|
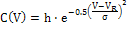
|
(1)
|
Наблюдаемый хроматографический пик, соответствующий хроматографической
зоне, изображен на рис. 1.2. Если измерять концентрацию веществ в растворе на
выходе колонки, то мы получим кривую, которая называется хроматограммой.
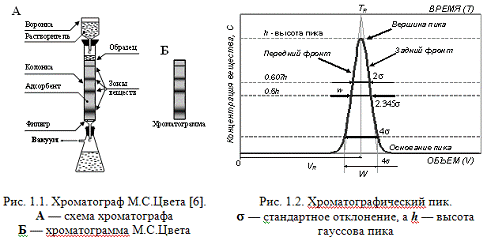
Таким образом, хроматограмма является функцией зависимости концентрации
вещества в растворе от объема, пропущенного через колонку растворителя или от
времени. Каждому веществу на хроматограмме соответствует свой пик. Начало
координат соответствует моменту ввода пробы (образца) в колонку. Абсцисса
вершины пика называется объемом удерживания вещества (VR), величина которого определяется
химическим строением этого вещества, составом подвижной фазы, свойствами
адсорбента (неподвижной фазы) и температурой. Когда говорят о времени
удерживания (TR), то имеют в виду, что 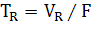 , где F - скорость потока растворителя через колонку. Мерой количества вещества, введенного в
колонку, является площадь хроматографического пика, равная
, где F - скорость потока растворителя через колонку. Мерой количества вещества, введенного в
колонку, является площадь хроматографического пика, равная
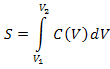
где V1 и V2 - "начало" и
"конец" хроматографического пика.
Концентрация вещества в подвижной фазе, вытекающей из колонки (элюат),
измеряется с помощью детектора, представляющего собой специальное устройство с
проточной измерительной ячейкой, выходной сигнал которого пропорционален
концентрации вещества в растворе. Устройство детектора может быть основано на
многих физико-химических принципах, но мы рассмотрим только фотометрический
детектор, который применяется в хроматографе "Милихром А-02" (ЗАО
"ЭкоНова", г. Новосибирск), который использовался в данной работе для
разделения смесей веществ.
Принцип работы фотометрического детектора показан на рис. 1.3:
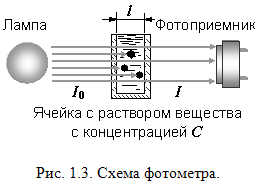
Пучок света от источника интенсивностью Io проходит через ячейку с раствором
вещества с концентрацией C и
попадает на фотоприемник. Так как часть света поглощается веществом,
интенсивность света на выходе I <
I0. Этот процесс описывается уравнением
Бугера-Ламберта-Бера:
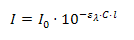
где
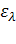 - коэффициент (коэффициент экстинкции),
характеризующий поглощение света с длиной волны λ раствором вещества с концентрацией С = 1 в кювете с
длиной оптического пути l = 1. В логарифмической форме это уравнение выглядит как:
- коэффициент (коэффициент экстинкции),
характеризующий поглощение света с длиной волны λ раствором вещества с концентрацией С = 1 в кювете с
длиной оптического пути l = 1. В логарифмической форме это уравнение выглядит как:
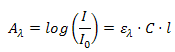
Важно,
что в уравнении (4) поглощение (оптическая плотность) раствора вещества при
длине волны  прямо пропорционально концентрации вещества. Величину
прямо пропорционально концентрации вещества. Величину
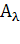 принято измерять в единицах оптической плотности
(е.о.п.).
принято измерять в единицах оптической плотности
(е.о.п.).
Длина
волны l фотометрического детектора выбирается, как правило
такой, чтобы обеспечить необходимую чувствительность анализа и определяется из
спектра поглощения раствора вещества. Спектральный диапазон детектора
хроматографа "Милихром А-02" равен 190÷360 нм (УФ область электромагнитного спектра), что
позволяет регистрировать концентрацию подавляющего количества веществ. Типичный
УФ-спектр показан на рис. 1.4:
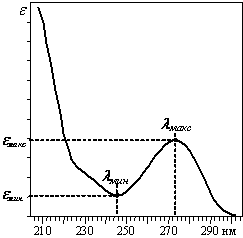
Рис.
1.4. УФ-спектр водного раствора кофеина.
Регистрация
оптической плотности при одной длине волны является простейшим способом
детектирования, при котором можно лишь определить количество вещества по площади
пика, но нельзя идентифицировать это вещество по его спектральным
характеристикам. Детектор хроматографа "Милихром А-02" может работать
в многоволновом режиме, быстро перестраивая монохроматор по циклической
программе (до восьми длин волн в цикле) так, что концентрация вещества в
элюате, протекающем через фотометрическую ячейку, успевает изменится лишь
незначительно. Хроматографический пик при многоволновом детектировании выглядит
так, как показано на рис. 1.5.
Кроме
того следует отметить, что существуют т.н. диодно-матричные детекторы [5]: в
них несколько фотоприемников объединены в матрицу (стоящую после рассеивающей
призмы) таким образом, что каждый из них фиксирует излучение на определенной
длине волны. Это создает большие трудности в процессе анализа, т.к. при малой
интенсивности излучения каждый приемник получает недостаточно света, что сильно
повышает уровень шумов. Если же использовать более мощный источник света, то
исследуемый образец может начать разлагаться, и на хроматограмме будет показан
спектр не самой пробы, а продуктов ее фотолиза. Кроме того, сильное излучение
вызывает сильный нагрев матрицы и, как следствие, ее расширение, в результате
чего фотоприемники смещаются и начинают фиксировать часть света на соседних
длинах волн. Чтобы избежать этого, матрицы приходится жестко термостатировать
(вплоть до −270°C).
Информационная
ценность многоволнового детектирования заключается в том, что кроме объема
удерживания для идентификации вещества можно использовать нормированные
величины R = Аln / Аlm (спектральные
отношения), измеренные в один момент времени, когда концентрация вещества в
растворе неизменная.
Исходя
из спектра вещества, легко показать, что
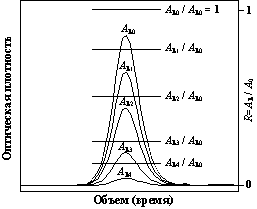
Рис.
1.5. Хроматографический пик, зарегистрированный при 5 длинах волн.
Таким
образом, набор спектральных отношений отражает "спектральный портрет"
вещества и может быть использован для идентификации вещества, а постоянство
значений R вдоль хроматографического пика является критерием его
"чистоты".
Если
мы начали с того, что определили хроматографию как метод разделения веществ, то
в современной терминологии этот метод включает в себя и измерение концентрации
веществ в образце, и их идентификацию по различным параметрам. Другими словами,
хроматография отвечает на вопросы:
1. Сколько различных веществ в образце?
. Есть ли в образце вещества x1, ..., xn из известного нам списка?
. Какова концентрация веществ x1, ..., xn в исходном образце?
Для того, чтобы получить ответы на вопросы 2 и 3, образцы стандартных
(эталонных) растворов веществ x1, ..., xn, хроматографируют в стандартных
условиях, регистрируя их объемы удерживания, площади пиков и спектральные
отношения, причем для каждого измеряемого параметра определяется погрешность
измерения [7]. Вся эта процедура называется калибровкой хроматографа. Если
калибровочные параметры стабильны во времени, то их сохраняют в виде баз
данных. Анализ исследуемой пробы проводят строго в тех же условиях. Полученные
данные обрабатывают, учитывая дрейф сигнала детектора, уровень его шумов,
неполное разделение пиков и т.п., а затем сравнивают экспериментальные данные
со стандартными, взятыми из базы данных. Разработка экспертной системы для
проведения сравнительного анализа образцов сложного состава, выполняемого с
помощью хроматографа "Милихром А-02", и будет являться нашей задачей.
Объектами хроматографического анализа могут быть экстракты различных растений,
экстракты крови и других биологических жидкостей, растворы комплексных
препаратов. Причем, одинаково ценными в таких анализах являются ответы
"эти образцы одинаковые" или "эти образцы разные" даже
тогда, когда вещества, входящие в состав образца нам неизвестны. Примером
такого сравнительного анализа, когда хроматограмма является "отпечатком
пальца" (англ. "chromatographic fingerprint")
образца, могут служить хроматограммы образцов зеленого чая, показанные на рис.
1.6.
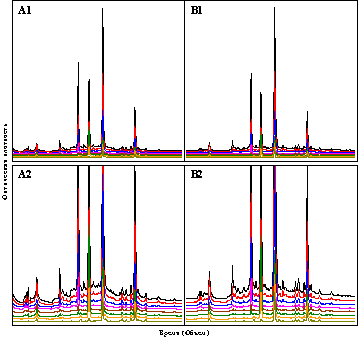
Рис.
1.6. Хроматограммы образцов А и В китайского зеленого чая при грубой (1) и
повышенной (2) чувствительности восьмиволнового детектирования.
·
1. ОСНОВНЫЕ
ПОДХОДЫ К ПРОБЛЕМЕ
Для решения задачи сравнения двух хроматограмм в мире применяются два
основных метода: изучение корреляции двух массивов данных и кластеризация этих
массивов и последующее сравнение отдельных пиков. Рассмотрим эти подходы
подробнее. В первом случае на основе записанных
данных вычисляется коэффициент корреляции двух хроматограмм - чаще всего применяются
методы Пирсона и Стьюдента. При значении коэффициента корреляции >0,99
хроматограммы считаются подобными. Коэффициент от 0,99 до 0,90 указывает на
некоторое сходство, но результат следует интерпретировать с осторожностью.
Значения ниже 0,9 подразумевают, что образцы различны. Данный метод достаточно
прост в реализации и имеет достаточно высокую эффективность, однако позволяет
ответить лишь на вопрос "идентичны представленные образцы или нет".
Кроме того, если возникнет необходимость сохранения хроматограммы
представленного образца в базу данных, потребуется запись всех имеющихся точек,
т.е. около 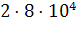 чисел с плавающей запятой. Второй метод подразумевает разбиение каждой хроматограммы на отдельные
пики для получения т.н. "хроматографических отпечатков" ("chromatographic fingerprints"). Этот процесс намного более
трудоемкий, однако имеющий два основных преимущества. Во-первых, при обработке
учитывается намного больше факторов: дрейф пиков и базовой линии, пересечение
двух и более пиков, и т.д. Во-вторых, метод позволяет описать каждый пик
набором из десятка чисел, что, с одной стороны значительно сокращает объем
данных, а с другой стороны дает возможность ответить на вопросы "идентичны
ли представленные образцы, и если нет - то чем они отличаются",
"содержится ли в представленном образце данное вещество", а при
наличии базы чистых веществ и на вопрос "из каких веществ состоит
представленный образец". Именно этот метод мы будем использовать в дальнейшем.
чисел с плавающей запятой. Второй метод подразумевает разбиение каждой хроматограммы на отдельные
пики для получения т.н. "хроматографических отпечатков" ("chromatographic fingerprints"). Этот процесс намного более
трудоемкий, однако имеющий два основных преимущества. Во-первых, при обработке
учитывается намного больше факторов: дрейф пиков и базовой линии, пересечение
двух и более пиков, и т.д. Во-вторых, метод позволяет описать каждый пик
набором из десятка чисел, что, с одной стороны значительно сокращает объем
данных, а с другой стороны дает возможность ответить на вопросы "идентичны
ли представленные образцы, и если нет - то чем они отличаются",
"содержится ли в представленном образце данное вещество", а при
наличии базы чистых веществ и на вопрос "из каких веществ состоит
представленный образец". Именно этот метод мы будем использовать в дальнейшем.
2. ФИЛЬТРАЦИЯ
ШУМОВ
Достоверность любого результата будет приближаться к нулю, если уровень
спектральных шумов будет иметь тот же порядок, что и максимальное поглощение на
тех же длинах волн. В литературе [3] описаны эксперименты, позволяющие с
уверенностью сказать, что относительный уровень шумов менее 2% не оказывает
существенного влияния на результат исследования.
Фильтрация может понизить уровень шумов спектра за счет их сглаживания.
Влияние того или иного фильтра на результат зависит от его вида. Например, при
использовании метода изучения коэффициента корреляции семиразрядный фильтр
Савицкого-Голея дает небольшое улучшение [3][13]. Его влияние возрастает при
расширении полосы фильтрации. Однако если брать фильтр со слишком широкой
полосой, то может потеряться тонкая структура.
Вообще, шум - т.е. нежелательный сигнал детектора - может быть по своей
природе как электронным, так и химическим [1]. Шум базовой линии, который мы
видим - это то, что остается после фильтрации и сглаживания, убирающих высокие
частоты. Разумеется, полностью избавиться от шума нельзя: если его частота
близка к частотам известных хроматографических пиков, то фильтры могут удалить
эти пики вместе с шумом.
Шум базовой линии размывает основание пика и затрудняет обнаружение
начала и конца и вычисление площади пика. Шум в районе вершины пика вызывает
ошибки при разделении методом долин, создавая "микропики".
Шум создает определенные ограничения на минимальное количество вещества,
которое может быть распознано как отдельный пик. Слишком маленькие пики будут
скрыты шумом базовой линии, и извлечь их оттуда будет невозможно. Минимальный
полностью распознанный пик позволяет ввести понятие отношения сигнал/шум,
которое демонстрирует связь между высотой пика и окружающим этот пик шумом.
Руководства ACS (American Cancer Society) 1980-го года определяют два предела:
предел детектирования (сигнал/шум = 3), описывающий наименьший пик, который
можно выделить из шума, и предел количественного определения (сигнал/шум = 10),
описывающий наименьший пик, параметры которого могут быть измерены с
достаточной точностью однако надо понимать, что понятие "достаточная
точность" довольно расплывчатое, и для каждого конкретного анализа может
потребоваться дополнительная корректировка пределов.
Кроме того, существует такое понятие, как "дрейф базовой линии"
[1] - изменение положения базовой линии с течением времени, вызванное
изменением температуры или другими причинами нестабильности работы
хроматографа. Если этот дрейф будет не постоянным в течение серии анализов, то
он может вызвать серьезные ошибки при измерении параметров пиков, таких как
высота, площадь, и коэффициент асимметричности.
3. РАЗБИЕНИЕ
НА ПИКИ
Одной из наиболее важных процедур является разбиение хроматограммы на
пики. Процедура поиска параметров пиков называется интегрированием.
Интегрирование включает в себя определение особых точек пиков (начало, конец,
вершина, долина), построение базовой линии, вычисление таких характеристик
пиков, как время удерживания (т.е. время выхода вершины пика), высота и
площадь. Обычно величиной, характеризующей содержание компонента в
анализируемой смеси, является именно площадь пика. Для ее вычисления необходимо
знать начало и конец пика и положение базовой линии.
Сверху пики ограничены хроматографической кривой, а снизу - базовой линией.
Слившиеся пики (неразделенные у основания) объединяются в группу, где конец
предыдущего пика совпадает с началом следующего (эта общая точка называется
долиной). В этом случае базовая линия начинается в точке, относящейся к началу
первого пика, и заканчивается в точке, относящейся к концу последнего пика в
группе и считается прямой. На данном этапе для разграничения смежных пиков
применяются два метода: "метод долин" (базовая линия проводится по
долинам группы) и "метод перпендикуляров" (смежные пики разграничиваются
вертикальной прямой, соединяющей хроматографическую кривую с базовой линией).
Каждый из этих методов имеет свои достоинства и недостатки, которые следует
рассмотреть более подробно.
Метод долин, с одной стороны, дает достаточно неплохое приближение,
однако имеет ряд существенных недостатков. Во-первых, пик, выглядящий как плечо
на склоне более высокого пика, не будет распознан. Во-вторых, может быть
проигнорирована большая площадь, лежащая ниже новой базовой линии [2].
Метод перпендикуляров, в свою очередь, дает возможность выделить
пики-наездники (пусть и с относительно низкой точностью) и сохранить для
дальнейшего изучения всю площадь под хроматографической кривой. Однако этот
метод хорошо подходит только для примерно равных по площади пиков: так как
начало второго пика оказывается в области первого, а конец первого - в области
второго, то ошибка будет минимальной при наименьшей разности площадей
перекрывающихся областей. Если же размер пиков сильно отличается (например, как
20:1), то большой пик будет лишь немного "испорчен" маленьким, в то
время как маленький получит большой кусок площади большого. В этом случае метод
долин может дать значительно более точный результат [2].
Для многоволновых же хроматограмм существует еще один метод, основанный
на изучении спектральных отношений. Дело в том, что для чистых веществ
оптическая плотность  на длине волны
на длине волны  прямо пропорциональна плотности на
другой длине волны
прямо пропорциональна плотности на
другой длине волны 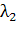 (согласно закону Бугера-Ламберта-Бера):
(согласно закону Бугера-Ламберта-Бера):
хроматограмма раствор концентрация фильтрация
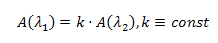
Любая примесь, элюирующаяся вместе с основным пиком, вызовет отклонение
линии относительного поглощения от горизонтальной прямой. Изучая поведение этой
линии (см. рисунок 4.1) можно с уверенностью сказать является ли пик чистым или
представляет собой смесь нескольких веществ [3].
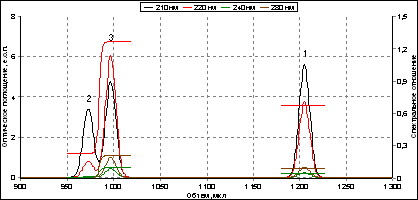
Рис. 4.1. Фрагмент смоделированной на компьютере хроматограммы раствора
3-х пептидов с детектированием при четырех длинах волн (210, 220, 240 и 280 нм)
В последнем случае, если два пика перекрывают друг друга так, что каждый
из них имеет фрагмент (начало первого и конец второго), свободные от примесей,
то, зная лишь сумму их сигналов, можно решить обратную задачу и разделить пики
точно:
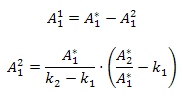
где 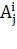 - оптическая плотность i-го пика на j-ой длине волны,
- оптическая плотность i-го пика на j-ой длине волны, 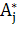 - сумма сигналов на j-ой длине волны,
- сумма сигналов на j-ой длине волны, 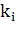 - спектральное отношение для i-го
пика (
- спектральное отношение для i-го
пика (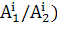 в начале первого пика и в конце
второго соответственно. Аналогично строятся выражения для трех, четырех и более
перекрывающихся пиков.
в начале первого пика и в конце
второго соответственно. Аналогично строятся выражения для трех, четырех и более
перекрывающихся пиков.
4.
МОДЕЛИРОВАНИЕ ПИКОВ
Моделирование хроматографического пика используется для построения на
экране хроматограммы по имеющемуся "отпечатку". В основе такой модели
всегда лежит функция Гаусса [1]:
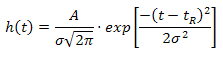
где 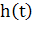 - высота пика в момент времени
- высота пика в момент времени  , - площадь пика,
, - площадь пика, 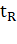 - время выхода вершины пика (время
удерживания),
- время выхода вершины пика (время
удерживания), 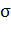 - стандартное отклонение, которое можно выразить как
- стандартное отклонение, которое можно выразить как 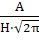 , где
, где 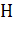 - высота пика. Эта формула позволяет
строить гауссовы пики с достаточно высокой точностью основываясь всего на трех
константах [11].
- высота пика. Эта формула позволяет
строить гауссовы пики с достаточно высокой точностью основываясь всего на трех
константах [11].
Однако не все пики можно описать одной лишь функцией Гаусса: если пик
асимметричен (т.е. 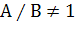 ), то потребуется модификация вышеуказанной формулы
экспоненциальной функцией. В результате асимметричный пик будет
аппроксимироваться следующей формулой:
), то потребуется модификация вышеуказанной формулы
экспоненциальной функцией. В результате асимметричный пик будет
аппроксимироваться следующей формулой:
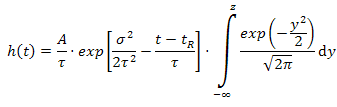
где - высота пика в момент времени , - площадь пика, - время удерживания, - стандартное отклонение,  - константа, характеризующая
асимметричность, и
- константа, характеризующая
асимметричность, и 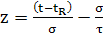 . Здесь добавляется еще одна константа, благодаря которой
теперь можно достаточно точно описать любой пик [1].
. Здесь добавляется еще одна константа, благодаря которой
теперь можно достаточно точно описать любой пик [1].
Константа может быть вычислена при помощи статистических моментов.
Если n-й момент 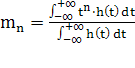 , то очевидно что
, то очевидно что 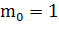 , однако намного удобнее будет
считать, что
, однако намного удобнее будет
считать, что 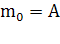 - это даст нам возможность нормировать последующие моменты.
Тогда нормированный первый момент
- это даст нам возможность нормировать последующие моменты.
Тогда нормированный первый момент 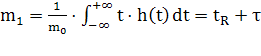 , нормированный второй момент
, нормированный второй момент 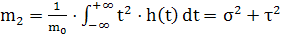 и нормированный третий момент
и нормированный третий момент 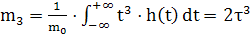 .
.
В процессе исследования для удобства изучения поведения спектральных
отношений была разработана программа "Хром-П" [16], позволяющая
моделировать подобным образом хроматограммы пептидов (последовательностей
аминокислот), все необходимые параметры которых были вычислены заранее [12].
5.
АРХИТЕКТУРА СИСТЕМЫ
Разработанная нами система имеет архитектуру, проиллюстрированную на
рис.6.1:
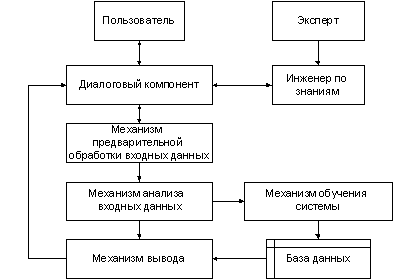
Рис.
6.1. Архитектура экспертной системы.
Далее
мы подробно рассмотрим механизмы предварительной подготовки и анализа входных
данных, а также механизмы вывода и обучения системы.
.1
Запись хроматограммы
На этом ("нулевом") этапе оператор должен выполнить запись
хроматограммы определенным образом подготовленного образца при определенных
условиях элюции (скорость потока, состав элюентов, объем пробы, настройки
термостата и т.д.). Для этого используется управляющая программа
"Альфахром 1.0". Далее полученный файл передается обрабатывающей
программе.
5.2
Предварительная
обработка и проверка условий анализа
Перед тем, как перейти к обработке и последующему анализу данных, система
выполняет проверку условий, при которых была записана хроматограмма, и
правильности хода самого процесса хроматографии - поведения графиков давления и
температуры. Если эти условия выполнены, то производится перевод содержимого
файла во внутренний формат с обязательным переходом к равномерной сетке с шагом
100 миллисекунд путем вычисления полиномов Лагранжа по 5 точкам. Далее следует
переход к следующему этапу.
.3
Алгоритм подготовки
хроматограммы
Мастер подготовки хроматограммы представляет собой 6 последовательно
появляющихся диалоговых окон, при помощи которых пользователь может пошагово
задать параметры обработки хроматограммы для последующей разметки пиков. На
каждом шаге имеется возможность предварительного просмотра результатов действий
с выбранными параметрами на экране.
Область шума
На первом шаге пользователю предлагается выделить на хроматограмме
область, не содержащую ни одного пика. Это позволит узнать порядок шумов,
который может меняться от анализа к анализу. Также эта область может быть
выделена автоматически как наиболее близкая к прямой линии.
Удаление
выбросов
Выбросы - ошибочные сигналы детектора, имеющие вид пиков шириной в 1
точку (до приведения к равномерной сетке) и высотой более 2 шумов. Такие скачки
подлежат сглаживанию. Этот шаг можно пропустить по желанию пользователя и сразу
перейти к следующему.
Фильтрация
шумов
На этом шаге пользователю предлагается выбрать способ фильтрации шума:
метод скользящего среднего, фильтр Савицкого-Голея (возможно расширение этого
списка) и ширину полосы фильтрации. Фильтры следует применять с осторожностью,
так как слишком сильное сглаживание может "сгладить" небольшие пики,
поэтому в некоторых случаях фильтры не следует применять вообще. Этот шаг можно
пропустить.
Выбор
диапазона разметки
Здесь указывается область на хроматограмме, которая подвергнется
дальнейшему анализу. Например, пользователь может исключить из рассмотрения
первые несколько минут, на которых нет никакой ценной информации, или вообще
выбрать только один интересующий его пик. По умолчанию анализируется вся
хроматограмма.
Минимальный
пик
Чаще всего пользователя интересуют только несколько достаточно крупных
пиков. Многочисленные небольшие пики, представляющие собой побочные примеси,
затрудняют дальнейший анализ и исключаются из рассмотрения. Критериями отсева
здесь служат минимальные допустимые высота и площадь пика, задаваемые на данном
шаге.
Максимальный
пик
В случае больших концентраций того или иного вещества в образце, могут
наблюдаться отклонения от закона Бугера-Ламберта-Бера, т.е. сигнал детектора
перестает быть линейной функцией от концентрации. Это делает невозможным
дальнейшее исследование спектральных отношений вещества, поэтому слишком
высокие (хотя бы на одной длине волны) пики должны быть исключены из
рассмотрения. Для хроматографа "Милихром А-02" верхняя граница
области линейности сигнала составляет 10 единиц оптической плотности. На данном
шаге пользователь может задать собственную верхнюю границу.
Маркеры
На последнем этапе подготовки можно указать один или несколько
пиков-маркеров (веществ, время выхода которых заранее известно) и
скорректировать таким образом сдвиг всех пиков по времени [10].
На этом мастер подготовки хроматограммы завершает свою работу и передает
обработанную хроматограмму мастеру анализа входных данных для выделения пиков,
вычисления их параметров и поиска в базе данных наилучшего кандидата.
.4
Алгоритм анализа и
разметки
Выделение
групп
На первом шаге выполняется разделение всей хроматограммы на отдельные
зоны (группы пиков), как это показано на рис. 6.2. Каждая такая зона
представляет собой участок между двумя соседними долинами, лежащими (с учетом
уровня шума) на базовой линии и имеющие между собой точки, высота которых
превышает минимальную высоту пика.
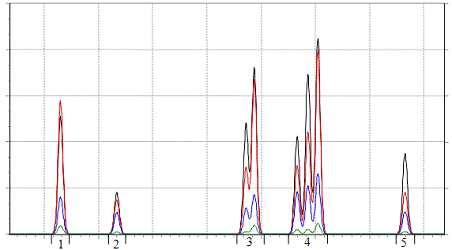
Рис. 6.2. Результат выделения групп пиков на хроматограмме,
смоделированной при помощи программы "Хром-П".
Поиск долин производится путем исследования поведения численной
производной (по всем длинам волн) вида 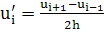 с 2-м порядком аппроксимации и
высоты соседних точек. Временная сложность этого алгоритма линейно зависит от
величины
с 2-м порядком аппроксимации и
высоты соседних точек. Временная сложность этого алгоритма линейно зависит от
величины 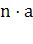 , где n - количество точек для каждой длины волны, a - количество длин волн.
, где n - количество точек для каждой длины волны, a - количество длин волн.
Наилучшие
спектральные отношения
На границах пиков (там, где оптическая плотность близка к нулевой линии),
спектральный шум многократно возрастает, т.к. спектральные отношения стремятся
к единице. Поэтому пользователю следует выбрать границы для рассмотрения
спектральных отношений. Обычно эти отношения рассматриваются на том участке,
где оптическая плотность превышает 10% высоты рассматриваемого пика, однако
возможно изменение этой планки от 2% до 80%.
Выделение
пиков
Для каждой группы, полученной на первом шаге мастера, возможны три
основных случая:
. Спектральные отношения всей группы близки к константе (в
пределах задаваемой погрешности), как это показано на рис. 6.3. Из этого
следует, что эта группа представляет собой единственный (а значит чистый) пик,
который сохраняется в промежуточный массив.
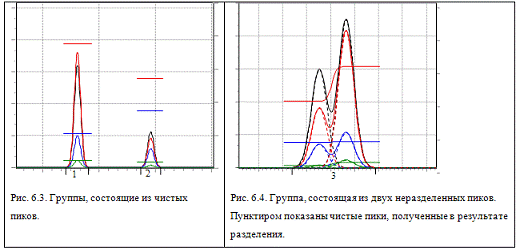
2. Спектральные отношения всей группы представляют собой S-образную кривую, как это показано на
рис. 6.4. Это означает, что эта группа состоит из двух неразделенных пиков.
Зная спектральные отношения чистых веществ (т.е. в начале первого пика и в
конце второго), группа точно разделяется на два пика (см. главу "Разбиение
на пики"), которые сохраняются в промежуточный массив.
. Спектральные отношения всей группы имеют более сложную форму.
Значит, эта группа состоит из трех или более пиков, и точное их разделение не
представляется возможным (т.к. о спектре пиков из середины неизвестно ничего).
Тогда к группе применяется метод перпендикуляров или долин (в зависимости от
высоты и положения смежных пиков), как это показано на рис. 6.5, и полученные
пики сохраняются в промежуточный массив. Метод перпендикуляров дает приемлемый
результат в том случае, когда площади разделяемых пиков соизмеримы - тогда
"загрязнения" пиков друг другом компенсируются. Если же большой пик
пересекается с маленьким (т.е. имеется "пик-наездник"), то
применяется метод долин с модифицированием базовой линии.
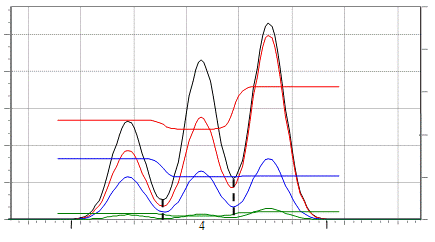
Рис. 6.5. Группа, состоящая из трех неразделенных веществ. Границы пиков,
полученные методом перпендикуляров, показаны пунктиром.
Проверка на
количество точек
Если получившийся пик описан менее чем 10 точками (до перехода к
равномерной сетке), то он исключается из рассмотрения из-за невозможности
достаточно точно предсказать его поведение между узлами изначальной сетки.
Число "10" было получено экспериментально.
Интегрирование
На этом шаге в промежуточном массиве остаются только интересующие нас
пики. Для них вычисляются площадь, время выхода, высота, коэффициент
асимметричности и 7 констант спектральных отношений.
.5
База данных
База данных (например, на основе MySQL или MS SQL) имеет три таблицы, схема которых
приведена на рис. 6.6:

Рис. 6.6. Схема таблиц в базе.
Первая (peaks) из них содержит в себе чистые пики,
т.е. имеет следующие поля:
· Время выхода вершины пика ()
· Стандартное отклонение ()
· Константа, характеризующая асимметричность ()
· 7 констант, описывающих спектральные отношения
· id пика (т.е. номер записи в таблице)
· Число экспериментов, проведенных для получения этих результатов
(необходимое для последующего уточнения полученных данных)
Вторая таблица (matters)
служит для хранения описаний сложных веществ (комбинаций пиков). Каждая запись
в такой таблице содержит id
некоторого пика, id и название
вещества, в котором он содержится, площадь A этого пика (т.е. концентрация компонента в смеси) и число
экспериментов, проведенных для получения этих результатов (аналогично таблице
пиков).
Третья таблица (groups)
служит для хранения описаний категорий сложных веществ (например, группа "черный
чай" содержит только компоненты, встречающиеся во всех сортах черного чая)
и устроена аналогично таблице matters,
за исключением параметров peak_A_min и peak_A_max, служащих для хранения минимальной и максимальной
возможной площади пика.
.6
Механизм вывода
Поиск
кандидатов
Сначала производится предварительный поиск по таблице peaks. Для каждого определяемого пика из
базы выбирается набор пиков-кандидатов, время выхода которых отличается от
времени выхода рассматриваемого пика не более чем на 10% (согласно методике №
ФР.1.31.2003.00951 [14]). Этот поиск осуществляется SQL-запросом вида
* FROM peaks WHERE peaks.tr
>= ctr*0.9 AND peaks.tr
<= ctr*1.1
где ctr - время выхода определяемого пика.
Выбор
наилучшего кандидата
Для всех найденных кандидатов пика производится сравнение их спектральных
отношений со спектральными отношениями этого пика следующим образом (согласно
методике № ФР. 1.31.2006.02966 [15]): спектральные отношения пика и кандидатов
записываются в виде двух векторов в 7-мерном пространстве. Пик-кандидат, для
которого угол между получившимися векторами минимален, признается наилучшим
кандидатом. Если этот угол не превышает 0,4°, то пик считается полностью
идентифицированным.
Определение
вещества
Далее, когда все пики идентифицированы (пусть их число равно N), производится определение вещества
по таблице matters. Для этого из нее выбираются все
записи о веществах, содержащих хотя бы один пик из обнаруженных (запросом вида SELECT * FROM matters WHERE matters.peak_id = peak_1_id OR matters.peak_id = peak_2_id OR ..., где peak_i_id - id i-го пика из определяемого набора). В результате создается
набор объектов Mi,
каждый из которых имеет два поля: Mi.Size - общее
количество компонент в веществе (т.е. SELECT COUNT(*) FROM matters WHERE matters.matter_id=M_id, где M_id - id
вещества Mi), и Mi.Matches - количество обнаруженных пиков, попавших в состав Mi. Этот разбивается на группы по Mi.Matches. Затем каждая группа сортируется по Mi.Size по возрастанию. Если в результате обнаруживается
вещество, у которого Mi.Size = Mi.Matches = N, то
процесс останавливается и найденное вещество считается искомым. В противном
случае производится дальнейший анализ для выделения веществ, наиболее похожих
на данный образец: сначала найденные вещества разбиваются на группы по
возрастанию величины |Mi.Size - Mi.Matches|, затем из каждой группы выбираются вещества с
максимальным значением Mi.Size, - они и будут считаться наиболее
похожими на образец. Пример такой выборки приведен на рис.6.7:
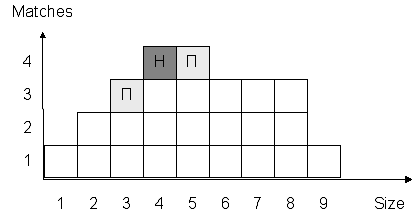
Рис. 6.7. Пример идентификации вещества, состоящего из 4 компонент.
Каждый квадрат является набором веществ Mi c соответствующими значениями Size и Matches.
Буквой "Н" выделено найденное вещество, буквами "П" -
наиболее похожие на него.
Работа с таблицей groups
производится аналогичным образом за исключением последнего шага: искомым
результатом будет считаться группа Gi с максимальным значением Gi.Size, содержащая в себе только пики из определяемого
набора. Если таких групп не обнаружено, то образец считается нераспознанным.
.7
Механизм обучения
системы
Внесение в таблицу новых веществ происходит следующим образом: эксперт
проводит несколько повторных анализов одного и того же образца и передает
получившиеся хроматограммы инженеру, который, в свою очередь, вводит их в
систему. Далее, хроматограммы разбиваются на пики, выделяются одинаковые
компоненты (механизм сравнения пиков был описан выше) и на основе этих данных
вычисляются средние времена удерживания, спектральные отношения и площади для
каждого пика (как среднее арифметическое соответствующего параметра этого пика
на всех хроматограммах). Пики, отсутствующие хотя бы на одной из хроматограмм
считаются случайными примесями и в рассмотрение не берутся. Результатом этого
процесса является набор пиков, характерный для данного образца и с временами
удерживания, спектром и площадями, максимально близкими к средним - chromatographic fingerprint [9]. Далее проверяется наличие
каждого пика из этого набора в таблице пиков. При отсутствии какого-либо пика в
таблице производится запись и присвоение id, в противном случае берется id уже внесенного в базу пика - это позволяет значительно
сэкономить объем хранимых данных и упрощает работу механизма вывода. В конце
полученный набор id и площадей
пиков вносится в таблицу веществ (под соответствующим matter_id).
Внесение данных в таблицу groups
отличается только тем, что в систему вводятся хроматограммы не одного и того же
вещества, а веществ, относящихся к определенной группе. Например, для создания
группы "черный чай" используются хроматограммы и китайского черного
чая, и цейлонского - таким образом создается fingerprint, позволяющий отличить черный чай от
зеленого, вне зависимости от его сорта. Кроме того, значение площади пика не
усредняется, а записывается как минимум и максимум возможного для последующего
определения попадания того или иного пика в этот диапазон.
Аналогично проводится уточнение данных для уже имеющихся в базе веществ:
для вычисления уточненного среднего значения того или иного параметра используются
данные из предложенной экспертом хроматограммы и данные из таблиц. Например,
время выхода какого-либо пика 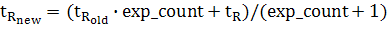 , где
, где  - новое (уточненное) время выхода,
- новое (уточненное) время выхода, 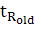 - время выхода, записанное в
таблице, - время выхода пика, при помощи
которого производится уточнение и
- время выхода, записанное в
таблице, - время выхода пика, при помощи
которого производится уточнение и 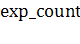 - количество экспериментов,
проведенное для получения .
- количество экспериментов,
проведенное для получения .
7.
ОЦЕНКА БЫСТРОДЕЙСТВИЯ
Было проведено несколько тестов быстродействия данной системы.
Исследования проводились на компьютере со следующими характеристиками: Intel
Core 2 Duo T5300 1,73 GHz, 2 GB RAM, Microsoft Windows 7 Professional 32bit.
Язык реализации системы - C# 3.0, СУБД -
Microsoft SQL Server 2005 Express Edition.
Результаты, полученные при тестировании, представлены в таблицах 6.1 и
6.2. Результаты поиска по базе данных в большей степени определяются скоростью
работы СУБД.
Таблица 6.1. Время, затраченное на выделение пиков на хроматограмме,
вычисление их параметров и сохранение данных в формате XML.
|
Количество компонент в
смеси
|
Средний результат из 1000
экспериментов
|
|
4
|
175,46 мсек
|
176,2 мсек
|
|
6
|
178,31 мсек
|
|
20
|
221,12 мсек
|
Таблица 6.2. Время, затраченное на определение 6 пиков и вещества по базе
данных.
|
Количество записей в базе
данных
|
Средний результат из 1000
экспериментов
|
|
1000 пиков и 100 веществ
(по 3-15 компонент)
|
150,23 мсек
|
|
10000 пиков и 1000 веществ
(по 3-15 компонент)
|
192,64 мсек
|
|
10000 пиков и 1000 веществ
(по 10-30 компонент)
|
218,67 мсек
|
ЗАКЛЮЧЕНИЕ
Следует отметить тот факт, что при составлении fingerprint не всегда известно, как называются
обнаруженные компоненты, однако мы можем с точностью сказать, является ли
предложенный образец тем, что мы предполагаем. Например, мы можем не знать, из
каких веществ состоит зеленый чай, однако всегда сможем отличить его от черного
чая или кофе.
Этот метод может найти применение во многих областях научной и
практической деятельности, в том числе в медицине при диагностике заболеваний.
К примеру, составив средние "отпечатки" образцов крови здорового
человека и человека, имеющего какое-то определенное заболевание и сравнив их,
мы можем выделить изменения в составе крови, характерные для этого заболевания.
Далее, сделав хроматограмму неизвестного образца крови, можно с высокой долей
вероятности утверждать, имеется ли данное заболевание у этого человека.
Однако простой покомпонентный анализ в связке с соответствующей базой
данных для чистых веществ может найти не меньшее количество применений в
реальной жизни, среди которых проверка состава лекарственных средств на предмет
соответствия стандартам, поиск пестицидов или иных вредных веществ в продуктах
питания, и т.д.
На текущий момент в мире существует достаточно много подобных программных
систем, реализующих те или иные алгоритмы сравнения хроматограмм и поиска
образцов в базе данных, среди которых ChemStation от компании Agilent, Empower от компании Waters, ChromQuest от компании Thermo Scientific и
десятки других. К сожалению, все они имеют ряд существенных недостатков:
· Идентификация веществ идет только по времени удерживания,
что, с учетом возможного дрейфа пиков, дает значительную погрешность в
результатах.
· В редких случаях исследуются спектральных отношения, но не
для идентификации пика, а лишь для проверки его чистоты.
· Эти программы могут анализировать лишь данные, полученные на
диодно-матричных хроматографах, тогда как многоволновые хроматограммы,
записанные на хроматографах с одним детектором, требуют предварительного
перехода к равномерной сетке.
· Разделение пиков проводится лишь при помощи методов долин и
перпендикуляров, что в некоторых случаях дает недостаточно точные результаты.
· Процесс анализа не автоматизирован, окончательное решение
должен принять оператор.
Некоторое исключение составляет программа "МультиХром" версии
1.5х-Е с дополнительным блоком "Спектр" [4], позволяющая производить
поиск по базе данных на основе спектральных отношений, однако, все же, и в этой
программе спектральные отношения для разделения пиков не применяются.
Таким образом, предложенное нами программное решение лишено большинства
известных недостатков, имеет принципиальное отличие от используемых программ, а
именно: для разделения пиков и для поиска наилучших кандидатов активно
применяются спектральные отношения, возможна обработка многоволновых
хроматограмм, полученных с хроматографов с одним детектором, процесс анализа
может быть полностью автоматизирован (т.е. не требует присутствия оператора), и
может найти практическое применение во многих областях прикладных наук.
СПИСОК
ЛИТЕРАТУРЫ
. N. Dyson, Chromatographic Integration
Methods, 2nd ed. (Royal Society of Chemistry, Letchworth, UK, 1998).
. J. Dolan, Integration Problems (LCGC North
America, Volume 27, Number 10, October 2009).
3. Л. Хубер, Применение диодно-матричного
детектирования в ВЭЖХ (Москва "Мир", 1993).
. МультиХром для Windows 9x & NT, версия 1.5x-Е.
Руководство пользователя. ("АМПЕРСЕНД" 1993-2009, "Эконова"
1997-2009).
5. Raymond P. W. Scott. Liquid Chromatography
Detectors / Library for Science, LLC, 2003.
6. Цвет М.С. О новой категории адсорбционных явлений и
о применении их к биохимическому анализу / Труды Варшавского общества естествоиспытателей,
1903, Том XIV, Отделение биологии, Протокол №6, с. 1-20.
7. Heyden Y.V. Extracting Information from
Chromatographic Herbal Fingerprints / LCGC Europe, September 2008, pp. 438-443.
8. Померанцев А.Л., Родионова О.Е. Хемометрика в
аналитической химии / Электронный ресурс, http://www.chemometrics.ru (свободный
доступ).
9. Zeng Z-D., Liang Y-Z., Xu C-J. Comparing
chemical fingerprints of herbal medicines using modified window target-testing
factor analysis / Anal. Bioanal.
Chem., 2005, Vol. 381, pp. 913-924.
10. Hansen P.W. Pre-processing method minimizing
the need for reference analyses / J.Chemom., 2001, Vol. 15, p. 123.
11. Померанцев А.Л. Методы нелинейного регрессионного
анализа для моделирования кинетики химических и физических процессов / Дис.
д-ра физ.-мат. наук, , Москва, ИХФ РАН, 2003.
. Азарова И.Н., Барам Г.И., Гольдберг Е.Л.
Предсказание объемов удерживания и УФ-спектров пептидов в обращенно-фазовой
ВЭЖХ / Биоорганическая химия, 2006, т.32, №1, с.56-63.
13. Savitzky A., Golay M.J.E. Smoothing and
differentiation of data by simplified least squares procedures / An. Chem., 1964.
. Свидетельство № 38-03 об аттестации МВИ.
Хроматографические и спектральные параметры УФ-поглощающих веществ. Методика
выполнения измерений методом высокоэффективной жидкостной хроматографии.
. Свидетельство № 67-06 об аттестации МВИ. Массовая
концентрация УФ-поглощающих веществ. Методика выполнения измерений методом
высокоэффективной жидкостной хроматографии.
. Программа "Хром-П" версии 1.1:
http://www.baram.ru/evgeny/chromp11.zip (свободный доступ).