Синтез и исследование алкилсалицилатных присадок
Министерство
образования и науки Украины
Национальный
технический университет Украины
"Киевский
политехнический институт"
Кафедра
органической химии и технологии органических веществ
Курсовая работа
Тема: "Синтез и исследование алкилсалицилатных присадок"
Выполнил:
студент 5-го курса
химико-технологического факультета
группы ХО - 11
Товмаченко Андрей Валентинович
Научные руководители:
к. х. н., с. н. с. Суховерхов В. Д.
к. х. н., доц. Черняев Б. В.
Киев - 2006
Реферат
В данной работе рассмотрены основные методы синтеза алкилсалицилатных
присадок Детерсол-50, Детерсол-140 и Детерсол-180.
Дана подробная характеристика каждого метода, приведён обзор патентов,
указаны особенности промышленного производства этих присадок. Проанализированы
механизмы ключевых стадий синтеза, указаны особенности применения
алкилсалицилатов. Кроме того, в работе приведена классификация
алкилсалицилатных присадок и описан механизм их действия. Экспериментальная
часть работы включает в себя лабораторный синтез Детерсолов, определение их
щёлочности и характеристику исходного сырья.
Введение
Масла, применяемые для смазки двигателей внутреннего сгорания, называют
моторными. В зависимости от назначения их подразделяют на масла для дизелей,
масла для бензиновых двигателей и универсальные масла, которые предназначены
для смазки двигателей обоих типов. Все современные моторные масла состоят из
базовых масел и улучшающих их свойства добавок, так называемых присадок,
представляющих собой химические соединения различных классов.
Моторные масла, в зависимости от назначения, должны выполнять следующие
функции:
образовывать устойчивую смазывающую плёнку, предотвращающую контакт и
износ трущихся деталей при любых условиях работы техники;
эффективно защищать детали двигателя от коррозии;
создавать уплотнение в зоне поршневых колец с целью сведения до минимума
проникновения продуктов сгорания в картер и масла в камеру сгорания;
обладать высокой устойчивостью к окислению при средних (80-120ºС) и высоких (250-300ºС) температурах;
предотвращать образование нагара на деталях цилиндро-поршневой группы
двигателя за счёт удаления с поверхности деталей смолистых образований, а также
шламов, образующихся в картере, за счёт их диспергирования в масле;
обладать вязкостно-температурной характеристикой, обеспечивающей
подвижность масла при минусовых температурах (возможность запуска двигателя) и
достаточной вязкостью при высоких температурах (для смазки верхнего поршневого
кольца);
иметь высокую стабильность против механической деструкции;
характеризоваться низкой испаряемостью;
защищать от ржавения детали двигателей при краткосрочной консервации;
не вспениваться.
Для обеспечения и улучшения перечисленных требований применяются
следующие присадки:
вязкостные (улучшают вязкостно-температурные свойства);
депрессорные (понижают температуру застывания);
антиокислительные (ингибируют окисление масла и образование смолообразных
продуктов этого процесса);
антикоррозионные (защищают цветные металлы подшипников от коррозионного
износа);
диспергирующие;
моющие (детергентные);
смазывающие (противоизносные и антизадирные);
антипенные;
антиржавейные.
Как указывалось выше, присадки представляют собой химические соединения
различных классов.
Так, антиокислительные присадки - это обычно пространственно
затруднённые алкилфенолы, производные дитиофосфорных кислот, аминосодержащие
соединения. Противоизносные присадки - это чаще всего серу-, азот-,
фосфорсодержащие соединения типа сульфидов, производных дитиофосфорных кислот,
дитиокарбаматы. Антикоррозионные присадки - чаще всего соединения
сульфонатного типа. Диспергирующие присадки - это высокомолекулярные
соединения полиалкилсукцинимидного типа или высокомолекулярные соединения
Манниха. Наиболее характерными моющими присадками являются
алкилсалицилаты; к таким присадкам также относятся металлсодержащие соединения
алкилфенольного и сульфонатного типов.
Целью данной работы является синтез и исследование зольных моющих
присадок алкилсалицилатного типа, обеспечивающих чистоту деталей двигателя при
высоких температурах.
Алкилсалицилаты являются важной составной частью моторных масел, потому
что эти вещества являются наиболее характерными представителями моющих
присадок. Кроме того, данные присадки нейтрализуют кислые соединения, которые
образуются при сгорании топлива в двигателе и попадают в картер в составе газов
сгорания. Тем самым алкилсалицилаты защищают детали двигателя от кислотной
коррозии.
Следует также отметить и то, что рассматриваемые присадки имеют
антиокислительное действие.
Высокие эксплуатационные свойства алкилсалицилатных присадок, их
стойкость к пресной и морской воде обеспечивают им широкую область применения,
в частности при создании масел для различных двигателей с тяжёлыми условиями
работы [54,55], для карбюраторных двигателей с высокой степенью сжатия, для высокофорсированных
дизелей, судовых дизелей с лубрикаторной смазкой, свободнопоршневых
дизель-компрессоров и ряда других объектов.
Литературный обзор
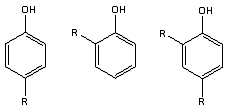
Алкилсалицилатные присадки являются солями или химическими производными алкилсалициловых
кислот. Исходными реагентами для синтеза этих кислот являются алкилфенолы,
имеющие хотя бы одно свободное орто-положение:
Введение карбоксильной группы в орто-положение ароматического ядра может
быть достигнуто несколькими путями.
Карбоксилирование ортометаллалкилфенолята диоксидом углерода
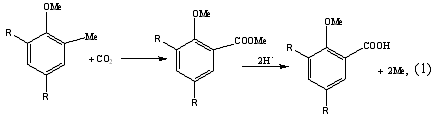
где Ме = Li, Na, K.
Взаимодействие с оксидом углерода:

Карбоксилирование алкилфенолятов металлов диоксидом углерода:
где Me = Na, K.
Алкилсалициловые кислоты можно получить и алкилированием салициловых
кислот [1]. Но из-за малого выхода продуктов алкилирования широкого применения
эта реакция не нашла.
Карбоксилирование ортометаллалкилфенолята диоксидом углерода
Металлирование органических соединений обычно проводят следующими
реагентами:
бутиллитием -C4H9Li,
амилнатрием -C5H11Na,
бутилкалием -C4H9K.
При этом могут замещаться на металл водород гидроксильной группы,
водороды ароматического кольца и алкильных групп.
Необходимо учитывать, что наличие заместителей в ароматическом ядре
изменяет его реакционную способность, причём она зависит как от химического
строения заместителя, так и от места замещения.
В литературе отсутствуют данные по металлированию алкилфенолятов, но
имеются данные по металлированию алкилбензолов [2,3], которые в известной мере
можно использовать при оценке металлирования алкилфенолятов. Установлено, что при металлировании литием или бутиллитием в
присутствии N, N, N`, N`-тетраметилэтилендиамина [4],
натрий- и калийорганическими соединениями [2,3] происходит замещение водорода
не только в ароматическом ядре, но и в алкильной цепи, так как металлированное
в кольцо производное подвергается перегруппировке в более стабильный
альфа-изомер. Карбоксилирование таких металлсодержащих соединений приведёт к
получению сложной смеси карбоновых кислот. В любом случае на металлирование
алкилфенолов будет расходоваться не менее двух молекул металлалкила. Если
учесть, что металлалкилы относительно дорогостоящие и малодоступные реагенты,
то становится очевидным, что синтез алкилсалициловых кислот через
металлорганические соединения в промышленном масштабе бесперспективен.
Взаимодействие с оксидом углерода
Реакция протекает с калиевыми соединениями и приводит к получению
п-оксибензойных, а не салициловых кислот. Условия реакции жёсткие: температура
230-260 ºС давление СО 8МПа, продолжительность
5 ч [5]. В зависимости от молекулярной массы алкилфенолов изменяется выход
алкилпараоксибензойных кислот от 80% от теории для крезолов до 20% от теории
для алкил(С14-С18)фенола.
Следовательно, данный метод получения оксибензойных кислот для крупного
промышленного производства является непреемлемлемым.
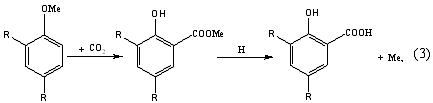
Карбоксилирование алкилфенолятов металлов диоксидом углерода
В ряде исследований [6-8] показано, что к преимущественному получению
салициловой кислоты приводит карбоксилирование фенолята натрия диоксидом
углерода. По имени авторов реакция названа реакцией карбоксилирования по
Кольбе-Шмидту. Эта реакция также лежит в основе получения алкилсалициловых
кислот. В связи с доступностью реагентов и умеренными условиями осуществления
реакции данный метод стал единственным в промышленном производстве
алкилсалициловых кислот.
Химизм и технологическое осуществление реакции Кольбе-Шмидта рассмотрены
ниже.
Классификация, состав и получение алкилсалицилатных присадок
Как уже было сказано ранее, алкилсалицилатные присадки подразделяются на
соли алкилсалициловых кислот и различные производные этих кислот. Рассмотрим
первый из этих классов в отдельности.
Соли алкилсалициловых кислот.
Этот класс присадок можно разделить на три группы, различающиеся по
химическому составу.
Группа 1. Низкощелочные ("нейтральные") соли
алкилсалициловых кислот, имеющие структуру:
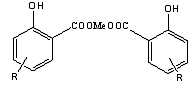
Они могут получаться любыми методами синтеза солей органических кислот,
но в основном по реакциям (4) и (5).

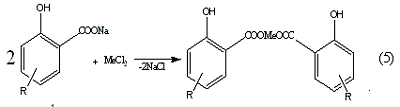
Так как алкилфенол содержится в исходных технических алкилсалициловых
кислотах, то он имеется и в готовой присадке.
Группа 2. Основные (среднещелочные) соли алкилсалициловых кислот плюс
другие металлсодержащие соединения имеют следующий химический состав:
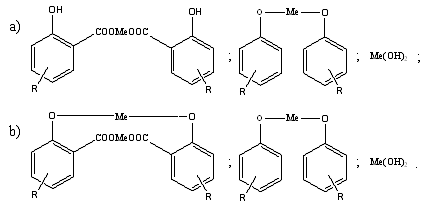
Добавочная щёлочность этих присадок достигается за счёт замещения
катионом металла гидроксильной группы либо алкилфенолов, либо и алкилфенола, и
алкилсалициловой кислоты. При этом в смеси остаётся некоторое количество
гидроксида металла.
Синтез этой группы присадок проводится, как правило, в две стадии. На
первой осуществляют либо реакцию (4), либо (5), а на второй - реакцию (6).
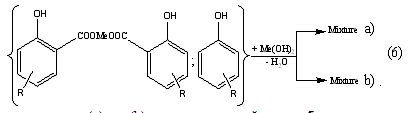
Получение состава (а) или (b)
зависит от условий синтеза.
Группа 3. Сверхосновные (высокощелочные) алкилсалицилатные присадки.
Их химический состав упрощённо можно представить в следующем виде:

Составы 1) и 2) получают совершенно различными методами синтеза, что
будет показано ниже.
В патентной литературе основность определяют по формулам либо (*) [9],
либо (**) [10]:
(а - в)/в*100%, (*)
где а - массовое содержание металла в неизвестной присадке; в
- массовое содержание металла в нейтральной соли.
Из этого следует, что основность нейтральной соли равна нулю.
((М/Е) - 1)*100%, (**)
где М - эквивалент металла; Е - эквивалент органической
кислоты на 100 г основной соли.
В отечественной литературе присадки оценивают по их щёлочности,
определяемой потенциометрически [11]. По этой методике устанавливают: общую
щёлочность, щёлочность, обусловленную алкилсалицилатом металла (нейтральной
соли), и щёлочность, обусловленную наличием в присадке гидроксида металла и его
углекислой соли. Следовательно, методика [11] более информативна.
Все описанные в патентах и изобретениях способы получения нейтральных,
основных и сверхосновных солей алкилсалициловых кислот приведены в табл. 1.
Применяются эти соли в виде 50-60%-ных масляных растворов для снижения вязкости
и улучшения растворимости в товарных маслах.
Получение нейтральных солей осуществляют элементарным взаимодействием
алкилсалициловых кислот с гидроксидом кальция или оксидом кальция при
температуре 80-100ºС в течение 30-90 мин. После завершения реакции легко
очищают присадку от механических примесей любым способом.
Основные алкилсалицилатные присадки, имеющие щёлочность выше суммы
щёлочности нормальных салицилатов и нормальных фенатов, получают из
карбоксилатов. Например [9] карбоксилат, растворённый в ксилоле и имеющий
температуру 75ºС, быстро смешивается с насыщенным
спиртовым раствором хлористого кальция, которого добавляют на 5%
больше, чем требуется для перевода натриевых соединений смеси в
соответствующие кальциевые соединения. Сразу же добавляется оксид кальция в
количестве на 10% больше, чем требуется для перевода нейтрального
алкилсалицилата кальция в основной, и смесь дополнительно разбавляется 96%-ным
этиловым спиртом. После непродолжительной выдержки при 75ºС температуру повышают до 100-105ºС и отгоняют спирт.
Реакционную смесь разбавляют бензином в два или в три раза, фильтруют,
удаляют бензин. В готовой присадке содержится 6,1% масс.кальция, тогда как в
нейтральной соли - 4,2% масс. Основность такого продукта равна ((6,1 -
4,2)/4,2)*100% = 45%. Напомним, что основность нейтральной соли равна нулю.
Таблица 1. Способы получения солей алкилсалициловых кислот.
|
Исходные реагенты
|
Технологические данные
|
Особенности способа
|
Ссылка на литер. источник
|
|
Алкилсалицилат-ное
соединение
|
Неорганическое соединение
|
Растворитель
|
Промотор
|
СО2
|
Т, ºС
|
Продолжит.,мин.
|
Число стадий
|
Способ ведения процесса
|
Способ очистки присадки
|
|
|
|
1
|
Алкил-салициловые
кислоты, полученные на хлорпарафина
|
Ме(ОН)n, где Ме -Al, Mg,
Na, Ca, Co
|
ROH
|
|
|
80
|
120
|
1
|
Периодический
|
Отстой в растворе светлого
минерального масла
|
|
[12-14]
|
|
2
|
Диизопропилсалициловая
кислота
|
Ме(ОН)n, где Ме - Cu, Mg,
Ca, Sr, Ba, Zn, Cd, Al, Sn, Pb, Mn, Ni, Co
|
Способ получения солей не
описан
|
В [12] испытан продукт
разложения после нагревания Zn
диизопропилсалицилата до 300ºС
|
[1517], [18,19]
|
|
3
|
Смесь: 75% масс. алкил(С8С20)
или алкил(С15С33)салицилата натрия плюс 25% масс.
соответствующих алкилфенолятов натрия
|
CaCl2,
CaO
|
Ксилол, CH3OH, C2H5OH,
бензин
|
|
|
75
|
60 - первая стадия, 240
вторая стадия
|
2
|
Периодический
|
Фильтрование
|
На первой стадии - обменная
реакция с CaCl2, на
второй - обработка CaO
|
[9]
|
|
4
|
Любое
|
Ca(OH)2,
CaO, Ba(OH)2*8H2O
|
Минеральное масло, керосин
|
СН3ОН
|
+
|
65 - на 1й стадии, 115132 -
на 2й стадии
|
25 - первая стадия, 10
вторая стадия
|
2
|
Непрерывный
|
Фильтрование через
фильтрующие порошки HiFlo, Super Sel 13 %
|
Вначале образование
комплекса оксида кальция с метанолом, затем взаимодействие с детергентом
|
[20]
|
|
5
|
Любое
|
CaO
|
Ксилольная фракция 120160
|
CH3OH
c 1% H2O
|
+
|
4957
|
2060
|
2
|
Непрерывный
|
Фильтрование через
фильтрующие порошки HiFlo, Super Sel 13 %
|
CaO диспергирован в метаноле. Первая стадия - быстрый
контакт реагентов, вторая - "дисперсионное твердение"
|
[21]
|
|
6
|
Калиевая соль полиизобутилен(С25)салицилата
в смеси с тетрапропиленсалициловой кислотой
|
CaCl2,
Ca(OH)2
|
Ксилол
|
Са(СH3COO)2*H2O
или 65/35=іC4H9OН/C5H11OH
|
+
|
60100
|
240300
|
4
|
Периодический
|
То же
|
Вначале обменная реакция с CaCl2 и затем три
последовательные карбонатации с промежуточным фильтрованием
|
[22]
|
|
6а
|
Тетрапропиленсалициловая
кислота
|
Ca(OH)2
|
Ксилол
|
СН3ОН
|
+
|
``
|
``
|
1
|
``
|
``
|
|
[22]
|
|
7
|
Натриевая соль алкил(С12С18)салициловой
кислоты
|
Алкилса-лицилат кальция, CaCl2, CaO
|
Ксилол, С2Н5ОН
|
|
|
70110
|
3060
|
2
|
Периодический
|
Фильтрование через фильтрующие
порошки HiFlo, Super Sel 13 %
|
На первой стадии обменная
реакция с CaCl2, на
второй - обработка активированной при 750ºС CaO
|
[23], [24],
[25], [26]
|
|
8
|
``
|
-``-
|
CH3OH
|
|
|
70110
|
3060
|
3
|
``
|
``
|
Третья стадия - кипячение в
метаноле 124 ч
|
[27]
|
|
9
|
Натриевая соль алкил(С12С18)салициловой
кислоты после повторного карбоксилиро-вания
|
-``-
|
``
|
|
|
70110
|
3060
|
3
|
``
|
``
|
``
|
[28]
|
|
10
|
Натриевая соль алкил(С12С18)салициловой
кислоты
|
CaCl2,
CaO, Na2CO3
|
CH3OH
|
|
|
70110
|
3060
|
3
|
``
|
``
|
Первая стадия - обработка Na2CO3,
вторая - обменная реакция с CaCl2,
третья - обработка CaO
|
[10], [29]
|
|
11
|
Натриевая соль алкил(С12С18)салициловой
кислоты
|
Na2CO3,
MgCl2, MgO
|
CH3OH
|
|
|
70110
|
3060
|
3
|
``
|
Любой
|
|
[30]
|
|
12
|
Алкил(С12С18)салициловая
кислота
|
Ca(OH)2
|
Ксилол
|
CH3OH
|
+
|
2060
|
3060
|
3
|
Периодический
|
Фильтрование через
фильтрующие порошки HiFlo, Super Sel 13 %
|
Первая стадия - получение
кальциевой соли, вторая - получение дисперсии CaCO3 в метаноле, третья - смешение и выдержка
|
[31]
|
|
13
|
Алкил(С12С18)салициловая
кислота
|
Ca(OH)2
|
Ксилол
|
CH3OH
|
+
|
2060
|
3060
|
4
|
Периодический
|
``
|
То же, но после смешения -
повторная карбонатация
|
[32]
|
|
14
|
Алкил(С3С18)салициловая
кислота
|
-``-
|
``
|
|
|
60100
|
60120
|
1
|
``
|
Любой
|
|
[19], [33]
|
|
15
|
Алкил(С12С20)салициловая
кислота
|
Ca(OH)2,
CaO
|
|
|
|
90180
|
|
|
Непрерывный
|
Любой
|
|
[34]
|
|
16
|
Алкил(С16С18)салициловая
кислота
|
Ca(OH)2
|
Ксилол
|
CH3OH
|
+
|
80
|
180
|
2
|
Периодический
|
Очистка на центробежных
машинах
|
Первая стадия - получение
кальциевой соли, вторая карбонатация
|
[35]
|
|
17
|
Натриевая соль алкил(С16С18)салициловой
кислоты
|
CaCl2,
Na2CO3
|
Ксилол, вода
|
|
|
90
|
120
|
``
|
``
|
``
|
Первая стадия - обработка Na2CO3,
вторая - обменная реакция с CaCl2
|
[36], [37], [38]
|
|
18
|
Алкилсалициловые кислоты,
полученные на базе крекинголефинов, фр. 240320ºС (С9С22)
|
Сa(OH)2
|
Бензин
|
CH3OH
|
+
|
4050
|
40
|
2
|
Периодический
|
Очистка на центробежных
машинах
|
Перед карбонатацией
добавляют NH3 или
соли аммония, карбонатация проводится под давлением 0,01 МПа
|
[39]
|
|
19
|
``
|
-``-
|
``
|
|
+
|
30
|
1
|
``
|
``
|
Карбонатация проводится под
вакуумом 730320 мм. рт. ст.
|
[40]
|
|
20
|
Аммонийная соль алкил(С9С22)салициловой
кислоты
|
-``-
|
``
|
``
|
+
|
3040
|
30
|
1
|
``
|
``
|
|
[41]
|
|
21
|
Алкил(С9С22)салициловая
кислота
|
Ba(OH)2*8H2O
|
``
|
|
|
60130
|
30300
|
2
|
``
|
``
|
Первая стадия -
барирование, вторая - обработка окисью алюминия или алюмосиликатом 13% масс.
|
[42]
|
|
22
|
``
|
Ca(OH)2
|
``
|
CH3OH
|
+
|
3040
|
30
|
1
|
``
|
Электрофорез
|
Перед очисткой добавляют
воду, а потом на присадку воздействуют неоднородным электростатическим полем
напряжённостью 0,15 кВт/см
|
[43]
|
Найдено, что в процессе обменной реакции конверсия алкилфенолята
натрия в очень сильной степени промотируется алкилсалицилатом кальция [25]. Как
правило, используют суспензию оксида или гидроксида кальция в этиловом или
метиловом спиртах (см. таблицу 1). Предложено [25] после обработки карбоксилата
хлоридом кальция в продукт добавлять NaOH, а затем CaCl2, в результате чего образуется
гидроксид кальция in situ в тонкодисперсном состоянии.
Допускается вариант обработки суспензией гидроксида кальция, добавленной в
насыщенный спиртовый раствор CaCl2. Очень
реакционноспособен CaO, полученный
прокаливанием Ca(OH)2 в течение 4-8 ч при 700-750 ºС.
Все основные алкилсалицилаты имеют пониженную стабильность [27] и при
стоянии из них выпадает гель. Для его разрушения и предотвращения выпадения
присадку обрабатывают метанолом [27] в количестве 15-50% на присадку при
температуре 70 ºС в течение 60-1440 мин. После обработки при стоянии
образуются две фазы: верхняя - метанольная, нижняя - углеводородная, содержащая
присадку. Фазы разделяют, из углеводородного слоя под вакуумом удаляют следы
метанола. Затем присадку разбавляют ксилолом, центрифугируют и, наконец,
отгоняют ксилол, получая готовый стабильный продукт.
Рассматриваемый метод имеет ряд преимуществ перед
современной схемой синтеза алкилсалицилатов: использование обменной реакции
между алкилсалицилатом натрия и солью щелочноземельного металла позволяет
исключить из процесса применение соляной кислоты, стадию разложения и промывки
и уменьшить количество механических примесей при получении присадок АСК и АСБ.
Однако "метод обменной реакции" имеет и свои недостатки: в частности,
полученная по нему присадка содержит в качестве примеси соли CaCl2 и NaCl,
что нежелательно.
Принципиальная возможность такого способа получения алкилсалицилатов
щелочноземельных металлов подтверждается рядом работ [56-58], в которых описано
получение нейтральных солей, т. е. продуктов, не имеющих избыточной щёлочности
и не обладающих вследствие этого необходимыми эксплуатационными свойствами.
Получение нейтральных солей объяснялось авторами как результат реакции двойного
обменного разложения солей. Более верным, на наш взгляд, является рассмотрение
этого процесса как ионного обмена на жидком ионите.
Проведёнными электронномикроскопическими исследованиями было установлено,
что раствор алкилсалицилата натрия в масле имеет коллоидный характер. В
процессе обменной реакции алкилсалицилата натрия с водным раствором соли
щелочноземельного металла должна происходить диссоциация полярных групп
алкилсалицилата натрия на поверхности раздела фаз углеводородный раствор -
водный раствор.
В отличие от растворов электролитов в данной системе подвижность
заряженных групп, связанных с углеводородным радикалом, будет ограничена.
Поэтому электростатическое отталкивание не может привести к равномерному
распределению ионов обоих знаков по всему объёму, как это происходит в растворе
обычных электролитов [59].
Коллоидный характер раствора алкилсалицилата натрия и наличие в его
молекуле способной к диссоциации функциональной группы позволяет отнести его к
классу жидких ионитов, что подтверждается и некоторыми литературными данными
[60].
Как известно, иониты способны не только к обмену противоионов молекулы,
но и к сорбции ионов и нейтральных молекул. В обменную реакцию могут вступать
как противоионы молекул ионита, так и сорбированные ионы, причём оба вида
обменных ионов продолжают удерживаться ионитом.
Таким образом, обменной реакцией можно получить соли алкилсалициловых
кислот с избыточной щёлочностью, если создать необходимый избыток щёлочности в
исходном алкилсалицилате натрия.
Эта возможность подтверждена экспериментальными данными [61].
Синтезированные образцы алкилсалицилата натрия с различной величиной
избыточной щёлочности в виде сорбированных молекул карбоната натрия
подвергались обменной реакции с хлоридами щелочноземельных металлов. В
результате получены алкилсалицилаты металлов, обладающие избыточной щёлочностью
в виде карбоната щелочноземельного металла.
Принципиально отличным путём увеличения щёлочности алкилсалицилатных
присадок является процесс карбонатации, который впервые детально
изложен английскими исследователями в 1957 году [31]. Cуть карбонатации состоит в получении устойчивой дисперсии
карбоната щелочноземельного металла в масле.
Обычно карбонатацию проводят так. В суспензию гидроксида кальция в смеси
масла, бензина (ксилола), метанола (или другого промотора) подают при температуре
20-60 ºС СО2. Основность продукта
при этом возрастает за счёт образования коллоидной дисперсии СаСО3,
достигает максимума, а затем резко падает.
Это явление получило название "перекарбонатация" [31,45] и
является нежелательным, поскольку приводит к выпадению геля, что проявляется в
снижении фильтруемости присадки. Вероятность перекарбонатации продукта снижает
избыточное количество Са(ОН)2. Эту задачу в [32] решают повторной
карбонатацией: в этом случае используют дополнительное количество Са(ОН)2
или СаО.
Химизм и технологическое осуществление процесса карбонатации рассмотрены
ниже.
В данной работе детально рассматриваются алкилсалицилатные
присадки Детерсол-50 (старое название - АСК
(алкилсалицилат кальция)), Детерсол-140 (старое название - МАСК
(многоосновный алкилсалицилат кальция)) и Детерсол-180.
Современное производство этих присадок является многостадийным процессом,
включающим следующие химические реакции:
1.) получение алкилфенолов алкилированием фенола олефинами;
2.) получение алкилфенолята натрия взаимодействием алкилфенола с
гидроксидом натрия;
.) получение алкилсалицилата натрия взаимодействием алкилфенолята
натрия с углекислым газом (реакция Кольбе-Шмидта);
.) получение алкилсалициловых кислот разложением алкилсалицилата
натрия минеральной кислотой;
.) получение алкилсалицилата кальция (присадки Детерсол-50);
.) получение высокощелочных алкилсалицилатов кальция (присадок
Детерсол-140 и Детерсол-180).
1.
Получение алкилфенолов.
Получение алкилфенолов осуществляют алкилированием фенола олефинами. В
качестве олефинов используют олефины фракции 240-320ºС термокрекинга парафинов и олигомеры
этилена состава С16-С18, получаемые олигомеризацией
этилена. Состав олефинов термокрекинга парафинов и олигомеров этилена состава С16-С18
приведён в таблицах 2 и 3.
Таблица 2.
Состав олефинов фракции 240-320ºС термокрекинга парафинов.
|
Углеводороды
|
Парафины
|
Олефины
|
|
|
Всего олефинов
|
С9
|
С10
|
С11
|
С12
|
С13
|
С14
|
С15
|
С16
|
С17
|
С18
|
С19
|
|
Содерж. в % масс.
|
39,1
|
60,9
|
0,1
|
0,6
|
0,9
|
2,4
|
6,3
|
11,4
|
13,0
|
11,4
|
8,0
|
4,8
|
2,0
|
Таблица 3.
Состав олигомеров этилена С16-С18.
|
Углеводороды
|
Парафины
|
Олефины
|
|
|
Всего олефинов
|
По длине
|
По строению
|
|
|
|
С16
|
С18
|
С14 и ниже
|
С20 и выше
|
α-олефины
|
Винили-деновые
|
С внутр. двойной связью
|
|
Содерж. в % масс.
|
4
|
96
|
57-67
|
28-38
|
До 2
|
До 3
|
До 40
|
До 40
|
До 20
|
Химизм процесса
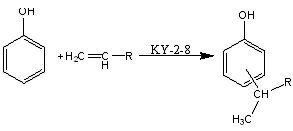
Реакция
алкилирования является реакцией электрофильного замещения и катализируется
катализаторами кислотного типа: минеральные и органические кислоты, галогениды
металлов, катионнообменные смолы, алюмосиликаты. В качестве катализаторов
алкилирования фенола высшими олефинами описаны также хлораты металлов,
триэтилаллюминий, молекулярный йод, фенолят алюминия и другие. Широкое
применение в производстве присадок, при алкилировании фенола высокомолекулярными
олефинами получила также бензолсульфокислота (далее БСК). Её каталитическая
активность близка к активности H2SO4 и AlCl3. Будучи сильной кислотой,
БСК не обеспечивает селективность процесса алкилирования. В её присутствии
образуется около 30% диалкилфенолов, главным образом 2,6-замещённых.
Существенным недостатком процесса алкилирования фенолов в присутствии
растворимых катализаторов (минеральных и органических кислот, галогенидов
металлов и их молекулярных соединений) является необходимость отмывки продуктов
реакции от катализатора. При этом образуется значительное количество фенольных
сточных вод и возникает проблема их очистки, имеют место повышенные потери
продукта. Применение нейтрализующих средств, например, аммиака и удаление
катализатора в виде аммонийных солей фугованием не всегда обеспечивает полную
очистку продуктов алкилирования от катализатора. Указанных недостатков лишён
метод алкилирования фенолов с применением гетерогенных катализаторов
(катионообменные смолы, алюмосиликаты, синтетические цеолиты и др.). К тому же
указанные катализаторы являются менее сильными кислотами, что в значительной
мере обеспечивает селективность процесса. В настоящее время алкилирование
фенола олефинами в промышленности осуществляют в реакторах колонного типа на кислотном
гетерогенном катализаторе сульфокатионите КУ-2-8, представляющим собой
сульфированный сополимер стирола и дивинилбензола. В результате набухания
полимера молекулы фенола и олефинов проникают внутрь зёрен, где у активных
центров (сульфогрупп) протекает алкилирование. Сульфокатионит в отсутствие
полярного растворителя (например, воды) не способен протонировать молекулы
олефинов, а следовательно, не может катализировать реакцию алкилирования. В
нашем случае среда безводная, однако роль полярного растворителя здесь играет
фенол. Молекулы фенола абсорбируются катионитом, и внутри зёрен последнего
происходит обмен протонами между SO3H-группой этого катионита и ОН-группой фенола. Именно
этот обмен протонами и делает возможным протонирование молекул олефинов, а
следовательно и протекание реакции алкилирования на сульфокатионите. Поскольку
катионит лучше сорбирует фенол, чем олефины, то внутри зёрен создаётся избыток
фенола, что обуславливает образование преимущественно
моноалкилфенолов.Количество функциональных групп на поверхности катионита
ничтожно мало, в связи с этим ионно-каталитические процессы происходят, главным
образом, внутри гранул, чему способствует набухание смол в реагирующих
компонентах. Повышение температуры увеличивает скорость алкилирования, что
связано с ростом скорости диффузии реагентов внутрь гранул катионитов и
увеличением подвижности протонов сульфогрупп. Так, при повышении температуры от
95 до 130 ºС скорость алкилирования увеличивается в 4 раза.
Показано, что при повышении температуры от 130 до 150 ºС выход алкилфенолов увеличивается от 64 до 81% с
повышением содержания дизамещённых. Количество последних зависит от соотношения
фенола и олефинов: с повышением доли фенола образование диалкилпроизводных
снижается. Продолжительность контакта реакционной смеси с катионитом определяет
глубину превращения. Алкилирование проводят при температуре 120-140 ºС и мольном соотношении реагентов фенол : олефины 1,5÷2 к 1. Перед проведением реакции фенол и олефины
должны быть тщательно обезвожены. Это необходимо потому, что при температуре
120-140 ºС вода гидролизует катионит с образованием серной
кислоты.  Выделившаяся серная кислота катализирует гомогенное
алкилирование, что приводит к снижению селективности процесса и образованию
кроме целевых моноалкилфенолов ещё и побочных продуктов: ди- и триалкилфенолов.
Поэтому гидролиз катионита нежелателен и реагенты должны быть максимально
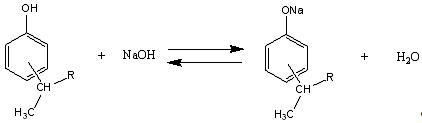
обезвожены. 2.Получение алкилфенолята натрия.Алкилфенолят натрия
получают взаимодействием алкилфенола с гидроксидом натрия. Как и всякая реакция
нейтрализации, эта реакция сопровождается выделением небольшого количества
тепла (до 2,4 ккал/моль).
Выделившаяся серная кислота катализирует гомогенное
алкилирование, что приводит к снижению селективности процесса и образованию
кроме целевых моноалкилфенолов ещё и побочных продуктов: ди- и триалкилфенолов.
Поэтому гидролиз катионита нежелателен и реагенты должны быть максимально
обезвожены. 2.Получение алкилфенолята натрия.Алкилфенолят натрия
получают взаимодействием алкилфенола с гидроксидом натрия. Как и всякая реакция
нейтрализации, эта реакция сопровождается выделением небольшого количества
тепла (до 2,4 ккал/моль).
Химизм процесса.
Процесс
проводится при температуре 100-125 ºС в реакторе с мешалкой. Используется 42%-ный водный раствор щёлочи.
Обезвоживание полученного алкилфенолята натрия осуществляют в двух роторных
плёночных испарителях, в которые подаётся азот (первая ступень обезвоживания),
а затем в циркуляционных плёночных реакторах, соединённых последовательно
(вторая ступень обезвоживания). Отпаривание влаги идёт при t=160 ºС и атмосферном давлении в токе азота, который
подаётся в нижнюю часть реактора. Полное обезвоживание достигается благодаря
многократной циркуляции алкилфенолята в каждом из реакторов.
3.Получение
алкилсалицилата натрия.
Алкилсалицилат
натрия получают карбоксилированием алкилфенолята натрия диоксидом углерода
(реакция Кольбе-Шмидта). Химизм процесса.Продуктом целевой
реакции Кольбе-Шмидта является в основном алкил-о-гидроксибензоат
(алкилсалицилат) натрия, образуются лишь следы пара-изомера. Однако если
реакцию проводить с алкилфенолятом калия, то основным продуктом является
алкил-п-гидроксибензоат. Поэтому в нашем производстве использование
алкилфенолята калия недопустимо. Предполагают, что преимущественное образование
алкил-о-гидроксибензоата при использовании алкилфенолята натрия связано с тем,
что образующееся в результате атаки орто-положения переходное состояние
стабилизировано в результате образования хелатного соединения в виде ионной
пары (см. ниже):
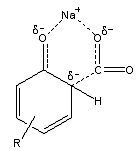
Размеры
катиона К+ больше, и, вероятно, он менее эффективен в качестве
хелатирующего агента, так что атака пара-положения становится поэтому более
предпочтительной [47]. Способность алкилфенолята натрия образовывать хелаты (σ-комплексы) с полярными соединениями (СО2, Н2О,
ацетон) обуславливает наличие в его молекуле группы -ONa. Реакция
образования хелата экзотермическая, её тепловой эффект равен 17,0 ккал/моль.
Так, при взаимодействии с СО2 при 20 ºС температура алкил(С9-С20)фенолята
натрия повышается на 20 ºС. При взаимодействии с СО2 фенолята натрия
выделяется тепло, равное 18,2 ккал/моль.Именно способность образовывать хелаты
вызывает необходимость максимального обезвоживания алкилфенолята. Влага прочно
удерживается из-за замыкания в кольцо координационными связями концевых атомов
воды. Образование таких структур препятствует протеканию реакции Кольбе-Шмидта,
потому что хелат алкилфенолята с водой более прочен, чем с СО2. В
итоге вода не даёт части молекул алкилфенолята вступить в реакцию, благодаря
чему снижается степень превращения его в алкилсалицилат и выход последнего. По
этой же причине снижается выход алкилсалициловых кислот при карбоксилировании в
полярных растворителях. Ацетон, например, способен количественно вытеснить СО2
из хелатов фенолята и алкилфенолята натрия. Летучие неполярные растворители
снижают выход незначительно из-за снижения парциального давления СО2
в системе. Нелетучие неполярные растворители, снижая вязкость алкилфенолята
натрия, способствуют увеличению степени превращения последнего в
алкилсалицилат. Природа растворителя оказывает значительное влияние на процесс
карбоксилирования незамещённых фенолятов натрия. Для высших алкилфенолятов это
влияние выражено не столь резко. Отмечается, что в присутствии карбоната
кальция реакцию между фенолом и двуокисью углерода проводят в присутствии воды.
Наиболее оптимальные условия проведения реакции Кольбе-Шмидта для
алкилфенолятов натрия таковы: температура 130 ºС, давление 0,5-2 МПа, продолжительность реакции - не
менее 4 ч. При этих условиях наблюдается минимальное образование побочных
продуктов при выходе алкилсалицилата натрия 60-75% от теории. Чем выше
температура реакции, тем выше должно быть давление СО2. В
промышленности процесс карбоксилирования алкилфенолята натрия обычно проводят
периодически в последовательно соединённых герметичных реакторах с интенсивным
перемешивающим устройством. В рубашки реакторов предусмотрена подача воды для
охлаждения. Описана непрерывная схема карбоксилирования в насадочной колонне с
последующей выдержкой карбоксилируемой смеси в пустотелой горизонтальной
ёмкости с рубашкой для обогрева.
Целевая
реакция:
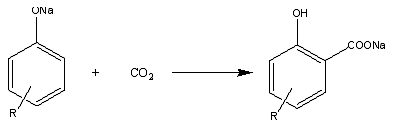
Побочные
реакции:

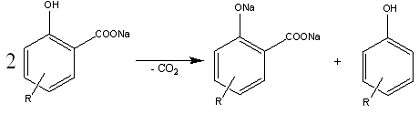
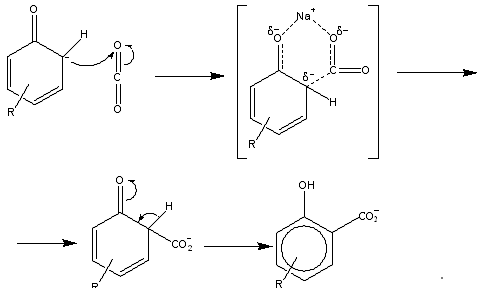
Механизм целевой реакции [47]:
Примечание. При протекании целевой реакции СО2
действует как электрофил, атакуя фенолят натрия.
4. Получение алкилсалициловых кислот разложением алкилсалицилата натрия
минеральной кислотой.
Как
правило, карбоксилат для снижения его вязкости перед разложением разбавляют
углеводородным растворителем (бензином) в соотношении 1 : 1. В качестве
минеральной кислоты чаще всего используют водный раствор соляной (реже серной)
кислоты.
Химизм процесса.
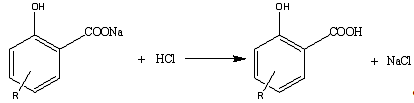
В
реальных условиях разложение алкилсалицилата натрия (далее АСН) осуществляют в
герметическом реакторе с интенсивным перемешивающим устройством. В этот реактор
подаётся АСН и вода в соотношении АСН : Н2О = 1 : 4 и при tº=70-90 ºС происходит перемешивание. Образующийся "раствор" АСН и
27-36%-ная водная соляная кислота насосом подаются в последовательно работающие
колонны, где идёт разделение водного и органического слоёв. После
карбоксилирования переход к алкилсалициловым кислотам можно осуществить также
обработкой карбоксилата катионообменной смолой в Н-форме с содержанием воды в
пределах 10-50% масс. при температуре 40-90 ºС. Для ускорения процесса обработку катионитом можно вести в
присутствии бензина. Н-форму катионита предложено восстанавливать
электрохимическим способом.
5. Получение алкилсалицилата кальция (присадки Детерсол-50).Получение присадки Детерсол-50 осуществляют
взаимодействием алкилсалициловых кислот с суспензией гидроксида кальция в масле
АС-6 при температуре 75-85 ºС в течение 60-90 мин при атмосферном давлении. Процесс
проводится периодически в реакторе с перемешивающим устройством. После
проведения процесса суспензия разбавляется бензином-растворителем для снижения
вязкости.
Химизм процесса.

6.Получение высокощелочных алкилсалицилатов кальция (присадок
Детерсол-140 и Детерсол-180).
Получение
присадок Детерсол-140 и Детерсол-180 осуществляется путём карбонатации
алкилсалициловых кислот.
Химизм процесса.
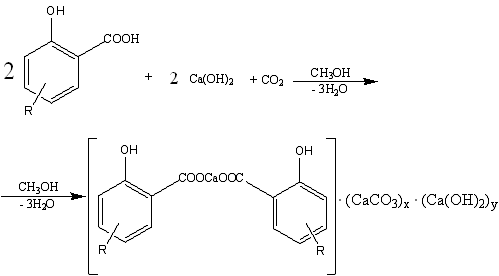
Карбонатация
осуществляется непрерывно в реакторе с гребным винтом. В низ реактора
карбонатации подаются алкилсалициловые кислоты в виде раствора в бензине и
суспензия гидроксида кальция в масле. Сюда же насосом подаётся метанол, который
является промотором реакции, вода и СО2. Температура реакции 40-50 ºС, давление 0,3 атм, продолжительность - 40-60 мин.
Отгон метанола и азеотропом с ним - части реакционной воды производится
непрерывно в испарителе при температуре 50-60 ºС и остаточном давлении 100-250 мм. рт. ст. После
этого для снижения вязкости в суспензию добавляется бензин-растворитель.
Процесс карбонатации может осуществляться также и периодически в реакторах с
мешалкой.Определение состава алкилсалицилатных присадок.Предложено [48]
содержание активного металлсодержащего компонента алкилсалицилатных присадок
определять методом диализа. Этим методом определено, что присадка АСК
содержит металлсодержащего компонента 41,6%, присадка МАСК - 38,5%, присадка
АСБ - 35,9%.Однако этот метод имеет малую точность. В связи с этим состав
алкилсалицилатов предложено [49,50] определять с помощью жидкостной
хроматографии. В процессе жидкостного хроматографического разделения
неорганическая часть присадки остаётся на адсорбенте, а элюируются с адсорбента
минеральное масло, свободные алкилфенолы и "нейтральные" соли
алкилсалициловых кислот. Данный метод нашёл широкое применение при контроле
качества товарных алкилсалицилатных присадок. Техническими требованиями
установлены нормы на содержание в этих присадках свободных алкилфенолов (не
более 20% масс.) и "нейтральных" алкилсалицилатов (активного вещества
не менее 25% масс.).Методом зонного электрофореза можно установить
наличие в средне- или высокощелочной алкилсалицилатной присадке алкилфенолятов
металлов [51]. В этом методе учитывается величина разделения при тонкослойном
электрофорезе, основанном на буферных системах (этанол-вода-борная
кислота-ацетат натрия) в сочетании с хроматографией. Предложено диализ сочетать
с методами жидкостной [52] и тонкослойной хроматографии [53].
Последний вариант позволяет качественно установить наличие алкилсалицилатов,
которые при проявлении окрашиваются в тёмно-голубой флюоресцирующий цвет, и
алкилфенолятов, окрашивающихся в чёрный цвет.
Экспериментальная часть
зольный моющий присадка алкилсалицилатный
В
качестве исходного продукта для получения алкилсалицилатных присадок
Детерсол-50, Детерсол-140 и Детерсол-180, были использованы технические
алкилсалициловые кислоты с алкильными радикалами С16 - С18
(олигомеров этилена), произведённые на основе алкилфенолов, которые в свою
очередь были получены алкилированием фенола олефинами на сульфокатионите КУ-2-8
чс.
Опыт
1. Определение активного числа исходных алкилсалициловых
кислот.
Исходные
или товарные (технические) алкилсалициловые кислоты представляют собой смесь собственно
алкилсалициловых кислот, алкилфенолов, не вступивших в реакцию
Кольбе-Шмидта, и углеводородов.
Эти
технические алкилсалициловые кислоты имеют кислотное число 103,8 мг КОН/г.
Идея
опыта состоит в том, чтобы отделить собственно алкилсалициловые
кислоты от алкилфенолов и углеводородов и определить
процентное содежание этих кислот в смеси. Отделение проводим при комнатной
температуре на хроматографической колонке, заполненной прокаленным при температуре
180 ºС в течение пяти часов оксидом алюминия (элюэнт -
смесь С2Н5ОН и бензола в объёмном соотношении 1:1). В
условиях опыта собственно АСК "сядут" на адсорбент и будут на
нём удерживаться, а алкилфенолы и углеводороды вымоются из
колонки элюэнтом. Достичь такого результата помогает этанол: он дезактивирует Al2O3 и таким образом не позволяет адсорбироваться на нём
обычной спиртовой ОН-группе. Таким образом С2Н5ОН не
позволяет осесть на адсорбенте алкилфенолам.
Методика
проведения опыта такова. Набиваем колонку оксидом алюминия и вносим в неё
навеску исходных алкилсалициловых кислот, предварительно растворённую в
гексане. Затем гексан уходит с низа колонки, а разделяемая смесь
"высаживается на адсорбент". Далее проводим вымывание элюэнтом алкилфенолов
и углеводородов из разделяемой смеси. Вымываем до тех пор, пока
вытекающая с низа колонки жидкая фаза не перестанет содержать вымываемых
веществ (проверяем по пробе на предметном стекле).
После
окончания вымывания полученный раствор смеси алкилфенолов и углеводородов
в спирто-бензольном элюэнте упаривают, получая при этом чистую смесь алкилфенолов
и углеводородов. Эту чистую смесь взвешивают и затем рассчитывают все
необходимые показатели.
Проводят
две пробы параллельно.
Результаты
параллельных проб приведены в таблице 4.
Таблица
4.
|
Показатели
|
Проба 1
|
Проба 2
|
|
Вес стакана без навески
|
9,3880 г
|
25,2127 г
|
|
Вес стакана с навеской
|
9,6738 г
|
25,5125 г
|
|
Вес навески
|
0,2858 г
|
0,2998 г
|
|
Вес стекла
|
34,5546 г
|
29,2005 г
|
|
Вес стекла с вымытым и
упаренным остатком
|
34,6580 г
|
29,3067 г
|
|
Вес вымытого и упаренного
остатка
|
0,1034 г
|
0,1062 г
|
|
Всего вымылось
|
(0,1034 г/0,2858 г)*100%
=36,179%
|
(0,1062 г/0,2998 г)*100% =
35,424%
|
|
Кислот в исходном образце
было
|
100% - 36,179% = 63,821%
|
100% - 35,424% = 64,576%
|
Сходимость проб = ((64,576% - 63,821%)/64,576%)*100% = (0,755%/64,576%)*100%
= 1,1692%.
Среднее по результатам двух параллельных опытов содержание алкилсалициловых
кислот в исходном образце = (63,821% + 64,576%)/2 = 64,199%.
Опыт 2. Получение присадки Детерсол-50.
В литровый трёхгорлый реактор с механической мешалкой, термометром и
обратным холодильником загружаем известь-пушонку, раствор алкилсалициловых
кислот в бензине, предварительно разбавленный маслом М-6. Включаем
перемешивание и нагрев. Нагреваем реакционную массу до tº=75-85ºC и перемешиваем при этой температуре
в течение 60-90 мин. По мере перемешивания реакционная смесь темнеет, что
свидетельствует о протекании реакции.
После окончания срока перемешивания реакционной смеси дают остыть, а
затем с целью очистки от непрореагировавшей пушонки её центрифугируют. После
этого от реакционной смеси отгоняют бензин. Делают это либо на роторном
испарителе, либо на приборе с одногорлым реактором, термометром и прямым
холодильником. Во втором случае отгон ведём до температуры 150ºC (именно при этой температуре бензин
отгоняется полностью).
После завершения отгона бензина получаем товарный Детерсол-50 в виде
тёмной вязкой жидкости.
Загрузка.
Известь-пушонка 5,6 г.
Раствор алкилсалициловых кислот в бензине 163,9 г.
Масло М-6 73,1 г.
Расчёт массы масла М-6, необходимой для разбавления раствора
алкилсалициловых кислот в бензине.
Обозначим алкилсалициловые кислоты как АСК.
На 100 г товарных АСК необходимо загрузить 5,6 г Са(ОН)2.
Используемый раствор АСК в бензине содержит 39% масс. бензина и 61% масс.
товарных АСК. Составляем пропорцию.
В 100 г раствора - 61 г товарных АСК.
В х г раствора - 100 г товарных АСК.
Отсюда х = (100 г * 100 г)/61 г = 163,9 г.
Значит, в реактор необходимо загрузить 163,9 г раствора АСК в бензине.
Масса бензина в этом растворе равна (163,9 г - 100 г) = 63,9 г.
Используемые товарные АСК имеют кислотное число, равное 103,85. Для
получения Детерсола-50 кислотное число (далее КЧ) АСК необходимо снизить до
значений 50-60. Для этого добавляют масло М-6.
Рассчитаем необходимое количество масла.
Разбавляем до КЧ = 60.
Пусть G - это общий вес раствора без учёта
растворителя.
= (Старое КЧ * масса чистых АСК в растворе) / Новое КЧ = (103,85 * 100 г
)/60 = 173,1 г.
Тогда общий вес раствора с учётом растворителя будет равен
+ масса бензина в растворе) = (173,1 г + 63,9 г) = 237 г.
Масса масла М-6 равна
(G - масса товарных АСК в растворе) =
(173,1 г - 100 г) = 73,1 г.
Значит, в реактор необходимо загрузить 73,1 г масла М-6.
Опыт 3. Получение присадки Детерсол-140.
В четырёхгорлый реактор с механической мешалкой, термометром и обратным
холодильником загружаем сначала известь-пушонку, затем раствор АСК в масле М-6.
После этого приливаем в реактор метанол (промотор реакции), затем воду, затем
бензин (растворитель).
Включаем перемешивание и нагрев, доводим температуру реакционной смеси до
63-64 ºС. Откручиваем вентиль подачи СО2,
не подсоединяя барботёр 2 к реактору, и таким образом продуваем реакционную
систему в течение нескольких минут. Это необходимо для того, чтобы перед
началом процесса СО2 полностью вытеснил весь воздух из реактора.
Установлено, что неполное вытеснение воздуха впоследствии приводит к
значительному уменьшению скорости поглощения СО2, а значит и
скорости карбонатации.
Затем подсоединяем барботёр 2 к реактору, начиная тем самым наш процесс.
Баллон с СО2 подсоединён к реактору через барботёр 1. Реактор
соединён с атмосферой через барботёр 2. Пробулькивание пузырьков в барботёре 1
свидетельствует о поступлении СО2 в реакционную систему, а отсутствие
пробулькивания в барботёре 2 - о том, что весь СО2, поступающий в
реактор, поглощается реакционной массой и не уходит в атмосферу. То есть
пробулькивание в барботёре 1 и отсутствие пузырьков в барботёре 2
свидетельствует о нормальном протекании процесса карбонатации.
Процесс карбонатации проводится до тех пор, пока реакционной системой не
поглотится нужный объём углекислого газа. Контроль расхода СО2
производится по манометру с использованием тарировочного графика. При этом мы
следим за тем, чтобы процесс протекал нормально. По мере продувания СО2
реакционная смесь темнеет, что свидетельствует о протекании карбонатации.
После поглощения системой нужного объёма углекислого газа (обычно на это
уходит 1-1,5 часа) вентиль подачи СО2 закручиваем, останавливая тем
самым карбонатацию.
После этого отгоняем от реакционной смеси метанол, воду и частично
бензин. Делаем это либо на роторном испарителе, либо на приборе с одногорлым
реактором и прямым холодильником. Полученный отгон состоит из двух слоёв:
верхний - бензин, нижний - раствор воды в метаноле. Отгонку продолжаем до тех
пор, пока нижний слой не перестанет увеличиваться при добавлении в систему
небольшого количества чистого бензина и последующем отгоне этого количества. То
есть отгоняем до тех пор, пока не отгоним всю воду.
После завершения отгонки реакционную смесь центрифугируем, чтобы очистить
её от непрореагировавшей пушонки.
После этого отгоняем из смеси бензин, получая товарную присадку
Детерсол-140 в виде чёрной вязкой жидкости.
Примечание. В данной методике в качестве растворителя вместо бензина
может использоваться гептан. Применять в качестве растворителя ксилол, как
рекомендовалось ранее [46], нецелесообразно, потому что он образует
трудноразделяемую смесь ксилол-метанол-вода.
Загрузка.
АСК в масле М-6 100 г (60 г товарных АСК, 40 г масла М-6).
Са(ОН)2 20 г.
СН3ОН 20 г.
Н2О 2,5 г.
СО2 5 г.
Бензин 60 г.
Опыт 4.
Получение присадки Детерсол-180.
Синтез осуществляется аналогично опыту 2, только промотором реакции
является не только метанол, но и уксусная кислота. Кроме того, используется
другое соотношение Са(ОН)2 : СО2.
Загрузка.
АСК в масле М-6 100 г (60 г чистых АСК, 40 г масла М-6).
Са(ОН)2 20 г.
СН3ОН 20 г.
Н2О 5 г.
СО2 5,5 г.
СН3СООН 0,3 г.
Бензин 30 г.
Опыт 5. Определение щелочного числа синтезированных алкилсалицилатных
присадок.
Определение щелочных чисел проводят по ГОСТ 11362-76.
Метод заключается в потенциометрическом титровании исследуемого образца,
растворённого в неводном растворителе, раствором соляной кислоты. Титрование
ведут до скачка потенциала или при отсутствии последнего до значений ЭДС,
установленных по буферным растворам.
За общее щелочное число принимают количество едкого кали в
миллиграммах, эквивалентное количеству соляной кислоты, израсходованной на
нейтрализацию всех основных соединений, содержащихся в 1 г анализируемого
продукта.
За щелочное число сильных оснований принимают количество едкого
кали в миллиграммах, эквивалентное количеству соляной кислоты, израсходованной
на нейтрализацию сильных оснований, содержащихся в 1 г анализируемого продукта.
Растворитель для анализируемого продукта готовят смешением (по объёму)
30% этилового спирта и 70% толуола (или бензола) или 50% изопропилового спирта,
49% толуола (или бензола) и 1% воды.
Приготовление буферных растворов.
Для приготовления 1000 мл щелочного буферного раствора (рН ~ 11)
27,80 ±0,01 г м-нитрофенола растворяют в 100 мл этилового спирта,
раствор количественно переносят в мерную колбу вместимостью 1000 мл, добавляют
при постоянном перемешивании 250 мл точно 0,2 н. спиртового раствора едкого
кали и доводят объём раствора спиртом до метки. Если раствор едкого кали не
точно 0,2 н., производят пересчёт объёма добавляемого раствора едкого кали.
Для приготовления 1000 мл кислого буферного раствора (рН ~ 4)
24,20 ±0,01 г γ-коллидина растворяют в 100 мл этилового спирта, переливают
раствор в мерную колбу вместимостью 1000 мл, добавляют при постоянном
перемешивании 750 мл точно 0,2 н. спиртового раствора соляной кислоты и доводят
объём раствора спиртом до метки. Если раствор соляной кислоты не точно 0,2 н.,
производят соответствующий пересчёт объёма добавляемого раствора соляной
кислоты.
При отсутствии γ-коллидина готовят кислый буферный раствор
бифталата калия (рН = 4). Для приготовления 1000 мл буферного раствора
10,20±0,01 г перекристаллизованного бифталата калия растворяют в 20-50 мл
свежепрокипяченной дистиллированной воды, раствор количественно переносят в
мерную колбу вместимостью 1000 мл и при постоянном перемешивании доводят объём
прокипяченной дистиллированной водой до метки. Допускается приготовление
буферного раствора из фиксанала бифталата калия для рН-метрии.
При приготовлении меньшего количества буферных растворов количество
реактивов соответственно уменьшают.
Определение значения ЭДС электродов в буферных растворах.
Для определения значений ЭДС в растворе γ-коллидина и м-нитрофенола в
стаканчик для титрования помещают 50 мл растворителя (см. выше) и добавляют из
пипетки 5 мл щелочного буферного раствора. Полученную смесь перемешивают в
течение 5 мин и измеряют значение ЭДС.
Для определения значения ЭДС в растворе бифталата калия в стаканчик для
титрования наливают 50 мл раствора бифталата калия (см. выше) и измеряют
значение ЭДС.
Проведение анализа.
В стаканчик для титрования берут навеску анализируемого продукта. Затем в
стаканчик с продуктом добавляют 50 мл растворителя. Если продукт не
растворяется полностью в этом растворителе, в стаканчик приливают ещё 10-15 мл
хлороформа. Если и в этом случае продукт не растворяется, то в составе
растворителя толуол или бензол заменяют на хлороформ и повторно взвешивают и
растворяют продукт.
Стаканчик устанавливают на титровальный стенд, опускают в раствор
электроды, включают мешалку и определяют начальную величину ЭДС. Если эта
величина меньше величины ЭДС, установленной в щелочном буферном растворе, то
это указывает на присутствие сильных оснований. Если начальная величина ЭДС
больше величины ЭДС, установленной в щелочном буферном растворе, то сильные
основания отсутствуют.
Определение щелочного числа сильных оснований.
Титрование ведут до величины ЭДС, установленной в щелочном буферном
растворе, или до первого скачка потенциала в этой области. В качестве титранта
применяют 0,1 н. раствор соляной кислоты, добавляя в один приём по 0,05-0,1 мл.
Если изменение ЭДС будет больше 15-20 мВ (0,25-0,35 рН), объём титранта,
добавляемого в один приём, уменьшают до 0,02 мл, и после каждой добавки
очередной порции титранта выдерживают пока потенциал установится, то есть
изменение его будет составляь не более 5 мВ (рН ~ 0.1) в минуту.
Определение общего щелочного числа.
После определения сильных оснований титрование продолжают до величины
ЭДС, установленной в кислом буферном растворе или до второго скачка потенциала
в этой области.
Титрование ведут сначала медленно, добавляя в один приём по 0,05-0,1 мл.
Если изменение величины ЭДС будет меньше 15-20 мВ (или 0,25-0,35 рН), объём
титранта увеличивают до 0,2-0,3 мл. Вблизи величины ЭДС буферного раствора
объём добавляемого в один приём титранта вновь уменьшают до 0,05 мл, и после
каждого добавления очередной порции титранта ожидают, пока потенциал
установится, то есть изменение его будет составлять не более 5 мВ (~ 0,1 рН) в
минуту.
Затем проводят контрольный опыт с тем же объёмом растворителя, но без
анализируемого продукта.
Титрованный раствор 0,1 н. соляной кислоты добавляют по 0,02-0,1 мл в
один приём.
Обработка результатов анализа.
Щелочное число сильных оснований (Щ1) в миллиграммах КОН
на 1 г продукта вычисляют по формуле
Щ1 = ((V1 - V0)*T)/m,
где V0 - объём 0,01 н. раствора соляной кислоты, израсходованный
на титрование контрольного опыта до величины ЭДС в щелочном буферном растворе
или до скачка потенциала в этой области, мл;
V1 - объём 0,01 н. раствора соляной
кислоты, израсходованный на титрование раствора исследуемого образца до
величины ЭДС в щелочном буферном растворе или до скачка потенциала в этой
области, мл;
Т - титр 0,01 н. раствора соляной кислоты, мг/мл КОН;
m -
масса анализируемого продукта, г.
Общее щелочное число (Щ2) в миллиграммах КОН на 1 г продукта вычисляют
по формуле
Щ2 = ((V3 - V2)*T)/m,
где V2 - объём 0,01 н. раствора соляной кислоты,
израсходованный на титрование контрольного опыта до величины ЭДС в кислом
буферном растворе или до скачка потенциала в этой области, мл;
V3 - объём 0,01 н. раствора соляной
кислоты, израсходованный на титрование раствора исследуемого образца до
величины ЭДС в кислом буферном растворе или до скачка потенциала в этой
области, мл.
За результат анализа принимают среднее арифметическое двух параллельных
определений щелочных чисел.
Результаты определения щелочных чисел исследуемых алкилсалицилатных
присадок приведены в таблице 5.
Таблица 5.
|
Исследуемая присадка
|
Щелочное число
|
|
Детерсол-50
|
64
|
|
Детерсол-140
|
165
|
|
Детерсол-180
|
214
|
Оценка качества синтезированных присадок.
Оценку проводим по следующим показателям (см. таблицу 6).
Таблица 6.
|
Показатель
|
Детер-сол-50
|
Детер-сол-140
|
Детер-сол-180
|
|
Вязкость кинематическая при
100 ºС, мм2/с, не более
|
40
|
35
|
60
|
|
Общее щелочное число, мг
КОН/г, в пределах
|
50-70
|
130-170
|
155-200
|
|
Температура вспышки,
определяемая в открытом тигле, ºС, не ниже
|
190
|
190
|
190
|
|
Массовая доля механических
примесей, %, не более
|
0,08
|
0,08
|
0,08
|
|
Массовая доля воды, %, не
более
|
0,1
|
0,1
|
0,1
|
|
Индукционный период
осадкообразования в приборе ДК-2,в масле М-11 с 6% присадки, ч, не менее
|
50
|
50
|
50
|
|
Массовая доля сульфатной
золы, %, не более
|
9,0
|
21,0
|
24,5
|
|
Массовая доля активного
вещества (алкилсалицилата и карбоната кальция), %, не менее
|
30
|
30,0
|
45
|
|
Массовая доля свободного
алкилфенола, %, не более
|
18,0
|
18,0
|
18,0
|
|
Растворимость в масле
|
Полная
|
Полная
|
Полная
|
1,0
|
1,0
|
1,0
|
|
Степень чистоты, мг на 100
г присадки, не более
|
700
|
800
|
-*
|
|
Цвет (при разбавлении
5:95), ед. ЦНТ, не более
|
7,5
|
7,5
|
7,5
|
|
Моющие свойства по ПЗВ
масла М-11 с 2,0% присадки, балл, не более
|
1,0
|
1,0
|
1,0
|
*
- Не нормируется. Определение обязательно.
Обсуждение результатов.
Процесс получения присадки Детерсол-50 проводился в безводной среде
бензина и масла. Это позволило нам избежать образования не растворимого в воде
циклического алкилсалицилата кальция.
Данное соединение образуется при взаимодействии алкилсалициловой кислоты
с Са(ОН)2 в водной среде:
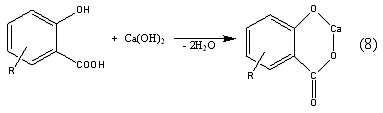
Благодаря тому, что мы проводили реакцию между исходными реагентами в
отсутствии воды, у нас вместо циклического алкилсалицилата кальция образовался
целевой продукт - Детерсол-50.
Таким образом, при взаимодействии алкилсалициловых кислот с гидроксидом
кальция в водной и в безводной средах образуются разные соли. Причиной этого
является влияние полярности растворителя, то есть способности растворителя
ионизировать растворённые в нём кислоты, основания, соли. Вода имеет высокую
полярность, а органическая среда (в данном случае бензин с маслом) - невысокую.
Поэтому в водной среде достаточно подвижным для отщепления в виде протона является
атом водорода не только карбоксильной группы, но и ОН-группы, связанной с
ароматическим кольцом. Поэтому оба этих атома водорода участвуют в
солеобразовании, вследствие чего и образуется циклическая соль (реакция (8)). А
в органической среде достаточно подвижным для отщепления в виде протона
является атом водорода только карбоксильной группы. Атом водорода ОН-группы,
связанной с ароматическим кольцом, подвижен недостаточно, и отщепляться в виде
протона не может. Поэтому в солеобразовании в данном случае участвует только
один атом водорода (карбоксильной группы), вследствие чего образуется
Детерсол-50 (реакция (7)).
По этой же причине в реакцию с Са(ОН)2 не вступает свободный
алкилфенол, содержащийся в технических алкилсалициловых кислотах (он является
продуктом одной из побочных реакций карбоксилирования алкилфенолята натрия). В
итоге присадка Детерсол-50 содержит кроме продукта реакции (7), ещё и побочные
продукты: алкилфенол, углеводороды (также содержащиеся в технических
алкилсалициловых кислотах (см. выше)) и масло-растворитель (см. опыт 2).
Точно такие же побочные продукты содержатся в Детерсоле-140 и в
Детерсоле-180.
При получении зольных моющих присадок Детерсол-140 и Детерсол-180
коллоидный раствор образуется в результате химической реакции, при которой два
вещества - гидроксид кальция и углекислый газ, растворимых в данной среде
(вода), образуют третье вещество - карбонат кальция - практически в ней не
растворимое.
Таким образом вода в реактор карбонатации загружается для того, чтобы
образовался карбонат кальция:

Нерастворимые молекулы СаСО3 образуют ядро коллоидной частицы
(мицеллы), которое не растворимо в дисперсионной среде - воде. Образовавшееся
ядро коллоидной степени дисперсности является носителем свободной энергии,
поэтому на его поверхности идёт адсорбционный процесс.
Согласно правилу "Пескова-Фаянса" на поверхности ядра мицеллы
обычно адсорбируются ионы, имеющиеся в составе ядра мицеллы. Поскольку ядро
частицы состоит из молекул СаСО3, то такими ионами могут быть Са2+
или СО32-.
Согласно тому же правилу "Пескова-Фаянса" из двух видов ионов,
находящихся в дисперсионной среде, адсорбируются те, которые находятся в
избытке. В нашем случае в избытке всегда имеются ионы Са2+,
поскольку при карбонатации Са(ОН)2 берётся в избытке, а СО2
дозируется [44]. Более того, при температурах карбонатации [45] растворимость в
воде Са(ОН)2 выше (0,128% масс.), чем СО2 (0,083% масс.).
Таким образом, на поверхности ядра коллоидной частицы адсорбируются ионы Са2+,
в результате чего частицы золя получают положительный заряд. Компенсирующие
ионы ОН- располагаются в адсорбционном (гельмгольцевском) и
диффузионном (гуи-чепменовском) слоях, завершая создание двойного
электрического слоя в целом нейтральной мицеллы (рис. 1).

Рис. 1.
Здесь А - ядро мицеллы, В - адсорбционный слой, С - диффузионный слой.
Брутто-формула образовавшейся мицеллы такова:
{СаСО3}|nCa2+ (2n - m)OH- |mOH-
Наличие Са2+ и ОН- в мицеллах высокощелочных
кальциевых детергентов было установлено экспериментально по количеству Са(ОН)2,
содержащегося в мицелле высокощелочного сульфоната кальция [45], и по
щёлочности, обусловленной гидроксидом кальция высокощелочного алкилсалицилата
кальция [46], что подтверждает правильность изложенного выше.
Дальнейшая стабилизация мицеллы в минеральном масле происходит за счёт
сольватации ионов ОН- диффузионного слоя полярными
поверхностно-активными веществами, которыми являются алкилсалицилаты кальция.
Не исключено, что сольватации подвергаются и ионы адсорбционного слоя. Лёгкость
прохождения сольватации отрицательно-заряженных ионов ОН- обуславливается,
во-первых, тем, что отрицательный заряд всегда сольватируется лучше
положительного (правило Хьюза-Ингольда), а во-вторых, наличием частичного
положительного заряда на атомах кальция полярных поверхностно-активных веществ.
Окончательно сформированная и стабилизированная в минеральном масле коллоидная
частица будет иметь следующее строение: рис. 2.
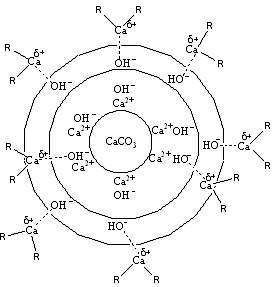
Рис. 2.
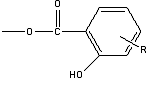
Здесь R - алкилсалицилатный остаток:
Мы предполагаем, что ядра мицелл высокощелочных алкилсалицилатов
образованы фатеритной кристаллической модификацией СаСО3. Такое
предположение подтверждается следующими экспериментальными фактами:
1.) получению высокощелочных детергентов способствует высокое
значение рН (10-12) реакционной среды [62];
2.) высокие значения рН способствуют образованию фатеритной
кристаллической модификации СаСО3 [63].
Предложенный механизм образования и строение коллоидных дисперсий
карбоната кальция при получении высокощелочных зольных моющих присадок
позволяют объяснить наблюдаемый исследователями эффект "перекарбонатации"
[31,45], приводящий к падению общей щёлочности присадок в случае не
контролируемой дозировки или подаче избыточного количества углекислого газа в
процессе карбонатации [46].
При стехиометрическом соотношении Са(ОН)2 и СО2, а
тем более при избытке последнего, в реакцию вступают ионы Са2+
и ОН- диффузионного и адсорбционного слоёв, вследствие чего
эти слои разрушаются. Исчезает двойной электрический слой и, как следствие,
разрушаются сольватные оболочки. Мицелла разрушается, наступает коагуляция
частиц карбоната кальция (ядер) и выпадение их в осадок. Щёлочность присадки,
обусловленная карбонатом и гидроксидом кальция, резко падает, доходя до
нулевой.
Вода загружается в реактор карбонатации в малом количестве для того,
чтобы её было не достаточно для эффективной сольватации мицеллы, изображённой
на рис. 1. Это необходимо для того, чтобы данную мицеллу сольватировала не
вода, а алкилсалицилат кальция (далее АК). Для этого нужно, чтобы мицелла,
образовавшаяся в водной фазе, смогла попасть в органическую фазу и там
просольватироваться алкилсалицилатом кальция. Эту задачу решает метанол: его роль
как промотора процесса карбонатации состоит в том, что он выступает посредником
между водной и органической фазами.
Возникает вопрос: почему мицелла, изображённая на рис. 1 сольватируется
именно АК, а не метанолом?
Дело в том, что в диффузионном слое мицеллы находятся анионы. Для
сольватации анионов нужен положительный заряд. А δ+ на атоме кальция в молекуле АК
больше, чем на атоме водорода ОН-группы молекулы метанола.
Поэтому в данном случае АК имеет большую сольватирующую способность, чем
метанол.
Механизм действия высокощелочных моющих присадок алкилсалицилатного типа
состоит в том, что благодаря наличию полярной части в алкилсалицилатных
остатках, торчащих наружу из мицелл присадок, эти мицеллы адсорбируются на
поверхности детали двигателя, и при этом своей массой вытесняют с этой
поверхности грязь, которая удерживается на поверхности менее прочно, чем эти
мицеллы. Наличие на поверхности детали двигателя адсорбированных мицелл не
препятствует работе детали, а наоборот, облегчает эту работу, благодаря тому,
что эти мицеллы оказывают смазывающее действие.
Механизм действия низкощелочных моющих присадок алкилсалицилатного типа в
целом аналогичен, только адсорбируются не мицеллы, а молекулы алкилсалицилата
кальция.
Ещё одна роль алкилсалицилатных присадок состоит в том, что они благодаря
своей щёлочности нейтрализуют сернистую кислоту, которая образуется при
сгорании в двигателе серусодержащих топлив, и тем самым защищают детали
двигателя от кислотной коррозии. Действительно, при сгорании топлива в двигателе
всегда образуется вода. Если же топливо содержало серу, то образуется ещё и SO2. В итоге происходит образование сернистой кислоты:

Чем
выше щёлочность присадки, тем большее количество сернистой кислоты она может
нейтрализовать. Действительно, если в топливе содержится немного серы, то для
нейтрализации Н2SO3
достаточно обычного количества среднещелочной присадки. Если же используемое
топливо является высокосернистым, то сернистой кислоты образуется много и для
нейтрализации такого её количества необходимо либо дополнительное количество
низкощелочной присадки, либо обычное количество, но высокощелочной присадки.
Последний вариант экономически выгоднее первого, потому что СаСО3 и
Са(ОН)2 дешевле, чем алкилсалицилат кальция. Поэтому применение
высокощелочных алкилсалицилатных присадок необходимо при использовании
высокосернистых топлив.
Следует
отметить, что наши планы на будущее связаны с разработкой процесса
карбонатации с целью получения алкилсалицилатов с щёлочностью выше 180 мг КОН
на г.
Выводы
В
настоящей работе детально описан процесс получения важных для моторных масел
присадок алкилсалицилатного типа. Заключительная стадия данного производства
была испытана в лабораторных условиях. Испытание заключалось в том, что для
проведения этой стадии использовались алкилсалициловые кислоты, полученные с
использованием олигомеров этилена состава С16 - С18
вместо олефинов фракции 240-320 ºС термокрекинга парафинов. Олефины термокрекинга парафинов содержат много
парафинов и кислородсодержащих соединений. Из-за этого процесс алкилирования
фенола с использованием данных олефинов протекает неэффективно, образуется
много отходов. Олигомеры этилена содержат намного меньше парафинов (см. таблицы
2 и 3) и кислородсодержащих соединений, и, кроме того, имеют меньший разброс по
длине углеродных цепей входящих в их состав углеводородов (см. те же таблицы).
Поэтому
целесообразно перейти на использование в производстве алкилсалицилатных
присадок олигомеров этилена состава С16 - С18 вместо олефинов
фракции 240-320 ºС термокрекинга парафинов.
Возможность
осуществления данного перехода и была проверена в ходе наших лабораторных
исследований. Полученные в результате проведённого испытания
Детерсолы отвечают требованиям научно-технической документации по основному
показателю общая щёлочность, что было проверено нами в ходе проведения опыта 5. Таким образом
результаты проведенных лабораторных исследований свидетельствуют о том, что
использование в производстве алкилсалицилатных присадок олигомеров этилена
состава С16 - С18 вместо олефинов фракции 240-320 ºС термокрекинга парафинов позволяет получать присадки
Детерсол-50, Детерсол-140 и Детерсол-180, отвечающие требованиям
научно-технической документации по основному показателю общая щёлочность.
Список литературы
1. Исагулянц В. И., Фёдорова Р. И., Кустанович З. Д. и др.
Алкилирование метилового эфира салициловой кислоты олефинами // Химия и
технология топлив и масел. - 1968. -№8. -С.15-19.
2. Broaddus C. D. Metallation of Toluene. The question of kinetic
v. s. Thermodynamic control // J. Amer. Chem. Soc. - 1966. - 88 , №18. - P. 4174-4178.
3. Benkeser R. A., Hooz J., Liston T. V. at al. Factors
governing orientationmetallation reactions. ׀׀. The metallation
of isopropylbenzene with n-amylsodium and n-amylpotassium // J. Amer. Chem. Soc. -
1963. - 85 , №24. -
P. 3984-3989.
4. Agami C. Les solvants aprotonique (|\/). Emploi de la
N,N,N`,N`-tetramethylethylenediamine en chimie organometallique // Bull. Soc. Chim. France.
-1970. №4. -P.1619-1624.s
. Турянчик И. Г., Лебедев Е. В., Скляр В. Т. и др. О карбоксилировании алкилфенольных соединений окисью углерода
в присутствии поташа // Нефтеперераб. и нефтехимия. -Киев: Наук. думка. -
1972. -Вып. 8. - С. 11-18.
. Kolbe H. Carboxylierung von Phenolen // Ann. Phys. R4. -1860. -B.113. -S.
125-128.
. Kolbe H. Ulber eine neue Darstellungsmethodeund einige
bemerken-swerthe Eigenschafften der salicylsaure // J. Pract. Chem. - 1874. -10,
№2. -S.
89-112.
8. Schmitt R.Beitrag zur Kenntniss der Kolbe`schen
salicylsauresynthese // J. Pract. Chem. - 1885. -B. 31, №1. -S. 397-411.
9. Pat. 2 744 069 USA, IC C 10 m 1/20. Compounded
lubricating compositions / M. Loon (Netherlands); Shell Development Company
(USA). -№ 350 525; Appl. 22.04.53; Publ. 1.05.56.
. Pat. 795 657 GB., IC C 07c. C 10 m. Basic polyvalent
metal salts of organic asids / J. Hartley, E. C. Lumb, J. C. Moseley (GB);
Shell Research Ltd. (GB). -№16347/55; Appl. 7.06.55; Publ. 28.05.58.
. Лященко А. Ф., Вязкова Е. А., Назарова К. Р. Раздельное
определение солей кальция в присадках потенциометрическим методом // Химия и
технология топлив и масел. -1964. -№ 9. -С. 62-65.
12. Pat. 2 197 837 USA, IC C 10 m. Mineral oil composition / O.
M. Reiff (USA); Socony Vacuum Oil Co. (USA). -No. 247001; Appl. 21.12.38; Publ.
23.04.40.
13. Pat. 2 252 662 USA, IC C 10 m. Metal salts of alkyl
substituted hydrohyaromatic carboxylic acids / O. M. Reiff (USA); Socony Vacuum
Oil Co. (USA). -No. 213 183; Appl. 11.06.38; Publ. 12.08.41.
14. Reiff O. M. Lubricating oil addition agents. Application of phenolic compounds
and their metal derivatives // Ind. Eng. Chem. - 1941. -№33. -P. 351-357.
15. Pat. 2 258 591 USA, IC2 10 M 1/24.
Lubricating oil compositions / F. R. Moser, A. S. Dijksman (Netherlands); Shell
Development Company (USA). -No. 306 330; Appl. 27.11.39; Publ. 14.10.41.
. Pat. 2 472 593 USA, IC2 10 M 1/54.
Lubricating oil compositions / J. L. van der Minne (Netherlands); Shell
Development Company (USA). -No. 708935; Appl. 9.11.46; Publ. 1.10.65.
. Pat. 2 477 913 USA, IC2 10 M 1/24.
Mineral lubricating oil / M. van Loon (Netherlands); Shell Development Company
(USA). -No. 659668; Appl. 4.04.46; Publ. 2.08.49.
. Pat. 495 270 Canada, IC. C 10 m. Lubricating oil
composition / Van der Minne J. L. (Netherlands); Shell Development Company
(USA). -No. 550 413; Appl. 9.11.46; Publ. 11.08.53.
. Pat. 496 510 Canada, IC. C 10 m. Lubricating oil
composition containing zinc salt / Van Loon M. (Netherlands); Shell Development
Company (USA). -No. 539 996; Appl. 3.04.46; Publ. 29.09.53.
. Pat. 3 318 809 USA, IC. C 07c 65/12. Counter current
carbonation process / U. B. Bray (USA); Bray Oil Company (USA). -No. 471 666;
Appl. 13.07.65; Publ. 9.05.67.
. Pat. 3 105 049 USA, IC. C 07c 65/12. Colloidal
dispersions of salts / V. Voorhees, J. Altos (USA); Bray Oil Company (USA).
-No. 46 459; Appl. 1.09.60; Publ. 24.09.63.
. Pat. 3 372 116 USA, IC2 C 10m 1/24,1/54.
Preparation of basic metal phenates and salicylates / N. A. Meinhardt (USA);
The Lubrizol Corp. (USA). -No. 631 144; Appl. 17.04.67; Publ. 5.03.68.
. Pat. 1 039 069 D E IC C10m. Verfanren zur
Herstellung von als Schmiermittelzusatzstoffe geegneten Gemisenen aus
Alkylphenolaten und Alkylsalizylaten mit einem neuen Gehaltan einem
meurwertigen Metall / M. van Loon (Netherlands); Bataafsche Petroleum
Maatschappij (Nederlande). -No. 1018-67; Anmeld. 27.10.54; Ausleg. 12.03.59.
. Pat. 1769670 D E IC. C07c. Schmiermittelgemisch / M.
van Loon A. Stran (Nederlande); Shell International Research Maatschappij B. V.
(Nederlande). -No. 29775-67; Anmeld. 26.06.68; Ausleg. 29.06.78.
. Pat. 729 376 G B, IC C 10 m. Improvements in or
relating to the preparation of basic metallic compounds of phenols / N. V. de
Bataafsche Petroleum Maatschappij (Netherlands). -No. 11 619/53; Appl.
29.04.52; Publ. 4.05.55.
. Pat. 790 471 G B, IC. C07b C 10 m. Improvements in
or relating to the preparation of polyvalent metal basic salts of organic acids
in hydrocarbon oil solution / Naamlooze Vennootschap de Bataafsche Petroleum
Maatschappij (Netherlands). -No. 16 030/54; Appl. 31.05.53; Publ. 12.02.58.
. Pat. 728 928 G B, IC C 10 m. Process for preparing
alkaline earth metal alkyl salicylates, or mixtures thereof with alkyl phenols
or alkyl phenolates, having improved properties as lubricating oil additives /
N. V. de Bataafsche Petroleum Maatschappij a Body Corp. (Netherlands). -No.
26581/53; Appl. 30.09.52; Publ. 19.10.55.
. Pat. 759 344 G B, IC C 10 m. Improvements in or
relating to the preparation of basic metallic compounds of alkyl phenols and
alkyl salicylic acids / N. V. de Bataafsche Petroleum Maatschappij Company
(Netherlands). -No. 729 376; Appl. 29.10.53; Publ. 17.10.56.
. Pat. 795 172 G B, IC. C 07c C 10 m. Improvements in
or relating to the preparation of basic oil-soluble polyvalent metal salts of
organic acids / A. Dewhurst, G. C. Eltenton (GB), Shell Research, Ltd. (GB).
-No. 10 259/55; Appl. 7.04.55; Publ. 21.05.58.
. Pat. 1 570 909 G B, IC3 C10m 1/20 C08k
11/00 C10L 1/10 C10m 3/14. Basic noncarbonated magnesium compositions and
fuels, lubricant and additive concentrate compositions containing the same / J.
L. Karn (USA); The Lubrizol Corp. (USA). - No. 25480/77; Appl. 17.06.77; Publ.
9.07.80.
. Pat. 786 167 G B, IC C10 m. Improvements in or
relating to the preparation of basic oil-soluble polyvalent metal salts of
organic acids and solutions of said basic salts in oils, and the resulting
salts / G. Ellis, J. Hartley, J. C. Moseley (GB); Shell Research Limited (GB).
-No. 27853/54; Appl. 27.09.54; Publ. 13.11.57.
. Pat. 2 097 417 A G B, IC C10 m 1/20. Preparation of
highly basic alcaline earth metal salts of organic acids and oil compositions
containing them / C. D. Mantinus Beverwijk, J. A. Ralt Waterhoff, G. de Lind
van Mijngaarden (Netherlands); Shell International Research Maatschappij B. V.
-No. 81131 08; Appl. 28.04.81; Publ. 3.11.82.
. A. OSV. 172 680 CSR IC. C 07 c 65/12; C 10 m 1/20.
Spǒsob prepravy alkylsalicylǎtov
kovov alcalických zemin / J. Kekeńǎk., L. Bartek., L. Salko., L.
Szũcs. (CSR., Bratislava). -No. 5532 - 74; Prihlásené 05.08.74; Vydané 15.05.78.
. A. OSV. 203277 ĊSR, IC3 C07 C51/00
C10M 1/24. Zariadenie pra kontinuálný pripravu
antioxidantudetergentu na báze kyseliny
alkylsalicylovej / M. Mataš, A.
Tkāč, J. Cvengroś (ĊSR, Bratislava). - No. 5336-77; Prihlásené 12.08.77; Vydané 15.12.82.
35. А. с. 151751 СССР МКИ С 10m; 23с. Способ получения солей алкилсалициловых кислот / В. Н.
Монастырский, Н. А. Дмитриева, Г. Г. Краснянская (СССР). - №733121/23-5; Заявл.
3.06.61; Опубл. 01.11.62; Бюл. № 22.
. А. с. 282567 СССР МПК С 10m 1/20. Способ производства высокощелочной многофункциональной
присадки к маслам / А. Я. Левин, В. Н. Монастырский, Н. А. Дмитриева (СССР). -
№1347864/23-4; Заявл. 12.07.69; Опубл. 28.09.70; Бюл. № 20.
. Куликов А. А., Суховерхов В. Д. Улучшение метода
жидкостного хроматографического анализа системы: масло-алкилфенол-кальциевая
алкилсалицилатная присадка // Нефтеперераб. и нефтехимия. -1982. - № 7. - С.
19-20.
. Тимошенко С. В. Хроматографический анализ коллоидных систем
детергентных присадок и пластичных смазок: Дис. канд. тех. наук. - Киев, 1984.
- 146 с.
. А. с. 352931 СССР МКИ С 10m 1/20. Способ получения алкилсалицилатной присадки к
смазочным маслам / О. Л. Главати, Р. В. Фиалковский, Н. Г. Мудрик и др. (СССР).
- № 1460549/28-4; Заявл. 16.07.70; Опубл. 29.09.72; Бюл. № 29.
. А. с. 476307 СССР МИК С 10m 1/20. Способ получения алкилсалицилатной присадки к
смазочным маслам / О. Л. Главати, Я. Е. Гарун, Н. Г. Мудрик и др. (СССР). - №
1922227/23-4; Заявл. 07.05.73; Опубл. 11.03.76; Бюл. № 25.
. А. с. 736625 СССР МКИ С 10m 1/20; С 07С 65/12. Способ получения алкилсалицилатной
присадки к смазочным маслам / В. Д. Суховерхов, А. С. Журба, Ю. Т. Гордаш и др.
- № 2667982/23-04; Заявл. 07.08.78; Опубл. не подлежит.
. А. с. 472148 СССР. МИК С 10m 1/20. Способ получения алкилсалицилатной бариевой присадки
// А. А. Гонор, О. Н. Добровольская, Т. А. Рогачевская и др. - № 2019289/23-4;
Заявл. 29.04.74; Опубл. 29.09.75; Бюл. № 20.
. А. с. 724562 СССР. МИК С 10m 1/08. Способ получения присадок к смазочным маслам / В. И.
Спицин, В. Ф. Хромых, В. Г. Сергиенко и др. - № 2502509/23-04; Заявл. 04.07.77;
Опубл. 02.04.80; Бюл. № 12.
. Суховерхов В. Д., Главати О. Л. и др. Изучение процесса
получения многоосновного алкилсалицилата кальция - присадки МАСК. - в кн:
Производство моющих и противоизносных присадок / Киев, апр. 1972 / Мат.
Всесоюзн. совещ. - Киев: Наукова Думка, 1972. Кн. 1, с. 139-143.
. Фиалковский Р. В. Исследование процесса карбонатации в
производстве высокощелочных детергентно-диспергирующих присадок к моторным
маслам. - Дис. канд. техн. наук. Киев: ВНИИПКнефтехим 1978. 237 с.
. Дмитриева Н. А., Краснянская Г. Г., Монастырский Б. Н.
Синтез солей алкилсалициловых кислот и изучение их свойств как присадок к
смазочным маслам. - в кн.: Присадки к маслам. - изд. "Химия" Москва,
1966, с. 115-121.
. П. Сайкс Механизмы реакций в органической химии. - Москва,
"Химия", 1991.
. Слободин Я. М., Малышева Т. Г. Выделение алкилсалицилатных
и сульфонатных присадок из масляных концентратов методом диализа // Химия и
технология топлив и масел. - 1969. - №6. - С. 27-29.
. Закупра В. А., Тимошенко С. В., Колосова Э. В. и др.
Исследование состава и контроль производства алкилсалицилатных присадок методом
жидкостной микрохроматографии // Совершенствование технологии производства
присадок. - Киев: Наук. думка. - 1976. - С. 218-227.
. Ляшенко А. Ф., Борисова В. И., Назарова К. Т. и др.
Определение содержания непрореагировавшего алкилфенола и масла-разбавителя в
алкилсалицилатных присадках // Улучшение качества смазочных масел и присадок. -
М. : ВНИИНП, 1976. - Вып. ХI\/. - С. 214-217.
51. Leighton B. D., Moody G. J., Thomas D. K. The zone Electrophoresis of
lubricating Oil Additives // The Analyst. - 1974. - V. 99. - P. 442-452.
52. Vergos T.,
Stepina V. Metódy analýzu smésných přisad a
stanoveni obsaha přisad v mazacich olejich / Ropa á Uhlie. - 1981.
- №7. - S. 393-408.
. Coates I. P. The analysis of lubricating oil and oil
additives by thin layer chromatography // J. Inst. Petr. - 1971. - V. 57. - №556., p.
209-218.
. Благовидов И. Ф. и др. - Нефтепереработка и нефтехимия,
вып. 3, 1967.
. Авт. свид. № 161848. - Бюлл. изобр., № 34, 1964.
. Пат. ФРГ 1039069, 1953.
. Пат. ФРГ 1016272, 1952.
. Голландский пат. 103.192.1962.
. Либинсон Г. С. Физико-химические свойства карбоксильных
катионитов. "Наука", М., 1969.
. Тремийон Б. Разделение на ионообменных смолах. ИЛ, М.,
1967.
. В. Н. Монастырский, А. Я. Левин, Н. А. Дмитриева, Г. Г.
Краснянская, Б. В. Грязнов Совершенствование технологии производства
алкилсалицилатных присадок. - В кн. "Производство моющих и противозадирных
присадок. Книга 1.", Киев, 1971.
. Главати О. Л., Фиалковский Р. В., Тарасенко П. В.
Совершенствование производства высокощелочных сульфонатных присадок //
Нефтеперераб. и нефтехимия. - М. : ВО Нефтехим. - 1973. - Вып. 5. - С. 27-39.
63. Johnstone J. H. E., Meerwin H. E., Williamson E. D. The
several forms of calcium carbonate // Amer. J. Sci. - 1916. - 41, №4. - P. 473-512.