3. Фотохромизм - явление
обратимого изменения пространственного или электронного строения молекул под
действием света, сопровождающееся изменением окраски вещества. На основе
фотохромных материалов изготовляются линзы с переменным светопропусканием,
оконные стекла, фотохромные системы на основе некоторых органических и
координационных соединений.
1. Классификация фотохимических процессов
Для характеристики
фотохимических реакций было введено понятие квантового выхода γ.
Квантовым выходом называется отношение числа прореагировавших молекул к числу
поглощенных квантов
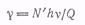
где N'
- число прореагировавших молекул; Q
- количество поглощенной све товой энергии; hv-
энергия одного светового кванта.
Для расчетов более удобно
пользоваться не числом молекул, а числом молей. Тогда квантовый выход запишется
в виде
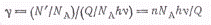
где п - число прореагировавших
молей вещества; NA-
число Авогадро.
Количество энергии, необходимое
для фотопревращения одного моля вещества при данной длине волны, равно 6,02∙1023hvи
носит название Эйнштейн. Таким образом

где 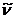 - волновое число.
- волновое число.
Числовое значение одного Эйнштейна
зависит от частоты колебаний.
Согласно закону эквивалентности
Эйнштейна квантовый выход должен быть равным единице. Однако, как показывает
опыт, все фотохимические реакции можно разделить по значениям квантового выхода
на четыре группы: 1) реакции, в которых квантовый выход γ = 1
(например, образование бромцикло-гексана, пероксида водорода, нитрозометана,
брома в результате реакции хлора с трихлорбромметаном, разложение сероводорода
в бензольном растворе и др.); 2) реакции, в которых квантовый выход γ< 1
(например, разложение аммиака, йодистого и бромистого метана, ацетона, уксусной
кислоты, образование гексабромбензола); 3) реакции, в которых квантовый выход γ > 1
(например, образование сульфурилхлорида, бромистого водорода, озона, разложение
бромистого водорода, диоксида азота, азометана, хлорноватистой кислоты и др.),
и 4) реакции, в которых квантовый выход γ>> 1 (например, реакция
взаимодействия хлора с водородом и оксидом углерода и др.).
Отклонение квантового выхода от
единицы не означает отклонения от закона эквивалентности Эйнштейна. Как
показывает опыт, фотохимический процесс слагается из первичного, протекающего в
результате поглощения светового кванта и обычно приводящего к диссоциации
молекулы с образованием свободных атомов и радикалов, и вторичных процессов,
протекающих в результате вступления в реакцию образовавшихся в первичном процессе
атомов и радикалов, или же дезактивации возникших в результате поглощения света
молекул, или рекомбинации атомов и радикалов. Первичные фотохимические
процессы, являющиеся истинно фотохимическими, всегда подчиняются закону
эквивалентности Эйнштейна. Таким образом, отклонение квантового выхода от
единицы указывает на наличие вторичных процессов, которые идут уже без участия
света.
Для реакций, протекающих в растворах
или в газах при очень малых давлениях, квантовый выход очень часто оказывается
меньше единицы.
В газах (если реакция идет с
участием возбужденных молекул) это может происходить вследствие того, что
возбужденные молекулы до столкновения с реагирующими молекулами дезактивируются
путем испускания света. Для растворов квантовый выход меньше единицы может быть
вследствие дезактивации возбужденных молекул молекулами растворителя или
вследствие рекомбинации возникших при фотодиссоциации атомов и молекул. Процесс
рекомбинации также облегчается молекулами растворителя, окружающими
рекомбинирующие частицы и образующими как бы "клетку", играющей роль
третьей частицы, которой передается избыточная энергия. Такое уничтожение
реакционноспособных частиц получило название эффекта "клетки".
Реакция рекомбинации в растворе при большой концентрации растворителя имеет
большую вероятность, чем в газе, следовательно, выход свободных атомов и
радикалов (и квантовый выход) в растворе меньше. Квантовый выход часто зависит
от температуры. Так, при изменении температуры фотолиза (разложения под
действием света)' аммиака от 20 до 500 °С квантовый выход изменяется от 0,2 до
0,5. Это объясняется следующими обстоятельствами. Первичный процесс поглощения
фотона сопровождается отщеплением одного из атомов водорода
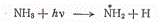
после чего могут произойти вторичные
процессы;
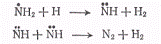
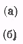
приводящие к образованию
водорода и азота. Однако существует возможность рекомбинационного процесса
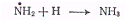 (в)
(в)
Его вероятность при комнатной
температуре в три раза больше вероятности процесса (б). Таким образом, три
четверти активных продуктов фотолиза возвращается в исходное состояние
(образуются молекулы аммиака). Этот процесс приводит к уменьшению квантового
выхода, который становится меньшим единицы. С увеличением температуры
вероятность рекомбинации несколько падает и соответственно увеличивается
квантовый выход.
В целом ряде случаев квантовый
выход оказывается больше единицы. Довольно часты случаи, когда квантовый выход
равен 2, или 3. Примером такого типа фотохимического процесса могут служить
реакции разложения в газовой фазе йодистого и бромистого водорода!
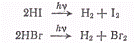
или реакция разложения
нитрозилхлорида
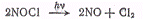
для которых квантовый выход
равен двум.
Реакции фотолиза бромистого и
йодистого водорода объяснены наиболее полно. Первичный процесс состоит в
диссоциации галогеноводорода на атомы
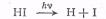
Для возникновения атомов Н и I
возможны следующие реакции (ΔН°
в кДж/моль):
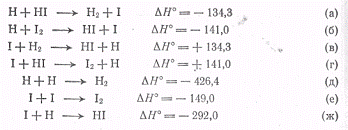
Для атомов водорода
существенной является только реакция (а), так как если I2
удаляется из сферы реакции, реакция (б), так же как и реакции (д) и (ж),
практически не имеет значения из-за малой концентрации партнеров. Для атомов I
возможна только реакция (е), так как из-за эндотермичности реакций (в) и (г) их
энергия активации очень велика, а реакция (ж) невозможна из-за малой
концентрации атомов Н, которые потребляются в основном по реакции (а). Поэтому
протекающая реакция состоит из совокупности следующих процессов:
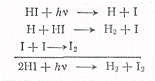
Таким образом, квантовый выход
реакции получается равным двум. Атомы I,
возникающие в элементарном фотохимическом процессе, по реакции (е)
рекомбинируют, образуя молекулярный йод. Добавление к йодистому водороду
молекулярного йода тормозит, вследствие реакции (б), процесс разложения
йодистого водорода.
Квантовый выход фотолиза HI
в газовой фазе, в конденсированном HI
и в 0,8 н. растворах приведен ниже:

Как видно, в растворе квантовый
выход меньше, хотя механизм фотолиза остается прежним.
Интересно, что с увеличением
длины волны квантовый выход растет; это объясняется уменьшением вероятности
возбуждения молекул растворителя с уменьшением энергии инициирующего распад
йодистого водорода излучения.
Для некоторых реакций квантовый
выход оказывается равным трем. Примером такой реакции может служить реакция
образования озона из кислорода (под давлением 4813 кПа), которая, по-видимому,
протекает по схеме
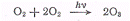
Иногда квантовый выход может
быть порядка нескольких десятков. Например, фотохимическая полимеризация
газообразного ацетилена идет с квантовым выходом γ=
9,2, а квантовые выходы реакций разложения пероксида водорода в воде лежат в
пределах от 7 до 500. Детальный механизм реакций с квантовым выходом больше
единицы во многих случаях неизвестен.
Примером реакции, квантовый
выход которой много больше единицы, является реакция соединения хлора с
водородом на свету. Квантовый выход для этой реакции γ=105,
т. е. одному поглощенному кванту соответствует около ста тысяч превратившихся
молекул хлора и водорода. Реакция хлора с водородом, инициируемая светом
протекает по схеме
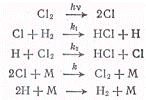
Скорость образования атомов
хлора равна 2φI
(где φ
- квантовый
выход первичного процесса, обычно лежит в пределах от 0 до 1; I
- число поглощенных квантов в единицу времени в единице объема, ее часто
называют интенсивностью поглощения света). Скорость исчезновения атомов хлора
при достаточно длинных цепях, когда реакции развития цепи можно не принимать во
внимание, будет равна k3[С1]2[М].
В стационарном состоянии
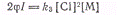 откуда
откуда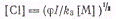
Скорость образования хлористого
водорода
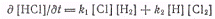
При достаточно длинных цепях
скорости возникновения и гибели атомов водорода и хлора (реакции развития цепи)
в стационарном состоянии будут одинаковыми, поэтому последнее выражение можно
записать в виде
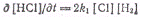
В итоге получим:
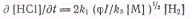
Норриш показал, что это уравнение
для скорости реакции согласуется с опытными данными, если исходная смесь газов
хорошо очищена от следов кислорода В присутствии следов кислорода происходят
реакции обрыва цепи по реакциям 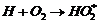 или
или 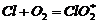 . Если учесть эти реакции обрыва,
скорость образования хлористого водорода окажется прямо пропорциональной не
. Если учесть эти реакции обрыва,
скорость образования хлористого водорода окажется прямо пропорциональной не  , а I и будет
уменьшаться с ростом, концентрации кислорода.
, а I и будет
уменьшаться с ростом, концентрации кислорода.
Таким образом, в случае цепной
реакции, инициируемой светом, если реакция обрыва будет первого порядка
относительно концентрации радикала, скорость реакции прямо пропорциональна
интенсивности поглощения света I. Обрыв цепи по реакции второго
порядка приводит к пропорциональности . Обрыв по второму порядку
происходит при больших концентрациях радикалов или больших значениях I. Обрыв по
первому порядку преобладает при малых значениях I и низких
концентрациях, когда имеет место быстрая диффузия.
Квантовый выход может зависеть от
концентрации исходных веществ. Такая зависимость наблюдается в хорошо изученной
фотохимической реакции димеризации антрацена. Если антрацен, растворенный в
бензоле, облучать светом, то происходит его ди-меризация и одновременно
наблюдается флуоресценция. С увеличением концентрации антрацена квантовый выход
димеров возрастает, а интенсивность флуоресценции падает. Уравнения
происходящих при этом процессовмогут быть записаны в виде схемы:
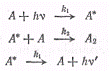
где А - молекула антрацена; А* -
молекула возбужденного антрацена; А2 - молекула димера; v - частота
излучения; v' - частота
флуоресценции.
Так как концентрация возбужденных
молекул, являющихся промежуточным продуктом реакции, мала, скорость изменения
их концентрации тоже мала и в некотором приближении может быть приравнена нулю.
Таким образом, концентрация возбужденных молекул на начальной стадии реакции
становится стационарной. В стационарном состоянии скорость образования активных
молекул должна равняться скорости их гибели. Скорость образования возбужденных
молекул равна φI. Скорость
исчезновения активных молекул, как видно из схемы реакции, равна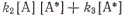 . Поэтому в
стационарном состоянии
. Поэтому в
стационарном состоянии
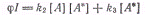 откуда :
откуда :
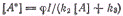
Скорость образования димера: 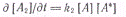
Получаем: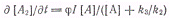
Квантовый выход димеров: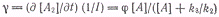
Из последнего выражения видно,
что при малых концентрациях антрацена [А] <k3/k2
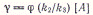
т. е. квантовый выход димеров
(в соответствии с данными опытов) растет с увеличением концентрации антрацена.
При высоких концентрациях антрацена [А] >k3/k2и
γ=φ,
т.
е. достигается предельное значение квантового выхода димеров.
К первичным фотохимическим
процессам близко стоят так называемые сенсибилизированные реакции, происходящие
не с теми молекулами, которые непосредственно поглощают лучистую энергию, а с
соседними, нечувствительными к излучению данной частоты и получающими энергию
непосредственно от поглощающих ее молекул. Примером такого процесса является
диссоциация молекулярного водорода в присутствии паров ртути, атомы которой
поглощают свет, соответствующий резонансной линии ртути с длиной волны λ
=
253,67 нм. В настоящее время известно большое число сенсибилизированных
реакций. Кроме сенсибилизации парами ртути известны реакции, в которых
сенсибилизаторами являются галогены, хлорофилл, ионы железа и др.
Как видно, во всех реакциях с
квантовым выходом, отличным от единицы, первичный фотохимический процесс
подчиняется закону эквивалентности Эйнштейна, а характер отклонения от этого
закона позволяет разобраться во вторичных нефотохимических процессах.
Для фотохимических реакций
характерна возможность смещения равновесия под действием света, так как
изменяется запас свободной энергии системы и соответственно константа равновесия.
Очевидно, что заметное изменение положения равновесия можно наблюдать только в
тех случаях, когда квантовый выход реакции близок к единице.
2. Различие кинетики фотохимических и темновых
реакций
Различия между кинетикой
фотохимических и обычных ("темновых") реакций хорошо могут быть
проиллюстрированы на примере образования бромистого водорода, Темновая реакция
Н2 + Вг2→2НВг была детально исследована в интервале
температур 200-300 °С и оказалась состоящей из следующих элементарных стадий (ΔН0в
кДж/моль)!
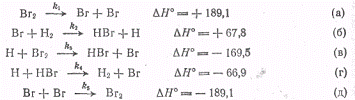
Из приведенной схемы вытекает,
что скорость образования бромистого водорода может быть выражена следующим
уравнением:
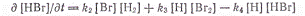
Применяя принцип стационарности
к реакциям возникновения и уничтожения атомов водорода и брома, получим
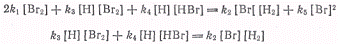
Откуда находим 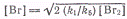 .
Подставляя
получим
.
Подставляя
получим
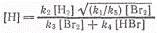 .
.
Подставляя концентрации атомов
брома и водорода получим окончательное уравнение, определяющее скорость
образования НВг
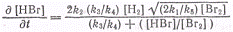
хорошо удовлетворяющее опытным
данным. Как видно, скорость реакции оказывается прямо пропорциональной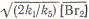 ,
т. е.концентрации атомов брома.
,
т. е.концентрации атомов брома.
Для температуры 246 °С
Боденштейн и Линд получили следующие значения констант:
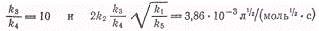
По смыслу принятой схемы k1/k5-
константа диссоциации молекулярного брома К, которая может быть вычислена с
использованием значения энергии диссоциации брома, равного 190 кДж/моль. Для
температуры 246°С эта константа оказывается равной K
= 3,26∙10-15 моль/л. Эти данные позволяют вычислить константу
скорости элементарной реакции
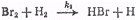
которая будет равна
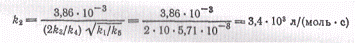
Полученное значение отношения k3/k4
показывает, что при температуре 246°С скорость взаимодействия атомов водорода с
молекулярным бромом в десять раз больше скорости взаимодействия с бромистым
водородом.
Фотохимическая реакция хорошо
идет уже при комнатной температуре при облучении смеси водорода и брома светом
с длиной волны, большей 300 нм. Скорость фотохимической реакции примерно в 300
раз превышает скорость темновой.
Фотохимическая реакция должна
отличаться схемой элементарных процессов образования атомов брома: термический
процесс (а) заменяется фотолизом молекулярного брома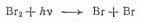 (а').
(а').
Очевидно, что при избытке
молекул брома каждый квант приводит к образованию двух атомов брома, и, таким образом,
скорость образования активных частиц зависит только от плотности излучения, т.
е. от концентрации фотонов [hv].
Замена процесса (а) процессом (а') приводит к кинетическому уравнению
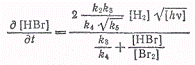
которое полностью описывает
фотохимическую реакцию. Отношение k3/k4оказалось
равным 10. Это указывает на полную однотипность вторичных процессов в обоих
случаях, разнящихся лишь по способу образования атомов брома.
Данные для фотохимической реакции
позволяют получить еще ряд сведений о кинетике элементарных вторичных
процессов. При [hv] = 9,8∙10-10
моль/(см3-с), парциальных давлениях =41,6 и
=41,6 и 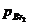 =16,5 кПа и
491 К отношение скоростей фотохимической и термической реакцией оказалось
равным 3,1 ∙102. При этой же температуре К = k1/k5=" 2,48∙10-17
моль/л.
=16,5 кПа и
491 К отношение скоростей фотохимической и термической реакцией оказалось
равным 3,1 ∙102. При этой же температуре К = k1/k5=" 2,48∙10-17
моль/л.
В итоге получаем
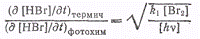
откуда может быть вычислена
константа скорости диссоциации брома при термической реакции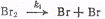 ,а
следовательно, и обратная ей кинетическая константа для обоих процессов.
,а
следовательно, и обратная ей кинетическая константа для обоих процессов.
Поскольку

(где 101 кПа - атмосферное
давление и 22,4∙103см3/моль стандартный объемгаза) получаем
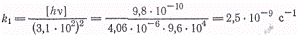
откуда
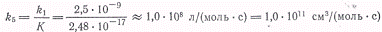
Сравним полученное значение с
тем, которое дается теорией столкновений при условии, что все столкновения
между атомами брома приводят к рекомбинации (диаметр столкновения примем равным
0,3 нм)
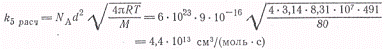
Таким образом

т. е. только одно из 440
столкновений атомов брома ведет к их соединению в молекулу. Поскольку энергия
активации рекомбинации равна нулю, этот результат указывает на необходимость
тройного столкновения для стабилизации получающейся молекулы брома.
3. Процессы, протекающие при фотовозбуждении
молекул
Молекулы реагирующего вещества
под действием света обычно переходят в электронно-возбужденное состояние.
Электронно-возбужденная молекула через некоторое время переходит в нормальное
состояние путем излучения поглощенного фотона (излучатель-, ный переход) или
путем превращения в тепло избыточной энергии в результате столкновений
(безызлучательная конверсия), или передачи ее другой молекуле, которая
вследствие этого диссоциирует (тушение). Кроме того, электронно-возбужденная
молекула может вступить в реакцию и тогда ее избыточная энергия переходит к
продуктам реакции.
Большинство химически
устойчивых молекул содержат четное число электронов. Эти электроны в молекулах
с ковалентными связями спарены, т. е. спины электронов антипараллельны и
результирующий спин равен 0. Если общий электронный спин равен S,
мультиплетность состояния равна 2S+
1. Поэтому при наличии в молекуле только спаренных электронов S
= 0, мультиплетность равна единице, т. е. система синглетна. Синглетное
состояние обозначают символом S.
Фотовозбуждение переводит один
электрон устойчивой молекулы на более высокий энергетический уровень. В этом
состоянии спин электрона может быть антипараллельным спину его партнера, тогда
состояние системы по-прежнему останется синглетным. Но возможны и такие
переходы, когда спин электрона, перешедшего на более высокий энергетический
уровень, становится параллельным партнеру, тогда результирующий спин будет
равен 1, а мультиплетность равна трем. Состояние системы будет триплетным и
обозначается символом Т. Схема физических процессов, вызванных возбуждением и
дезактивацией молекулы, показана на рис. 1. Как видно из схемы,
электронно-возбужденная молекула может разными путями (излучательными и
безызлучательными) возвращаться в основное состояние. Безызлучательные переходы
каскадного типа могут происходить как в синглетном состоянии, так и в
триплетном. Молекула постепенно переходит из одного колебательного состояния в
другое до более низкого электронного уровня. Энергия, выделяющаяся при этом,
передается безызлучательным процессом другим молекулам среды. Безызлучательные
переходы между состояниями одинаковой мультиплетности называются внутренней
конверсией.
Вслед за поглощением фотонов,
переводящим молекулу на более высокий электронный уровень, могут последовать
переходы на низшие уровни, в результате чего происходит испускание излучения.
Излучательные переходы между синглетными состояниями называются флуоресценцией.
Если испускание является единственным способом дезактивации
электронно-возбужденной молекулы, то (так как процесс может быть рассмотрен в
качестве реакции первого порядка) величина, обратная константе скорости
высвечивания, называется естественным временем жизни τ0
возбужденного состояния. Естественное время жизни флуоресценции лежит в
интервале от 10-9 до 10-6 с.
Переход из состояния одной
мультиплетности в состояние с другой мультиплетностью (интеркомбинационый
переход)согласно правилам отбора запрещен. Но так как реальные состояния не
являются чисто синглетными или триплетными, то эти переходы происходят, но с
меньшей вероятностью (в 103-106 раз), чем переходы между
состояниями одинаковой мультиплетности, и играют большую роль в дезактивации
электронно-возбужденных молекул. Интеркомбинационный переход обычно совершается
с одного электронного синглетного уровня на колебательный уровень той же
энергии триплетного состояния. Этот процесс является адиабатическим. Такой
безызлучательный переход называют межсистемным, или интеркомбинационной
конверсией.
Триплетные состояния молекул
являются метастабильными. Времена пребывания в этих состояниях больше, чем в
синглетных состояниях, и излучательный переход из триплетного уровня в
синглетный уровень может длиться 10-3 с, а иногда даже до 10 с.
Излучательные переходы между
состояниями разной мультиплетности называются фосфоресценцией. В органических
молекулах фосфоресценция осуществляется с низшего колебательного уровня
триплетного состояния на колебательный уровень основного (синглетного)
электронного состояния.
Времена жизни возбужденных
состояний (в секундах) для конденсированных (жидких) систем показаны на
следующей схем:

Где А - молекула в основном
низшем энергетическом состоянии; 1А и 3А - молекулы в
синглетном и триплетном состояниях соответственно; Акол - молекулы с
избыточной колебательной энергией; А* - молекула в низшем электронном
возбужденном состоянии (синглетном или триплетном); А** - молекула в высшем
электронном возбужденном состоянии.
Переход электрона с одного
уровня на другой можно рассматривать как переход с одной молекулярной орбитали
на другую. Для органических молекул характерны три типа одноэлектронных
орбиталей:, π- и n-орбитали.
π-
и
σ-Орбитали
бывают связывающими и разрыхляющими (последние обозначают индексом *), n-орбиталь
называется несвязывающей.
σ-Орбиталь
соответствует ординарной, простой химической связи; электроны, локализованные
на этой орбитали, называются σ-электронами.
Они обычно являются сильно связывающими электронами и в значительной мере
локализованы. π-Орбиталь
соответствует одной из связей в атомной группе с кратной связью. Каждой π-
и
σ-орбиталям
соответствуют разрыхляющие (свободные в основвом состоянии)-*- и σ*-орбитали.
n-Орбиталью
называется атомная орбиталь, занимаемая неподеленной парой электронов
гетероатома, не участвующих в образовании химической связи. Неподеленная пара
электронов может быть локализована на n-орбитали
(например, в карбонильных соединениях) или может участвовать в сопряжении с π-электронами
(например, в анилине, фуране). Обычно связывающие σ-орбитали
имеют более низкую энергию, чем π- и
n-орбитали, а
разрыхляющие σ*-орбитали обладают
более высокой энергией, чем π*-орбитали.
Электронные переходы в молекуле
обычно обозначают, указывая орбитали, между которыми переходит электрон: n→π*π→π*σ→π*
и т. д. Возбужденное состояние, реализующееся в результате таких переходов,
называется состоянием типа соответствующего перехода, например, синглетное
состояние nπ*-типа
- Snπ*-Типы
электронных переходов можно установить по электронным спектрам поглощения и
излучения. Возбужденное состояние, как видно из рис. 1, может быть или
синглетным, или триплетным.
Электронно-возбужденные
состояния в фотохимии играют огромную роль. Молекула в возбужденном состоянии
обладает большей энергией и иным распределением электронов, чем в основном,
поэтому должна являться более реакционноспособной.
Однако реакционноспособными
могут оказаться возбужденные молекулы, находящиеся только в синглетном или
только триплетном состоянии, а также молекулы, находящиеся на высоких
колебательных уровнях основного состояния .
Если разница в энергиях
синглетного и триплетного состояний невелика, то в химических реакциях
возбужденные синглетные и триплетные молекулы могут вести себя одинаково, т. е.
иметь одинаковую реакционную способность, хотя по своим физическим свойствам
молекулы будут отличаться. Молекулы, находящиеся в триплетном состоянии,
парамагнитны, молекулы в синглетном состоянии - диамагнитны. Некоторые
молекулы, находящиеся в триплетном состоянии, могут быть вполне устойчивыми.
Типичным примером является молекула кислорода. Кислород парамагнитен и имеет
пару электронов с параллельными спинами. Эти электроны находятся на двух
орбиталях одинаковой энергии, но одна из них вырождена.
В радикале метилена :СН2
два электрона могут иметь антипараллельные спины, тогда состояние радикала
будет синглетным, или параллельные спины, тогда состояние радикала будет
триплетным. Метилен, полученный фотолизом диазометана в газовой фазе по реакции
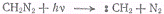
находится в синглетном
возбужденном состоянии, которое переходит в более стабильное триплетное с
выделением энергии. Последняя может быть передана третьим частицам, например
молекулам инертного газа, которые добавлены к диазометану:
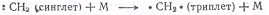
В результате температура
реакционной смеси возрастает.
Метилен в возбужденном
синглетном состоянии стереоспецифичен и легко присоединяется к цис- и
транс-бутену по месту двойной связи, образуя два стереоизомера:
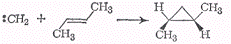

Присоединение метилена в
триплетном состоянии к олефинам нестероспецифично. Например, цис-бутен-2 дает
два изомерных диметилциклопропана по схеме:
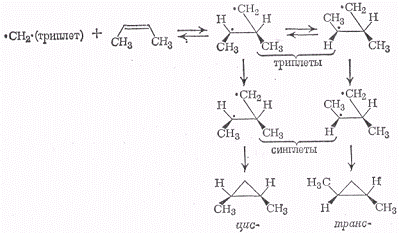
При присоединении метилена,
находящегося в синглетном состоянии, по месту двойной связи электроны с
противоположными спинами соединяются в пары, образуя циклопропан, а в случае
метилена, находящегося в триплетном состоянии, как видно из приведенной схемы
реакции, образуется бирадикал в триплетном состоянии, который переходит в
синглетное состояние, и только после этого образуются конечные устойчивые
продукты - стереоизомеры замещенного циклопропана.
Обладающий меньшей энергией
метилен - триплет легче вступает в реакции простого присоединения по месту
двойной связи нежели в реакции включения, когда СН2-группа просто
внедряется в С-Н-связь. Последняя реакция более характерна для метилена,
находящегося в синглетном состоянии.
Исследование фотохимических
процессов в органических молекулах показало, что при поглощении фотона
молекулой наибольшую вероятность имеет переход молекулы или в самое нижнее
возбужденное синглетное состояние S1*
или в самое нижнее триплетное состояние T1.
4. Полные и локальные скорости фотохимических
реакций
Согласно закону Ламберта -
Бера, ослабление интенсивности - dlсвета,
прошедшего через слой толщиной dl,
прямо пропорционально толщине слоя, концентрации молекул с, интенсивности света
I:
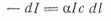
где α
- коэффициент
поглощения света, зависящий от природы вещества и от длины волны поглощаемого
света.
Функция α(λ)
количественно
описывает спектр поглощения света. Интегрирование этого уравнения от I0-
интенсивности света, падающего на кювету, когда l
= 0, до интенсивности Ilна
расстоянии l от начала кюветы
дает

Количество поглощенных квантов
будет равно
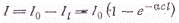
Если происходит заметное
поглощение света, точисло поглощенных квантов будет разным у передней и задней
стенок реакционного сосуда. Поэтому скорости зарождения радикалов и
возбужденных молекул, концентрации этих частиц и скорости их реакций не будут
одинаковыми по всему объему сосуда.
Это приводит к тому, что при
исследовании фотохимических реакций измеренные полные или средние скорости
реакций не всегда соответствуют локальным скоростям, к которым и применимы все
рассмотренные теоретические представления. Равенство полных и локальных
скоростей наблюдается только в тех случаях, когда а) поглощение света
равномерно по всему объему реакционного сосуда; б) времена жизни радикалов и
других активных частиц достаточно велики, так что в результате диффузии
создаются одинаковые концентрации по всему объему; в) скорости образования
конечных продуктов прямо пропорциональны интенсивности поглощенного света.
Полная или средняя измеряемая
скорость связана с локальной скоростью очевидным соотношением
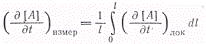 ,
,
где [А] - концентрация продуктов
реакции.
Для локальной скорости в общем
виде можно записать
где β
- функция, в которую входят концентрации реагирующих веществ и константы
скоростей; Iа-
интенсивность поглощенного света в данном сечении реакционного сосуда.
Подставляя выражения, получим:
 (1)
(1)
Пользуясь законом Ламберта -
Бера, можно найти Iаследующим
образом:
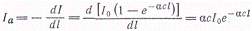 (2)
(2)
Если пучок света параллелен и
имеет одинаковую интенсивность по сечению, полностью охватывающую кювету, в
которой диффузией радикалов из данного элемента объема в другой можно
пренебречь, то βне зависит
от l и Iа.
Тогда, интегрируя (1) после подстановки (2), можно найти полную скорость

Отношение измеренной скорости к
локальной будет равно
 (3)
(3)
Из этого выражения видно, что
измеряемая полная и локальная скорости становятся равными только при условии с→0,
l→0, α→0
или n=1. Отношения
полной и локальной скоростей при различных долях поглощенного света,
рассчитанные по (3) для случая п = 1/2, показаны ниже:
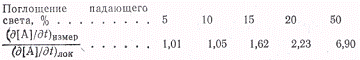
Нетрудно заметить, что если
поглощается до 10 % падающего света, то отношения измеряемых и локальных
скоростей не сильно отличаются друг от друга. Но чем больше процент
поглощенного света, тем эти скорости сильнее различаются. Строгое применение
рассмотренного уравнения и обработка полученных экспериментальных данных
затруднены из-за отсутствия данных о скоростях диффузии радикалов в реакционной
среде. Если скорость диффузии радикалов велика, то различие между измеряемой и
локальной скоростями будет меньше даже при больших долях поглощаемой световой
энергии. Если образуются очень активные радикалы, то они легко гибнут в
заданном линейном объеме, и различие полной и локальной скорости будет
приближаться к теоретически предсказанному выражением (3). Поэтому закон
скоростей всегда необходимо проверять при нескольких значениях долей
поглощенной световой энергии.
5. Зависимость скорости фотохимических реакций
от температуры
Скорость фотохимической реакции
не зависит от температуры, если конечные продукты реакции образуются
непосредственно из возбужденных частиц, если лимитирующей стадией образования
конечных продуктов является фотохимический процесс, или энергия активации
"темновых" реакций очень мала. Энергия электронного возбуждения
молекул обычно имеет более высокое значение, чем прирост средней тепловой
энергии при нагревании от комнатной до повышенных температур. Например,
повышение температуры ацетальдегида от 20 до 220°С приводит к увеличению
средней тепловой энергии на 12,5 кДж/моль,в то время как энергия электронного
возбуждения молекулы ацетальдегида составляет около 418,4 кДж/моль. Поэтому
направление и эффективность фотолиза мало зависят от температуры. Зависимость
скорости первичного фотохимического процесса от температуры может быть
значительной, если энергии поглощенного кванта не хватает для разрыва связи.
Однако независимость скорости
фотохимической реакции от температуры еще не говорит о том, что продукт
образуется в первичном процессе. Это может происходить и вследствие того, что в
реакции участвуют "горячие" радикалы или вторичные реакции протекают
с малыми энергиями активации.
Если считать, что избыток
энергии при фотодиссоциации переходит только в кинетическую энергию
поступательного движения (нет колебательно-, враодательно- и
электронно-возбужденных радикалов), то полный избыток кинетической энергии
частиц А и В, образующихся по реакции АВ + hv→A
+ В будет
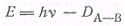
где Da-b- энергия
диссоциации молекулы АВ.
Эта избыточная энергия
распределяется между радикалами таким образом, что радикал, имеющий меньшую
массу, приобретает большой избыток энергии (и становится "горячим".
Его участие в последующих реакциях характеризуется энергией активации близкой к
нулю.
Фотолиз исходных молекул обычно
приводит к появлению вращательно-, колебательно- или электронно-возбужденных
радикалов, имеющих, кроме того, и повышенную кинетическую энергию
поступательного движения. Энергия активации реакций с их участием также будет
близка к нулю.
6.
Кинетика
флуоресценции, фосфоресценции и интеркомбинационной конверсии
Количественное изучение
флуоресценции и фосфоресценции позволяет определить ряд важных величин,
характеризующих фотохимический процесс; время жизни возбужденных молекул,
скорость интеркомбинационной конверсии, число и природу возбужденных состояний,
эффективные сечения тушения молекул, эффективность переноса электронной
энергии, первичный квантовый выход. Рассмотрим процесс, который слагается из
стадий первичного возбуждения исходных молекул светом и последующих процессов
флуоресценции или фосфоресценции, конверсии энергии электронного возбуждения в
энергию теплового движения и химического превращения возбужденных частиц, т. е.
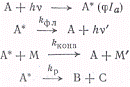
где Iа
- число квантов, поглощенных в единицу времени в единице объема, которое
принимается равным числу молекул, возбужденных в единицу времени в единице
объема (это соответствует φ = 1).
Скорость, накопления
возбужденных частиц запишется в виде
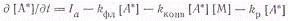
Из условия стационарности ∂[А*]/∂t=0
вытекает:

Пользуясь этим выражением,
легко найти первичный квантовый выход распада А* на вещества В и С:
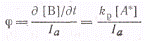
Подставляя выражение, получим
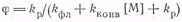
Принимая, что число излучаемых
квантов в единицу времени из единицы объема равно
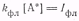
из условия стационарности
находим
После подстановки в него Iфл
получим
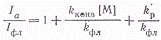 (4)
(4)
Как видно из выражения (4),
зависимость Iа/Iфл
от [М] является линейной. Наклон прямой равен kконв/kфл,
а отрезок по оси ординат составляет 1 + kр/kфл.
Отсюда
вытекает, что определяя независимым путем kфлможно
из этих данных вычислить величины kконвиkфл.
Предположение о том, что Iа
иIфл
одинаковы по воему объему, не всегда верно. Учет изменения I
в объеме возможен и приводит к более сложным зависимостям.
7. Хемилюминесценция
Свечение, сопровождающее
некоторые химические реакции, возникает в результате образования
электронно-возбужденных продуктов реакции. Схема реакции, сопровождающейся
хемилюминесценцией

Согласно схеме
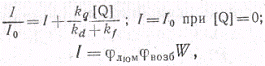
где W-
скорость реакции А с В; φвозб
-выход возбуждения; φлюм
-
квантовый выход люминесценции продукта.
В присутствии активатора Dдополнительно
идут реакции:

Заключение
В большинстве фотохимических
реакций получаемая молекулой энергия превышает типичные энергии активации
темновых (термических) реакций и может даже превосходить энергию разрыва
химических связей. Тем не менее многие фотохимические реакции отличаются
высокой избирательностью, что связано с особенностями электронного строения
возбужденного состояния.
В результате реакции
фотодиссоциации происходит разрыв химических связей с образованием свободных
радикалов Образующиеся атомы и радикалы обладают высокой реакционной
способностью и вступают в быстрые темновые реакции, часто цепные. В результате
квантовый выход суммарной реакции может стать значительно больше единицы. С
помощью реакций фотодиссоциации можно осуществлять различные процессы
радикальной полимеризации. Подобные процессы применяются в производстве
интегральных схем; с помощью фоторезистов на кремниевой подложке обозначаются
участки, на которых в последующем образуются элементы будущей микросхемы. В зубоврачебной
технике фотополимеризация используется для отверждения современных
пломбировочных материалов, также используется в промышленно важном процессе
получения - капролактама - исходного вещества для производства капрона.
Реакции фотодиссоциации с
разрывом химических связей могут идти в разнообразных полимерных материалах под
действием видимого или ультрафиолетового света. Образующиеся при этом радикалы
могут приводить к разрыву связей С-С в углеродных цепочках полимера. Эти
процессы приводят к так называемой фотодеструкции полимеров, которые ускоряются
под действием агрессивных компонентов атмосферы - кислорода, озона, оксида
азота(IV). В результате ухудшаются механические свойства полимера. Они особенно
заметны на полиэтиленовой пленке парника, которая в течение многих месяцев
подвергалась действию прямых солнечных лучей. Поэтому очень важны меры по
фотостабилизации полимеров; ее можно осуществить введением ингибиторов
радикальных реакций. С другой стороны, для упаковочных полимерных материалов,
наоборот, желательно быстрое их разрушение после использования, чтобы избежать
загрязнения этими полимерами окружающей среды. Такие полимеры намеренно делают
светочувствительными; под действием солнечных лучей они рассыпаются в тонкий
порошок.
Возбужденные светом молекулы
могут также вступать в реакции переноса электрона - фотоокисление и
фотовосстановление. Так, возбужденные карбонильные соединения в присутствии
восстановителя могут превращаться в спирты, а возбужденные молекулы красителей,
реагируя с кислородом, превращаются в бесцветные соединения. На
фотовосстановлении солей железа(III) органическими реагентами (щавелевая или
лимонная кислота) основаны методы светокопирования чертежей: образующиеся при
фотовосстановлении Fe3+ ионы Fe2+, реагируя с красной
кровяной солью, дают синий осадок.
Под действием света могут идти
реакции фотоприсоединения. Так, при возбуждении молекул с двойной связью
возможен ее разрыв с образованием бирадикала, например, Н2С=СН2+
Н2С.-.СН2. Присоединение к нему
второй молекулы этилена дает циклобутан. Такое циклоприсоединение с участием
двух молекул алкенов происходит только под действием ультрафиолетового света;
при нагревании эта реакция не идет, поскольку она запрещена так называемым
правилом Вудворда - Хоффмана. В карбонильных соединениях с двойной связью С=О
также возможен ее частичный разрыв с образованием реакционноспособного
бирадикала. К нему может присоединиться молекула алкена с образованием
четырехчленного циклического соединения - оксетана. Оксетановый цикл входит в
состав некоторых антибиотиков; подобные соединения часто могут быть получены
только фотохимически.
Особую группу составляют
реакции фотосенсибилизации, в которых возбужденные светом атомы или молекулы
передают избыточную энергию другим молекулам, которые и реагируют. Перенос
энергии от возбужденных молекул красителей - основа процессов, делающих
фотографические материалы чувствительными к зеленым, желтым и красным лучам
спектра.
Под действием света идут также
разнообразные реакции фотоизомеризации. Например, транс-стильбен
(1,2-дифенилэтилен) под действием света превращается в цис-стильбен.
Цис-транс-изомеризация ретиналя (вещества, родственного ретинолу - витамину А)
под действием света приводит в результате ряда последующих процессов к
возникновению зрительного сигнала в сетчатке глаза.
Фотохимическим хлорированием бензола
получают инсектицид гексахлорциклогексан. Фтотохимическим хлорированием,
сульфохлорированием (одновременная реакция с SO2 и Cl2) и
сульфоокислением алканов получают растворители, моющие средства и средства для
химической чистки. Большое практическое значение имеют фотохимические реакции в
зеленых растениях (фотосинтез
<http://www.krugosvet.ru/articles/116/1011698/1011698a1.htm>).
Обратимые превращения веществ под
действием света в ряде случаев приводят к интересному явлению - фотохромизму.
Оно заключается в приобретении или изменении окраски под действием света.
Обратная реакция может идти как в темноте, так и под действием света с другой
длиной волны. Скорости прямой и обратной реакции могут быть различными. Часто
прямая фотохимическая реакция идет сравнительно быстро, а обратная темновая -
медленно. Фотохромизм наблюдается как у неорганических, так и у органических
соединений. Механизм явления может быть разным. В случае кристаллических
соединений действие света может сводиться к перемещению электронов или атомов
из одних узлов кристаллической решетки в другие. Так, светозащитное фотохромное
стекло содержит около 0,5% хлорида или бромида серебра, сплавленного с
боросиликатами щелочных металлов. Под действием света происходит перенос
электронов от ионов галогена к ионам серебра; образовавшиеся атомы серебра
делают стекло непрозрачным. Обратная реакция может идти под действием света с
другой длиной волны или в темноте. Такое стекло используется для изготовления
солнечных очков, окон зданий и автомобилей; оно само регулирует пропускаемый
световой поток, делая его оптимальным.
Фотохромизм находит практическое
применение. Для изготовления фотохромных галогенсеребряных стекол,
иллюминаторов самолетов используют пластиковые стекла, содержащие фотохромный
краситель, который темнеет на ярком солнечном свету, а при слабом освещении
восстанавливает свою прозрачность. Если в прозрачную пластмассу ввести всего
0,1% гексакарбонила хрома Cr(CO)6, то при облучении бесцветное
вещество окрашивается в интенсивный желтый цвет в результате отщепления одной
молекулы СО. В темноте при комнатной температуре примерно в течение 4 ч
происходит обратная рекомбинация СО и Cr(СО)5, и цвет исчезает.
Изобретены даже "загорающие" куклы, при изготовлении которых
применяют краситель, дающий обратимо коричневую окраску на солнечном свету.
Недостатком всех известных фотохромных материалов, прежде всего органических,
является их постепенное разрушение под действием тепла и света с утратой фотохромных
свойств после определенного числа циклов.
Список литературы
. Денисов Е.
Т. Кинетика гомогенных химических реакций: Учеб. пособие для химических
специальностей ун-тов. - М.: Высш. школа, 1987. - 367 с., ил.
. Панченков
Г. М., Лебедев В. П. Химическая кинетика и катализ. Учебное пособие для вузов.
- 3-е изд. испр. и доп. - М., Химия, 1985. - 592 с., ил.