического анализа
На различных этапах фармацевтического анализа в
зависимости от поставленных задач имеют значение такие критерии, как
избирательность, чувствительность, точность, время, затраченное на выполнение
анализа, израсходованное количество анализируемого препарата (лекарственной
формы).
Избирательность метода очень важна при
проведении анализа смесей веществ, поскольку дает возможность получать истинные
значения каждого из компонентов. Только избирательные методики анализа
позволяют определять содержание основного компонента в присутствии продуктов
разложения и других примесей.
Требования к точности и чувствительности
фармацевтического анализа зависят от объекта и цели исследования. При испытании
степени чистоты препарата используют методики, отличающиеся высокой
чувствительностью, позволяющие устанавливать минимальное содержание примесей.
При выполнении постадийного контроля
производства, а также при проведении экспресс-анализа в условиях аптеки важную
роль имеет фактор времени, которое затрачивается на выполнение анализа. Для
этого выбирают методы, позволяющие провести анализ в наиболее короткие
промежутки времени и вместе с тем с достаточной точностью.
При количественном определении лекарственного
вещества используют метод, отличающийся избирательностью и высокой точностью.
Чувствительностью метода пренебрегают, учитывая возможность выполнения анализа
с большой навеской препарата.
Мерой чувствительности реакции является предел
обнаружения. Он означает наименьшее содержание, при котором по данной методике
можно обнаружить присутствие определяемого компонента с заданной доверительной
вероятностью. Термин ''предел обнаружения" введен вместо такого понятия,
как "открываемый минимум", им пользуются также взамен термина
"чувствительность". На чувствительность качественных реакций
оказывают влияние такие факторы, как объемы растворов реагирующих компонентов,
концентрации реактивов, рН среды, температура, продолжительность опыта. Это
следует учитывать при разработке методик качественного фармацевтического
анализа. Для установления чувствительности реакций все шире используют
показатель поглощения (удельный или молярный), устанавливаемый
спектрофотометрическим методом. В химическом анализе чувствительность
устанавливают по величине предела обнаружения данной реакции. Высокой
чувствительностью отличаются физико-химические методы анализа. Наиболее
высокочувствительны радиохимические и масс-спектральный методы, позволяющие
определять 10-8-10-9% анализируемого вещества, полярографические и
флуориметрические 10-6-10-9%; чувствительность спектрофотометрических методов
Ю-3-10-6%, потенциометрических 10-2%.
Термин "точность анализа" включает
одновременно два понятия: воспроизводимость и правильность полученных
результатов. Воспроизводимость характеризует рассеяние результатов анализа по
сравнению со средним значением. Правильность отражает разность между
действительным и найденным содержанием вещества. Точность анализа у каждого
метода различна и зависит от многих факторов: калибровки измерительных
приборов, точности отвешивания или отмеривания, опытности аналитика и т.д.
Точность результата анализа не может быть выше, чем точность наименее точного
измерения.
Так, при вычислении результатов титриметрических
определений наименее точная цифра - количество миллилитров титранта,
израсходованного на титрование. В современных бюретках в зависимости от класса
их точности максимальная ошибка отмеривания около ±0,02 мл. Ошибка от натекания
тоже равна ±0,02 мл. Если при указанной общей ошибке отмеривания и натекания
±0,04 мл на титрование расходуется 20 мл титранта, то относительная ошибка
составит 0,2%. При уменьшении навески и количества миллилитров титранта
точность соответственно уменьшается. Таким образом, титриметрическое
определение можно выполнять с относительной погрешностью ±(0,2-0,3)%.
Точность титриметрических определений можно
повысить, если пользоваться микробюретками, применение которых значительно
уменьшает ошибки от неточного отмеривания, натекания и влияния температуры.
Погрешность допускается также при взятии навески.
Отвешивание навески при выполнении анализа
лекарственного вещества осуществляют с точностью до ±0,2 мг. При взятии обычной
для фармакопейного анализа навески 0,5 г препарата и точности взвешивания ±0,2
мг относительная ошибка будет равна 0,4%. При анализе лекарственных форм,
выполнении экспресс-анализа такая точность при отвешивании не требуется,
поэтому навеску берут с точностью ±(0,001-0,01) г, т.е. с предельной
относительной ошибкой 0,1-1%. Это можно отнести и к точности отвешивания
навески для колориметрического анализа, точность результатов которого ±5%.
.2 Общие принципы испытаний подлинности
лекарственных веществ
Испытание на подлинность - это подтверждение
идентичности анализируемого лекарственного вещества (лекарственной формы),
осуществляемое на основе требований Фармакопеи или другой
нормативно-технической документации (НТД). Испытания выполняют физическими,
химическими и физико-химическими методами. Непременным условием объективного
испытания подлинности лекарственного вещества является идентификация тех ионов
и функциональных групп, входящих в структуру молекул, которые обусловливают
фармакологическую активность. С помощью физических и химических констант
(удельного вращения, рН среды, показателя преломления, УФ- и ИК-спектра)
подтверждают и другие свойства молекул, оказывающие влияние на
фармакологический эффект. Применяемые в фармацевтическом анализе химические
реакции сопровождаются образованием окрашенных соединений, выделением
газообразных или нерастворимых в воде соединений. Последние можно
идентифицировать по температуре плавления.
.3 Общие требования к испытаниям на чистоту
Оценка степени чистоты лекарственного препарата
- один из важных этапов фармацевтического анализа. Все лекарственные препараты
независимо от способа получения испытывают на чистоту. При этом устанавливают
содержание примесей. Их можно разделить на две группы: примеси, оказывающие
влияние на фармакологическое действие лекарственного препарата, и примеси,
указывающие на степень очистки вещества. Последние не влияют на
фармакологический эффект, но присутствие их в больших количествах снижает
концентрацию и соответственно уменьшает активность препарата. Поэтому фармакопеи
устанавливают определенные пределы этих примесей в лекарственных препаратах.
Таким образом, основной критерий
доброкачественности лекарственного препарата - наличие допустимых пределов
физиологически неактивных примесей и отсутствие токсичных примесей. Понятие
отсутствие условно и связано с чувствительностью способа испытания.
Общие требования, которые предъявляются к
испытаниям на чистоту, - чувствительность, специфичность и воспроизводимость
используемой реакции, а также пригодность ее применения для установления
допустимых пределов содержания примесей.
Для испытаний чистоты избирают реакции с такой
чувствительностью, которая позволяет определить допустимые пределы примесей в
данном лекарственном препарате. Эти пределы устанавливают предварительной биологической
проверкой с учетом возможного токсического воздействия примеси.
Определить максимальное содержание примесей в
испытуемом препарате можно двумя путями (эталонным и безэталонным). Один из них
основан на сравнении с эталонным раствором (стандартом). При этом в одинаковых
условиях наблюдают окраску или помутнение, возникающие под действием
какого-либо реактива. Второй путь - установление предела содержания примесей по
отсутствию положительной реакции. При этом используют химические реакции,
чувствительность которых ниже, чем предел обнаружения допустимых примесей.
Для ускорения выполнения испытаний на чистоту,
их унификации и достижения одинаковой точности анализа в отечественных
фармакопеях использована система эталонов. Эталон представляет собой образец,
содержащий определенное количество открываемой примеси. Установление наличия
примесей производят колориметрическим или нефелометрическим методом,
сравнивания результаты реакций в растворе эталона и в растворе препарата после
добавления одинаковых количеств соответствующих реактивов. Достигаемая при этом
точность вполне достаточна, чтобы установить, больше или меньше, чем допустимо,
содержится примесей в испытуемом препарате.
При выполнении испытаний на чистоту необходимо
строго соблюдать общие указания, предусмотренные фармакопеями. Вода и
используемые реактивы не должны содержать ионов, наличие которых устанавливают;
одинакового диаметра и бесцветными должны быть пробирки; навески должны
отвешиваться с точностью до 0,001 г; реактивы следует добавлять одновременно и
в одинаковых количествах как к эталонному, так и к испытуемому раствору;
образующуюся опалесценцию наблюдают в проходящем свете на темном фоне, а
окраску - в отраженном свете на белом фоне. Если устанавливают отсутствие
примеси, то к испытуемому раствору прибавляют все реактивы, кроме основного;
затем полученный раствор делят на две равные части и к одной из них прибавляют
основной реактив. При сравнении не должно быть заметных различий между обеими
частями раствора.
Следует иметь в виду, что последовательность и
скорость прибавления реактива влияют на результаты испытаний на чистоту. Иногда
необходимо также соблюдать интервал времени, в течение которого следует вести
наблюдение за результатом реакции.
Источником примесей при производстве готовых
лекарственных форм могут служить плохо очищенные наполнители, растворители и
другие вспомогательные вещества. Поэтому степень чистоты этих веществ должна
подвергаться тщательному контролю перед использованием их в производстве.
фармацевтический анализ
лекарственный анальгин
2. Виды и методы контроля качества
.1 Методы анализа
Химические методы
Эти методы используются для установления
подлинности лекарственных веществ, испытаний их на чистоту и количественного
определения.
Для целей идентификации используют реакции,
которые сопровождаются внешним эффектом, например изменением окраски раствора,
выделением газообразных продуктов, выпадением или растворением осадков.
Установление подлинности неорганических лекарственных веществ заключается в
обнаружении с помощью химических реакций катионов и анионов, входящих в состав
молекул. Химические реакции, применяемые для идентификации органических
лекарственных веществ, основаны на использовании функционального анализа.
Чистота лекарственных веществ устанавливается с
помощью чувствительных и специфичных реакций, пригодных для определения
допустимых пределов содержания примесей.
Количественные методы химического анализа
подразделяют на гравиметрический и титриметрическии. Гравиметрический метод
основан на взвешивании осажденного вещества в виде малорастворимого соединения
или отгонки органических растворителей после извлечения лекарственного
вещества. Метод точен, но длителен, так как предусматривает такие операции, как
фильтрование, промывание, высушивание (или прокаливание) до постоянной массы.
Наибольшее применение получил титриметрическии
метод. Название происходит от слова "титр" (фр.) - концентрация.
Основная операция метода--титрование, заключающаяся в постепенном приливании к
раствору анализируемого вещества титрованного раствора до точки
эквивалентности. По измеренному объему титрованного раствора рассчитывают
количественное содержание вещества.
Титриметрический метод анализа получил широкое
распространение потому, что он позволяет использовать разнообразные химические
реакции и определять вещества, учитывая их свойства и строение. Он выполняется
быстро, с большой степенью точности, не нуждается в сложном оснащении и может
использоваться как в лабораториях, так и в аптеках.
Для количественного определения лекарственного
вещества титриметрическим методом необходимы титрованный (стандартный) раствор,
набор простой лабораторной посуды (бюретки, пипетки, мерные колбы для
титрования) и средств фиксации точки эквивалентности (конечной точки
титрования). Последнюю фиксируют как с помощью индикаторов, так и с помощью
физико-химических методов, измеряя приборами физическую константу системы
(потенциометрическое, амперометрическое титрование и др. способы). Однако не
всякая химическая реакция может быть применима для процесса титрования. К
реакциям, используемым в титриметрическом методе, предъявляются следующие
требования:
возможность фиксировать точку эквивалентности
(конечную точку титрования);
количественное протекание реакции, т. е. в
реакцию должно вступить 100% анализируемого вещества.
Для этого необходимо строго соблюдать
определенные условия титрования:
реакция должна протекать быстро;
не допускаются побочные реакции.
Физические и физико-химические методы
К ним относятся: определение температур
плавления и затвердевания, а также температурных пределов перегонки;
определение плотности, показателей преломления (рефрактометрия), оптического
вращения (поляриметрия); спектрофотометрия - ультрафиолетовая, инфракрасная;
фотоколориметрия, эмиссионная и атомно-абсорбционная спектрометрия,
флуориметрия, спектроскопия ядерного магнитного резонанса, масс-спектрометрия;
хроматография - адсорбционная, распределительная, ионообменная, газовая,
высокоэффективная жидкостная; электрофорез (фронтальный, зональный,
капиллярный); электрометрические методы (потенциометрическое определение рН,
потенциометрическое титрование, амперометрическое титрование,
вольтамперометрия).
Кроме того, возможно применение методов,
альтернативных фармакопейным, которые иногда имеют более совершенные аналитические
характеристики (скорость, точность анализа, автоматизация). В некоторых случаях
фармацевтическое предприятие приобретает прибор, в основе использования
которого лежит метод, еще не включенный в Фармакопею (например, метод
романовской спектроскопии - оптический дихроизм). Иногда целесообразно при
определении подлинности или испытании на чистоту заменить хроматографическую
методику на спектрофотометрическую. Фармакопейный метод определения примесей
тяжелых металлов осаждением их в виде сульфидов или тиоацетамидов обладает
рядом недостатков. Для определения примесей тяжелых металлов многие
производители внедряют такие физико-химические методы анализа, как
атомно-абсорбционная спектрометрия и атомно-эмиссионная спектрометрия с
индуктивно связанной плазмой.
В некоторых частных статьях ГФ X рекомендуется
определять температуру затвердевания или температуру кипения (по ГФ XI -
«температурные пределы перегонки») для ряда жидких ЛC. Температура кипения
должна укладываться в интервал, приведенный в частной статье. Более широкий
интервал свидетельствует о присутствии примесей.
Во многих частных статьях ГФ X приведены
допустимые значения плотности, реже вязкости, подтверждающие подлинность и
доброкачественность ЛC.
Практически все частные статьи ГФ X нормируют такой
показатель качества ЛC, как растворимость в различных растворителях.
Присутствие примесей в ЛB может повлиять на его растворимость, снижая или
повышая ее в зависимости от природы примеси.
Критериями чистоты являются также цвет ЛB и/или
прозрачность жидких лекарственных форм.
Определенным критерием чистоты ЛC могут служить
такие физические константы, как показатель преломления луча света в растворе
испытуемого вещества (рефрактометрия) и удельное вращение, обусловленное
способностью ряда веществ или их растворов вращать плоскость поляризации при
прохождении через них плоскополяризованного света (поляриметрия). Методы
определения этих констант относятся к оптическим методам анализа и применяются
также для установления подлинности и количественного анализа ЛС и их
лекарственных форм.
Биологические методы.
Биологические методы контроля качества ЛС весьма
разнообразны. Среди них испытания на токсичность, стерильность,
микробиологическую чистоту.
.2 Виды внутриаптечного контроля
Контроль качества. Изготовляемые в аптеках ЛС в
настоящее время контролируются в соответствии с требованиями,
регламентированными Государственной Фармакопеей и действующими нормативными
документами. Эти требования распространяются на все аптеки, включая
гомеопатические, независимо от форм собственности и ведомственной
принадлежности. Изготовление ЛС по индивидуальным прописям, в виде
внутриаптечной заготовки, а также концентратов и полуфабрикатов считается
законченным только после оценки качества их изготовления и правильности оформления.
Независимо от источника поступления
лекарственные средства подвергаются приемочному контролю. Лекарственные
средства, изготовленные в аптеках по индивидуальным рецептам или требованиям
лечебных учреждений, подвергаются внутриаптечному контролю: письменному,
органолептическому и контролю при отпуске - обязательно; опросному и
физическому - выборочно, а также химическому контролю.
Провизор-аналитик обязан владеть всеми видами
внутриаптечного контроля. Впервые назначенный на должность, провизор-аналитик
проходит обязательную стажировку в территориальной контрольно-аналитической
лаборатории. Провизор-аналитик, назначенный на должность для выполнения
контроля качества гомеопатических лекарственных средств, изготовляемых в
аптеке, проходит стажировку на факультетах повышения квалификации провизоров,
имеющих образовательную лицензию.
Для проведения химического контроля качества ЛС,
которые изготавливают в аптеке, выделяется специально оборудованное рабочее
место. На нем размещается типовое оборудование, приборы и реактивы.
Провизор-аналитик обеспечивается нормативными документами и справочной
литературой.
Результаты, полученные при контроле качества
лекарственных средств, регистрируются в журналах, которые хранятся в аптеке в
течение года. Один раз в год отчет о работе по контролю качества ЛС,
изготовленных в аптеке, направляется в территориальную контрольно-аналитическую
лабораторию или центр контроля качества ЛС.
Приемочный контроль. Такой контроль проводится в
целях предупреждения поступления в аптеку некачественных лекарственных средств.
Приемочный контроль заключается в проверке поступающих ЛС на соответствие
требованиям по показателям «Описание», «Упаковка», «Маркировка»; в проверке
правильности оформления расчетных документов (счетов), а также наличия сертификатов
качества (паспортов) производителя и других документов, подтверждающих качество
ЛС, в соответствии с действующими приказами и инструкциями.
Письменный контроль. При изготовлении ЛФ по
рецептам и требованиям лечебных учреждений заполняются паспорта письменного
контроля. В паспорте должны быть указаны; дата изготовления, номер рецепта
(номер больницы, название отделения), наименования взятых ЛВ и их количества,
число доз, подписи изготовившего, расфасовавшего и проверившего ЛФ. В случае
изготовления ЛФ практикантом ставится подпись ответственного за
производственную практику. Ведение паспортов письменного контроля, если ЛФ
изготавливаются и отпускаются одним и тем же лицом, также является
обязательным. В этом случае паспорт заполняется в процессе изготовления ЛФ.
Все расчеты должны производиться до изготовления
ЛФ и записываться на обратной стороне паспорта. Паспорт заполняется немедленно
после изготовления ЛФ по памяти на латинском языке в соответствии с
последовательностью технологических операций. При заполнении паспорта на
гомеопатические ЛФ указываются гомеопатические названия последовательно взятых
лекарственных средств.
Опросный контроль. Этот вид контроля применяется
выборочно. Проводится после изготовления фармацевтом не более пяти
лекарственных форм.
При проведении опросного контроля
провизор-технолог называет вещество, входящее в ЛФ первым, а в ЛФ сложного
состава указывает также его количество. После этого фармацевт называет все
взятые ЛВ и их количества. При использовании полуфабрикатов (концентратов)
фармацевт называет также их состав и концентрацию.
Органолептический контроль. Проверка ЛФ
проводится по следующим показателям: внешний вид («Описание»), запах,
однородность, отсутствие механических включений (в жидких ЛФ). На вкус
проверяются выборочно ЛФ, предназначенные для детей.
Однородность порошков, гомеопатических
тритураций, мазей, пилюль, суппозиториев проверяется до разделения массы на
дозы в соответствии с требованиями действующей ГФ. Проверка осуществляется
выборочно у каждого фармацевта в течение рабочего дня с учетом видов ЛФ.
Физический контроль. Физический контроль
заключается в проверке общей массы или объема ЛФ, количества и массы отдельных
доз (не менее трех доз), входящих в данную ЛФ. При проверке ЛФ контролируется
также качество укупорки.
Химический контроль. Химический контроль
заключается в оценке качества изготовления ЛС по показателям «Подлинность»,
«Испытания на чистоту и допустимые пределы примесей» (качественный анализ) и
«Количественное определение» (количественный анализ) ЛВ, входящих в его состав.
Выборочно качественный химический анализ
проводят для ЛФ, изготовленных по индивидуальным рецептам и требованиям
лечебных учреждений (у каждого фармацевта в течение рабочего дня, но не менее
10 % от общего количества изготовленных ЛФ).
Контроль при отпуске. Данному виду контроля
подвергаются все изготовленные в аптеках ЛС (в том числе гомеопатические).
При этом проверяется соответствие:
упаковки ЛС физико-химическим свойствам входящих
в них ЛВ;
указанных в рецепте доз ядовитых, наркотических
или сильнодействующих ЛВ возрасту больного;
номера на рецепте номеру на этикетке;
соответствие фамилии больного на квитанции, на
этикетке и рецепте или его копии;
копий рецептов прописям рецептов;
оформления ЛС действующим требованиям.
Предупредительные мероприятия внутриаптечного
контроля качества ЛС. При изготовлении ЛС в аптеке должны строго соблюдаться
санитарные нормы и правила, противоэпидемический режим в соответствии с
действующими нормативными документами, инструкциями и приказами.
В аптеке должны быть обеспечены условия и сроки
хранения ЛС в соответствии с их физико-химическими свойствами и требованиями
действующей ГФ, действующих приказов и инструкций. В помещениях хранения аптеки
на всех штангласах с ЛС должны быть указаны: номер серии
предприятия-изготовителя, номер анализа контрольно-аналитической лаборатории
(центра контроля качества ЛС), срок годности, дата заполнения и подпись
заполнившего штанглас.
.3 Особенности экспресс-анализа лекарственных
форм в условиях
аптеки
Экспресс-метод химического внутриаптечного
контроля предусматривает быстрое выполнение анализа лекарств и минимальный
расход исследуемых объектов и реактивов.
Для ускоренного определения подлинности веществ
в лекарствах обычно используют капельные реакции, которые выполняются в
пробирках, на предметных или часовых стеклах, в фарфоровых чашках, на
фильтровальной бумаге, пропитанной соответствующими реактивами. Для проведения
реакций используют 1 - 5 капель жидких ЛФ, 0,01 - 0,03 г порошков, 0,05 - 0,1 г
мазей и суппозиториев.
Количественный экспресс-анализ в условиях аптеки
предусматривает определение содержания ингредиентов в JIC с применением методов
объемного титрования и рефрактометрии. При титровании используют такое
количество ЛВ, чтобы уходило 2 - 3 мл титранта. Жидкие Л С отбирают пипетками
на 1, 2 или 5 мл. Массу порошков - 0,05, 0,1 или 0,2 г - определяют на ручных
аптечных весах. Точность определения массы составляет 0,01 г. Массу мазей или
суппозиториев, помещенных на заранее взвешенную пергаментную бумагу, определяют
на аптечных весах и вместе с бумагой помещают в пробирку, колбу или склянку для
анализа. Добавляют воду или органический растворитель, индикатор и титруют из
микробюретки с ценой деления 0,02 мл.
3. Принципы проведения экспресс-анализа
.1 Качественный экспресс-анализ
Качественный экспресс-анализ лекарственных форм
отличается от макроанализа только тем, что на его выполнение расходуется
меньшее количество вещества и реактива. Анализ растворов и порошков выполняют
без предварительного выделения лекарственных веществ, когда наполнители не
мешают выполнению качественных реакций. Для выделения лекарственного вещества
из таблеток, драже, мазей, суппозиториев достаточно перемешивания или
растирания с растворителем.
Для выполнения качественного экспресс-анализа
используют цветные или осадочные химические реакции на соответствующие катионы,
анионы неорганических или функциональные группы органических веществ. Анализ
выполняют капельным методом, при котором расходуется от 0,001 до 0,01 г порошка
или 1-5 капель жидкости.
Цветные реакции выполняют на фильтровальной
бумаге или в фарфоровых чашках, а осадочные - на часовых стеклах.
Чувствительность реакций, выполняемых на фильтровальной бумаге, можно повысить,
используя такие физические явления, как поверхностное натяжение, капиллярность,
адсорбция, диффузия. Так, например, за счет различия в скорости диффузии
растворенных компонентов лекарственной смеси можно одновременно
идентифицировать без разделения два и даже три лекарственных вещества. Они
образуют с реактивом окрашенные кольца, отличающиеся по цвету и расположенные
на различном расстоянии от центра. Избирательность цветных реакций можно также
повысить обработкой полосок фильтровальной бумаги парами летучих веществ.
Для экспресс-анализа многокомпонентных жидких и
твердых лекарственных форм представляют интерес и другие методы, позволяющие
идентифицировать компоненты смеси без их разделения. Иногда можно одним
реактивом обнаружить два ингредиента. Например, действуя окислителями, можно
последовательно открывать бромиды и иодиды, раствором хлорида железа (III) -
бензоаты и салицилаты и т. д. Можно подобрать реактив, который с одним
лекарственным веществом, содержащимся в смеси, образует окрашенное соединение
(растворимое или нерастворимое в воде), а с другим выделяет газообразный
продукт. Такого результата достигают, действуя концентрированной серной
кислотой на смесь, содержащую гидрокарбонаты и алкалоиды.
Если не удается выполнить анализ без разделения
компонентов, то используют те же принципы разделения, что и при макроанализе.
Они основаны на различии в растворимости лекарственных веществ. С помощью воды,
этилового спирта, ацетона, хлороформа можно разделять смесь, состоящую из
веществ, растворимых и нерастворимых в указанных растворителях. Растворы кислот,
щелочей, буферные растворы позволяют последовательно извлекать из смеси
вещества, различающиеся по кислотно-основным свойствам. Идентификацию
выделенных индивидуальных лекарственных веществ осуществляют теми же реакциями,
которыми испытывают на подлинность субстанции.
Качественный экспресс-анализ лекарственных
веществ, содержащихся в мазях, суппозиториях и пастах, можно выполнять
смешиванием и растиранием на стеклянной пластинке с соответствующим реактивом.
Если такой способ не дает положительных результатов, то небольшую массу мази
(пасты) предварительно обрабатывают спиртом, бензолом, эфиром или хлороформом
для растворения основы (жиров, вазелина). Можно также извлекать из мази (пасты)
лекарственное вещество водой или растворами кислот и щелочей при слабом
подогревании. Иногда сочетают оба эти способа, а затем отделяют полученное
извлечение (декантацией, фильтрованием) от мазевой основы, растворенной в
органическом растворителе. Если компоненты, содержащиеся в мази, нерастворимы в
воде (растворах кислот, щелочей, в органических растворителях), то мазевую
основу растворяют в эфире, бензине или хлороформе. Затем фильтруют и остаток на
фильтре растворяют, подбирая для этого соответствующий растворитель, в котором
растворяется компонент мази. Полученные экстракты анализируют соответствующими
реактивами теми же методами, что и сухие или жидкие лекарственные формы.
Качественный экспресс-анализ в условиях аптеки
осуществляют не только химическими, но и физическими или физико-химическими
методами.
Поляриметрия позволяет сделать заключение о
подлинности лекарственного вещества в растворе по значению удельного вращения,
рефрактометрия - по показателю преломления раствора определенной концентрации.
Доступным для использования во внутриаптечном
контроле является метод флуориметрии. По характеру флуоресценции кристаллов или
растворов можно, например, осуществлять идентификацию препаратов некоторых
алкалоидов, витаминов и др. Для возбуждения флуоресценции на растворы
испытуемых веществ воздействуют ультрафиолетовым излучением с длиной волны
365-366 нм. Некоторые лекарственные вещества сами не флуоресцируют, но при
взаимодействии с рядом реактивов образуют флуоресцирующие продукты.
Для выделения анализируемого лекарственного
вещества из многокомпонентной лекарственной формы используют хроматографию.
Особенно перспективно применение для экспресс-анализа распределительной
хроматографии на бумаге и тонкослойной хроматографии. После выделения
лекарственного вещества из лекарственной формы выполняют химические реакции на
ионы или функциональные группы, причем эти реакции могут быть выполнены прямо
на хроматограмме.
Для качественного экспресс-анализа настоек,
экстрактов, настоев и отваров может быть применено сочетание адсорбционной
хроматографии и люминесцентного анализа. Вначале, используя различие в
адсорбционной способности, компоненты лекарственных форм разделяют на отдельные
зоны в колонках из оксида алюминия. Полученные хроматограммы идентифицируют в
ультрафиолетовом излучении или с помощью групповых реакций на алкалоиды,
гликозиды, сапонины, дубильные и другие вещества.
.2 Количественный экспресс-анализ
Количественный экспресс-анализ может быть
выполнен титриметрическими или физико-химическими методами.
Титриметрический экспресс-анализ отличается от
макрометодов расходом меньших количеств анализируемых лекарственных форм
(0,05-0,1 г порошка или 1-3 мл раствора). Это позволяет анализировать
лекарственную форму без изъятия, т. е. контролировать качество того лекарства,
которое отпускается больному. На выполнение количественного экспресс-анализа
затрачивается минимальное время, так как используются методики, как правило, не
требующие процессов извлечения, выпаривания, фильтрования. Навеску порошка или
объем жидкой лекарственной формы (растворы, глазные капли и др.) берут с таким
расчетом, чтобы на определение расходовалось не более 2 мл 0,1М титрованного
раствора. Из твердых лекарственных форм вначале получают раствор. При
необходимости жидкие лекарственные формы предварительно разбавляют. Навеску
мази, если основа не мешает определению, растворяют в 3-5 мл этанола или эфира,
а затем титруют. Для уменьшения расхода анализируемого вещества и реактивов в
количественном экспресс-анализе обычно используют не только 0,1М, но и 0,02 и
0,01М титрованные растворы. Чтобы упростить расчеты, титрованные растворы
готовят точной нормальности (из фиксаналов).
Из титриметрических методов для количественного
экспресс-анализа хлоридов, бромидов, иодидов используют аргентометрию или
меркуриметрию. Соли цинка, магния, кальция определяют комплексоно-метрически.
Кислоты и соли органических оснований (гидрохлориды, гидробромиды, гидроиодиды,
сульфаты, фосфаты) титруют алкалиметрически. Растворы, аммиака и щелочей,
органические основания (амидопирин, гексаметилентетрамин) определяют
ацидиметрически; иодометрию применяют для определения растворов иода,
хлорамина, формальдегида и др.
Для количественного экспресс-анализа двух- и
трехкомпонентных лекарственных форм сочетают несколько методов. При выборе
методик анализа используют те же способы, что и при обычном анализе
многокомпонентных смесей титриметрическими методами.
Расчеты в количественном экспресс-анализе можно
упростить, пользуясь факторами титрования (Ф), значения которых вычисляют в
процентах по формуле
Ф= Т*100/а,
и в граммах по формуле
Ф = Т*b/а
Таким образом, фактор титрования включает
навеску (а) и титр исследуемого вещества (Т). Последующий расчет концентрации
или массы сводится к вычислению произведения ФVК, а для титрованных растворов с
К = 1 - к произведению ФV.
Ответственным этапом внутри аптечного контроля
является оценка качества концентрированных растворов (концентратов).
Концентраты подвергаются обязательному количественному анализу во всех аптеках.
Они содержат одно лекарственное вещество и анализируются как обычный водный
раствор высокой концентрации, который перед выполнением определения разбавляют.
Для облегчения расчетов результатов титриметрического экспресс-анализа
концентратов разработаны специальные таблицы. Пользуясь этими таблицами, можно
по объему затраченного титранта судить о соответствии содержания лекарственного
вещества допустимым нормам отклонений.
Из физико-химических методов для количественного
экспресс-анализа лекарственных форм применяют рефрактометрию. Этот метод
пригоден для определения лекарственных веществ в концентратах, жидких и
растворимых в воде сухих лекарственных формах, содержащих один или два
ингредиента, а также для определения концентрации этилового спирта в
водноспиртовых растворах. Обязательным условием выполнения рефрактометрических
определений является соблюдение температурного режима. Обычно определения
выполняют при 20°С. При небольших отклонениях температуры (на 5-7°) можно
вводить поправку с помощью несложных расчетов.
Рефрактометрию применяют для количественного
экспресс-анализа глюкозы, кислот (борной, аскорбиновой, никотиновой); солей
неорганических и органических кислот (калия и натрия бромиды, хлориды, иодиды;
кальция хлорид и глюконат, калия ацетат, натрия тетраборат, тиосульфат,
гидрокарбонат, цитрат, бензоат, салицилат); водорастворимых натриевых солей
сульфаниламидов (стрептоцида, норсульфазола, этазола, сульфацила). Более точные
результаты достигаются, если концентрация лекарственного вещества выше 5%.
Иногда при анализе многокомпонентных смесей рефрактометрию сочетают с
титриметрическими методами.
Расчет концентрации лекарственного вещества в
лекарственной форме производят также с помощью фактора показателя преломления
который вычисляют по формуле
= F0 + K*c,
где F0 - начальный фактор (прирост показателя
преломления при переходе от воды к раствору с бесконечно малой концентрацией);
К - вторая производная (величина, соответствующая среднему изменению фактора
при повышении концентрации препарата на 1%); с - концентрация раствора.
Значения показателей преломления и факторов для
различных концентраций лекарственных веществ приведены в таблицах, которые
имеются в руководствах по внутриаптечному контролю. Использование таблиц
значительно упрощает расчеты.
Концентрацию веществ в однокомпонентных
растворах (1) вычисляют по формуле
= (п - n0)/F
где п - показатель преломления анализируемого
раствора; n0 - показатель преломления растворителя при той же температуре.
В двух- и трехкомпонентных смесях одно или два
лекарственных вещества определяют титриметрическим методом, а содержание
третьего компонента (X) устанавливают измерением п раствора смеси. Расчет
проводят по формуле

где с и c1 - концентрации ингредиента смеси,
установленные титриме- трически, %; F и F1 - соответствующие им факторы; F2 -
фактор показателя преломления компонента, определяемого рефрактометрически.
Для определения небольших (0,1-2%) концентраций
препаратов в лекарственных формах используют интерферометричес- кий метод,
отличающийся от титриметрических небольшим расходом испытуемого объекта.
Интерферометрия основана на измерении показателей преломления растворов. Но в
отличие от рефрактометрии измеряется разность показателей преломления п
испытуемого вещества и эталона с известной величиной n0.
Расчет концентраций X в интерферометрическом анализе
выполняют по калибровочным графикам по формуле
= (n - p) / K,
где п - показатель интерферометра; р - поправка;
К - удельный коэффициент смещения интерференционной картины.
С помощью интерферометрии анализируют
растворимые в воде сухие двухкомпонентные лекарственные формы. Для этого
готовят раствор лекарственной формы, а также растворы каждого из двух
компонентов, входящих в ее состав в равных концентрациях. Затем измеряют
величины смещения интерференционной картины раствора смеси относительно раствора
с меньшим значением показателя преломления (Δn1). После
этого делают аналогичные измерения раствора второго компонента относительно
раствора смеси (Δn2). Концентрацию
первого (X1) и второго (X2) компонентов вычисляют по формулам
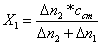
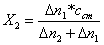
Интерферометрию применяют для количественного
экспресс-анализа неорганических веществ (натрия хлорид, цинка сульфат, борная
кислота), гидрохлоридов алкалоидов (пилокарпина, эфедрина, папаверина) и
органических оснований (новокаина, дикаина, димедрола, дибазола) и др.
Для определения концентрации веществ, обладающих
флуоресценцией, используют количественное флуориметрическое определение. Оно
включает такие операции, как подготовка анализируемой пробы и стандартного
раствора; измерение интенсивности флуоресценции стандартного раствора,
анализируемого раствора и контрольной пробы (если она имеет флуоресценцию).
Флуориметрию применяют для количественного
экспресс-анализа лекарственных форм, включающих витамины, алкалоиды,
антибиотики и др. Содержание лекарственного вещества в одном порошке или
таблетке (X) можно вычислить по формуле

где с0 - концентрация раствора стандартного
образца, г/мл или %; A - интенсивность флуоресценции анализируемого раствора;
A1 - интенсивность флуоресценции контрольной пробы; А2 - интенсивность
флуоресценции стандартного раствора; а - навеска, г; b - средняя масса порошка
(таблетки), г.
Для количественного экспресс-анализа могут быть
использованы фотометрические методы.
Фотоколориметрию или визуальную колориметрию
применяют для количественного определения аптечных концентрированных растворов
(концентратов). Фотоколориметрическое определение раствора фурацилина 0,02%
основано на цветной реакции с раствором гидроксида натрия, а раствора
этакридина лактата 0,1% - на образовании окрашенного диазосоединения.
Особенно перспективен дифференциальный
фотометричеcкий метод с использованием заменителей растворов сравнения. При
разработке методик дифференциального анализа вместо раствора сравнения,
содержащего стандартный образец, можно использовать нейтральные светофильтры,
матовые стекла, целлофановые пленки, имеющие стабильную оптическую плотность.
Предварительно по калибровочному графику, построенному с помощью стандартного
образца, необходимо установить, какой концентрации анализируемого
лекарственного вещества соответствует по своей оптической плотности заменитель
раствора сравнения. В последующем, пользуясь этим заменителем как стандартным
образцом, можно экспресс-методом на том же приборе определять концентрацию
данного лекарственного вещества в различных лекарственных формах.
4. Методики экспресс-анализа
.1 Анализ неорганических лекарственных веществ
При анализе таких веществ в большинстве случаев
приходится иметь дело с водными растворами электролитов, поэтому анализ
сводится к определению не растворенного вещества в целом, а отдельных ионов
(катионов и анионов).
. Аммония хлорид
,01 г + 1 к Н2О + 1 к р-ва Несслера →
красно-бурый осадок
,01 г + 1 к разв. НNО3 + 1 к р-ра AgNО3 →
белый осадок;
. Висмута нитрат
,01 г + 1 к разв. НС1 + 1 к р-ра КІ →
черный осадок + 5 к р-ра КІ → оранжевая окраска;
,01 г + 2 к р-ра дифениламина в конц. Н2SO4 →
синее окрашивание.
. Калия ацетат
,05 г + 10 к Н2О + 1 к р-ра FеС13 →
красно-бурая окраска;
. Калия бромид
,05 г + 1 к Н2O + 1к р-ра Na3[Со(NO2)6) →
золотисто-желтый осадок
,05 г + 0,01 г СuSO4 + 5 к конц. H2SO4 →
чёрная окраска, исчезающая от прибавления 5 к Н2O
,001 г + 0,5 мл разв. HCl + 0,5 мл р-ра КМnО4 +
0,5 мл хлороформа, взбалтывают → бурая окраска хлороформного слоя
. Калия йодид
,01 г + 3 к Н2O + 2 к р-ра (СНзСОО)2РЬ →
желтый осадок:
,005 г + 10 к Н2О + 0,5 мл разв. HCl + 0,5 мл
р-ра NaNO2 + 0,5 мл хлороформа, взбалтывают → фиолетовая окраска
хлороформного слоя
,05 г + 1 к Н2O + 1к р-ра Na3[Со(NO2)6) →
золотисто-желтый осадок
. Кальция глицерофосфат
,01 г + 3 к р-ва Несслера, нагревают →
черный осадок;
,01 г + 2 к разв. СН3СООН + 2 к р-ра (СН3СОО)2РЬ
→ белый осадок
,01 г + 2 к разв. СН3СООН + 2 к р-ра (NH4)2C2O4 →
белый осадок
. Кальция хлорид
,01 г + 2 к разв. СН3СООН + 2 к р-ра (NH4)2C2O4 →
белый осадок
,01 г + 1 к разв. НNО3 + 1 к р-ра AgNО3 →
белый осадок;
. Квасцы алюминиево-калиевые
,05 г + 20 к Н2O + 5 к р-ра ВаС12 → белый
осадок:
,05 г + 20 к H2O + 5 к NaOH → студенистый
осадок, растворимый в избытке NaOH, от прибавления р-ра NH4C1 → белый
осадок
. Кислота борная
,01 г + 20 к С2Н5ОН, зажигают → пламя с
зеленой каймой
. Кислота хлористоводородная
к препарата 1 к 5 %-ного р-ра NaHCO3 →
пузырьки СО2
,01 г + 1 к разв. НNО3 + 1 к р-ра AgNО3 →
белый осадок;
. Магния окись
,005 г + 20 к разв. HCl + 5 к р-ра NH4Cl + 5 к
р-ра Na2HPO4 + 20 к р-ра NH4OH → белый осадок
. Магния сульфат
,001 г + 10 к Н2O + 1 к р-ра ВаС12 → белый
осадок (сульфаты)
,005 г + 20 к разв. HCl + 5 к р-ра NH4Cl + 5 к
р-ра Na2HPO4 + 20 к р-ра NH4OH → белый осадок
. Меди сульфат
,005 г + 5 к Н2O + 1 к р-ра NH4OH →
голубой осадок от прибавления 3 к р-ра NН4ОН темно-синее окрашивание
,001 г + 10 к Н2O + 1 к р-ра ВаС12 → белый
осадок (сульфаты)
. Натрия бромид
крупинка препарата окрашивает бесцветное пламя
горелки в желтый цвет.
,001 г + 0,5 мл разв. HCl + 0,5 мл р-ра КМnО4 +
0,5 мл хлороформа, взбалтывают → бурая окраска хлороформного слоя
. Натрия гидрокарбонат
,01 г + 5 к разв. НС1 → пузырьки СO2
крупинка препарата окрашивает бесцветное пламя
горелки в желтый цвет.
. Натрия йодид
,05 г + 1 к Н2O + 1к р-ра Na3[Со(NO2)6) →
золотисто-желтый осадок
крупинка препарата окрашивает бесцветное пламя
горелки в желтый цвет.
. Натрия нитрит
,01 г + 3 к разв. НС1 → бурые пары;
,01 г + 0,01 г антипирина + 2 к разв. НС1 →
зеленая окраска
крупинка препарата окрашивает бесцветное пламя
горелки в желтый цвет.
. Натрия сульфат
,005 г + 1 к Н2O + 1 к разв. HCl + 1 к р-ра ВаСl
→ белый осадок
крупинка препарата окрашивает бесцветное пламя
горелки в желтый цвет.
. Натрия тиосульфат
,01 г + 10 к Н2O + 1 к разв. HCl →
помутнение, запах SО2
,01 г + 10 к Н2О + 2 к р-ра AgNО3 → белый
осадок, переходящий в желтый, бурый, черный.
. Натрия хлорид
,01 г + 1 к разв. НNО3 + 1 к р-ра AgNО3 →
белый осадок;
крупинка препарата окрашивает бесцветное пламя
горелки в желтый цвет.
. Перекись водорода
к препарата 1 к разв H2SO4 + 1 мл эфира + 1 к
р-ра K2Сг2О7 → синяя окраска эфирного слоя
. Свинца ацетат
,005 г + 5 к Н2О + 1 к р-ра KI желтый осадок
,05 г + 10 к Н2О + 1 к р-ра FеС13 →
красно-бурая окраска;
. Серебра нитрат
,01 г + 3 к Н2О + 1 к 5 %-ного р-ра амидопирина →
фиолетовая окраска
,01 г + 1 к Н2О + 1 к разв. HCl → белый
осадок;
,005 г + 2к р-ра дифениламина → синяя
окраска.
. Цинка сульфат
,005 г + 2 к Н2О + 2 к р-ра К4 [Fe(CN)6] →
белый студенистый осадок
,005 г + 1 к Н2O + 1 к разв. HCl + 1 к р-ра ВаСl
→ белый осадок
.2 Анализ органических лекарственных веществ
Поскольку большинство лекарственных средств
органического происхождения не являются электролитами, для их идентификации не
могут быть применены реакции ионного типа (кроме солей органических кислот,
которые диссоциируют в водных растворах и определяются по соответствующим
анионам и катионам). Для анализа органических лекарственных веществ используют
специфические реакции, основанные на химических свойствах, содержащихся в этих
веществах функциональных групп.
. Атропина сульфат
,005 г + 1 к Н2О + 1 к р-ра ВаС12 → белый
осадок (сульфат-ион)
. Гоматропина гидробромид
,01 г + 2 к р-ва Фреде (р-р молибдата аммония в
конц. Н2SО4 → желтая окраска,
,005 г + 0,01 г СuSO4 + 2 к конц. Н2SO4 →
черный осадок (бромиды) и фиолетовая окраска жидкости.
. Диазолин
,005 г + 2 к конц. Н2SO4 + 0,005 г NaN02 →
фиолетовая окраска.
. Дибазол
,01 г + 10 к Н2О + 1 к разв. НС1 + 1 к 0,1H р-ра
I2 → красновато-серебристый осадок:
. Димедрол
,005 г + 1 к конц. H2SO4 → желтая окраска
+ 5 к Н2О → окраска исчезает:
,005 г + 1 к Н20 + 1 к 0,1М р-ра HCl + 1 к р-ра
CuSO4 + 2 к 1 %-ного р-ра NH4CNS → коричневая окраска.
. Кодеин
,005 г + 1к р-ва Марки (р-р формальдегида в
конц. H2SO4 → сине-фиолетовое окрашивание.
,002 г + 1 к р-ва Фреде → желто-зеленая
окраска, переходящая в синюю.
. Кодеин фосфат
,005 г + 1к р-ва Марки (р-р формальдегида в
конц. H2SO4 → сине-фиолетовое окрашивание.
,002 г + 1 к р-ва Фреде → желто-зеленая
окраска, переходящая в синюю.
,01 г + 1 к Н2О + 1 к р-ва (NH4)2MoО4 + 1 к р-ра
бензидина + 1 к р-ра CH3COОNa → синяя окраска (фосфат-ион)
. Кофеин
,01 г + 1 мл Н2О, нагревают, охлаждают, после прибавления
2 к 0,1H I2 → осадка нет, после прибавления 2-3 к разв. HCl → бурый
осадок
. Кофеин-бензоат натрия
,01 г + 20 к Н2О + 1к р-ра FeCl3 →
розово-желтый осадок:
,01 г + 1 мл Н2О, нагревают, охлаждают, после
прибавления 2 к 0,1H I2 → осадка нет, после прибавления 2-3 к разв. HCl →
бурый осадок
. Папаверина гидрохлорид
Реакции окрашивания: 0,001 г + 1 к конц. HNO3,
нагревают → желтая окраска
,05 г + 10 к Н2О + 1 к р-ра FеС13 →
красно-бурая окраска;
,01 г + 1 к разв. НNО3 + 1 к р-ра AgNО3 →
белый осадок;
. Пахикарпина гидройодид
,005 г + крист. молибдата аммония + 1 конц.
H2SО4 → коричневая окраска, от прибавления 1 к Н2О → темно-синяя.
,01 г + 3 к Н2O + 2 к р-ра (СНзСОО)2РЬ →
желтый осадок:
,05 г + 1 к Н2O + 1к р-ра Na3[Со(NO2)6) →
золотисто-желтый осадок
. Пилокарпина гидрохлорид
,005 г + 1 к разв. H2SО4 + 5 к Н2О2 + 1 к р-ра
К2Сr2О7 + 1 мл хлороформа, взбалтывают → сине-фиолетовая окраска
хлороформного слоя.
,01 г + 1 к разв. НNО3 + 1 к р-ра AgNО3 →
белый осадок;
. Платифиллина гидротартрат
,005 г + 2 к конц. H2SО4 + 0,01 г β-нафтола,
нагревают → темно-зеленая флюоресценция
,05 г + 10 к H2О + 0,02 г KCl + 5 к С2Н5ОН →
белый кристаллический осадок.
. Прозерин
,01 г + 10 к конц. HNO3, кипятят + 3 мл Н2О + 1
мл р-ра ВаС12 → белый осадок
,002 г + 10 к Н2О + 1 к 0,1M р-ра KMnO4 + 2 мл
смеси хлороформа с ацетоном (2:1) → розово-фиолетовая окраска
хлороформного слоя.
. Резерпин
,0001 г + 20 к разв. CH3COOH + 10 к р-ра КIO3,
нагревают → желтая окраска.
. Скополамина гидробромид
,002 г + 1 к р-ва Фреде → желтая быстро
исчезающая окраска.
,05 г + 0,01 г СuSO4 + 5 к конц. H2SO4 →
чёрная окраска, исчезающая от прибавления 5 к Н2O
,001 г + 0,5 мл разв. HCl + 0,5 мл р-ра КМnО4 +
0,5 мл хлороформа, взбалтывают → бурая окраска хлороформного слоя
. Теобромин
,05 г + 1 мл 0,1 и. р-ра NaOH, встряхивают 2
мин, фильтруют, приливают к фильтрату 2 к р-ра CoCl2 → фиолетовая
окраска, переходящая в серо-голубой осадок.
. Теофиллин
,01 г + 5 к Н2О + 1 к р-ра Со(NO3)2 + 1 к р-ра
NH4OH → фиолетовая окраска.
. Хинина гидрохлорид
,005 г + 20 к, Н20 + 1 к разв. H2SO4 →
голубая флюоресценция.
,01 г + 1 к разв. НNО3 + 1 к р-ра AgNО3 →
белый осадок;
. Этилморфина гидрохлорид
,002 г + 1 к р-ва Марки → сине-фиолетовая
окраска.
,005 г + 5 к р-ра NaOH + крист. I2 нагревают →
запах йодоформа;
,001 г + 1 к р-ва Фреде → желтая окраска,
переходящая в зеленую:
. Эуфиллин
,01 г + 5 к Н2О + 1 к р-ра СuSO4 →
фиолетовая окраска.
,01 г 2 к ацетона + 2 к р-ра нитропрусс. натрия →
постепенно появляется фиолетовая окраска.
. Эфедрина гидрохлорид
,01 г + 4 к Н2О + 1 к р-ра СuSO4 + 5 к р-ра NaОН
+ 5 мл смеси спирта с хлороформом (1:1), встряхивают → розово-фиолетовая
окраска хлороформного слоя.
,01 г + 20 к Н2О + 0,02 г К3[Fе(СN)6], нагревают
→ запах бензальдегида
,01 г + 1 к разв. НNО3 + 1 к р-ра AgNО3 →
белый осадок;
. Кислота никотиновая
,01 г + 2 мл Н2О, нагревают + 5 к р-ра
(СН3СОО)2Сu → сине-голубой осадок.
,005 г + 1 крист. молибдата аммония + 1 к Н2О +
1 к конц. H2SO4 → темно-синяя окраска.
. Пиридоксина гидрохлорид
,01 г + 10 к Н2О → голубая флюоресценция
р-ра в УФ-свете.
,001 г + 3 к Н2О + 2 к р-ра FeCl3 →
красная окраска, при добавлении разв. H2SO4 окраска исчезает.
. Рибофлавин
,001 г + 2 к конц. H2SO4 → красная
окраска, переходящая в желтую при добавлении 5 к Н2О.
. Тиамина бромид
,005 г 10 к Н20 + 10 к р-ра K3[Fe(CN)6] + 10 к
0,1Y р-ра NaOH + 2 мл C2H5OH → синяя флюоресценция.
,005 г + 1 к р-ва Несслера → желтая
окраска, переходящая в черную.
,05 г + 0,01 г СuSO4 + 5 к конц. H2SO4 →
чёрная окраска, исчезающая от прибавления 5 к Н2O
,001 г + 0,5 мл разв. HCl + 0,5 мл р-ра КМnО4 +
0,5 мл хлороформа, взбалтывают → бурая окраска хлороформного слоя
. Анестезин
,01 г + 1 к разв. HCl на газетн. бумаге →
оранжевое пятно
,01 г + 3 к Н2О + 0,01 г I2 + 1 к р-ра NaOH,
нагревают → запах йодоформа.
. Дикаин
,005 г + 2 мл р-ра КIO3} + 10к разв. H3РО4
нагревают → фиолетовая окраска.
,005 г + 1 к конц. HNO3, нагревают →
желтая окраска.
,003 г + 2 к разв. HCl + 2 к р-ва Майера →
белый осадок.
. Кислота фолиевая
,005 г + 1 мл 0,1H р-ра NaOH + 1 к р-ра FeCl3 →
красно-оранжевая окраска.
,005 г + 1мл 0,1H р-ра NaOH + 1к р-ра CuSÖ4
→ зеленый
осадок.
. Натрия парааминосалицат
,005 г + 10 к Н2О + 1 к разв. HCl + 1 к р-ра
FeCl3 → фиолетовая окраска.
,005 г + 2мл Н2О + 3 к разв. HCl + 3 к р-ра
NaNO2 + 2 мл р-ра β-нафтола →
красная окраска.
. Новокаин
,005 г + 1 к разв. HCl на газетн. бумаге →
оранжевое пятно.
,005 г + 1 к Н20 + 1 к р-ра AgNO3 → белый
осадок.
,005 г + 1 к разв. H2SO4 + 1 к 0,1М р-ра КМnО4 →
обесцвечивание р-ра.
. Норсульфазол
,05 г + 3 мл 0,1М р-ра NaOH, взбалтывают 2 мин,
фильтруют, к 0,5 мл фильтрата приливают 2 к р-ра CuSO4 →
грязно-фиолетовый осадок.
,5 мл фильтрата + 2 к р-ра CoCl2 →
сиреневый осадок.
. Норсульфазол-натрий
,005 г + 10 к Н2О + 1к р-ра CuSO4 →
грязно-фиолетовый осадок.
,005 г + 10 к Н2О + 1 к р-ра СоС12 →
сиреневый осадок.
. Сульфацил-натрий
,01 г + 5 к Н20 + 1 к р-ра СuSО4 →
голубовато-зеленоватый осадок.
,01 г + 1 к разв. НС1 на газетн. бумаге →
оранжевое пятно.
. Стрептоцид
,01 г + 1 к разв. НС1 на газетн. бумаге →
оранжевое пятно.
. Сульфадимезин
,05 г + 1 мл 0,1М р-ра NаОН, взбалтывают 2 мин,
фильтруют, к фильтрату добавляют 5 к р-ра СuSО4 → желтовато-зеленый
осадок, переходящий в коричневый.
. Тримекаин
,001 г + 5 к р-ва Марки, нагревают 10 мин →
красная окраска, при добавлении 10 к Н2О голубая флюоресценция.
. Этазол
,05 г + 1 мл 0,1М р-ра NаОН, взбалтывают 2 мин,
фильтруют, к фильтрату добавляют 2 к р-ра СuSО4 → травянисто-зеленый
осадок.
. Фенацетин
,01 г + 1 к конц. НNO3 желтая окраска.
,01 г кипятят 3 мин с 2 мл разв. HС1 →
сине-фиолетовая окраска.
. Кислота аминокапроновая
,03 г + 2 мл Н2О + 10 к р-ра нингидрина,
нагревают → сине-фиолетовая окраска.
. Кислота глютаминовая
,01 г + 20 к Н2О + 5 к р-ра нингидрина,
нагревают → сине-фиолетовая окраска.
,005 г + 2 мл Н2О + 2 к р-ра CuSO4 → синее
окрашивание (контр. р-р.: 2 мл Н2О + 2 к р-ра CuS04 → бледно-голубая
окраска).
. Метионин
,005 г + 1 к р-ра Н2О + 2 к р-ра CH3COONa + 1 к
р-ра CuS04 → сиреневато-голубой осадок.
,002 г + 0,5 мл С2Н5ОН + 2 к р-ра Со(NО3)2 + 2 к
р-ра NH4OH → желто-бурая окраска.
. Цистеин
,005 г + 1 к Н2О + 1-2 к р-ра FeCl3 →
быстро исчезающая сине-фиолетовая окраска.
. Левомицетин
,01 г + 2 мл р-ра NаОН, нагревают → желтое
окрашивание, которое усиливается при кипячении, появляется запах NН3; после
подкисления разв. НNО3 осадок фильтруют и добавляют 5 к р-ра АgNО3 →
белый осадок. При дальнейшем кипячении со щелочью выделяется аммиак, а
ковалентный хлор переходит в ионное состояние (хлорид-ион); обнаруживается
реакцией с р-ром АgNO3.
. Фурадонин
,001 г + 1 мл ацетона 4-5 к 0,5М спирт, р-ра КОН
→ буро-зеленая окраска, быстро переходящая в черный осадок
. Фуразолидон
,001 г + 1 мл ацетона + 5 к 0,5М спирт, р-ра КОН
→ фиолетовая окраска, при стоянии буреет
. Фурацилин
,001 г + 1 мл ацетона + 5 к 0,5М спирт, р-ра КОН
→ устойчивая ярко-красная окраска.
к 0,02 %-ного р-ра + 1 к р-ра NаОН →
оранжево-красная окраска.
. Глицерин
к + 2 к СuSО4 + 1 к р-ра NаОН → синяя
окраска.
. Глюкоза
,05 г 1 мл р-ва Фелинга-I + 1 мл р-ва
Фелинга-II, нагревают → кирпичио-красный осадок.
. Маннит
,05 г + 10 к Н2О + 5 к р-ра СuSО4 + 2 к р-ра
NН4ОН → голубой осадок.
. Мезатон
,01 г + 5 к Н2О + 1 к р-ра FеС13 →
фиолетовая окраска.
,001 г + 1 к р-ва Марки → розовая окраска.
,002 г + 10 к разв. НNО3, нагревают →
желтая окраска.
. Пиридоксина гидрохлорид
,01 г + 10 к Н2О → голубая флюоресценция
р-ра в УФ-свете;
,001 г + 5 к Н2О + 2 к р-ра FеС13 →
красная окраска, при добавлении разв. Н2SO4 исчезает,
. Рибофлавин
,001 г + 2 к конц, Н2SO4 → красная
окраска, переходящая в желтую при добавлении 5 к Н2O.
. Сахар
,05 г + 2 к Н20 + 1 к р-ра Со(NO3)2 + 1 к р-ра
NаОН → фиолетовая окраска.
,01 г + 3 к разв. НС1 + 0,01 г резорцина,
нагревают → розовая окраска.
. Терпингидрат
,01 г + 2 к конц. Н2SO4 → оранжевая
окраска и запах терпенола
. Эфедрина гидрохлорид
,01 г + 4 к Н2О + 1 к р-ра СuSO4 + 5 к р-ра NaОН
+ 5 мл смеси спирта с хлороформом (1:1), встряхивают → розово-фиолетовая
окраска хлороформного слоя.
,01 г + 20 к Н2О + 0,02 г К3[Fе(СN)6], нагревают
→ запах бензальдегида
,01 г + 1 к разв. НNО3 + 1 к р-ра AgNО3 →
белый осадок;
. Гексаметилентетрамин
,02 г + 0,02 г салицил. к-ты + 2 к конц. Н2SО4,
нагревают → красная окраска
,01 г + 2 мл разв. Н2SО4, кипятят → запах
формальдегида
. Пентоксил
,01 г + 2 к конц. Н2SО4 + 0,01 г β-нафтола
+ 3 к С2Н5ОН→ зеленая флюоресценция.
,01 г + 2 к Н2О + 5 к конц. Н2SО4, + 0,01 г
салициловой к-ты → красная окраска.
. Хлоралгидрат
,01 г + 10 к р-ра NаОН + 0,01 г резорцина,
нагревают → малиново-красная окраска.
. Цитраль,
Органолептическое определение - по запаху
. Адреналина гидрохлорид или гидротартрат
к + 1 к р-ра FеС13 → изумрудно-зеленая
окраска.
к + 1 к р-ра NаNО2, нагревают → розовая
окраска.
. Кислота салициловая
,001 г + 20 к Н2О + 1 к р-ра FeCl3 → сине-фиолетовая
окраска.
. Рутин
,005 г + 5 к Н2О + 1 к p-pа FeCl3 →
зеленая окраска.
,005 г + 5 мл 1H NaOH → желто-оранжевая
окраска
. Таннин
,01 г + 5 к Н2О + 1 к р-ра FеСl3 →
черно-синяя окраска.
. Фенилсалицилат
,01 г + 5 к С2Н5ОН + 1 к р-ра FеСl3 → фиолетовая
окраска.
. АТФ
,01 г + 10 к Н2О + 20 корцинового р-ва →
сине-зеленая окраска.
,01 г + 10 к Н2О + 10 к р-ра NH4С1 + 10 к р-ра
NH4ОН + 5 к р-ра МgSО4 → белый кристаллический осадок.
. Кислота ацетилсалициловая
,005 г + 3 к р-ва Марки, нагревают → розовая
окраска.
,01 г + 1 мл Н2О, кипятят, охлаждают, приливают
р-р FеС13 → фиолетовая окраска.
. Кислота фолиевая
,005 г + 1 мл 0,1Н р-ра NaОН + 1 к р-ра FеС13 →
красно-оранжевая окраска.
,005 г + 1 мл 0,1М р-ра NаОН + 1 к р-ра СuSO4 →
зеленый осадок.
. Кальция глюконат
,005 г + 5 к горячей воды + 2 к р-ра (NH4)2С2О4 →
белый осадок.
,02 г + 20 к Н2О + 2 к р-ра FеС13 →
зеленовато-желтая окраска.
. Кальция лактат
,005 г + 5 к горячей воды + 2 к р-ра (NH4)2С2О4 →
белый осадок.
,02 г + 10 к разв. Н2SО4 + 5 к р-ра КМnО4,
нагревают → запах ацетальдегида
. Натрия цитрат
,01 г индик. смеси мурексида + 3 к Н2О + 1 к
р-ра CuSО4 + р-р 0,01 г цитрата в 5 к Н2О →фиолетовая окраска.
. Кислота аскорбиновая
,005 г + 1 к Н2О + 1 к р-ра AgNО3 → черный
осадок
,002 г + 1 к р-ра ванадата аммония в конц. H2SO4
→ голубая окраска.
. Анальгин
,01 г + 1 К разв. HCl + 1 к р-ра FeCl3 →
синяя окраска.
б)0,01 г + 2 к конц. НNО3 → синяя окраска,
переходящая в желтую.
. Антипирин
,005 г + 1 к р-ра FeCl3 → кроваво-красная
окраска
,005 г + 1 к р-ра NаNO2 + 1 к разв. Н2SО4 →
изумрудно-зеленая окраска
. Бутадион
,01 г + 1 к. конц. Н2SО4 + 0,01 г NaNO2 →
оранжевая окраска, переходящая в вишнево-красную
,05 г + 1 мл 0,1 М р-ра NаОН, взбалтывают,
фильтруют, к фильтрату добавляют 3 к р-ра FеС13 → желтый осадок
. Барбитал-натрий
,01 г 2 к р-ра Н2О + 2 к Со(NО3)2 →
фиолетовый осадок, переходящий в синий.
. Гексамидин
,02 г + 2 мл р-ра NаОН, нагревают + 5-6 к р-ра
нитропрусс. натрия + 0,5 мл ледяной СН3СООН → зеленая окраска.
. Метилурацил
,005 г + 5 к С2Н5ОН, нагревают, охлаждают,
добавляют 2 к спирт. р-ра Со(NO3)2 + 1 к р-ра NН4ОН → фиолетовая окраска.
. Пeнтоксил
,01 г + 2 к конц. Н2SО4 + 0,01 г β-нафтола
+ 3 к С2Н5ОН → зеленая флюоресценция.
,01 г. + 2 к Н2О + 5 к конц. Н2SО4, + 0,01 г
салициловой кислоты → красная окраска.
. Фенобарбитал
,01 г + 5 к С2Н5ОН + 1 к р-ра СоС12 + 1 к р-ра
NН4ОН → фиолетовая окраска
. Этаминал натрия
,01 г + 3 к С2Н5ОН + 1- 2 к р-ра СuS04 + 1 к
р-ра СuС12 → сине-фиолетовая окраска.
5. Экспериментальная часть
.1 Методика анализа
Таблетки анальгина 0,5 г
Состав на одну таблетку.
Анальгина - 0,5 г
Вспомогательных веществ - достаточное количество
Описание. Таблетки белого или слегка желтоватого
цвета, горьковатого вкуса.
Подлинность. К 0,2 г порошка растертых таблеток
приливают 5 мл спирта, подкисляют 1 мл разведенной соляной кислоты, взбалтывают
и фильтруют. Прибавляют 5 мл 0,1 н. раствора йодата калия; раствор окрашивается
в малиновый цвет, при дальнейшем добавлении реактива окраска усиливается и
выделяется бурый осадок.
,2 г порошка растертых таблеток взбалтывают с 10
мл воды, фильтруют. Нагревают с 2 мл разведенной соляной кислоты; сначала
ощущается острый запах сернистого ангидрида, а затем формальдегида.
Количественное определение. Около 0,5 г (точная
нареска) порошка растертых таблеток помещают в мерную колбу емкостью 50 мл,
прибавляют 10 мл воды и взбалтывают в течение 1 минуты, затем доводят объем
раствора спиртом до метки, тщательно перемешивают и фильтруют; 25 мл фильтрата
вносят в коническую колбу емкостью 100 мл и титруют 0,1Н раствором йода до
появления желтой окраски раствора, неисчезающей в течение 30 секунд.
мл 0,1 н. раствора йода соответствует 0,01757 г
С13Н16N3NаO4S * Н2O, которого должно быть 0,475-0,525 г, считая на средний вес
одной таблетки.
5.2 Результаты анализа
|
Название
и лекарственная форма препарата
|
Пока-затель
|
№
п/п
|
Методика
исследования
|
Результат
|
|
Таблетки
анальгина 0,5 г
|
Подлин-ность
|
1
|
К
0,2 г порошка растертых таблеток приливают 5 мл спирта, подкисляют 1 мл
разведенной соляной кислоты, взбалтывают и фильтруют. Прибавляют 5 мл 0,1 н.
раствора йодата калия; раствор окрашивается в малиновый цвет, при дальнейшем
добавлении реактива окраска усиливается и выделяется бурый осадок.
|
Соответ-ствует
|
|
Таблетки
анальгина 0,5 г
|
Подлин-ность
|
2
|
0,2
г порошка растертых таблеток взбалтывают с 10 мл воды, фильтруют. Нагревают с
2 мл разведенной соляной кислоты; сначала ощущается острый запах сернистого
ангидрида, а затем формальдегида.
|
Соответ-ствует
|
|
Таблетки
анальгина 0,5 г
|
Количес-твенное
опреде-ление
|
3
|
Около
0,5 г (точная нареска) порошка растертых таблеток помещают в мерную колбу
емкостью 50 мл, прибавляют 10 мл воды и взбалтывают в течение 1 минуты, затем
доводят объем раствора спиртом до метки, тщательно перемешивают и фильтруют;
25 мл фильтрата вносят в коническую колбу емкостью 100 мл и титруют 0,1Н
раствором йода до появления желтой окраски раствора, неисчезающей в течение
30 секунд. 1 мл 0,1 н. раствора йода соответствует 0,01757 г С13Н16N3NаO4S *
Н2O, которого должно быть 0,475-0,525 г, считая на средний вес одной таблетки
|
Соответ-ствует
|
Выводы
Одна из наиболее важных задач фармацевтической
химии - это разработка и совершенствование методов оценки качества
лекарственных средств.
Для установления чистоты лекарственных веществ
используют различные физические, физико-химические, химические методы анализа
или их сочетание. ГФ предлагает следующие методы контроля качества ЛC.
Физические и физико-химические методы. К ним
относятся:
определение температур плавления и
затвердевания, а также температурных пределов перегонки;
определение плотности,
определение показателей преломления
(рефрактометрия),
определение оптического вращения (поляриметрия);
спектрофотометрия (ультрафиолетовая,
инфракрасная);
фотоколориметрия,
эмиссионная и атомно-абсорбционная
спектрометрия,
флуориметрия,
спектроскопия ядерного магнитного резонанса,
масс-спектрометрия;
хроматография (адсорбционная, распределительная,
ионообменная, газовая, высокоэффективная жидкостная);
электрофорез (фронтальный, зональный,
капиллярный);
электрометрические методы (потенциометрическое
определение рН, потенциометрическое титрование, амперометрическое титрование,
вольтамперометрия).
Кроме того, возможно применение методов,
альтернативных фармакопейным, которые иногда имеют более совершенные
аналитические характеристики (скорость, точность анализа, автоматизация). В
некоторых случаях фармацевтическое предприятие приобретает прибор, в основе
использования которого лежит метод, еще не включенный в Фармакопею (например,
метод рамановской спектроскопии - оптический дихроизм). Иногда целесообразно
при определении подлинности или испытании на чистоту заменить
хроматографическую методику на спектрофотометрическую. Фармакопейный метод
определения примесей тяжелых металлов осаждением их в виде сульфидов или
тиоацетамидов обладает рядом недостатков. Для определения примесей тяжелых
металлов многие производители внедряют такие физико-химические методы анализа,
как атомно-абсорбционная спектрометрия и атомно-эмиссионная спектрометрия с
индуктивно связанной плазмой.
Важной физической константой, характеризующей
подлинность и степень чистоты ЛC, является температура плавления. Чистое
вещество имеет четкую температуру плавления, которая изменяется в присутствии
примесей. Для веществ, которые плавятся с разложением, обычно указывается
температура, при которой вещество разлагается и происходит резкое изменение его
вида.
В некоторых частных статьях ГФ X рекомендуется
определять температуру затвердевания или температуру кипения (по ГФ XI -
«температурные пределы перегонки») для ряда жидких ЛC. Температура кипения
должна укладываться в интервал, приведенный в частной статье. Более широкий
интервал свидетельствует о присутствии примесей.
Во многих частных статьях ГФ X приведены
допустимые значения плотности, реже вязкости, подтверждающие подлинность и
доброкачественность ЛC.
Практически все частные статьи ГФ X нормируют
такой показатель качества ЛC, как растворимость в различных растворителях.
Присутствие примесей в ЛB может повлиять на его растворимость, снижая или повышая
ее в зависимости от природы примеси.
Критериями чистоты являются также цвет ЛB и/или
прозрачность жидких лекарственных форм.
Определенным критерием чистоты JIC могут служить
такие физические константы, как показатель преломления луча света в растворе
испытуемого вещества (рефрактометрия) и удельное вращение, обусловленное
способностью ряда веществ или их растворов вращать плоскость поляризации при
прохождении через них плоскополяризованного света (поляриметрия). Методы
определения этих констант относятся к оптическим методам анализа и применяются
также для установления подлинности и количественного анализа ЛС и их
лекарственных форм.
Важным критерием доброкачественности целого ряда
ЛС является содержание в них воды. Изменение этого показателя (особенно при
хранении) может изменить концентрацию действующего вещества, а, следовательно,
и фармакологическую активность и сделать ЛС не пригодным к применению.
Химические методы. К ним относятся: качественные
реакции на подлинность, растворимость, определение летучих веществ и воды,
определение содержания азота в органических соединениях, титриметрические
методы (кислотно-основное титрование, титрование в неводных растворителях,
комплексонометрия), нитритометрия, кислотное число, число омыления, эфирное
число, йодное число и др.
Биологические методы. Биологические методы
контроля качества ЛС весьма разнообразны. Среди них испытания на токсичность,
стерильность, микробиологическую чистоту.
Экспресс-метод химического внутриаптечного
контроля предусматривает быстрое выполнение анализа лекарств и минимальный
расход исследуемых объектов и реактивов.
Для ускоренного определения подлинности веществ
в лекарствах обычно используют капельные реакции, которые выполняются в
пробирках, на предметных или часовых стеклах, в фарфоровых чашках, на
фильтровальной бумаге, пропитанной соответствующими реактивами. Для проведения
реакций используют 1 - 5 капель жидких ЛФ, 0,01 - 0,03 г порошков, 0,05 - 0,1 г
мазей и суппозиториев.
Количественный экспресс-анализ в условиях аптеки
предусматривает определение содержания ингредиентов в JIC с применением методов
объемного титрования и рефрактометрии. При титровании используют такое
количество ЛВ, чтобы уходило 2 - 3 мл титранта. Жидкие Л С отбирают пипетками
на 1, 2 или 5 мл. Массу порошков - 0,05, 0,1 или 0,2 г - определяют на ручных
аптечных весах. Точность определения массы составляет 0,01 г. Массу мазей или
суппозиториев, помещенных на заранее взвешенную пергаментную бумагу, определяют
на аптечных весах и вместе с бумагой помещают в пробирку, колбу или склянку для
анализа. Добавляют воду или органический растворитель, индикатор и титруют из
микробюретки с ценой деления 0,02 мл.
Список использованной литературы
1.
Фармацевтична хімія. Підручник для студентів вищ. фармац. начальних закладів і
фарм. фак. вищих мед. навчальних закладів III-IV рівня акредитації / За заг.
ред. П.О. Безуглого. - Вінниця: Нова книга, 2008. - 560 с.
.
Арзамасцев А.П. Фармакопейный анализ - М.: Медицина, 1971.
.
Беликов В.Г. Фармацевтическая химия. В 2 частях. Часть 1. Общая
фармацевтическая химия: Учеб. для фармац. ин-тов и фак. мед. ин-тов. - М.:
Высш. шк., 1993. - 432 с.
.
Глущенко Н.Н. Фармацевтическая химия: Учебник для студ. сред. проф. учеб.
заведений / Н.Н. Глущенко, Т.В. Плетенева, В.А. Попков; Под ред. Т.В.
Плетеневой. - М.: Издательский центр «Академия», 2004. - 384 с.
.
Драго Р. Физические методы в химии - М.: Мир, 1981
.
Кольтгоф И.М., Стенгер В.А. Объемный анализ В 2 томах - М.: Государственное
научно-техническое издательство химической литературы, 1950
.
Коренман И.М. Фотометрический анализ - М.: Химия, 1970
.
Коростелев П.П., Фотометрический и комплексометрический анализ в металлургии -
М.: Металлургия, 1984, 272 с.
.
Логинова Н. В., Полозов Г. И. Введение в фармацевтическую химию: Учеб. пособие
- Мн.: БГУ, 2003.-250 с.
.
Мелентьева Г.А., Антонова Л.А. Фармацевтическая химия. - М.: Медицина, 1985.-
480 с.
.
Мискнджьян С.П. Кравченюк Л.П. Полярография лекарственных препаратов. - К.:
Вища школа, 1976. 232 с
.
Фармацевтическая химия: Учеб. пособие / Под ред. Л.П. Арзамасцева. - М.:
ГЭОТАР-МЕД, 2004. - 640 с.
.
Фармацевтический анализ лекарственных средств / Под общей редакцией В.А.
Шаповаловой - Харьков: ИМП «Рубикон», 1995
.
Фармацевтичний аналіз: Навч. посіб. для студ. вищ. фармац. навч. закл. III-IV
рівнів акредитації/ П.О. Безуглий, В.О. Грудько, С.Г. Леонова та ін.; За ред.
П.О. Безуглого,- X.: Вид-во НФАУ; Золоті сторінки, 2001.- 240 с.
.
Халецкий A.M. Фармацевтическая химия - Ленинград: Медицина, 1966
.
Эшворт М.Р. Титриметрические методы анализа органических соединений кн.1,2 -
М.: Химия, 1972.