Влияние растворителя на кинетику органических реакций
Курсовая работа
Влияние растворителя на кинетику
органических реакций
Содержание
Введение
.
Качественные теоретические оценки влияния растворителей на скорости реакций
.1
Правила Хьюза-Ингольда
.
Теоретическая количественная оценка влияния растворителей на скорости реакций
.1
Реакции между нейтральными аполярными молекулами
.2
Реакции между биполярными молекулами
.3
Реакции между нейтральными молекулами и ионами
.4
Реакции между ионами
.
Влияние специфической сольватации на скорости химических реакций
.1
Роль водородной связи в химической кинетике
Заключение
Список
литературы
Введение
Природа растворителя оказывает большое влияние на скорость и порядок
гомогенных химических реакций. Это впервые отметили Бертло и Пеан де Сен-Жиль в
1862 г. в ходе работ по изучению этерификации уксусной кислоты этанолом: «…ход
этерификации нарушается, а её скорость снижается при добавлении нейтральных
растворителей, не участвующих в реакции». Ещё в 1890 г. Меншуткин изучал
кинетику реакции триэтиламина с иодэтаном в 23 растворителях; в этой
классической работе было показано, что скорость реакции в большей степени
зависит от выбора растворителя.
Так, в диэтиловом эфире, бензоле, метаноле и бензиловом спирте скорость
образования четвертичной соли была выше, чем в н-гексане, соответственно в 4,
36, 280 и 742 раза. Следовательно, путём подбора соответствующего растворителя
можно существенно ускорит или замедлить химическую реакцию. Очевидно, что этот
факт чрезвычайно важен как в лабораторных исследованиях, так и в химической
промышленности. В отдельных случая путём простой замены растворителя удавалось
повысить скорость реакции примерно в 109 раз. В этой связи
становится понятной важность эмпирических правил и теорий, позволяющих выбрать
наиболее подходящий для планируемой реакции растворитель[1].
Растворитель может оказывать влияние как на скорость, так и на механизм
реакций. Иногда растворитель изменяет скорость процесса без изменения его
механизма, но встречаются случаи, когда растворитель влияет на механизм, а скорость
остаётся неизменной. Растворитель может влиять на скорость без изменения
механизма реакции, вследствие изменения сил, действующих между реагирующими
частицами. Такое явление иллюстрируется влиянием диэлектрической проницаемости
среды на электростатические силы, действующие между реагирующими частицами. От
вязкости растворителя зависит скорость диффузии и, следовательно, частота
соударений реагирующих частиц, что может вызвать изменение скорости реакций,
лимитируемых диффузией. Многие другие свойства растворителя, такие, как
сольватация реагентов, нуклеофильность, электрофильность, когезия, способность
давать водородную связь и т.д., также оказывают влияние на скорость и
механизм[2].
Зависимость скорости реакции от среды можно изучать, во-первых, сравнивая
скорости реакции в газовой фазе и растворе и, во-вторых, сравнивая скорости
реакции в различных растворителях.
Несмотря на огромное число работ, посвящённых исследованию влияния среды
на скорость химических реакций, до сих пор не существует строгой количественной
теории, которая могла бы «универсально», с помощью одного уравнения, описать
зависимость константы скорости реакции от природы среды, в которой происходит
реакция.
Отсутствие общей теории влияния среды на кинетику химических реакций
объясняется тем, что изменение растворителя может не только влиять на скорость
процесса, но и зачастую усложнять механизм реакции. Учёт удельного вклада
каждого из факторов чаще всего достаточно сложен, требует глубокого и
всестороннего понимания свойств среды и реагирующих частиц, так как только
полные сведения об этих свойствах позволят количественно учесть все типы
взаимодействия реагирующих частиц со средой.
1.
Качественные теоретические оценки влияния растворителей на скорости реакций
В зависимости от природы промежуточного активированного комплекса
органические реакции грубо можно разбить на три группы: реакции с биполярным,
изополярным или свободно-радикальным переходным состоянием.
По разделению зарядов или делокализации заряда биполярные комплексы
существенно отличаются от реагентов. Реакции с биполярным переходным
состоянием, на которые природа растворителя оказывает очень большое влияние,
можно обнаружить среди реакций ионизации, замещения, элиминирования и
фрагментации, например:
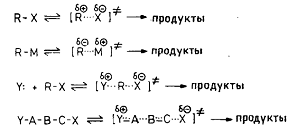
По характеру распределения зарядов изополярные активированные комплексы
очень мало или вообще не отличаются от реагентов. Реакции с изополярным
переходным состоянием, на которые растворитель оказывает небольшое или даже
пренебрежительно малое влияние, можно найти среди перициклических процессов,
например реакция циклоприсоединения Дильса - Альдера и перегруппировка Коупа
гексадиенов-1,5:
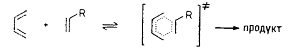
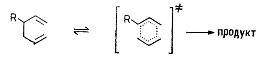
Свободнорадикальные активированные комплексы с неспаренными электронами
образуются при гомолитическом разрыве связей. К реакциям со
свободно-радикальным переходным состоянием, на которые растворитель оказывает
небольшое или пренебрежительно малое влияние, относятся образование радикальных
пар и перенос атома, например,
.1 Правила
Хьюза-Ингольда
Влияние растворителей на реакции алифатического нуклеофильного замещения
изучали Хьюз и Ингольд. Для этой цели они применили простую качественную модель
сольватации, учитывающую только электростатические взаимодействия между ионами
(или бимолекулярными молекулами) и молекулами растворителя как в начальном, так
и в переходных состояниях. В зависимости от того, являются ли реагирующие
частицы нейтральными, отрицательно или положительно заряженными, все реакции
нулеофильного замещения и элиминирования можно отнести к трём типам. Далее
можно достаточно обоснованно предположить, что степень сольватации
непосредственно связана с характером электрического заряда реагирующей частицы,
а именно: степень сольватации а) возрастает при повышении величины заряда; б)
понижается при делокализации заряда; в) при нейтрализации заряда снижается в
ещё большей степени. Отсюда следует, что общий эффект растворителя на реакции с
участием нейтральных, положительно или отрицательно заряженных частиц можно
суммировать следующим образом:
а) повышение полярности растворителя приведёт к росту скорости реакций, в
которых активированный комплекс имеет большую плотность заряда, чем исходная
молекула (или молекулы);
б) повышение полярности растворителя приведёт к снижению скорости
реакций, в которых плотность заряда в активированном комплексе меньше, чем в
исходной молекуле (или молекулах);
в) изменение полярности растворителя практически не будет влиять на
скорости реакций, в которых при переходе от реагента (или реагентов) к
активированному комплексу плотность заряда остаётся постоянной или изменяется
незначительно.
Другими словами, замена менее полярного растворителя на более полярный
приведёт к повышению или снижению скорости реакции в зависимости от того,
является ли активированный комплекс более или менее полярным, чем реагенты.
Здесь под «полярностью растворителя» понимают его способность сольватировать
находящиеся в растворе заряженные частицы, причём считается, что такая
способность растворителя повышается при увеличении дипольного момента его
молекул и снижается при повышении степени экранирования зарядов биполярной молекулы.
Например, реакция между одноименно заряженными ионами будет
сопровождаться повышением плотности заряда на стадии образования
активированного комплекса. Следовательно, при замене менее полярного
растворителя на более полярный скорость такой реакции возрастёт. Напротив,
реакция между ионами с зарядами противоположных знаков в хорошо сольватирующих
ионы полярных растворителях будет замедляться, поскольку в этом случае в
активированном комплексе плотность заряда снижается по сравнению с исходными
ионами. Кроме того, полярность растворителя должна оказывать большее влияние на
скорость реакций, в которых заряд возникает или нейтрализуется на стадии
активации, сопровождающиеся только делокализацией заряда. Действительно, замена
воды на этанол приводит к изменению скорости реакций замещения с возникновением
или нейтрализацией заряда в 103 - 104 раз, тогда как SN-реакции
нуклеофильного замещения, сопровождающиеся делокализацией заряда, при переходе
от этанола к воде ускоряются только в 3 - 10 раз.
Приведённые выше правила, называемые правилами Хьюза - Ингольда,
позволяют качественно оценить влияние полярности растворителя на скорость любой
гетеролитической реакции, если известен её механизм. Для реакций нуклеофильного
замещения типа (1) и (2)
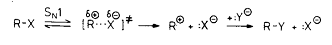 (1)
(1)
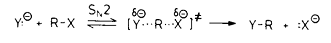 (2)
(2)
Соответствующие следствия из этих правил суммированы в табл. 1, где в
трёх средних графах указано, что происходит с зарядами при образовании
активированного комплекса, а в последней указан ожидаемый эффект растворителя.
Таблица 1. − Предсказываемое правилами Хьюза - Ингольда влияние
растворителей на скорости нуклеофильного замещения [1]

Следует подчеркнуть, что замена одного растворителя на другой может
изменять не только скорость реакции, но и её механизм. Так, реакции с участием
некоторых галогеналканов в водном растворе этанола проходят по механизму SN1,
а в ацетоне - по механизму SN2. С другой стороны, реакции с участием
галогенметанов, протекающие в водном этаноле по механизму SN2, могут
осуществляться по механизму SN1 в растворителях с большей
ионизирующей способностью, например в муравьиной кислоте.
В отношении стадий, определяющих скорость реакции, механизм
нуклеофильного замещения весьма близок к механизму β-элиминирования. Так, скорости
мономолекулярных SN1 и Е1-реакций контролируются одной и
той же стадией, а у бимолекулярных SN2 и Е2-реакций
аналогичны стадии переноса электрона от реагента к уходящей группе: они
различаются лишь тем, что в реакциях элиминирования электроны проходят по
большей цепи атомов углерода. В этой связи неудивительно, что для описания
влияния растворителей на мономолекулярные и бимолекулярные реакции β-элиминирования с различной судьбой
зарядов при активации Хьюз и Ингольд предложили правила, аналогичные правилам,
используемым для оценки эффектов растворителей в SN1-реакциях (табл.
2).
Согласно правилам Хьюза - Ингольда, Е2- и SN2-реакциях
заряд делокализуется в большей степени, чем в Е1- и SN1-реакциях,
поэтому в общем случае повышение полярности растворителя будет
благоприятствовать реакциям, осуществляющимся по Е2- и SN2-механизмам.
Отсюда, в частности, следует, что замена одного растворителя на другой может
изменять не только скорость, но и механизм реакции.
Хотя правила Хьюза - Ингольда впервые были продемонстрированы на примере
реакций нуклеофильного алифатического замещения и β-элиминирования, они должны быть
применимы и для любых других гетеролитических реакций в растворах, в которых
образование активированного комплекса связано с возникновением, делокализацией
или нейтрализацией заряда [1, 2, 3].
Таблица 2. − Правила, описывающие влияние растворителей на скорости
реакций β-элиминирования [1]

2.
Теоретическая количественная оценка влияния растворителей на скорости реакций
В отличие от довольно простой теории реакций в газовой фазе расчёт
констант скоростей реакций в жидкой фазе чрезвычайно сложен. Причина этого
заключается в первую очередь в сложности многочисленных возможных межмолекулярных
взаимодействий между растворителем и растворённым веществом. При изучении
кинетики реакций в растворе всегда возникает проблема выбора конкретного
свойства растворителя, которое можно было бы использовать в математических
выражениях, связывающих это свойство со скоростью реакции. Другая проблема
заключается в выборе характеристик реагирующих молекул, которые необходимо
учитывать при определении влияния растворителя на их реакционную способность.
Количественная оценка влияния растворителей на константу скорости k элементарной реакции сводится к
нахождению функции типа
k=f(a, b, c, … m, n, o, …) (3)
Здесь a, b и c - параметры, характеризующие
свойства реагентов, а m, n и o - параметры, характеризующие
свойства среды. Найденная функция должна правильно описывать зависимость
константы скорости от свойств среды. В этой связи возникает задача нахождения
параметров реагентов и растворителя, ответственных за наблюдаемые зависимости,
т.е. таких параметров, которые должны быть включены в уравнение (3).
В рамках теории абсолютных скоростей реакций проблема количественной
оценки индуцированного растворителем изменения константы скорости реакции A+B↔(AB)≠→C+D сводится к вычислению разности между энергиями Гиббса
сольватации активированного комплекса (AB)≠ и реагентов А и В [уравнение (4)].
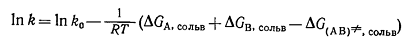 (4)
(4)
Здесь k0 -
константа скорости в стандартном растворителе или в газовой фазе,
k - константа скорости реакции в изучаемом растворителе.
Если учесть все возможные взаимодействия между растворителем и
растворённым веществом, то попытки найти корреляции между константой скорости
реакции и параметрами среды в общем случае приведут к настолько сложным
уравнениям, что их экспериментальная проверка будет невозможна. Поэтому
уравнения, связывающие константу скорости со свойствами среды, обычно
разрабатывают на основе теоретически более или менее обоснованных моделей,
учитывающих лишь ограниченное число важнейших типов взаимодействия. Если
принятая модель правильно отражает доминирующие взаимодействия между
растворителем и растворённым веществом, то с помощью соответствующих уравнений
обычно удаётся количественно удовлетворительно описать экспериментальные
данные. В связи с этим целесообразно подразделить все реакции на следующие
четыре типа:
1) реакции между нейтральными аполярными молекулами (с участием
изополярных активированных комплексов);
2) реакции между нейтральными биполярными молекулами (с участием
биполярных активированных комплексов);
) реакции между нейтральными биполярными молекулами и ионами (с
участием биполярных и заряженных активированных комплексов);
) реакции между ионами (с участием биполярных и/или заряженных
активированных комплексов) [1].
2.1
Реакции между нейтральными аполярными молекулами
В любой реакции, проходящей в растворе, в растворителе должны
образовываться полости, в которых располагаются реагенты, активированный
комплекс и продукты реакции. Лёгкость возникновения таких полостей,
образующихся за счёт увеличения расстояния между молекулами растворителя,
является важным фактором, определяющим растворимость конкретного вещества в
данном растворителе. Кроме того, поскольку растворимость часто тем или иным
образом связана с реакционной способностью, то силы взаимодействия между
молекулами растворителя также должны влиять на скорость реакций.
Для возникновения полостей необходимо преодолеть суммарные силы
притяжения между молекулами растворителя. Степень когезии можно оценить,
определив поверхностное натяжение, но более надёжной и точной мерой когезии
служит количество энергии, необходимой для отделения молекул растворителя друг
от друга на бесконечно большое расстояние. Соответствующий параметр называют когезионным
давлением с (или плотностью энергии когезии). Когезионное давление
определяют как энергию испарения ∆Uυ,,отнесённую к
молярному объёму Vm [уравнение (5)].
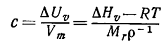 (5)
(5)
В смесях, представляющих собой регулярные растворы, взаимная
растворимость компонентов смеси зависит от когезионного давления, поэтому
Гильдебранд назвал квадратный корень из с параметром растворимости δ [уравнение (6)].
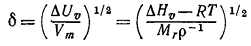 (6)
(6)
Допустив, что в системе растворитель - растворённое вещество действуют
только силы Ван-дер-Ваальса, что все отклонения от идеального поведения
обусловлены теплотой смешения и что энергия взаимодействия между растворителем
и растворённым веществом равна среднегеометрическому от энергий взаимодействий
растворитель - растворитель и растворённое вещество - растворённое вещество,
Гильдебранд и Скэтчард предложили следующее уравнение для зависимости
коэффициента активности fi неэлектролита i, растворённого в растворителе s, от параметров чистого жидкого
вещества i (но не при бесконечном разбавлении):
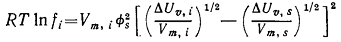 (7)
(7)
Здесь Vm,i - молярный объём растворённого вещества i (в виде чистой жидкости), ∆Uυ,i - молярная энергия испарения
растворённого вещества (в виде чистой жидкости), Vm,s и ∆Uυ,i - такие же параметры растворителя s, φs - объёмная доля растворителя
s (равная единице при бесконечном
разбавлении).
Поскольку реакции обычно проводят в разбавленных растворах, то уравнение
(7) можно упростить:
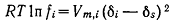 (8)
(8)
Константу скорости k бимолекулярной реакции A+B↔(AB)≠→C+D можно описать
или уравнением (4), или через коэффициенты активности уравнением
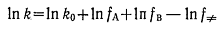 (9)
(9)
Здесь k0 -
константа скорости реакции в идеальном растворе.
Подставив уравнение (8) в (9), получим следующее выражение для скорости
реакции между неполярными реагентами А и В:
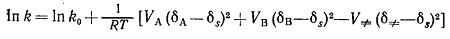 (10)
(10)
Здесь VA,
VB, V≠ - молярные объёмы А, В и
активированного комплекса соответственно. Таким образом, константа скорости
реакции зависит не только от разности молярных объёмов реагентов и
активированного комплекса [называемой активационным объёмом ∆V≠=V≠-(VA+VB)], но и от относительного когезионного давления
реагентов, активированного комплекса и растворителя. Если реагенты лучше
растворимы в данном растворителе, чем активированный комплекс, то скорость
реакции в этом растворителе будет ниже, чем в идеальном растворе. Обратная
взаимосвязь наблюдается в том случае, когда растворимость активированного
комплекса превышает растворимость реагентов.
Неудобное для расчётов уравнение (10) можно преобразовать в линейное
уравнение:
 (11)
(11)
Ещё в 1929 г. Ричардсон и Соупер, а позднее также Гласстон предложили
правила, позволяющие учитывать влияние когезии реагентов, продуктов реакции и
растворителя на скорость реакции. Они обнаружили, что реакции, продукты которых
имеют большую (или меньшую) когезию, чем реагенты, обычно ускоряются (или
замедляются) в растворителях с высокой когезией; напротив, на скорость реакций,
в которых реагенты и продукты характеризуются близкой когезией, растворитель
обычно оказывает небольшое влияние.
Концепция об определяющей роли когезионного (или внутреннего) давления
полезна только при изучении реакций между нейтральными неполярными молекулами в
неполярных растворителях, поскольку в таких случаях можно пренебречь другими свойствами
растворителей, в том числе их сольватирующей способностью и полярностью. В
реакциях между биполярными молекулами и ионами растворитель взаимодействует с
реагентами и активированным комплексом за счёт неспецифической и специфической
сольватации настолько эффективно, что вклад когезионного давления в ln k
становится очень небольшим. Следует подчеркнуть, что когезионное, или
внутреннее, давление не является мерой полярности растворителя. Полярность
отражает способность растворителя взаимодействовать с растворённым веществом, в
то время как когезионное давление, будучи характеристикой структуры
растворителя, служит мерой количества энергии, необходимой для создания в
данном растворителе полостей, способных вместить молекулы растворённого
вещества. Таким образом, полярность и когезионное давление - это
комплементарные параметры и скорость реакции зависит от каждого из них [1, 2,
4, 5].
2.2
Реакции между биполярными молекулами
Скорости реакций между нейтральными биполярными молекулами должны в
первую очередь определяться электростатическими взаимодействиями между
растворителем и растворённым веществом, например диполь-дипольными
взаимодействиями. Для описания электростатических эффектов растворителей
необходимо рассмотреть взаимодействие между точечными зарядами, разделёнными
изотропной непрерывной диэлектрической средой, воспользовавшись для этой цели
функциями диэлектрической проницаемости. В общем случае для описания скорости
реакций с участием бимолекулярных молекул можно использовать уравнение Кирквуда,
которое устанавливает связь между энергией Гиббса идеального диполя в
непрерывной среде, обладающей диэлектрической проницаемостью εr, с энергией Гиббса в аналогичной
среде, диэлектрическая проницаемость которой равна единице (εr =1).
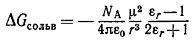 (12)
(12)
В этом уравнении μ - постоянный дипольный момент находящейся в растворе
биполярной молекулы радиусом r, ε0 - диэлектрическая проницаемость вакуума, NA
- число Авогадро. Это важное уравнение связывает изменение энергии Гиббса сольватации
биполярного растворённого вещества как с дипольным моментом и радиусом его
молекул, так и с диэлектрической проницаемостью растворителя; следует
подчеркнуть, что здесь учитываются электростатические взаимодействия только
между молекулами растворителя и растворённого вещества.
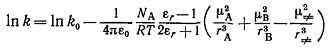 (13)
(13)
Здесь k -
константа скорости реакции в среде с диэлектрической проницаемостью εr, а k0 - константа скорости той же реакции в
конденсированной фазе с диэлектрической проницаемостью, равной единице.
Уравнение (13) показывает, что скорость реакции будет возрастать при повышении
диэлектрической проницаемости среды, если активированный комплекс более
биполярен, чем реагенты.
Основываясь на тех же допущениях, но слегка модифицировав модель
электростатического взаимодействия Кирквуда, Лейдлер и Ландскронер предложили
сходное уравнение (14), также описывающее скорость реакции между двумя
биполярными соединениями в среде с диэлектрической проницаемостью εr.
 (14)
(14)
Согласно уравнению (14), зависимость ln(k/k0) от 1/εr графически должна выражаться прямой,
наклон которой s определяется уравнением (15), т.е. величинами радиусов
и дипольных моментов реагентов и активированного комплекса.
 (15)
(15)
Если дипольный момент активированного комплекса μ≠ больше, чем дипольный момент μA и μB, то при повышении диэлектрической
проницаемости среды в соответствии с уравнением (14) константа скорости реакции
возрастёт. Причина этого заключается в том, что большая диэлектрическая
проницаемость среды благоприятствует образованию любых сильно биполярных
частиц, в данном случае активированного комплекса.
Другим подходом к расчёту влияния электростатической эффективности
растворителей на скорости реакций между биполярными молекулами воспользовался
Амис [2]. Он применил устанавливаемую известным уравнением Аррениуса
зависимость между константой скорости реакции и энергией активации k=A∙exp(-Ea/RT).
Согласно Амису, влияние диэлектрической проницаемости среды на скорость реакции
можно описать уравнением:
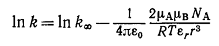 (16)
(16)
В этом уравнении k - константа скорости реакции в любой среде с диэлектрической
проницаемостью εr, k∞ - константа скорости реакции в среде с бесконечно большой
диэлектрической проницаемостью, а μA и μB - дипольные моменты двух биполярных
реагентов А и В в вакууме. В соответствии с уравнением (16) зависимость lnk от 1/εr выражается прямой, наклон которой
может служить мерой r=rA+rB, т.е. мерой расстояния
между двумя биполярными молекулами А и В, при котором между ними может начаться
реакция.
Впервые описываемая уравнением (13) зависимость константы скорости
реакции Менщуткина была обнаружена Гласстоном, Лейдлером и Эрингом, которые, в
частности, нашли, что в бинарной смеси растворителей бензол - этанол существует
линейная зависимость между ln(k/k0) и (εr-1)/(2εr+1), а в смеси бензол - нитробензол
наблюдается монотонное отклонение от линейной зависимости.
На рис. 1 представлена типичная зависимость ln(k/k0) от параметра Кирквуда (εr-1)/(2εr+1) для реакции Меншуткина между
триэтиламином и йодэтаном в бинарных смесях растворителей.
Бинарные смеси растворителей обладают тем преимуществом, что изменение их
состава отражается преимущественно на электростатических взаимодействиях между
растворителем и растворённым веществом, в то время как неэлектростатические и
специфические взаимодействия в целом ряде растворителей остаются практически на
постоянном уровне и поэтому могут не учитываться. Представленные на рис. 1
данные показывают, что уравнение Кирквуда применимо к таким системам
растворителей, но наклон прямых (характеризующих различные бинарные смеси)
заметно различается, хотя в соответствии с уравнением (13) все прямые должны
иметь один и тот же угол наклона, т.е. совпадать.
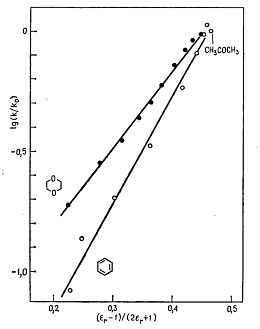
Рис. 1. − Корреляция между ln(k/k0) и функцией Кирквуда (εr-1)/(2εr+1) для реакции Меншуткина между
триэтиламином и йодэтаном при 40°С в бинарных смесях ацетон - бензол и ацетон -
1,4-диоксан (в качестве стандартного растворителя выбран ацетон, константа
скорости в котором принята равной единице) [1]
Как показывает изучении реакции Меншуткина, характер взаимодействия между
растворителем и растворёнными реагентами на самом деле более сложен, чем это
описывается уравнением (13). Очевидно, что одной функцией диэлектрической
проницаемости можно охарактеризовать зависимость скорости реакции от природы
растворителя только в некоторых особых случаях, например при проведении реакции
в бинарных смесях растворителей. По-видимому, при оценке эффектов растворителей
в реакции Меншуткина нужно учитывать не только электростатические, но и другие
взаимодействия, в том числе дисперсионные и обусловленные образованием
водородных связей [1, 6, 7].
2.3 Реакции
между нейтральными молекулами и ионами
Многие органические реакции относятся к ион-дипольным взаимодействиям.
Для константы скорости реакции между ионом А с зарядом zAe и
нейтральной биполярной молекулой В с дипольным моментом μВ, протекающей, согласно уравнению AzAe+В↔(АВ)≠
zAe→C+D, в среде с нулевой ионной силой,
Лейдлер и Эйринг предложили уравнение:
 (17)
(17)
Здесь k и k0 - константы скорости
реакции в среде с диэлектрической проницаемостью εr и 1 соответственно, а NA - число Авогадро.
Это уравнение должно особенно точно описывать скорость реакции в смесях двух
растворителей, в которых диэлектрическую проницаемость можно регулировать путём
изменения относительного содержания компонентов. Поскольку r≠ должно быть больше rA, то скорость реакции должна
несколько возрастать в среде с более низкой диэлектрической проницаемостью.
Для описания зависимости константы скорости реакции 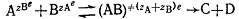 (в таких реакциях определяющую роль
играют электростатические взаимодействия) от диэлектрической проницаемости
среды Лейдлер и Ландскронер предложили несколько модифицированное выражение:
(в таких реакциях определяющую роль
играют электростатические взаимодействия) от диэлектрической проницаемости
среды Лейдлер и Ландскронер предложили несколько модифицированное выражение:

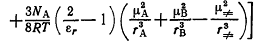 (18)
(18)
Если реакция осуществляется между двумя биполярными соединениями,
имеющими нулевой результирующий заряд, стоящее в квадратных скобках первое
слагаемое становится равным нулю; тогда эффект среды полностью описывается
вторым слагаемым и уравнение (18) преобразуется в уравнение (14). Если же в
реакции участвуют ион и биполярная молекула (или две биполярных заряженных
частицы), то необходимо учитывать оба слагаемых. Если рассматривается
простейшая реакция между однозарядным бесструктурным ионом А с зарядом zAe
(μA=0) и нейтральной молекулой В (zВe =0) с
дипольным моментом μВ, то уравнение (18) преобразуется в выражение
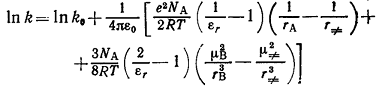 (19)
(19)
Другое уравнение, описывающее зависимость ln k от
диэлектрической проницаемости среды с помощью энергии электростатического
взаимодействия, предложил Амис [2]:
 (20)
(20)
Уравнение (20) применяли для анализа реакций биполярных соединений как с
положительными, так и отрицательными ионами. Типичными примерами сферы
применения уравнений (19) и (20) может служить изучение щелочного гидролиза
метилпропионата в водном ацетоне и SN2-реакция между азид-ионом и
1-бромбутаном в чистых полярных растворителях-НДВС [1, 2, 8].
2.4
Реакции между ионами
Реакции между простыми неорганическими ионами обычно протекают очень
быстро и скорость таких реакций зависит только от скорость диффузии ионов.
Существуют, однако, многочисленные реакции между ионами, включающие образование
и расщепление ковалентных связей или перенос электрона; скорость таких реакций
может не превышать скорости реакций между нейтральными соединениями. Здесь
будут рассматриваться только реакции второго типа.
При описании общих эффектов природы ионных реагентов и диэлектрической
проницаемости растворителя оказывается полезной электростатическая теория. В
соответствии с уравнением (21) изменение энергии Гиббса, сопровождающееся
образованием ионной пары из ионов А и В в стандартной среде с диэлектрической
проницаемостью εr, равно электростатической энергии сближения двух
точечных зарядов zAe и zBe на расстояние rAB (NA - число Авогадро).
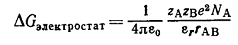 (21)
(21)
Из этого уравнения, применимого только в условиях бесконечного
разбавления, следует уравнение (22), описывающее зависимость константы скорости
реакции от диэлектрической проницаемости среды при нулевой ионной силе:
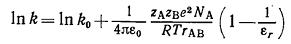 (22)
(22)
Здесь k и k0 - константы скорости реакции в среде
с диэлектрической проницаемостью εr и 1 (т.е. в газовой фазе)
соответственно, а rAB -
расстояние между ионами A и B, обеспечивающее их взаимодействие;
можно принять, что rAB=rA+rB. Согласно уравнению (22), впервые
предложенному Скэтчардом, между ln k и 1/εr существует линейная зависимость,
причём наклон соответствующей прямой положителен, если ионы имеют разноименные
заряды, и отрицателен, если заряды ионов А и В одноименны.
Лейдлер и Эйринг использовали несколько иную модель распределения зарядов
в активированном комплексе и для описания зависимости константы скорости
реакции AzAe+ВzВe↔(АВ)≠
(zA+zВ)e→C+D от диэлектрической проницаемости среды предложили уравнение
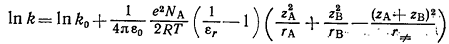 (23)
(23)
Уравнения (22) и (23) были проверены экспериментально, в частности, путём
изучения кинетики некоторых ионных реакций в ряду бинарных смесей растворителей
с различной диэлектрической проницаемостью. В целом эти уравнения хорошо
описывают зависимость скорости от свойств растворителя, хотя иногда, особенно в
средах с низкой диэлектрической проницаемостью, наблюдаются большие отклонения,
обусловленные, очевидно, ассоциацией ионов с разноименными зарядами. Отклонения
от предсказываемой теорией линейной зависимости можно объяснить также принятыми
при выводе этих уравнений серьёзными упрощениями, избирательной сольватацией
ионов в бинарных смесях растворителей и пренебрежением такими явлениями, как
взаимная поляризация ионов или бимолекулярных молекул и связанные с этим
индукционные взаимодействия и специфическая сольватация.
Так как коэффициенты активности реагентов зависят от ионной силы раствора
I, величина которой определяется молярными концентрациями ci
ионов i с зарядом zi 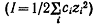 , то скорость реакции в растворе
должна зависеть и от его ионной силы. Описываемое уравнением (24) влияние
ионной силы раствора на константу скорости реакции называют первичным солевым
эффектом.
, то скорость реакции в растворе
должна зависеть и от его ионной силы. Описываемое уравнением (24) влияние
ионной силы раствора на константу скорости реакции называют первичным солевым
эффектом.
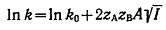 (24)
(24)
В этом уравнении А - константа, характеризующая данный растворитель при
данной температуре, а k0 - константа скорости реакции
при бесконечном разбавлении, определяемая путём экстраполяции. Уравнение
впервые было предложено Бренстедом, Бьеррумом и Христиансеном, которые для
решения проблемы влияния нейтральных солей на скорости реакций в растворах
воспользовались теорией Дебая - Хюккеля. Поскольку уравнение Дебая - Хюккеля
применимо только к разбавленным растворам, в которых ассоциацией ионов можно
пренебречь, то уравнение (24) оказывается справедливым только для растворов с
концентрацией 1-1-электролита не выше 10-2 моль∙л-1.
В заключение следует подчеркнуть, что электростатическая теория эффектов
растворителей оказалась очень полезной при изучении и расчёте кинетики
разнообразных реакций в растворах. Однако, несмотря на некоторые достижения
такого подхода, ему всё же присущ один принципиальный недостаток, обусловленный
пренебрежением множеством других типов взаимодействия растворителя с
растворёнными веществами, в том числе взаимной поляризацией ионов или
бимолекулярных молекул, специфической сольватацией и другими, а также
возможностью отклонения локальной микроскопической диэлектрической
проницаемости в непосредственном окружении реагирующих частиц от
макроскопической диэлектрической проницаемости среды [1, 5, 9, 10].
3. Влияние
специфической сольватации на скорости химических реакций
растворитель органический реакция кинетика
Одним из наиболее распространённых типов сольватации является
специфическая сольватация. Она осуществляется за счёт различного типа
комплексообразования между реагирующими частицами и молекулами растворителя.
Сюда относится образование водородных связей между реагирующими частицами и
растворителем, образование π-комплексов и других
донорно-акцепторных комплексов.
Такое определение специфической сольватации оказывается чрезвычайно ёмким
и, по существу, сближает сольватацию с областью гомогенного катализа. Так что
зачастую трудно провести грань между действием данного компонента химического
процесса как гомогенного катализатора или как растворителя, влияющего на
скорость реакции и её механизм за счёт простого комплексообразования.
Если на ранних стадиях развития представлений о роли среды в кинетике
химических реакций многие химики-исследователи пытались в основном приписать
все изменения констант скоростей реакций изменению полярности среды при
проведении реакции в различных растворителях, то в настоящее время оказалось,
что очень многие растворители влияют на процесс именно за счёт специфической
сольватации реагирующих частиц.
Способность растворителя образовывать водородные связи и π-комплексы чаще всего является более
важным фактором, чем полярность среды и, тем более, ванн-дер-ваальсово
взаимодействие.
Важным шагом в развитии представлений о роли среды являются работы [9], в
которых, в частности, на примере ряда реакций сольволиза трет-бутилхлорида
показано, что если механизм влияния двух растворителей на скорость реакции
одинаков, то константа скорости реакции в смешанном растворителе оказывается
связанной с соответствующими константами в индивидуальных растворителях
следующим соотношением:
lg kAB = N lgkA + (1-N) lg kB, (25)
где kAB, kA и kB - константы скорости реакции в
смешанном растворителе и в растворителях соответственно, а N - молярная доля компонента А в системе.
3.1 Роль
водородной связи в химической кинетике
Водородная связь оказывает огромное влияние на реакционную способность
соединений и в целом на механизм химических реакций. В качестве примера рассмотрим
данные по ингибирующему действию эфиров в реакциях алкилирования фенолов.
Падение скорости алкилирования фенола трет-бутилхлоридом, наблюдаемое при
введении в раствор диоксана и пропорциональное концентрации диоксана, можно
объяснить в предположении, что в системе образуются комплексы фенол-диоксан
(2:1), связанные водородной связью, и что фенол, входящий в комплекс, неактивен
и не подвергается алкилированию.
Можно сформулировать следующее правило, описывающее влияние растворителя
на скорость химической реакции вследствие образования водородных связей: если
активный цент, участвующий в реакции, блокируется водородной связью (или за
счёт другого взаимодействия с растворителем), то скорость реакции в этом
растворителе будет меньше, чем в отсутствие растворителя; если же растворитель
за счёт образования водородных связей (или иным путём) вызывает сдвиг
электронов, способствующий данной реакции, то скорость процесса в этом
растворителе будет увеличиваться.
В большинстве случаев, однако, реакционная способность соединений при
образовании водородных связей с растворителем уменьшается [1, 9, 11].
Заключение
Имеется ряд теоретических соотношений, описывающих влияние растворителя
на скорость химических реакций. Некоторые из этих соотношений описывают влияние
диэлектрической проницаемости растворителя на скорость реакций, другие -
влияние вязкости и иных свойств растворителя. Имеется также много эмпирических
уравнений, связывающих свойства растворителя со скоростями химических реакций в
этом растворителе.
В работе были рассмотрены некоторые качественные и количественные оценки
влияния растворителя на скорости реакций, а также роль сольватации в химической
кинетике.
Теоретические расчёты кинетики реакций в жидкой фазе затруднены в силу
того, что пока ещё не существует единой универсальной теории жидкого состояния.
До сегодняшнего дня не удалось точно определить характер сил взаимодействия,
объяснить перенос заряда между реагирующими молекулами и обусловленное этими
взаимодействиями изменение реакционной способности, а также роль реальной
структуры растворителя. В настоящее время единственной достаточно разработанной
теорией кинетики химических реакций в растворах является теория абсолютных
скоростей реакций, несмотря на то, что ей свойственны некоторые ограничения. В
соответствии с этой теорией при анализе эффектов растворителей необходимо
рассматривать индуцированную сольватацией относительную стабилизацию реагентов
и активированного комплекса.
Список
литературы
1. Райхардт
К. Растворители и эффекты среды в органической химии. Пер. с англ. - М.: Мир,
1991. - 763 с.
2. Амис
Э. Влияние растворителя на скорость и механизм химических реакций. Пер с англ.
/ Под ред. М.И. Кабачкина. - М.: Мир, 1968. - 328 с.
. Фадеев
Г.Н. Химические реакции: Пособие для учащихся. - М.: Просвещение, 1980. - 176
с.
. Сайкс
П. Механизмы реакций в органической химии. 4-е изд. Пер. с англ./ Под ред. В.Ф.
Травеня. - М.: Химия, 1991. - Пер. изд.: Великобритания, 1986. - 448 с.
. Лейдлер
К. Кинетика органических реакций. Пер. с англ. - М.: Мир, 1966.
. Пальм
В.А. Основы количественной теории органических реакций. - Изд. 2-е, перераб. и
доп. - Л.: Химия, 1977. - 360 с.
. Органическая
электрохимия: В двух книгах: Кн. 1 / Под ред. М. Бейзера и Х. Лунда. - Пер. с
англ./ Под ред. В.А. Петросяна и Л.Г. Феоктистова. - М.: Химия, 1988. - 469 с.
. Семиохин
И.А. Кинетика химических реакций: Учеб. пособие. / И.А. Семиохин, Б.В. Страхов,
А.И. Осипов. - М.: Изд-во МГУ, 1995. - 351 с.
. Эммануэль
Н.М. Роль среды в радикально-цепных реакциях окисления органических соединений.
/ Н.М. Эммануэль, Г.Е. Заиков, З.К. Майзус. - М.: Наука, 1973. - 279 с.
. Асланов
Л.А., Яценко А.В. Лекции по общей химии. Растворы и химическая кинетика. - М.:
Хим. фак. МГУ им. М.В. Ломоносова, 2004.
. Фиалков
Ю.Я. Растворитель как средство управления химическим процессом. - Л.: Химия,
1990. - 240 с.